Back to Journals » Journal of Inflammation Research » Volume 16
SARS-CoV-2 N Protein Triggers Acute Lung Injury via Modulating Macrophage Activation and Infiltration in in vitro and in vivo
Authors Lai D ![]() , Zhu K, Li S
, Zhu K, Li S ![]() , Xiao Y
, Xiao Y ![]() , Xu Q, Sun Y
, Xu Q, Sun Y ![]() , Yao P, Ma D
, Yao P, Ma D ![]() , Shu Q
, Shu Q
Received 23 January 2023
Accepted for publication 15 April 2023
Published 28 April 2023 Volume 2023:16 Pages 1867—1877
DOI https://doi.org/10.2147/JIR.S405722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Dengming Lai,1,* Kun Zhu,2,* Sisi Li,3,* Yi Xiao,1 Qi Xu,4 Yisheng Sun,5 Pingping Yao,5 Daqing Ma,6,7 Qiang Shu7
1Department of Neonatal Surgery, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, People’s Republic of China; 2Department of Pathology, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, People’s Republic of China; 3School of Medicine, Hangzhou City University, Hangzhou, People’s Republic of China; 4School of Laboratory Medicine and Bioengineering, Hangzhou Medical College, Hangzhou, People’s Republic of China; 5Key Laboratory of Vaccine, Prevention and Control of Infectious Disease of Zhejiang Province, Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou, People’s Republic of China; 6Division of Anaesthetics, Pain Medicine & Intensive Care, Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, Chelsea & Westminster Hospital, London, UK; 7Department of Thoracic and Cardiovascular Surgery, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiang Shu, Department of Thoracic and Cardiovascular Surgery, Children’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310052, People’s Republic of China, Tel +86-13906500193, Email [email protected] Dengming Lai, Department of Neonatal Surgery, Children’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310052, People’s Republic of China, Tel +86-13567164728, Email [email protected]
Background: SARS-CoV-2-induced acute lung injury but its nucleocapsid (N) and/or Spike (S) protein involvements in the disease pathology remain elusive.
Methods: In vitro, the cultured THP-1 macrophages were stimulated with alive SARS-CoV-2 virus at different loading dose, N protein or S protein with/without TICAM2-siRNA, TIRAP-siRNA or MyD88-siRNA. The TICAM2, TIRAP and MyD88 expression in the THP-1 cells after N protein stimulation were determined. In vivo, naïve mice or mice with depletion macrophages were injected with N protein or dead SARS-CoV-2. The macrophages in the lung were analyzed with flow cytometry, and lung sections were stained with H&E or immunohistochemistry. Culture supernatants and serum were harvested for cytokines measurements with cytometric bead array.
Results: Alive SARS-CoV-2 virus or N protein but not S protein induced high cytokine releases from macrophages in a time or virus loading dependent manner. MyD88 and TIRAP but not TICAM2 were highly involved in macrophage activation triggered by N protein whilst both inhibited with siRNA decreased inflammatory responses. Moreover, N protein and dead SARS-CoV-2 caused systemic inflammation, macrophage accumulation and acute lung injury in mice. Macrophage depletion in mice decreased cytokines in response to N protein.
Conclusion: SARS-CoV-2 and its N protein but not S protein induced acute lung injury and systemic inflammation, which was closely related to macrophage activation, infiltration and release cytokines.
Keywords: SARS-CoV-2, S protein, N protein, macrophage, cytokine, acute lung injury
Introduction
SARS-CoV-2-induced disease is still a huge healthy burden worldwide. Although the latest variant causes less severe cases, acute respiratory distress syndrome (ARDS) or even multi-organ failure or death due to this virus infection can still be seen daily.1–3 The SARS-CoV-2 induced acute lung injury, as an initial organ injury, is characterized with diffuse alveolar damage in early phase and epithelial cell losing repairing ability towards respiratory failure in late phase in severe cases.3,4 The complex pathophysiological process suggests that SARS-CoV-2 infection dysregulates the host response and induces immuno-inflammatory and parenchymal derangements, but the mechanistic basis of lung injury remains incompletely understood.
The SARS-CoV-2 infects host cells via spike (S) protein binding to host cell’s surface angiotensin-converting enzyme 2 (ACE2).5 The S1 subunit of S protein is involved in attaching to host cells whilst S2 subunit is involved in cell membrane fusion, indicating that targeting S epitopes can be elicited upon vaccination.6 The nucleocapsid (N) protein of SARS-CoV-2 plays a significant role in regulation of viral gene transcription and mediating cytokine responses or organ injury.7 S and N proteins have been reported as the two key molecular in the infection and invasion of SARS-CoV-2; however, their role in mediating acute lung injury and underlying mechanisms remain elusive.
SARS-CoV-2 infection followed acute lung injury has been repeatedly reported to be a progressing detrimental immune-dysfunction condition, and monocytes and macrophages were considered as critical mediators of dysregulated immune response,3,4,8,9 causing consequent heart and lung injuries and even multiple organ failure.10,11 Monocytes and macrophages are sentinel cells that sense SARS-CoV-2 and leading to potent systemic inflammation that contributes to COVID-19 pathogenesis through virus interaction with Fcγ receptors, ACE2 and TMPRSS2.12–14 Moreover, histone methyltransferase MLL1/KMT2A in monocytes also drove inflammation in SARS-CoV-2 infection.15 Monocyte-derived macrophages accumulate in the lung and a special subset of macrophages was found to trigger lung fibrosis in COVID-19.2 The cell entry mechanisms of SARS-CoV-2 have been fully clarified, but the mechanisms of how SARS-CoV-2 triggers macrophages activation and the role of monocyte/macrophage in mediating cytokine releases following SARS-CoV-2 infection lead to inflammatory lung damage remain unknown.
In the current study, the SARS-CoV-2 and macrophage on host immunity and pathological changes by comparison between pathogenic S and N proteins of SARS-CoV-2 were investigated in human macrophage cell line and mice.
Materials and Methods
In vitro Study
Cell Culture and Treatment
The human monocytic cell line (THP-1) (NCACC, Shanghai, China) was cultured in RPMI medium 1640 supplemented with 10% FBS and 50 μg/mL penicillin/streptomycin in the incubator at 37°C with 5% CO2 balanced with air. THP-1 monocytic cells were treated with 100 ng/mL phorbol myristate acetate (PMA) (Solarbio, Beijing, China) for 24 h to induce macrophage phenotype.
siRNAs targeting human MyD88 (5’-CGGAGGAGAUGGACUUUGAdTdT; 5’-UCAAAGUCCAUCUCCUCCGdTdT), TICAM2 (5’-GGAAGAGAUGUUUGAAGAAdTdT; 5’-UUCUUCAAACAUCUCUUCCdTdT), TIRAP (5’-UCAAAGUCCAUCUCCUCCGdTdT; 5’-UCUGUUUCUAGCUUCACUCdTdT) and negative control siRNAs were purchased from HanBio Biotechnology (Shanghai, China). THP-1 cell suspension in 500 μL (containing 3.5×104 cells) in antibiotic-free medium was seeded in each well of a 24-well plate. Transfection complexes were generated by vortexing and incubating 100 nM siRNA with 3 μL of transfection reagent (RNAfit) at room temperature for 10 min in 100 μL serum and antibiotic-free medium. The complex was used to transfect each well. The plates were incubated at 37°C for 6 h, after which the medium was replaced with a fresh antibiotic-free complete medium. Transfection complexes were not cytotoxic to THP-1 cells, as assessed by the trypan blue exclusion assay. Cells were used for further experiments 48 h after transfection.
They were then treated with cultured alive SARS-CoV-2 (supplied by Zhejiang Center for Disease Control and Prevention center), N protein or S protein (Purity>93%, Endotoxin free, GenScript, Nanjing, China) for 3, 24 or 48 h. The culture supernatants were collected for cytokine measurements (see below) and cells were collected for Western blotting assessment.
Western Blotting
THP-1 macrophages lysates were separated by 5% and 10% SDS-polyacrylamide gel electrophoresis and then were transferred onto polyvinylidene difluoride membranes. The membranes were blocked with blocking buffer (LI-COR Biosciences, Lincoln, USA) for 1 h at room temperature and incubated with the anti-TICAM2 antibody (1:1000; mAbcam77169; Abcam, Cambridge, USA), anti-TIRAP antibody (1:1000; LS-C747701; Lifespan, Seattle, USA), anti-MyD88 antibody (1:1000; EPR590; Abcam) and anti-GAPDH antibody (1:2000; 60004-1-1G; Proteintech, Chicago, USA) at 4°C for 4 h. After washing, membranes were incubated with secondary antibodies (LI-COR Biosciences) for 1 h. Protein bands were visualized using the Odyssey System (LI-COR Biosciences).
In vivo Study
Mice and Treatment
All experiments with virus exposure were performed in the P3-level laboratory of Zhejiang Provincial Center for Disease Control and Prevention (CDC), Zhejiang, China. Virus was also provided by this Center. All animal experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committees of Zhejiang University followed the Laboratory animal-Guideline for ethical review of animal welfare (GB/T 35892-2018), Zhejiang, China.
Adult C57BL/6J male mice (8 weeks old) were maintained at the Animal Facility of Zhejiang University. Mice were intratracheally injected with recombinant N protein (4 mg/kg) and dead SARS-CoV-2 (supplied by Zhejiang CDC center) (106.25 TCID50/mL) in 80 μL PBS/mouse or equal volume of PBS, and their blood and lung samples from different cohorts were harvested at day 1, 3 or 5 after treatment.
Macrophage Depletion
Other mice cohorts were injected with 200 μL of clodronate-conjugated liposomes or control liposomes (LIPOSOMA, Amsterdam, the Netherlands) through tail vein. After 48 h, they were then intratracheally injected with the dose described above of N protein, dead SARS-CoV-2 or equal volume PBS. Their blood and lung samples were then harvested at day 5 after treatment. The macrophage depletion in the lung was assessed with flow cytometry analysis of lung suspension cells.
Histopathology and Lung Injury Score
The harvested lung tissue was fixed and embedded in paraffin wax and then sectioned at 5 µm in thickness. Sections were stained with hematoxylin and eosin (H&E) and the lung injury score was computed.16 Images of lung sections were taken with an Olympus VS120 microscope (Shinjuku, Tokyo, Japan).
Lung Suspension Cell Isolation
Lung suspension cell isolation was done as described previously.17 In brief, after culled under CO2, their lungs were harvested and then filled with digestion medium [Liberase TM (50 μg/mL, Sigma-Aldrich, St. Louis, USA) and DNase I (1 μg/mL, Sigma-Aldrich)] by intratracheal injection for 45 min at 37°C in a water bath. The tissue was mashed through 70 μm cell strainers and washed again with DMEM media supplemented with 10% FBS and 1% Penicillin/Streptomycin (Thermo Fisher Scientific, Pittsburgh, USA). Red blood cells were lysed by RBC lysis buffer (Thermo Fisher Scientific) and resuspended by PBS for further experiments.
Flow Cytometry Analysis
Lung suspension cells were then incubated with Alexa Fluor 700-conjugated anti-CD45 antibody (30-F11, eBioscience, San Diego, USA), phycoerythrin (PE)-conjugated F4/80 (BM8, eBioscience) and fluorescein isothiocyanate-conjugated CD11c (N418, eBioscience) for 30 min. After washing, cells were then analyzed by flow cytometry (BD Biosciences, San Jose, USA) and the data were analyzed with BD FACSDiva and FlowJo software (BD Biosciences).
Cytokine Measurement
Cell culture media and blood samples from above experiments plasma cytokines were used to measure cytokines (TNF-α, IL-1β, IL-6, IL-10, IL-12, IFN-γ and MCP-1) with cytometric bead array (BD Biosciences) according to manufacturer’s instructions.
Statistical Analysis
Data are presented as mean ± SEM unless indicated otherwise. GraphPad Software was used to analyze data with two-tailed unpaired Student’s t-tests or one-way analysis of variance followed by post hoc Dunn’s analysis as appropriate. A P value less than 0.05 was considered to be of statistical significance.
Results
Cytokine Releases from Activated Macrophage Induced by SARS-CoV-2 N Protein
We analyzed the cytokines release from human macrophages induced by alive SARS-CoV-2 virus. TNF-α, IL-1β, IL-6 and MCP-1 level were increased at 24 h and 48 h when cultured macrophages were challenged with SARS-CoV-2 in a virus loading-dependent manner (Figure 1A–D). In line with previous reports,10,18 we demonstrated a potent inflammatory response of macrophage after alive SARS-CoV-2 virus infection. Moreover, in order to gain the mechanism of SARS-CoV-2 triggers cytokine secretion from macrophage. We analyzed that the cytokines released from macrophage after stimulated with the recombinant N protein and S protein. We found that N protein, but not S protein, induced the increases of TNF-α, IL-1β, IL-6 and MCP-1 at 24 h and 48 h (Figure 1E–H). Interestingly, IL-6 and MCP-1 releases were also increased when cells exposed to S protein at 48 h with the peak level that was much lower than that after N protein stimulation. Collectively, these results indicated that SARS-CoV-2-induced macrophage activation was mainly due to N protein.
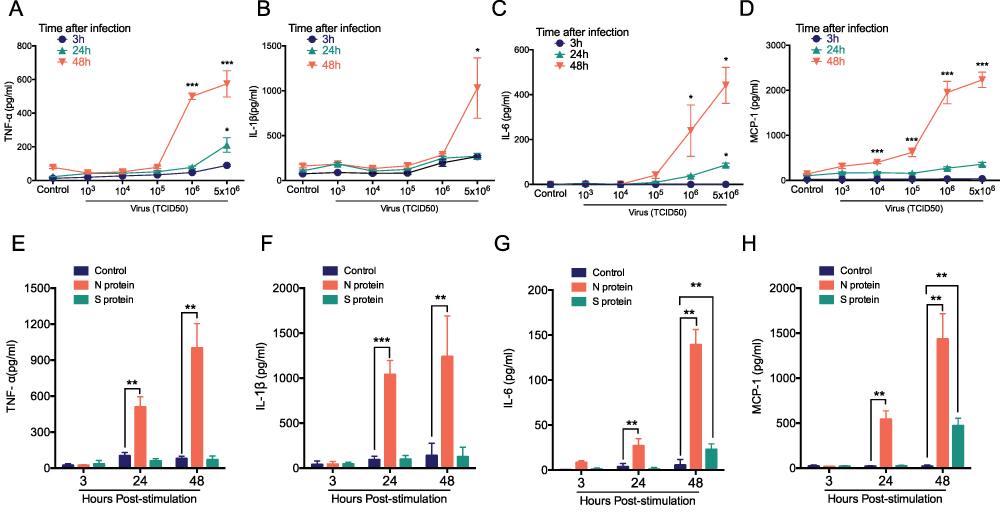 |
Figure 1 Cytokine release from macrophage after treated with alive SARS-CoV-2, S, or N protein. THP-1 macrophages were stimulated with the different load of SARS-CoV-2 from 5x106, 106, 105, 104, 103, 0 TCID50 at 3, 24 and 48 h. (A) TNF-α, (B) IL-1β, (C) IL-6 and (D) MCP-1 were detected with cytometric bead array. THP-1 macrophages were challenged with N protein (4μg/mL), S protein (4μg/mL) and PBS control for 3, 24 and 48h. (E) TNF-α, (F) IL-1β, (G) IL-6 and (H) MCP-1 were measured with cytometric bead array. Data are expressed as mean ± SEM, n=3-6; *P<0.05, **P< 0.01, ***P< 0.001, NS=not significance. |
MyD88 and TIRAP Pathway Mediated by N Protein Induced Macrophage Activation
Myeloid Differentiation Primary Response 88 (MyD88), Toll-interleukin 1 receptor domain-containing adaptor protein (TIRAP) and TIR-containing adaptor molecule-2 (TICAM2) were involved in cytokine response after SARS-CoV-2 S protein stimulation situations,19,20 but the role in SARS-CoV-2 N protein remains unclear. We next investigated whether the N protein induced macrophage activation through MyD88, TIRAP and TICAM2. Thus, the expression of MyD88, TIRAP and TICAM2 in macrophages after N protein treatment was determined. MyD88 and TIRAP but not TICAM2 expressions were increased at 6 h, reached to the peak at 12 h after treatment (Figure 2A). Moreover, TNF-α and IL-1β in culture media of macrophages challenged with N protein for 24 h and 48 h were significantly decreased after the inhibition with TIRAP and MyD88 siRNA, respectively (Figure 2B and C). IL-6 was also significantly decreased after MyD88 siRNA treatment at 24 h and 48 h but this was not the case after treated with the TIRAP siRNA at 24 h (Figure 2D). Interestingly, MCP-1 was not changed by any siRNA inhibitions of MyD88, TIRAP or TICAM2 (Figure 2E). Overall, our data demonstrated that N protein caused macrophage activation and subsequently released cytokine through MyD88 and TIRAP modulation.
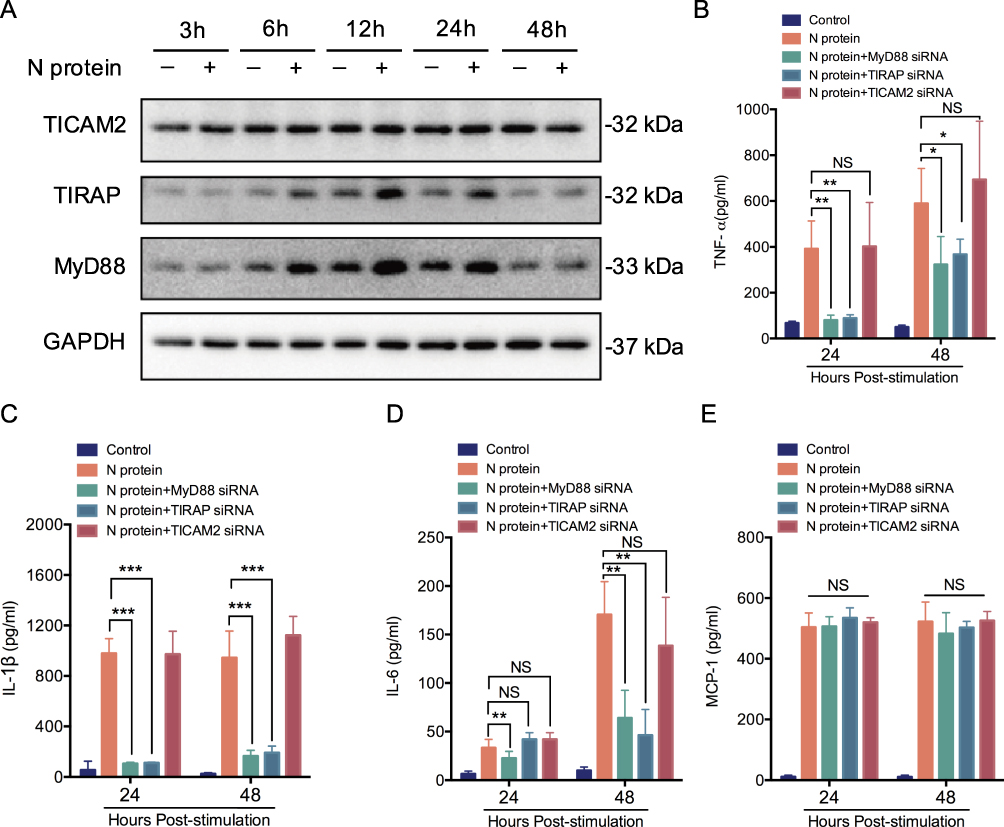 |
Figure 2 MyD88 and TIRAP pathway mediates N protein induced macrophage activation. (A) TICAM2, TIRAP and MyD88 in whole-cell THP-1 macrophage lysates at 3, 6, 12, 24 and 48h after N protein stimulation (4 μg/mL) (n=3). (B-E) TNF-α, IL-1β, IL-6 and MCP-1 release from the siRNA of TICAM2, TIRAP or MyD88 inhibited THP-1 macrophages under 24 and 48 h N protein challenge (n=3-6). Data are expressed as mean ± SEM; *P<0.05, **P< 0.01, ***P< 0.001, NS=not significance. |
N Protein Induced Systemic Inflammation in Mice
To determine the role of N protein in inducing systemic inflammation via airway administration, the dead SARS-CoV-2 or N protein was injected intratracheally in mice. N protein intensively induced systemic inflammation in mice at post-injection day 3 and 5; However, the inactivated SARS-CoV-2 only caused moderate responses at day 5 (Figure 3A–F). These results suggested that SARS-CoV-2 N protein is a potent activator in inducing systemic inflammation responses of COVID-19.
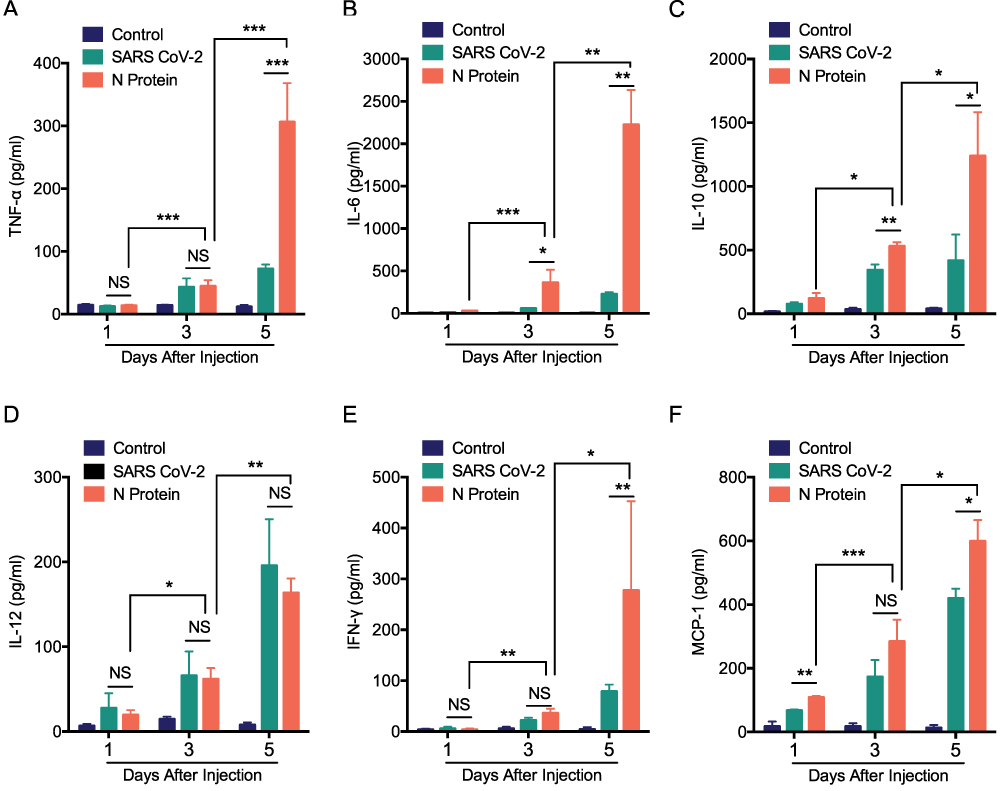 |
Figure 3 N protein induced systemic inflammation in mice. Serum cytokines in mice after challenged with N protein, dead SARS-CoV-2 and PBS control. (A) TNF-α, (B) IL-6, (C) IL-10, (D) IL-12, (E) IFN- γ and (F) MCP-1 were detected with cytometric bead array. Data are expressed as mean ± SEM, n=3-6; *P<0.05, **P< 0.01, ***P< 0.001, NS=not significance. |
N Protein Induced Systemic Inflammation Accompanied with Macrophage Accumulation in the Lung
MCP-1, a chemokine for macrophage migration,21 was increased in mice after N protein and dead SARS-CoV-2 stimulation (Figure 3F). Macrophage accumulation in the lung was found at day 1 up to day 5 after N protein challenge and dead SARS-CoV-2 administration, respectively (Figure 4B). Moreover, this change was much higher with N protein stimulation than that with dead SARS-CoV-2 challenge (Figure 4C). Acute lung injury and macrophage accumulation induced by N protein were evident with H&E and immunohistochemistry staining assessment (Figure 4D). N protein induced acute lung injury indicated with the injurious scores were also readily seen (Figure 4E).
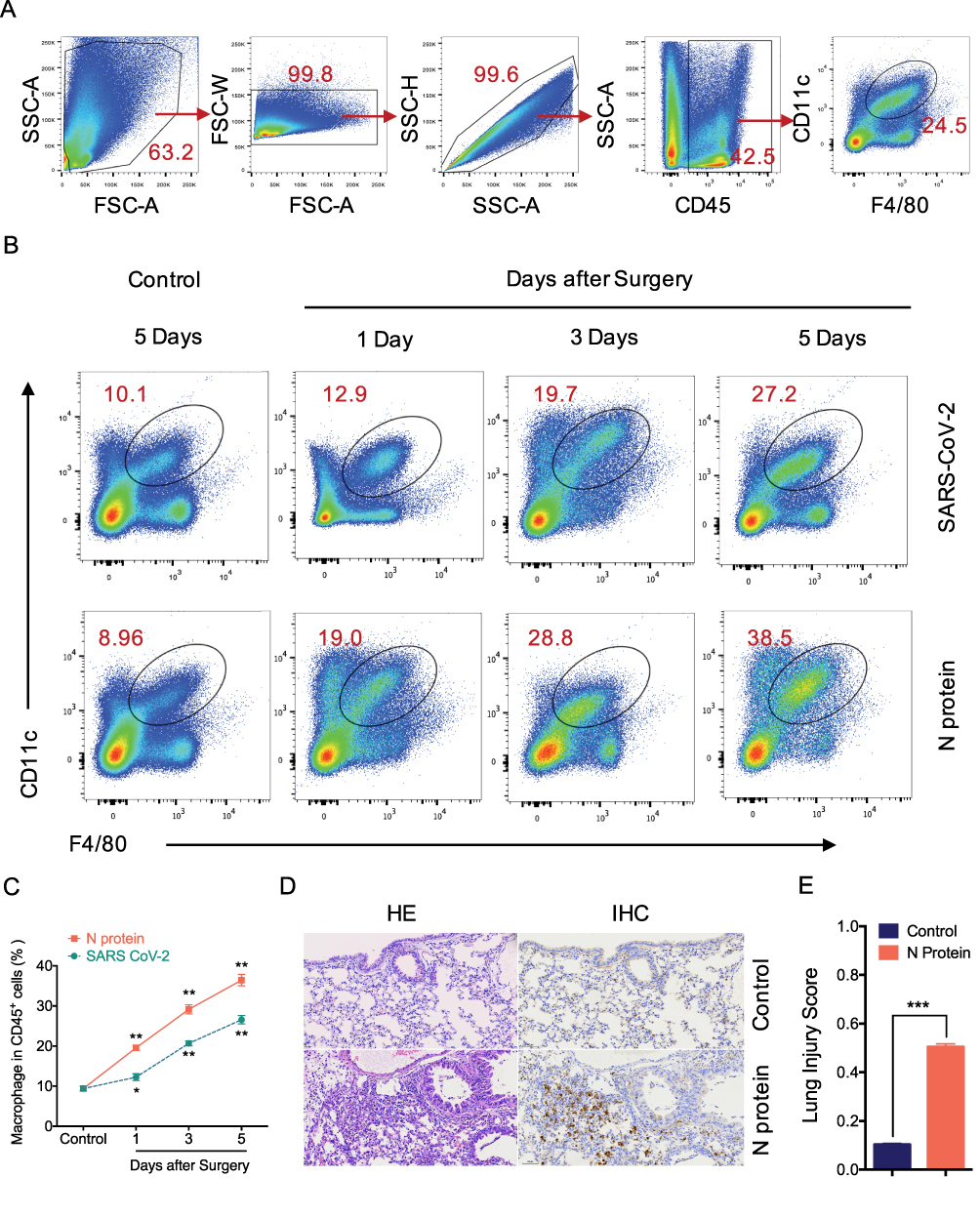 |
Figure 4 N protein induced systemic inflammation and macrophage accumulation in the lung. (A) Representative flow cytometry plots and gating strategy of lung macrophages. Macrophage was defined as CD45+F4/80+CD11c+ cells. The lung single suspension cells were gated, and F4/80+CD11c+ cells were selected in CD45+ cells. (B) Representative flow cytometry plots showing the percentages of macrophages in the lung of mice after N protein or PBS treatment for 1, 3 and 5 days. (C) Line graph showing lung macrophages frequency of CD45+ cells at time points up to 5 days after treatments. (D) Lung sections were stained with H&E for lung injury analysis and immunohistochemistry (IHC) stained macrophage with F4/80 were visualized at ×200 magnification. (E) Lung injury score were quantified in the N protein or control group. Data are expressed as mean ± SEM; n=3-6. NS=not significance, *P<0.05, **P< 0.01, ***P< 0.001. |
In addition, to test the role of macrophage in the systemic inflammation of mice, clodronate liposomes was used to deplete macrophages. The cytokines of TNF-α, IL-6, IL-10, IL-12, IFN-γ and MCP-1 were significantly reduced in mice after macrophage depletion (Figures 5A-5F).
 |
Figure 5 N protein inducing systemic inflammation were attenuated after macrophage depletion in mice. The serum cytokines of macrophages in the depleted mice after challenged with N protein or PBS control at day 5. (A) TNF-α, (B) IL-6, (C) IL-10, (D) IL-12, (E) IFN- γ and (F) MCP-1 were detected with cytometric bead array. Data are expressed as mean ± SEM; n=3; *P<0.05, **P< 0.01, ***P< 0.001, NS=not significance. |
Discussion
In this study, we found cytokine releases from activated macrophage induced by SARS-CoV-2 and its N protein, but not S protein. These cytokine responses were mediated through MyD88 and TIRAP cellular signalling, but not TICAM2. Along with the activated role of N protein on macrophage demonstrated in vitro, we also noted a pronounced expansion of macrophage in the lung after N protein stimulation and a decreased cytokine release in response to N protein stimulation after macrophage depletion in mice. Our work suggested that macrophage activation and subsequent infiltration mediated by SARS-CoV-2 virus and its N proteins may be the main driver of COVID-19 lung injury and even multiple organ dysfunction and/or failure.
The S spike protein of SARS-CoV-2 mediates the cell entry.11,22 Acute respiratory distress syndrome and multiple organ dysfunction/failure with high mortality were closely related to inflammatory responses.18,23 Our data reveal that dead SARS-CoV-2 virus and recombinant N protein but not S protein triggered severe systemic inflammation. Moreover, the cytokine release was increased rapidly after N protein stimulation within 24 h, which led to the further lung tissue damage. We found that SARS-CoV-2 derived N protein but not S protein is likely the main mediator in inducing hyperinflammation. Although the mixture effects of N or S proteins in inducing inflammatory cytokine responses have been reported through activating TLR2 receptor or NLRP3 inflammasome,19,20,24 the potent pro-inflammatory response due to N protein was concluded in our current study. Interestingly, the S1 subunit of S protein has been found to alter lung vascular permeability and induce hyperinflammation,25 but it is not the case under our experiment conditions and further study into this is needed. Moreover, previous studies also reported that immune dysregulated response rather than hyperimmune response, namely “cytokine storm syndrome”, drove SARS-CoV-2 virus-induced lung injury.26 All these indicate the complexity of COVID-19 but one can argue that patient’s body lacks response in severe cases at the disease “later stage” cannot be also excluded per se.
We demonstrated that macrophages were exposed to alive SARS-CoV-2 or N protein to be activated and subsequently released cytokines. These findings suggest that the hyperinflammation after SARS-CoV-2 infection may mainly be due to activating macrophages. Indeed, activated macrophages were reported to be associated with “cytokine storm” and also lung fibrosis after SARS-CoV-2 infection clinically.2,10 Massive cytokines released from SARS-CoV-2-infected macrophages and monocytes led to inflammation-mediated injurious cascades causing multi-organ injury/failure.11,27 MyD88, TIRAP and TICAM2 cellular signaling have been demonstrated to be involved in cytokine release from macrophage.28–30 However, in our study, we only found that MyD88 and TIRAP but not TICAM2 were involved in macrophage activation. MyD88 has been reported to be activated by SARS-CoV-2 S protein,19 but we found that it was mainly due to N protein stimulation. The discrepancy was likely due to the different experimental settings but warrants further study.
Macrophage, lymphocytes and other cell infiltration in the lung were evident in COVID-19 patients.10 Accordingly, BALF collected from patients with mild or severe COVID-19 revealed as much as 80% of mononuclear phagocyte of total BALF cells compared with only approximately 60% and 40% in patients with mild disease or healthy controls.31 A dysregulated macrophage response may damage the host and even led to death after SARS-CoV-2 infection.14 Our data also support this conclusion as the depletion of macrophages in lungs directly attenuated the systemic inflammation after N protein injection shown in our study. In contrast, rapid macrophage accumulation in the lung was found at day 1 challenge of N protein. Hence, these findings may suggest that blocking N protein may be a potential therapeutic strategy to dampen macrophage activation for COVID-19 patients.
The strength of our study is the first to demonstrate the damaging role of macrophage activation induced by live or dead SARS-CoV-2 and N protein challenge in vitro and in vivo, respectively, and the interaction between SARS-CoV-2 and macrophage may be mainly due to SARS-CoV-2 derived N protein. However, due to the high risk using alive SARS-CoV-2 virus infection in mice, the underlying molecular mechanisms and impact under this condition are required further study.
Conclusion
Our data suggested that SARS-CoV-2 N protein may be the main mediator to induce lung macrophage activation and infiltration and subsequently result in dysregulated cytokine responses to further cause acute lung injury and multiple organ dysfunction and failure. Our work implied that blocking N protein may be a potential therapeutic strategy to dampen macrophage activation to tackle COVID-19 disease whilst developing vaccines to protect body through against both SARS-CoV-2 S and N proteins may be more effective than those mRNA vaccines currently used clinically which are mainly targeted on S protein.
Abbreviations
ACE2, Angiotensin-converting enzyme 2; N protein, Nucleocapsid protein; S protein, Spike protein; MyD88, Myeloid Differentiation Primary Response 88; TICAM2, TIR-containing adaptor molecule-2; TIRAP, Toll-interleukin 1 receptor domain-containing adaptor protein.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article. The original data will be made available for assessment upon request by the editors.
Ethics Approval
All animal experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committees of Zhejiang University, Zhejiang, China.
Author Contributions
Dengming Lai, Kun Zhu and Sisi Li share first authorship. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Supported by Zhejiang University special scientific research fund for COVID-19 prevention and control (Q.S.); National Natural Science Foundation of China 82272191 (Q.S.), 81901989 (D.L.); the Fundamental Research Funds for the Central Universities 226-2022-00060 (Q.S.); Natural Science Foundation of Zhejiang Province LY21H150005 (D.L.); Foundation for The Top-Notch Youth Talent Cultivation Project of Independent Design Project of National Clinical Research Center for Child Health Q21B0007 (D.L.); Special Fund for the Incubation of Young Clinical Scientist, The Children’s Hospital of Zhejiang University School of Medicine CHZJU2022YS002 (D.L.); Natural Science Foundation of Zhejiang Province LQY19H190001 (Q.X.); Health Commission of Zhejiang Province 2019KY063, 2021KY133 (Q.X.), 2022485642, 2013KYA107 (S.L.); the Scientific Research Foundation of Zhejiang University City College J-202103(S.L.).
Disclosure
The authors have no conflicts of interest or disclosures relevant to this work.
References
1. Osuchowski MF, Winkler MS, Skirecki T, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med. 2021;9:622–642. doi:10.1016/S2213-2600(21)00218-6
2. Wendisch D, Dietrich O, Mari T, et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. 2021;184:6243–61 e27. doi:10.1016/j.cell.2021.11.033
3. Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–45 e9. doi:10.1016/j.cell.2020.04.026
4. Mehta P, McAuley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi:10.1016/S0140-6736(20)30628-0
5. Papanikolaou V, Chrysovergis A, Ragos V, et al. From delta to Omicron: S1-RBD/S2 mutation/deletion equilibrium in SARS-CoV-2 defined variants. Gene. 2022;814:146134. doi:10.1016/j.gene.2021.146134
6. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–92 e6. doi:10.1016/j.cell.2020.02.058
7. Wang W, Chen J, Hu D, et al. SARS-CoV-2 N protein induces acute kidney injury via Smad3-dependent G1 cell cycle arrest mechanism. Adv Sci. 2022;9:e2103248. doi:10.1002/advs.202103248
8. Webb BJ, Peltan ID, Jensen P, et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: a cohort study. Lancet Rheumatol. 2020;2:e754–e63. doi:10.1016/S2665-9913(20)30343-X
9. Del Valle DM, Kim-Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26:1636–1643. doi:10.1038/s41591-020-1051-9
10. Wang C, Xie J, Zhao L, et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine. 2020;57:102833. doi:10.1016/j.ebiom.2020.102833
11. Iwasaki M, Saito J, Zhao H, Sakamoto A, Hirota K, Ma D. Inflammation triggered by SARS-CoV-2 and ACE2 augment drives multiple organ failure of severe COVID-19: molecular mechanisms and implications. Inflammation. 2021;44:13–34. doi:10.1007/s10753-020-01337-3
12. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80 e8. doi:10.1016/j.cell.2020.02.052
13. Junqueira C, Crespo A, Ranjbar S, et al. FcgammaR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature. 2022;606:576–584. doi:10.1038/s41586-022-04702-4
14. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:355–362. doi:10.1038/s41577-020-0331-4
15. Sharma SB, Melvin WJ, Audu CO, et al. The histone methyltransferase MLL1/KMT2A in monocytes drives coronavirus-associated coagulopathy and inflammation. Blood. 2022;2022:245.
16. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi:10.1165/rcmb.2009-0210ST
17. Lai DM, Tang J, Chen LS, et al. Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis. 2018;9. doi:10.1038/s41419-018-0412-5
18. Ramasamy S, Subbian S. Critical determinants of cytokine storm and type I interferon response in COVID-19 pathogenesis. Clin Microbiol Rev. 2021;34:154.
19. Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife. 2021;10. doi:10.7554/eLife.68563
20. Zheng M, Karki R, Williams EP, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22:829–838. doi:10.1038/s41590-021-00937-x
21. Abid S, Marcos E, Parpaleix A, et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J. 2019;54:e15.
22. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2022;23:3–20. doi:10.1038/s41580-021-00418-x
23. Levy M, Recher M, Hubert H, et al. Multisystem inflammatory syndrome in children by COVID-19 vaccination status of adolescents in France. JAMA. 2022;327:281–283. doi:10.1001/jama.2021.23262
24. Pan P, Shen M, Yu Z, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun. 2021;12:4664. doi:10.1038/s41467-021-25015-6
25. Colunga Biancatelli RML, Solopov PA, Sharlow ER, Lazo JS, Marik PE, Catravas JD. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Kappa18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2021;321:L477–L84. doi:10.1152/ajplung.00223.2021
26. Ocal S. SARS-CoV-2 and lung injury: dysregulation of immune response but not hyperimmune response as in “cytokine storm syndrome”. Clin Respir J. 2022;16:13–16. doi:10.1111/crj.13455
27. Xia J, Tang W, Wang J, et al. SARS-CoV-2 N protein induces acute lung injury in mice via NF-kB activation. Front Immunol. 2021;12:791753. doi:10.3389/fimmu.2021.791753
28. Akbarpour M, Lecuona E, Chiu SF, et al. Residual endotoxin induces primary graft dysfunction through ischemia/reperfusion-primed alveolar macrophages. J Clin Invest. 2020;130:4456–4469. doi:10.1172/JCI135838
29. Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi:10.1038/nature01180
30. Hsieh WY, Zhou QD, York AG, et al. Toll-like receptors induce signal-specific reprogramming of the macrophage lipidome. Cell Metab. 2020;32:128–43 e5. doi:10.1016/j.cmet.2020.05.003
31. Xu G, Qi F, Li H, et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020;6:73. doi:10.1038/s41421-020-00225-2
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Antrodia cinnamomea May Interfere with the Interaction Between ACE2 and SARS-CoV-2 Spike Protein in vitro and Reduces Lung Inflammation in a Hamster Model of COVID-19

Li LH, Chiu HW, Wong WT, Huang KC, Lin TW, Chen ST, Hua KF
Journal of Inflammation Research 2023, 16:4867-4884
Published Date: 26 October 2023