Back to Journals » Journal of Inflammation Research » Volume 16
Sarcoplasmic Myxovirus Resistance Protein A: A Study of Expression in Idiopathic Inflammatory Myopathy
Authors Waisayarat J ![]() , Wongsuwan P, Tuntiseranee K, Waisayarat P
, Wongsuwan P, Tuntiseranee K, Waisayarat P ![]() , Dejthevaporn C
, Dejthevaporn C ![]() , Khongkhatithum C, Soponkanaporn S
, Khongkhatithum C, Soponkanaporn S
Received 24 August 2023
Accepted for publication 31 October 2023
Published 20 November 2023 Volume 2023:16 Pages 5417—5426
DOI https://doi.org/10.2147/JIR.S433239
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Jariya Waisayarat,1 Phumin Wongsuwan,1 Kiarttiyot Tuntiseranee,2 Phu Waisayarat,3 Charungthai Dejthevaporn,4 Chaiyos Khongkhatithum,5 Sirisucha Soponkanaporn6
1Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 2Department of Anatomy, Faculty of Science, Mahidol University, Bangkok, Thailand; 3Faculty of Medicine, King Mongkut’s Institute of Technology Ladkrabang, Bangkok, Thailand; 4Division of Neurology, Department of Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 5Division of Neurology, Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 6Division of Rheumatology, Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Jariya Waisayarat, Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Ratchathewi, Bangkok, 10400, Thailand, Email [email protected]
Background: Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of autoimmune diseases affecting primarily proximal muscles. Major subtypes include dermatomyositis, polymyositis, inclusion body myositis, immune-mediated necrotizing myopathy and antisynthetase syndrome. Overexpression of sarcoplasmic myxovirus-resistance protein A (MxA) has been observed in muscle biopsy specimens of dermatomyositis but is rarely seen in other subtypes of IIM and other myopathies.
Objective: We evaluate the expression of sarcoplasmic MxA and its diagnostic value in IIM and other myopathies.
Methods: One hundred and thirty-eight muscle biopsy specimens with the diagnosis of IIM and other myopathies from 2011 to 2020 were reviewed and stained for MxA by immunohistochemistry. The difference of the expression of MxA between IIM and other myopathies was analyzed by Fisher’s exact test, and the sensitivity and specificity of MxA immunohistochemistry in the diagnosis of IIM were assessed.
Results: MxA protein was positive in 16/138 (11.6%) specimens. All 12 dermatomyositis specimens positive for MxA protein were positive in perifascicular area pattern. Only dermatomyositis specimens had a significantly higher percentage of positive sarcoplasmic MxA expression than specimens of other subtypes of IIM (p< 0.001). Sarcoplasmic MxA expression for dermatomyositis diagnosis had a sensitivity of 46.15% (95% CI 26.59– 66.63%) and a specificity of 94.44% (95% CI 81.34– 99.32%) with the positive and negative likelihood ratio of 8.31 (95% CI 2.03– 34.01) and 0.57 (95% CI 0.40– 0.82), respectively.
Conclusion: The MxA immunohistochemistry is highly specific for dermatomyositis and should be added to a routine inflammatory panel of muscle biopsy. MxA expression should be cautiously interpreted to avoid pitfalls.
Keywords: dermatomyositis, idiopathic inflammatory myopathies, muscle biopsy, myxovirus resistance protein A
Introduction
Idiopathic inflammatory myopathies (IIM) compose a heterogeneous group of rare autoimmune disorders initially characterized by skeletal muscle inflammation and muscle weakness.1 Extramuscular involvement (eg skin, lungs, and malignancy) affects a number of patients, making classification more challenging.2,3 Muscle-specific autoantibodies (MSAs) have been discovered over recent years and are correlated with specific clinicopathological subgroups.4 IIM classifications have integrated clinical manifestation, MSA, and pathologic features to categorize IIM into five subgroups: dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), immune-mediated necrotizing myopathy (IMNM), and antisynthetase syndrome (ASS).5–9
Among all subgroups, DM has the widest range of manifestations, and 20% to 38.3% was negative for MSA,10–13 demonstrating the role of muscle biopsy in aiding the diagnosis. Perifascicular atrophy, considered as a definitive DM muscle biopsy finding in the European Neuromuscular Centre (ENMC) diagnostic criteria,5 has a sensitivity of 36% to 59% and specificity of 92% to 98%.14–16 Gene expression profiling studies showed distinct type 1 interferon-inducible genes in DM, including myxovirus-resistance protein A (MxA) and interferon-stimulated gene 15 (ISG15).17,18 Subsequent studies have shown that the immunohistochemical stain for MxA is a sensitive and specific marker for DM with a sensitivity of 57% to 77% and specificity of 98% to 100%.16,17,19,20 Thus, the 2018-ENMC diagnostic criteria adopted MxA immunopositivity in perifascicular pattern, as a pathological criterion to make a definitive diagnosis for DM as well as perifascicular atrophy.5 However, there are limited data on the expression of MxA protein on muscle biopsy samples from patients with other subtypes of IIM.
This study aims to evaluate the expression of sarcoplasmic MxA in muscle biopsy samples from patients with IIM and the diagnostic value of MxA immunohistochemistry (IHC) among subtypes of IIM.
Materials and Methods
Patients and Muscle Biopsy Specimens
We enrolled all 381 patients who had muscle biopsy specimens in Ramathibodi Hospital from 2010 to 2020. Inclusion criteria were patients with diagnosis of IIMs, overlap myositis (OM), paraneoplastic myopathy, muscular dystrophies (MD), and unspecified myopathies (UM). The diagnosis of IIMs including DM, PM, IBM, IMNM, and ASS was based on the Bohan and Peter criteria,21,22 the 2017 European League against Rheumatism (EULAR) and the American College of Rheumatology (ACR) classification criteria for IIMs and the 2018 ENMC criteria.5,23 OM was defined as follows: 1) patients who fulfill both IIMs and other connective tissue disease criteria or 2) patients who have myositis plus 1 or more clinical overlap features and / or the presence of myositis-associated autoantibodies.24 Most MD cases were classified based on clinical information and dystrophic muscle biopsy including IHC study.25 Several equivocal muscle biopsies of limb-girdle muscular dystrophy (LGMD), Becker muscular dystrophy (BMD), and oculopharyngeal muscular dystrophy (OPMD) were included based on the genetic analysis. For paraneoplastic myopathy, the diagnosis is based on the presence of necrotic fibers without other characteristic features of IIM with pathologically proven internal malignancy.26 UM was defined based on clinical diagnoses of questionable myopathies, including unexplained weakness, muscle pain, hyperCKemia, and irritating pattern on EMG. Muscle biopsies in this group showed minimal or nonspecific change.
Patients with incomplete clinical data and unavailable muscle specimens were excluded. Medical records including clinical diagnosis, myositis autoantibody status, muscle biopsy pathologic report and available glass slides were reviewed. After applying the inclusion and exclusion criteria, 138 patients were included in this study (Figure 1a).
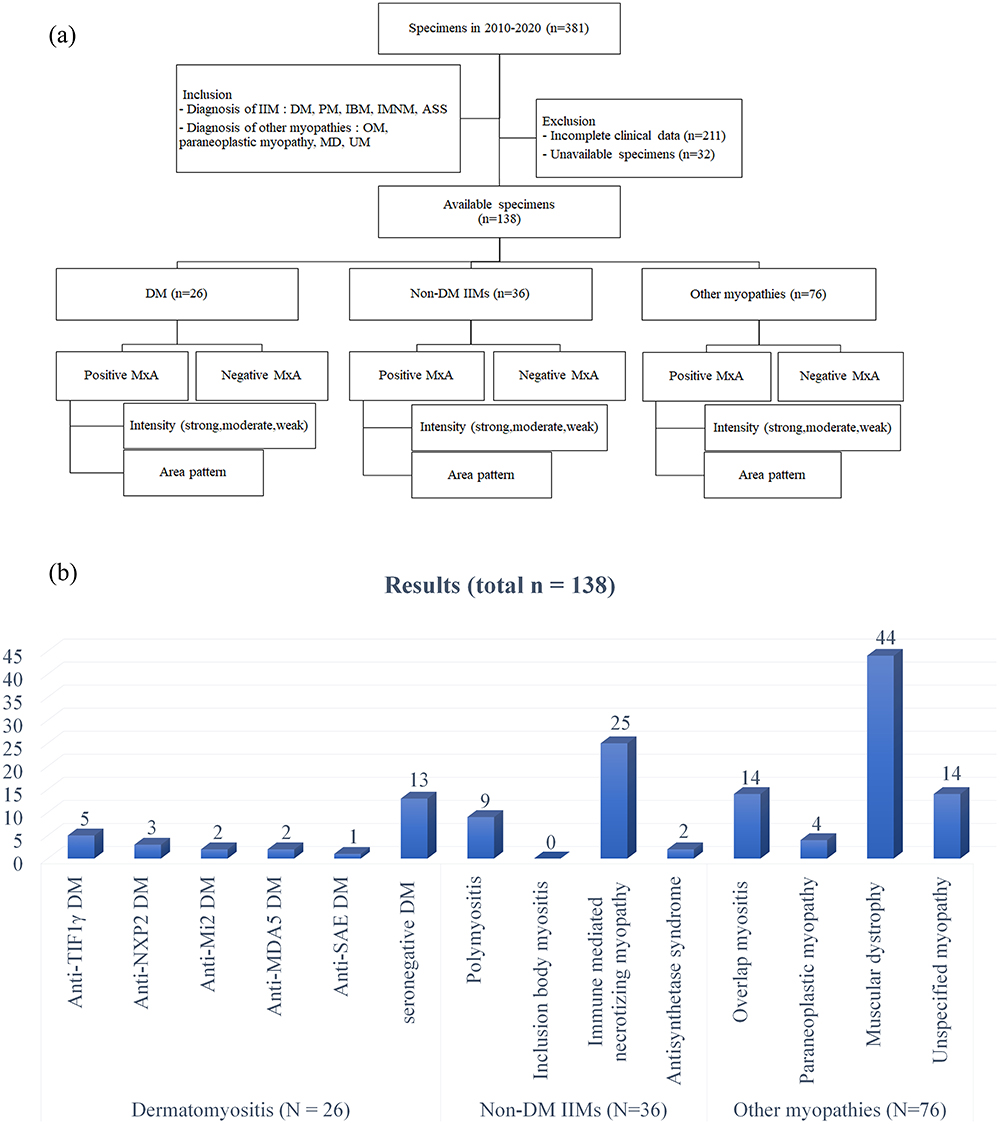 |
Figure 1 (a) Study flow chart. (b) Patients categorization chart. Abbreviations: ASS, anti-synthetase syndrome; DM, dermatomyositis; IBM, inclusion body myositis; IIM, idiopathic inflammatory myopathy; IMNM, immune-mediated necrotizing myopathy; MD, muscular dystrophy; OM, overlap myositis; PM, polymyositis; UM, unspecified myopathy. |
All enrolled patients were categorized as 1) DM-IIM (N=26), 2) non-DM-IIMs (N=36) including PM (N=9) IBM (N=0), IMNM (N=25) and ASS (N=2), 3) other myopathies (N=76) including OM (N=14), paraneoplastic myopathy (N=4), MD (N=44), and UM (N=14).
For DM-IIM, we categorized subtypes according to the MSAs as followed; anti-transcription intermediary factor 1 gamma (anti-TIF1γ DM, N=5), anti-nuclear matrix protein 2 (anti-NXP2 DM, N=3), anti-complex nucleosome remodeling histone deacetylase (anti-Mi2 DM, N=2), anti-melanoma differentiation-associated gene 5 (Anti-MDA5 DM, N=2), anti-small ubiquitin-like modifier activating enzyme (anti-SAE DM, N=1) and seronegative DM (N=13). The immunoblot technique was used to detect myositis autoantibodies.
Among OM patients, there were systemic lupus erythematosus (N=10), systemic sclerosis (N=3), and Sjögren’s syndrome (N=1) (Figure 1b).
Biopsy Site
The biopsy sites were determined by clinicians. All muscle biopsy specimens were snap-frozen in isopentane (the freezing point at −160°C) and liquid nitrogen (the boiling point at −196°C) and were stored in an ultra-low temperature freezer at −80°C until use. Routine histochemical and selected immunohistochemical staining were performed and evaluated.
MxA-Immunohistochemistry Preparation
The stored specimens thawing from the freezer were put in the cryostat for 45–60 min to equilibrate with the cryostat temperature before sectioning to prevent the tissue shattering. Cryostat sections, 6 µm thick, were cut and dried on the superfrost glass slides at room temperature, for 45–60 min. We included unspecified myopathy as a negative control.
The primary antibodies used to detect MxA protein were mouse anti‐human anti‐MxA monoclonal antibodies (clone: M143, 1:200 dilution; Merck Millipore, CA, USA). Horseradish peroxidase and DAB (Dako, Cambridgeshire, UK) were used as secondary antibodies and chromogen, respectively, under BenchMark ULTRA (Ventana Medical System, Tucson, AZ, USA). The IHC protocol was as follows: briefly after blocking the endogenous peroxidase by I–View Inhibitor (Ventana), samples were incubated at 36°C for 1 hr with anti-MxA. Antigen–antibody reactions were visualized using Ventana OptiView Amplification kit (Optiview HQ Linker for 12 min and Optiview HRP Multimer for 12 min). Counterstaining was obtained using Ventana Hematoxylin II for 16 min, followed by a bluing reagent for 4 min. Finally, all slides were removed from the stainer, dehydrated in 70% alcohol (twice), 95% alcohol (twice), 100% alcohol (twice), and xylene (twice). It is then mounted with coverslip for microscopic examination under optical microscopy.
Microscopic Interpretation of MxA Staining
The glass slides were interpreted under a traditional light microscope by two independent pathologists, one general and one muscle consultant (PW and JW) without clinical information. If there were any discrepancies between the evaluations, a slide review with discussion, and a consensus were made.
The expression was evaluated as positive by unequivocal sarcoplasmic staining in only intact fiber, not a regenerating or degenerated/necrotic fiber. The three-tier intensity; strong, moderate, and weak, and the pattern; either perifascicular area or non-perifascicular area, were recorded when one or more fibers were strongly, moderately, and faintly cytoplasmic stained. Perifascicular area pattern was defined by positivity at peripheral area of muscle fascicle, and the non-perifascicular area pattern was defined by a single fiber, scattered fibers, patchy, or diffuse staining. When no intact fiber was stained, a negative result was recorded (Figure 2).
 |
Figure 2 Representative immunohistochemical staining of MxA in muscle biopsies. Positive expression was defined by unequivocal sarcoplasmic staining in only intact fiber, not a regenerating or degenerated/necrotic fiber. (a) MxA negativity. (b) Moderate MxA positivity and perifascicular area pattern. (c) Strong MxA positivity and perifascicular area pattern (b and c in dermatomyositis patients). (d) Weak MxA positivity and non-perifascicular area pattern, positive fibers labeled with black arrow (in overlap myositis patient). |
Statistical Analysis
The difference between sarcoplasmic MxA expression of DM and other non-DM IIMs with perifascicular area pattern or non-perifascicular area pattern was evaluated by Fisher’s exact test. The p-value less than 0.05 was considered statistically significant. Sensitivity and specificity with 95% CI for a diagnosis of myopathies, which correlated with sarcoplasmic MxA expression were calculated. Data were analyzed using STATA version 17 (STATACorp LLC, Texas, USA).
Ethics Approval and Consent
This study was approved by the Institutional Review Board of Faculty of Medicine Ramathibodi Hospital, Mahidol University, in full compliance with International Guidelines for Human Research Protection such as the Declaration of Helsinki, The Belmont Report, CIOMS Guidelines, and the International Conference on Harmonization in Good Clinical Practice (ICH-GCP). The informed consent obtained from the study participants had been reviewed by the Institutional Review Board of Faculty of Medicine Ramathibodi Hospital, Mahidol University, prior to study commencement (COA. MURA2020/1547 Ref.3562). A copy of the approved ethic form is available for review at the institute.
Results
The demographic data of 138 patients is shown in Table 1. There was no sex preference. Median age (interquartile range, IQR) at the enrollment age was 42.5 (IQR 12–59). About 70% of patients were over 16 years old. Among patients with DM, about half of them had negative MSA. The most common MSA was anti-TIF1- γ (38%), followed by anti-NXP2 (23%). For IMNM patients, anti-signal recognition particle (SRP) and anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) were negative in all patients.
 |
Table 1 Demographic Data, Diagnosis and Myositis-Specific Autoantibodies of 138 Patients |
The details of MxA staining on muscle biopsy specimens of patients with different diagnoses are summarized in Table 2. Nearly half of patients with DM (46%) had sarcoplasmic MxA expression, and all of them had perifascicular pattern. Among DM patients with positive MSAs, patients with anti-NXP2 (67%) tended to have a higher percentage of sarcoplasmic MxA expression than other MSAs followed by anti-TIF1- γ (40%), whereas about 60% of DM patients with negative MSA had sarcoplasmic MxA expression on muscle samples. Although, the intensity of MxA expression on muscle samples of DM patients with anti-NXP2 and anti-TIF1- γ was moderate to strong degree, patients with negative MSA had moderate to weak degree of MxA expression.
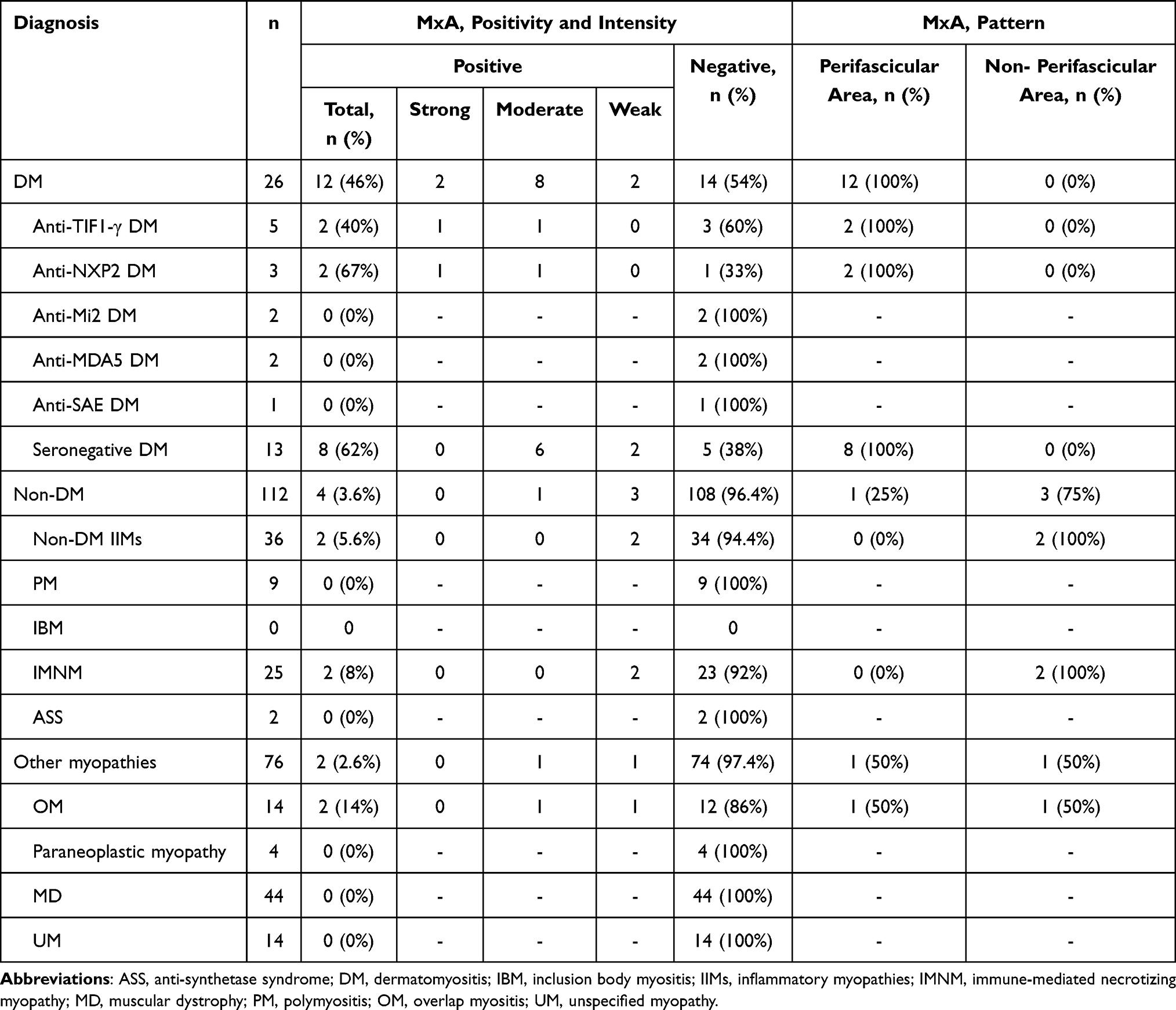 |
Table 2 The Details of MxA Staining on Muscle Biopsy Specimens of Patients with Different Diagnosis |
Moreover, there was a weak MxA staining on few fibers of muscle samples of two patients with IMNM (8%), and both were stained in a non-perifascicular pattern. One IMNM showed scattered equivocal MxA staining fibers but were confirmed as degenerated/necrotic fibers by other stains.
Also, there was sarcoplasmic MxA expression in 2 OM (14%), one OM patient with Sjögren’s syndrome and the other with scleroderma. OM patient with Sjögren's syndrome had moderate intensity with perifascicular pattern, and the other with scleroderma had weak intensity with non-perifascicular pattern. No MxA expression was observed in all OM patients with SLE. Additionally, all patients with MD and UM had no MxA expression on muscle biopsy samples.
Among IIMs, all DMs (not specified subtypes) showed a statistically significant higher percentage of positive sarcoplasmic MxA expression on muscle samples than patients with non-DM-IIMs (p-value <0.001). Interestingly, the strongly positive intensity was observed only in the DM (2 out of 16 positive MxA, 12.5%). Sarcoplasmic MxA expression in perifascicular and non-perifascicular areas for DM diagnosis among IIMs patients has a sensitivity of 46.15% (95% CI 26.59–66.63%) and a specificity of 94.44% (95% CI 81.34–99.32%) with the positive and negative likelihood ratio of 8.31 (95% CI 2.03–34.01) and 0.57 (95% CI 0.40–0.82), respectively.
Discussion
This study demonstrated the important role of MxA expression on muscle biopsy samples in diagnosing DM. MxA expression was found in a significantly higher proportion of DM patients when compared to other IIMs. Moreover, the pattern and intensity of MxA staining, which is a perifascicular pattern and moderate to strong intensity, might help diagnose DM. MxA expression also showed a moderate sensitivity of 46.15% and a high specificity of 94.44% in diagnosing DM.
The two pathological hallmarks in muscle biopsy specimens for diagnosis of definitive DM are perifascicular atrophy (47% to 51% sensitivity and 98% specificity) and capillary membrane attack complex (MAC, C5b-9) (35% to 81% sensitivity and 89% to 93% specificity).14–16,20,27,28 Although, they have quite high specificity, sensitivity is low and varied.
MxA, a type 1 interferon-inducible protein, is crucial to the pathophysiology of DM. Since 2005, Greenberg SA et al have addressed sarcoplasmic MxA staining, including capillary MxA staining in DM.17 Higher sensitivity and specificity were reported by Uruha A et al, 2017 and 2019.16,20 Soponkanaporn S et al 2019 showed high sensitivity and specificity of sarcoplasmic MxA associated with clinical severity.19 The mentioned studies showed a similar MxA immunohistochemistry with high sensitivity and specificity for DM, 57% to 77% sensitivity and 98% to 100% specificity.16,17,19,20 Thus, the 2018-ENMC diagnostic criteria adopted MxA immunopositivity in a perifascicular pattern, as a pathological criterion to make a definitive diagnosis as well as perifascicular atrophy.5
Our study revealed a high specificity but moderate sensitivity of sarcoplasmic MxA expression to DM compared with previous studies, which showed slightly higher specificity and sensitivity. This might be explained by the heterogeneity of MxA in expression; not all fibers expressed the protein, and it may be expressed in variable area and intensity. Different techniques and protocols in laboratory processes may result in different interpretations. Moreover, different subsets of DM based on MSAs may cause different results between studies.
In Uruha et al’s study, 2019, 10 out of 10 cases (100%) of Anti-TIF1-γ DM expressed MxA sarcoplasmic expression, while 2 out of 5 cases (40%) of Anti-TIF1-γ DM in our study showed MxA positivity. Thirteen out of 17 cases (76%) of seronegative DMs expressed in MxA, while 8 out of 13 of our cases (61.5%) of seronegative DM were positive for MxA. Anti-NXP2 DM and seronegative DM in our study showed the same results of MxA positivity when compared to the study.20 There was no MxA expression in anti-Mi2 DM or anti-SAE DM. Interpreting these relationships was difficult due to the limited number of cases.
In amyopathic DM, especially with anti-MDA5 antibody positive, perifascicular pathology was rarely identified.29 However, the pathomechanism of anti-MDA5 DM was activation of the type 1 interferon signaling pathway, evidenced by high serum levels of type 1 interferons.30 Uruha A et al, 2019, and Soponkanaporn S et al, 2018, demonstrated that anti-MDA5 DM and ASS had no or weak MxA expression on muscle samples, similar to our study.19,20 In our study, all two patients with anti-MDA5 DM presented with mild weakness, skin lesions, and interstitial lung disease and were both negative for sarcoplasmic MxA. Uruha et al also mentioned the decreased sensitivity of MxA expression on muscle samples from anti-MDA5 DM. However, our study could not conclude this is due to the minimal number of our anti-MDA5 DM.
The present study shows MxA positivity in two OM (including systemic sclerosis and Sjögren syndrome) and two IMNM. These results are similar to previous studies by Uruha A et al.16
In summary, sarcoplasmic MxA expression could be found in non-DM IIMs. Thus, the diagnosis of dermatomyositis specimens should still be based on clinical presentation, laboratory investigation, and muscle biopsy findings.
Sarcoplasmic MxA expression interpretation should be rigorous. Comparing the sarcoplasmic MxA expressed fibers with routine tinctorial, histochemical, enzymatic, and IHC stains is essential to avoid false positivity. In one IMNM case in the present study, a 4-year-old girl presented with subacute symmetrical proximal weakness and dropped neck without any skin sign on physical examination. Her muscle biopsy revealed scattered necrotic fibers, scant cellular reaction and an absence of perifascicular atrophy. On MxA, several scattered equivocal sarcoplasmic expressions were initially interpreted as positive but confirmed as degenerated/necrotic fibers on the corresponding stains (Figure 3). The interpretation of MxA in the patient reflected the pitfalls of the IHC study. The authors advise caution in interpreting the results of MxA staining.
 |
Figure 3 Necrotic fibers features on MxA and other IHCs. Sarcoplasmic MxA expression interpretation should be rigorous. (a) Five atrophic/shrinkage fibers with pale cytoplasmic stain were identified on H&E, 20X (black arrows). (b) The fibers revealed equivocal sarcoplasmic expression on MxA (black arrows). (c) The corresponding fibers revealed strong/clumping stained on MAC, 20X (black arrows). (d) Strong sarcoplasmic utrophin upregulation, 20X (black arrows). All stains represented that they were degenerated/necrotic fibers. |
In addition, all cases of MD and UM in the present study were negative for MxA expression on muscle samples, considering that it may have no type I interferon association in the pathogenesis of the diseases.
Limitation
There are several limitations to this study. First, there were only a few cases in each DM subgroup, resulting in the insufficiency to indicate the statistical correlation. Second, between 2010 and 2020, we had no definite IBM with characteristic clinical and typical rimmed vacuoles in the filing system to include in this study. There were only two overlap myositis with rimmed-liked vacuoles. Finally, our institute used only the immunoblot technique to detect muscle-specific antibodies and myositis-associated autoantibodies. There were ancillary methods, including immunoprecipitation or enzyme-linked immunosorbent assay (ELISA), which might be specific to some antibodies.
Conclusions
This study shows a strong association between sarcoplasmic MxA expression and DM, among other IIMs. The MxA immunohistochemistry was highly specific for DM and should be included as a routine immunohistochemistry for diagnostic evaluation.
Acknowledgments
We gratefully acknowledge Ms Umaporn Udomsubpayakul of the Department of Clinical Epidemiology and Biostatistics, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, for statistical analysis. We are grateful for the slide preparation by Ms. Natha Thubthong, Ms. Sasithorn Foyhirun, and Ms. Suda Sanpapant of the Department of Pathology. We thank Chayanont Netsawang, M.D., and Anechisa Sakulnurak, M.D. (Department of Pathology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand) for their technical support. This work was funded by a Ramathibodi Research Grant from the Faculty of Medicine Ramathibodi Hospital, Mahidol University (grant no. RF64059).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Plotz PH, Rider LG, Targoff IN, et al. Myositis: immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med. 1995;122(9):715–724. doi:10.7326/0003-4819-122-9-199505010-00010
2. Dimachkie MM, Barohn RJ, Amato AA. Idiopathic inflammatory myopathies. Neurol Clin. 2014;32(3):595–628, vii. doi:10.1016/j.ncl.2014.04.007
3. Cox S, Limaye V, Hill C, et al. Idiopathic inflammatory myopathies: diagnostic criteria, classification and epidemiological features. Int J Rheum Dis. 2010;13(2):117–124. doi:10.1111/j.1756-185X.2010.01472.x
4. Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280(1):8–23. doi:10.1111/joim.12451
5. Mammen AL, Allenbach Y, Stenzel W, et al. 239th ENMC International Workshop: classification of dermatomyositis, Amsterdam, the Netherlands, 14-16 December 2018. Neuromuscul Disord. 2020;30(1):70–92. doi:10.1016/j.nmd.2019.10.005
6. Mariampillai K, Granger B, Amelin D, et al. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. 2018;75(12):1528–1537. doi:10.1001/jamaneurol.2018.2598
7. Allenbach Y, Mammen AL, Benveniste O, et al. Immune-Mediated Necrotizing Myopathies Working Group. 224th ENMC International Workshop: clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord. 2018;28(1):87–99. doi:10.1016/j.nmd.2017.09.016
8. Rose MR; ENMC IBM Working Group. 188th ENMC International Workshop: inclusion Body Myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044–1055. doi:10.1016/j.nmd.2013.08.007
9. De Bleecker JL, De Paepe B, Aronica E, et al. 205th ENMC International Workshop: pathology diagnosis of idiopathic inflammatory myopathies part II 28-30 March 2014, Naarden, The Netherlands. Neuromuscul Disord. 2015;25(3):268–272. doi:10.1016/j.nmd.2014.12.001
10. Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology. 2009;48(6):607–612. doi:10.1093/rheumatology/kep078
11. Hozumi H, Fujisawa T, Nakashima R, et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir Med. 2016;121:91–99. doi:10.1016/j.rmed.2016.10.019
12. Watanabe E, Gono T, Kuwana M, et al. Predictive factors for sustained remission with stratification by myositis-specific autoantibodies in adult polymyositis/dermatomyositis. Rheumatology. 2020;59(3):586–593. doi:10.1093/rheumatology/kez328
13. Betteridge Z, Tansley S, Shaddick G, et al. Frequency, mutual exclusivity and clinical associations of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathy patients. J Autoimmun. 2019;101:48–55. doi:10.1016/j.jaut.2019.04.001
14. Suárez-Calvet X, Gallardo E, Pinal-Fernandez I, et al. RIG-I expression in perifascicular myofibers is a reliable biomarker of dermatomyositis. Arthritis Res Ther. 2017;19(1):174. doi:10.1186/s13075-017-1383-0
15. Scola RH, Werneck LC, Prevedello DM, et al. Diagnosis of dermatomyositis and polymyositis: a study of 102 cases. Arq Neuropsiquiatr. 2000;58(3B):789–799. doi:10.1590/s0004-282x2000000500001
16. Uruha A, Nishikawa A, Tsuburaya RS, et al. Sarcoplasmic MxA expression: a valuable marker of dermatomyositis. Neurology. 2017;88(5):493–500. doi:10.1212/WNL.0000000000003568
17. Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57(5):664–678. doi:10.1002/ana.20464
18. Salajegheh M, Kong SW, Pinkus JL, et al. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol. 2010;67(1):53–63. doi:10.1002/ana.21805
19. Soponkanaporn S, Deakin CT, Schutz PW, et al. Expression of myxovirus-resistance protein A: a possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol Appl Neurobiol. 2019;45(4):410–420. doi:10.1111/nan.12498
20. Uruha A, Allenbach Y, Charuel JL. Diagnostic potential of sarcoplasmic myxovirus resistance protein A expression in subsets of dermatomyositis. Neuropathol Appl Neurobiol. 2019;45(5):513–522. doi:10.1111/nan.12519
21. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–347. doi:10.1056/NEJM197502132920706
22. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292(8):403–407. doi:10.1056/NEJM197502202920807
23. Bottai M, Tjärnlund A, Santoni G, et al. EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups: a methodology report. RMD Open. 2017;3(2):e000507. doi:10.1136/rmdopen-2017-000507
24. Nuño-Nuño L, Joven BE, Carreira PE, et al. Overlap myositis, a distinct entity beyond primary inflammatory myositis: a retrospective analysis of a large cohort from the REMICAM registry. Int J Rheum Dis. 2019;22(8):1393–1401. doi:10.1111/1756-185X.13559
25. Bushby K, Finkel R, Birnkrant DJ, et al.; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. doi:10.1016/S1474-4422(09)70271-6
26. Levin MI, Mozaffar T, Al-Lozi MT, Pestronk A. Paraneoplastic necrotizing myopathy: clinical and pathological features. Neurology. 1998;50:764–767. doi:10.1212/WNL.50.3.764
27. Sakuta R, Murakami N, Jin Y, et al. Diagnostic significance of membrane attack complex and vitronectin in childhood dermatomyositis. J Child Neurol. 2005;20(7):597–602. doi:10.1177/08830738050200071201
28. Jain A, Sharma MC, Sarkar C, et al. Detection of the membrane attack complex as a diagnostic tool in dermatomyositis. Acta Neurol Scand. 2011;123(2):122–129. doi:10.1111/j.1600-0404.2010.01353.x
29. Allenbach Y, Leroux G, Suárez-Calvet X, et al. Dermatomyositis with or without anti-melanoma differentiation-associated gene 5 antibodies: common interferon signature but distinct NOS2 expression. Am J Pathol. 2016;186(3):691–700. doi:10.1016/j.ajpath.2015.11.010
30. Horai Y, Koga T, Fujikawa K, et al. Serum interferon-α is a useful biomarker in patients with anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive dermatomyositis. Mod Rheumatol. 2015;25(1):85–89. doi:10.3109/14397595.2014.900843
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Significance of Abnormal Serum LGALS3BP Expression in Patients with Idiopathic Inflammatory Myopathies
Huang L, Huang X, Zhou W, Jiang Y, Zhu H, Lao Y, Deng Z, Tang Y, Wang J, Li X
Journal of Inflammation Research 2024, 17:9697-9710
Published Date: 25 November 2024