")
Back to Journals » Journal of Inflammation Research » Volume 14
Role of Pyroptosis in Diabetes and Its Therapeutic Implications
Authors Al Mamun A , Wu Y, Nasrin F, Akter A, Taniya MA, Munir F, Jia C, Xiao J
Received 10 November 2020
Accepted for publication 14 January 2021
Published 24 May 2021 Volume 2021:14 Pages 2187—2206
DOI https://doi.org/10.2147/JIR.S291453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Abdullah Al Mamun,1 Yanqing Wu,2 Fatema Nasrin,3,4 Afroza Akter,5 Masuma Afrin Taniya,6 Fahad Munir,7 Chang Jia,8 Jian Xiao1
1Department of Hand Surgery and Peripheral Neurosurgery, The First Affiliated Hospital and School of Pharmaceutical Sciences, Wenzhou, Zhejiang Province, 325035, People’s Republic of China; 2Institute of Life Sciences, Wenzhou University, Wenzhou, Zhejiang Province, 325035, People’s Republic of China; 3Institute of Health and Biomedical Innovation, Translational Research Institute, Brisbane, Australia; 4School of Clinical Sciences, Queensland University of Technology, Brisbane, Australia; 5Department of Microbiology, Noakhali Science and Technology University, Noakhali, Bangladesh; 6Department of Life Sciences, School of Environment and Life Sciences, Independent University, Bangladesh, Dhaka, 1229, Bangladesh; 7Department of Hepatobiliary Surgery, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, Zhejiang Province, People’s Republic of China; 8Pediatric Research Institute, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, 325027, Zhejiang Province, People’s Republic of China
Correspondence: Jian Xiao
Molecular Pharmacology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang Province, 325035, People’s Republic of China
Email [email protected]
Abstract: Pyroptosis is mainly considered as a new pro-inflammatory mediated-programmed cell death. In addition, pyroptosis is described by gasdermin-induced pore formation on the membrane, cell swelling and rapid lysis, and several pro-inflammatory mediators interleukin-1β (IL-1β) and interleukin-18 (IL-18) release. Extensive studies have shown that pyroptosis is commonly involved by activating the caspase-1-dependent canonical pathway and caspase-4/5/11-dependent non-canonical pathway. However, pyroptosis facilitates local inflammation and inflammatory responses. Current researches have reported that pyroptosis promotes the progression of several diabetic complications. Emerging studies have suggested that some potential molecules targeting the pyroptosis and inflammasome signaling pathways could be a novel therapeutic avenue for managing and treating diabetes and its complications in the near future. Our narrative review concisely describes the possible mechanism of pyroptosis and its progressive understanding of the development of diabetic complications.
Keywords: diabetes, pyroptosis, GSDMD, NLRP3, caspase-1, IL-1β, IL-18
Introduction
Diabetes mellitus (DM) is an immunological disease characterized by metabolic dysregulation and chronic inflammation.1 However, hyperglycemia is commonly associated with the generation of ROS and the formation of AGEs.2–4 Chronic and low-grade inflammation has been related to hyperglycemia in diabetes. Programmed cell death plays a significant role in the hosts’ advancement and survival in the body’s defensive mechanism against several microbial infections.5–8 Apoptosis, necrosis, pyroptosis, ferroptosis, autophagy, and efferocytosis are the most common cell death mechanisms.9–14 Apoptosis is the most widely studied, while the research of pyroptosis is neither comprehensive nor concrete. Apoptosis-mediated cell death regulates multicellular organism development.9 Notably, the apoptosis-mediated cell death mechanism predominantly impairs single cells and appears without any inflammatory responses.15 Apoptosis is induced by the cell surface death-receptor stimulation, exogenous, mitochondrial, and ER stress-regulated signaling pathways facilitating caspases initiation.16–19 In apoptosis-mediated cell death, cells shrink and are fragmented into apoptotic bodies engulfed by surrounding macrophages, leading to cell death’s non-inflammatory nature.20 Inhibiting the apoptotic caspase-8 due to pro-apoptotic stimulus initiates necroptosis-regulated cell death through the receptor-interacting serine/threonine-protein kinase 1 (RIPK1)–RIPK3-mixed lineage kinase domain-like (MLKL) axis.21 Mounting evidence shows that RIPK3-phosphorylated MLKL changes to the membrane to accomplish membrane rupture, promoting cell swelling as well as lysis.22–25
Pyroptosis is programmed necrosis, which regulates a common innate immune effector mechanism in invertebrates.26,27 Significantly, the innate immune response depends on PRRs to sense and recognize several microbial products or endogenous danger signals.28 Stimulating cytokine transcription and inducing PRRs cause pyroptosis to promote inflammatory response.29 Pyroptosis’s immune defense functions are increased by disrupting the pathogen replication niche and directly invading intracellular bacteria through pore-stimulated intracellular traps.30,31 For a long time, pyroptosis has been described as a caspase-1-dependent monocyte cell death mechanism.32 Caspase-1 belongs to the inflammatory caspase family group, which processes pro-IL-1β and IL-18 into mature IL-1β and IL-18 pro-inflammatory factors.33,34 These pro-inflammatory factors nature discriminate pyroptosis from apoptosis despite the dependency on caspases.35,36 Indeed, pyroptosis is lytic, attributing cell rupture and swelling and many bubbles blowing on the plasma membrane.28,37,38
Our narrative review highlights the current understanding and knowledge of the molecular mechanism of pyroptosis-mediated cell death and the potential role of inflammasome and pyroptosis in diabetes mellitus and its complications. More in-depth research of pyroptosis-mediated cell death may produce novel insight into the pathogenesis and development of diabetes and its complications.
History of Pyroptosis
In 1986 Friedlander AM first notated cellular components’ rapid release in mouse macrophages after treatment with lethal anthrax toxin.39 Furthermore, Zychlinsky et al in 1992 observed Shigella flexneri induced mouse macrophage cell death through an apoptosis-mediated cell mechanism.40 Regrettably, both pieces of research demonstrated the secretion of cellular contents that are stimulated by classical apoptosis. Cookson performed a combination of Greek roots to establish a novel term, “pyroptosis.” The novel term pyroptosis “pyro” and “ptosis” indicate fire and falling, respectively, to describe a newly discovered inflammatory-mediated programmed cell death.41 Pyroptosis was first described to be a caspase-1-mediated pro-inflammatory programmed cell death.42 Many researchers have identified the GSDMD protein molecule is the primary substrate of caspase-1 and caspase-4/5/11.43,44 In addition, accumulating studies have documented that gasdermin-mediated proteins commonly induce pyroptosis, and the gasdermin family groups can be cleaved by inflammatory caspases, which are essential for the activation of pyroptosis-mediated cell death.11 Shao et al have suggested that pyroptosis is a gasdermin family-mediated programmed necrotic cell death.45 The Nomenclature Committee on Cell Death (NCCD) in 2018 recommended that pyroptosis is considered a programmed cell death characterized by the formation of gasdermin mediated pores on the membrane, often but not always as the results of inflammatory caspases induction.46
Canonical Inflammasome Signaling Pathways
Pyroptosis is a novel pro-inflammatory mediated-programmed cell death mechanism that involves innate immune response.32 The innate immune response depends on PRRs to recognize PAMPs and DAMPs.47,48 The PRR family members compromise NLRs and membrane-bound TLRs, mainly observed in immune and inflammatory cells, including monocytes, macrophages, and neutrophils.47 PAMPs or DAMPs activate PRRs, leading to the initiation of inflammatory response, which secretes pro-inflammatory factors, including IL-1β and IL-18.49 DAMPs-induced inflammation regulates inflammatory diseases.50 Inflammasomes are multi-molecular complexes containing PRRs family members, including TLRs, NLRs, and AIMs like receptors.51,52 Under several intracellular and extracellular stimulations, inflammasomes regulate the molecular signal platform to trigger caspase-1, leading to the stimulation of cell death and inflammatory responses.51 Mounting research has demonstrated several stimuli, including bacterial flagellin, lethal anthrax toxin, double-stranded DNA activates NLRs (NLRP4, AIM2, NLRP1, and Pyrin), which trigger the canonical inflammasome pathways.53–56 Then the canonical inflammasomes covert pro-caspase-1 to caspase-1, which stimulates GSDMD protein molecule. GSDMD induces pores formation on the cell membrane and secretes the inflammatory cytokines, including IL-1β, IL-18, and HGRB1, leading to pyroptosis-mediated cell death initiation.57,58 Inflammasomes, including NLRP1, NLRP3, NLRP4, AIM2, and pyrin, can form inflammasome complex with the adaptor protein ASC to caspase initiation and recruitment domain ASC and stimulate caspase-1.35,59,60 NLRP3 inflammasome components include sensor molecules, such as NLRP3, ASC, and pro-caspase-1.52,61
Extensive studies have shown the following two steps activate the NLRP3 inflammasome. The first is regarded as the initiation phage, and the second one is the inflammasomes initiation phage.62,63 The initiation phage triggers NLRP3 and pro-IL-1β and IL-18 expression in response to several microbial molecules or endogenous factors. Accumulating evidence suggests that three types of stimulators activate the NLRP3 inflammasome.59,62,64,65 ROS produced by mitochondria activates the NLRP3 inflammasome.66 In addition, lysosomal membrane disruption and lysosomal devastation can induce NLRP3 inflammasome. Growing evidence reports that phagocytosis of particles, including crystals, devastates the lysosomal membrane.67 Besides, ion efflux, such as K+ efflux and Ca2+ mobilization, promotes NLRP3 inflammasome activation.68
Current reports have documented that miRNAs stimulate the NLRP3 inflammasome.69,70 MiRNAs are commonly regarded as endogenous single-stranded non-coding RNA molecules involving 21–23 nucleotides and regulate RNA silencing.71,72 In addition, further research indicates that miRNAs induce the NLRP3-mediated pyroptosis signaling pathway.73
Non-Classical Inflammasome Signaling Pathways
In-classical inflammasome signaling pathways, LPS activates pro-caspase-4/5/11 (Figure 1).74 Then, the pro-caspase-4/5/11 initiates pyroptosis-mediated cell death.75 Caspase-4/5/11 binds to LPS and can cleave the 53-kDa precursor form of GSDMD protein molecule to form the N-terminal of mature GSDMD p30 fragment, leading to pore formation on the cell membrane.76 Caspase-4/5/11 also stimulates NLRP3-mediated caspase-1 initiation by the cleaves of GSDMD and secretion of IL-1β/IL-18.75 Previous research has demonstrated that caspase-11 stimulates non-canonical caspase-1-mediated IL-1β and IL-18 pro-inflammatory factors secretion with NLRP3 and ASC inflammasome, indicating caspase-4/5/11 triggers the non-classical NLRP3 inflammasome under several microbial-induced LPS stimulations.77,78 Surprisingly, activated caspase-4/5/11 can cleave pannexin-11 to promote potassium efflux and ATP release. Then, the potassium ion efflux activates NLRP3-mediated pyroptotic cell death. Simultaneously, ATP-gated P2X7 receptors are initiated by new stimulators, inducing pore formation on the plasma membrane. Russo et al have reported that the non-canonical inflammasome triggers several secretory pathways, which play a significant role in Gram-negative bacterial infection.79 Growing research has indicated that ROS can induce the non-canonical NLRP3 inflammasome by the JNK signaling pathway promoting C. rodentium infection-mediated colitis.80 These findings strongly suggest that the non-canonical inflammasome signaling pathway’s activation plays a critical role in multiple diseases, including cardiovascular, renal, spinal cord injury, inflammatory, and infectious diseases.
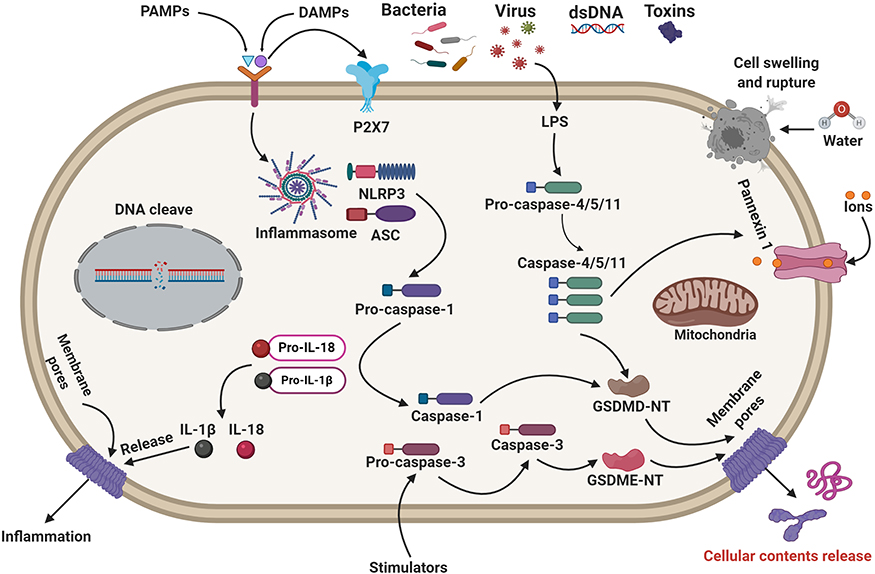 |
Figure 1 Molecular mechanisms of pyroptosis-regulated cell death. Pyroptosis-signaling pathways are generally activated by the stimulation of DAMPs or PAMPs triggering several inflammasomes. The stimulated inflammasome components trigger the cleavage of caspase-1. Caspase-1 can significantly cleave GSDMD to form GSDMD N-fragment and plasma membrane pores formation, leading to pyroptosis-regulated cell death. Stimulated caspase-1 leads to the maturation and secretion of IL-1β and IL-18 inflammatory factors. Besides, LPS binds to the caspase-4/5/11 precursor, leading to the activation of pyroptosis-regulated cell death. Caspase-3/GSDME can also lead to pyroptosis-regulated cell death. Notably, mitochondrial and death receptor pathways can trigger caspase-3. Then, the activated caspase-3 cleaves GSDME to form GSDME N-fragments, leading to plasma membrane pores formation, cell swelling, and pyroptosis. |
Other Signaling Pathways
Recently, emerging studies have reported that caspase-3 activation can cleave GSDME to promote a necrotic GSDME-N portion, causing pore formation on the plasma membrane.81 Particularly, caspase-3 can cleave GSDME protein molecules into N-terminal and C-terminal fragments. The N-terminal part of GSDME (GSDME-N) is generally similar to the GSDMD-N component, can stimulate pyroptosis-regulated cell death. It has been traditionally believed that caspase-3 promotes apoptosis-mediated cell death mechanisms.82 Moreover, the formation of the gasdermin-mediated pores promotes mitochondrial apoptotic pathways to trigger cytochrome c release. Notably, GSDME-N permeabilizes the mitochondrial membrane, which releases cytochrome c and initiates the apoptosome, indicating GSDMD-N has a similar activity.82 Intriguingly, Jing et al suggest that caspase-1, 3, and 7 can also cleave GSDME, leading to pyroptosis.83 This finding indicates that caspase-1, 3, and 7-induced GSDME can create a prospective investigation of the pyroptosis-signaling pathways. A current study has documented that caspase-8 activates the NLRP3 inflammasome, demonstrating caspase-8 can also induce pyroptosis.84 Recently, Oring and co-workers claimed that the tak-1 kinase suppression by the Yersinia effector protein molecule yopj induced caspase-8-mediated pyroptosis, illustrating caspase-8 could be another initiator of pyroptotic cell death.84 The authors also showed that caspase-8 activation could significantly lead to GSDMD-mediated pores formation on the plasma membrane. In addition, Mandal et al have demonstrated that caspase-8 activation triggers an inflammatory response.85
Role of Pyroptosis and Inflammasomes in the Development of Diabetes and Its Complications
The innate immune system plays a critical role in diverse pathological situations, such as autoimmune diseases. However, T1DM is an autoimmune disorder described by the destruction of the β-cell functions and reducing insulin levels. Insulin resistance and disrupted insulin secretion from pancreatic β-cells are the critical pathological conditions of T2DM. Growing research has reported that pyroptosis in β-cells promotes insulin resistance pathogenesis.86–88 In addition, pyroptosis plays a crucial role in developing obesity-associated inflammation and insulin resistance.89 Recently, Ma et al also noted that the activation of pyroptosis-regulated cell death promotes β-cell destruction.90 Carlos et al have indicated that mitochondrial DNA stimulates the NLRP3 inflammasome and predisposes T1D in a murine model.91 The authors showed that diabetic mice elevated NLRP3 and pro-IL-1β mRNA expression levels in pancreatic lymph nodes. The NLRP3, ASC, and pro-IL-1β mRNA expression levels were increased by multiple low doses of STZ administration in the C57BL/6 mice model.91 Elevated IL-1β expression levels at day 7 and day 15 were notably found in the pancreatic tissue of diabetic mice. Furthermore, after STZ treatment, diabetic mice demonstrated the enhanced positive caspase-1 macrophages in the PLNs, which were remarkably downregulated in the NLRP3-gene knockout mice model. Interestingly, NLRP3- and IL-1R-gene knockout mice could tremendously improve insulin levels and downregulate hyperglycemia. NLRP3-mediated pyroptosis promotes the pathogenesis and progression of T1D.91 Recently, emerging research has reported that NLRP3 inflammasome activation promotes T2D progression and insulin resistance.92,93 Kursawe et al suggest that elevated NLRP3 inflammasome promotes inflammation in adipose tissues and reduces insulin sensitivity.94 Previous studies have demonstrated the down-regulation of the NLRP3 inflammasome can considerably attenuate or prevent diet-treated insulin resistance and metabolic abnormalities.95,96 Kim et al have documented that overexpression of the NLRP3 inflammasome facilitates the progression of T2D in db/db mice.97 Downregulation of the NLRP3 inflammasome can reduce immune cell accumulation to the white adipose tissue and preserve insulin sensitivity. The potential role of NLRP3 inflammasome in the pathogenesis of insulin resistance was verified in human studies.98,99 TXNIP activates the caspase-1 inflammasome and regulates innate immune responses in pancreatic β-cell and macrophages.100 Recent reports suggest that diabetes-mediated pyroptosis and inflammation are associated with myeloid or no-myeloid cells, which play a significant role in diabetic complications.101 Muller cells seem to be a prime candidate for this form of inflammation-driven cell death. Feenstra et al have documented that hyperglycemia leads to Muller cell death by introducing pyroptosis-like mechanisms.102 Therefore, these findings indicate that pyroptosis-regulated cell death plays a crucial role in the pathogenesis and development of diabetes and its complications.
Diabetic Cardiomyopathy
Diabetes cardiomyopathy (DCM) is a crucial complication of long-term chronic diabetes mellitus (DM) and is described by myocardial inflammation, fibrosis, and myocardial hypertrophy.103 Growing evidence reports that DCM is closely related to pyroptosis associated with inflammation.104,105 Pyroptosis-regulated cell death plays a significant role in DCM development. Previous research has documented that pyroptosis-related proteins, including GSDMD, caspase-1, NLRP3 inflammasome, and IL-1β expression levels are augmented in cardiac tissues.106 Furthermore, immunohistochemistry staining reported that GSDMD-N, NLRP3, caspase-1, and IL-1β expression levels were also augmented in diabetic mice models’ cardiac tissues. A prior study also reported that NLRP3, caspase-1, and IL-1β mRNA expression levels were substantially upregulated in the DM group.107 Fu et al have documented that lncRNA Kcnq1ot1 regulates pyroptosis in the diabetic heart tissues.106 Western blotting, qRT-PCR, and immunofluorescence staining confirm that kcnq1ot1 and pyroptosis are initiated in HG-stimulated cardiac fibroblasts. Luo et al have claimed the increased NLRP3, caspase-1, and IL-1β expressions are closely associated with the pathogenesis of DCM progression.108 Increasing research reports that non-coding RNAs (ncRNAs) play a significant role in DCM progression.109 Non-coding RNAs can induce pyroptosis in DCM.110,111 Increased caspase-1 expression promotes DN progression via regulating miR-214- 3p expression in cardiac fibroblasts. The authors transfected cardiac fibroblasts with si-Kcnq1ot1 with or without AMO-214-3p in HG conditions and found that HG promotes NLRP3 and IL-1β expression levels.106 Intriguingly, co-transfection with siKcnq1ot1 and AMO-214-3p could promote NLRP3 and IL-1β expression levels. Yang et al identified increased Kcnq1ot1 expression levels in diabetic patients, HG-stimulated cardiomyocytes, and diabetic cardiac tissues.111 Silencing Kcnq1ot1 alleviates pyroptosis by targeting miR-214-3p and attenuating caspase-1 activation. Besides, silencing Kcnq1ot1 can significantly hinder cell death, cytoskeletal structure abnormalities, and higher calcium levels in vitro and improve cardiac functions and histopathological scores in the diabetic animal model. Increasing evidence reports that pyroptosis facilitates myocardial inflammation in DCM.107 Wei et al have documented that hyperglycemia promotes cardiomyocyte pyroptosis via activating the AMPK-TXNIP signaling pathway in a high-fat diet-treated T2D mice model.112 TXNIP overexpression leads to pyroptosis-mediated cardiomyocyte cell death.113 Therefore, TXNIP activation can play a significant role in the development of DCM. Prior research has reported that upregulation of the mir-30d can significantly promote pyroptosis in cardiomyocytes by enhancing caspase-1, IL-1β, and IL-18 expression levels.114 The fundamental role of miRNAs regulating pyroptosis in DCM remains unknown. The authors evaluated the regulatory role of microRNA-30d on cardiomyocyte pyroptosis by directly targeting FoxO3a in STZ-induced diabetic rats and HG-stimulated cardiomyocytes.114 Cao et al have noted that NLRP3 inflammasome and pyroptosis promote HG-treated H9C2 cardiac cell injury.115 The author and colleagues have recently documented that NLRP3 inflammasome-dependent pyroptosis promotes the pathogenesis of non-ischemic DCM. Prior research has also reported that pyroptosis promotes DCM progression in DIC-induced cardiac dysfunction.116 The inflammasome markers, including TLR4 and NLRP3 inflammasome, pyroptotic indicators including caspase-1, IL1-β, IL-18, and cell signaling proteins p-JNK, p-P38, and MyD88, M1 macrophages, and TNF-α mediator are significantly augmented in DCM experimental mice model.116 However, spleen tyrosine kinase-stimulated JNK-dependent NLRP3 inflammasome promotes DCM progression.117 Therefore, inflammasome-mediated cell death is a key player in the pathogenesis of DCM progression. Dargani et al have documented that pyroptotic indicators, such as caspase-1, IL-1β, caspase-11, and GSDMD-N expression levels are markedly higher in doxorubicin (Dox)-induced H9c2 cell.118 Macrophage-derived miR-155-containing exosomes can increase cardiomyocyte pyroptosis and uremic cardiomyopathy changes, such as cardiac fibrosis and hypertrophy by directly targeting FoxO3a in uremic mice. Wang et al have shown that AIM2 expression levels are elevated in diabetic heart tissues than in the control rats group.119 The authors have also shown that AIM2-gene silencing attenuates DCM progression in the T2D rat model. AIM2 promotes cell death and fibrosis via GSDMD pathway activation in HG-stimulated and ROS-mediated DCM.119 A recent study suggests that pyroptosis facilitates DCM progression via enhancing caspase-1 and NLRP3 inflammasome and IL-1β pro-inflammatory mediator release.107 Kar et al have documented that pyroptosis-mediated cell death can trigger cardiac dysfunctions in high-fat diet-treated diabetic C57BL6 mice.120 Therefore, these reports illustrate that pyroptosis plays a significant role in contributing to the pathogenesis of DCM. Further exploring the essential role of pyroptosis in DCM can create a novel therapeutic avenue for treating DCM in the future.
Diabetic Nephropathy (DN)
Diabetic nephropathy (DN) is a severe complication of DM and is regarded as a sterile inflammatory disease. However, growing research suggests that pyroptosis and subsequent inflammation play a crucial role in DN progression.121 The exact molecular mechanism of pyroptosis in DN remains elusive. Zhang et al claimed that pyroptosis-regulated cell death facilitates DN’s pathogenesis and progression.122 Previous studies also documented that pyroptosis-related proteins, including caspase-1, NLRP3, and IL-1β expression levels, were considerably augmented in the STZ-treated diabetic rats.123 The authors claimed that pyroptosis is another key player to DN progression via activating the NLRP3 inflammasome.123 NLRP3 inflammasome activation promotes DN development in the diabetic mice model.124 Shahzad et al have demonstrated that caspase-1-deficiency could hinder inflammasome activation and protect against DN in diabetic mice model, indicating pyroptosis contributes to DN progression. A recent study has documented that pyroptosis-associated proteins, including IL-1β, caspase-1, GSDMD, and NLRP3 inflammasome expression levels are elevated in HFD/STZ diabetic mice model.125 Numerous investigations have also reported that the NLRP3 inflammasome senses and recognizes endogenous risk signals, leading to the initiation of caspase-1 and IL-1β, which promotes the inflammatory cascade induced-DN progression.124 Immunofluorescence findings illustrated that pyroptosis-associated proteins, including GSDMD, caspase-1, and NLRP3 expression levels, were augmented in diabetic renal tissues.125 The authors evaluated the possible role of NLRP3 inflammasome and pyroptosis-signaling pathways in DN.125 The Western blotting analysis showed that pyroptosis-associated proteins, such as GSDMD-N, NLRP3, cleave-caspase-1, p-NF-κB, ASC, and cleaved-IL-1β expression levels were tremendously augmented in high-fat diet/STZ diabetic mice and HG-induced podocytes.126 In addition, the TUNEL assay and cell viability test confirm that pyroptosis-regulated cell death promotes DN progression. Extensive studies show that podocyte loss promotes DN progression.127,128 Cheng et al have reported that podocytes are nearly associated with pyroptosis in the diabetic kidneys.122 The author and colleagues have demonstrated that the caspase-11 and GSDMD-N expression levels in podocytes are augmented, escorted by decreased expression of the podocyte markers nephrin and podocin, loss and fusion in podocyte foot processes, promoted pro-inflammatory factors NF-κB, IL-1β, and IL-18, macrophage accumulation, glomerular matrix expansion and higher urinary albumin to creatinine ratio (UACR). Podocyte pyroptosis is characterized by inflammasome activation, which plays a crucial role in DN progression. These findings strongly indicate that pyroptosis and inflammasome activation play significant roles in podocyte loss, eventually leading to the pathogenesis of DN progression. Ding et al have recently documented that miR-21-5p in macrophage-derived EVs promote pyroptosis in podocytes by regulating A20 in DN.129 The authors investigated whether miR-21-5p in macrophage-derived EVs could largely impact podocyte injury.129 Flow cytometry and the Western blotting analysis indicated that pyroptosis-mediated cell death is involved in DN progression.129 Intriguingly, caspase-11/GSDMD gene knockout can prevent pyroptosis-regulated cell death.122 Furthermore, the Western blotting analysis demonstrated that caspase-11, caspase-4, GSDMD-N, NF-κB, IL-1β, and IL-18 protein expression levels were considerably upregulated in HG-stimulated human and mouse podocytes.122 Caspase-4 or GSDMD knockdown by siRNA can considerably prevent notable changes.122 These findings report that caspase-4/11 and GSDMD-dependent pyroptosis under hyperglycemia promotes podocyte loss and DN progression. With DN research progression, the Trx-TXNIP signaling system’s potential role is progressively acknowledged.130 Current research has reported that TXNIP and inflammasome-related pathway activation promotes DN progression and the complex of NLRP3, and TXNIP regulates the formation of NOX4-mediated NLRP3 inflammasome in hyperhomocysteinemia-stimulated glomerular damages.131,132 Accumulating studies report that NLRP3 inflammasome activation via TXNIP overexpression leads to DN progression (Figure 2).133 The author and colleagues demonstrated severe metabolic abnormalities, disorganized ultrastructure, renal inflammation, fibrosis, cell death, and excessive stimulation of NLRP3, ASC, pro-caspase-1, activated caspase-1, and mature IL-1β were noticeably found in diabetic rats. The author also showed the caspase-1-mediated pyroptotic cell death in vitro experiment. Long non-coding RNA MALAT1 has also been documented to be contributed to DN progression.134 The authors evaluated the potential role of MALAT1 and miR-23c and its target gene ELAVL1 in renal tubular epithelial cells. They showed that MALAT1 expression was significantly enhanced, but miR-23c expression was lower levels in STZ-treated diabetic rats and HG-stimulated HK-2 cells.135 Upregulation of MALAT1 or downregulation of miR-23c expression could significantly hinder pyroptosis in HK-2 cells.135 Further research has documented that LncRNA MALAT1 can promote HG-stimulated pyroptosis in renal tubular epithelial cell by sponging miR-30c and targeting NLRP3 inflammasome.136 The authors revealed the downregulated MALAT1 and miR-30c expression levels in HG-induced HK-2 cells, leading to the activation of NLRP3 inflammasome and pyroptosis.136 Intriguingly, MALAT1 knockdown or miR-30c overexpression could protect HK-2 cells from HG-stimulate pyroptosis.136 In contrast, the authors found MALAT1 expression could significantly promote the NLRP3 inflammasome by sponging miR-30c through dual-luciferase reporter assay. Furthermore, sh-MALAT1 and miR-30c inhibitor’s co-transfection could considerably invert the protective effects of the sh-MALAT1 on HG-stimulated pyroptosis. These notable findings verified that MALAT1 stimulated HK-2 cell pyroptosis by suppressing miR-30c and targeting NLRP3 inflammasome, facilitating to a better understanding of DN pathogenesis. Zhu et al have reported that lncRNA KCNQ1OT1 promotes oxidative stress and pyroptosis in HG-treated renal tubular epithelial cells.137 The authors have performed RT-qPCR analysis to determine the KCNQ1OT1 expression levels in serum with DN and HG-treated HK-2 cells, analyze the GSDMD-N, NLRP3, cleaved-caspase-1, pro-caspase-1, IL-1β, and p-IL-1β expression in HG-treated HK-2 cells, and verify the transfection effects. ELISA analysis detected the increased TNF-α, IL-6, and MCP-1 serum levels. These findings indicate that pyroptosis-mediated inflammation promotes DN pathogenesis. Song et al have reported long non-coding RNA LINC00339 can significantly facilitate renal tubular epithelial pyroptosis by regulating the miR-22-3p/NLRP3 signaling axis in calcium oxalate-stimulated diabetic kidney stone.138 The author and colleagues determined the pyroptosis-regulated cell death by lactate dehydrogenase release and active caspase-1-propidium iodide double staining.138 They performed luciferase reporter to confirm whether miR-22-3p could bind to LINC00339 or NLRP3 inflammasome. The authors reported enhanced LINC00339, downregulated miR-22-3p, NLRP3 inflammasome stimulation, and promoted cell pyroptosis in COM-induced HK-2 cells.138 Furthermore, LINC00339 and NLRP3 overexpression can significantly promote pyroptosis in COM-induced HK-2 cells, whereas miR-22-3p mimics and NLRP3 knockdown provide the contrary effects. These findings suggest that NLRP3 inflammasome mediated pyroptosis death plays a significant role in DCM progression. Qiao et al have established the Syk/JNK/NLRP3 signaling axis might play a crucial role in DN.139 A recent study documented that decreased GAS5 expression could promote TNF-α, IL-6, MCP-1, ROS expression levels in HG-stimulated renal tubular cells.140 Besides, reduced GAS5 expression can significantly promote NLRP3, caspase-1, IL-1β, and GSDMD-N expression levels, and the results of immunofluorescence confirmed the critical findings.140 Notably, the miR-452-5p disruption could promote similar changes as GAS5 overexpression for HG-treated HK-2 cells, and GAS5 suppression could lead to opposite the potential effects of miR-452-5p disruption. Emerging research has reported increased pyroptosis-regulated proteins, including GSDMD, caspase-1, IL-1β, and IL-18 expression and lowered serum levels of IL-1β and IL-18, and enhanced catheter activities B and cathepsin L can promote DN progression in the diabetic SD rats.141
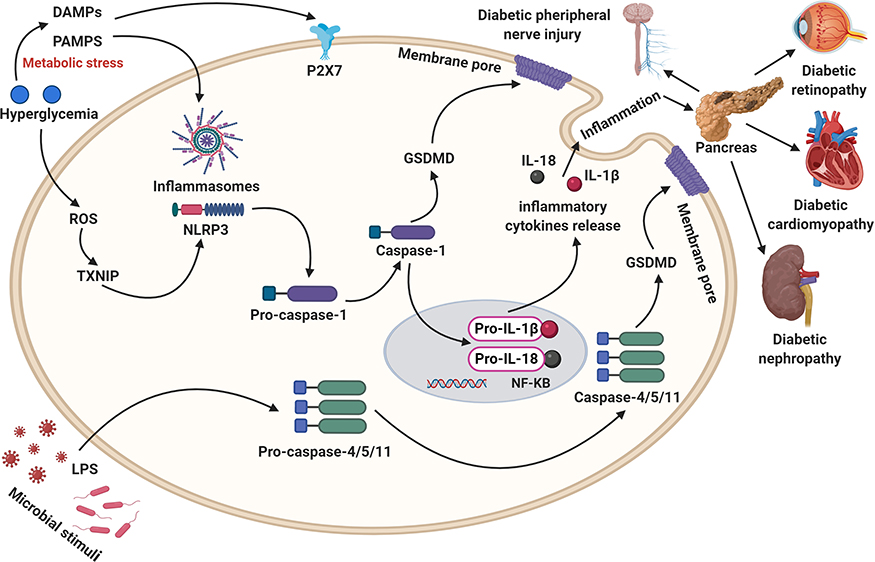 |
Figure 2 Activating the caspase-1/4/5/8/11 signaling pathways, inflammatory factors including IL-1β and IL-18 release, and ROS generation, leading to GSDMD/GSDME-mediated pore formation as well as cell lysis and promote the development of diabetic complications. |
Diabetic Retinopathy
Diabetic retinopathy (DR) is one of the most critical causes of visual damage and accumulated blindness among adults worldwide. Retinal microvascular pericyte deficiency is one of the earliest pathological alterations associated with DR. However, long noncoding RNA MIAT regulates DR’s microvascular dysfunctions. Accumulating research has demonstrated that pyroptosis and inflammasome activation leads to DR progression. The potential role of pyroptosis in retinal pericytes loss in DR remains elusive. Yu et al investigated pyroptosis’s role in primary HRPCs.142 The authors implemented advanced glycation end product modified bovine serum albumin (AGE-BSA) to induce DR progression and demonstrated that AGE-BSA triggered the cleavage of caspase-1 and GSDMD, release of IL-1β, IL-18, and LDH, and decreased cell viability.142 Notably, Immunofluorescence staining indicated pyroptosis’s phenotypic features, such as pyknosis, swelling, cellular disruption, and hyperpermeability in plasmalemma. The authors claimed that pyroptosis plays a critical role in DR progression.142 Loukovaara et al have reported that caspase-1, ASC, and IL-18 expression levels are increased in PDR eyes.143 NLRP3 inflammasome activation can be associated with the pathogenesis of PDR.143 Immunohistochemistry staining and RT-PCR analysis indicate that NLRP3, ASC, and cleaved-caspase-1 expression levels are upregulated in STZ-induced diabetes’ retinas rats.144 These notable findings indicate that inflammasomes-induced pyroptosis promotes DR progression. Previous studies have reported that HG can significantly induce the NLRP3-caspase-1-GSDMD axis and pore formation in the cell membrane in a dose- and time-dependent manner.145 Furthermore, the GSDMD, GSDMD-N, NLRP3, caspase-1, cleaved-caspase-1, and cleaved-IL-1β expression levels were remarkably augmented in lysates of HRPs. Immunofluorescence staining also confirms the caspase-1-dependent pyroptosis promotes human ARPE-19 cell death.146 Plasmids overexpressing human GSDMD-N with a GFP tag or pEGFP-C1 control plasmid were designed to measure the potential role of GSDMD-N overexpressed in HG-induced inflammation and HRPs pyroptosis cell death.145 Immunofluorescence staining indicates increased GSDMD-N expression levels in HRPs. Fluorometric plots also demonstrate PI uptakes over time to show the kinetics of pore appearance in HRPs. These findings indicate that GSDMD-mediated HRPs cell death plays a significant role in DR progression. Chien et al have reported that caspase-1, IL-1β, and IL-18 mRNA expression levels are considerably higher in MCMV-infected eyes of MAIDS-10 mice.147 The authors have also documented that pyroptosis participates simultaneously in MAIDS-related MCMV retinitis. Growing research demonstrates that NLRP3 inflammasome activation promotes bystander cone-photoreceptor cell death in a P23H rhodopsin model of retinal degeneration.148 Elevated NLRP3 and caspase-1, mature IL-1β and IL-18 expression levels, and enhanced cell survival with N-acetylcysteine treatment suggest inflammasome activation leads to cone cell death.148 P2RX7 receptors induce the NLRP3 inflammasome and promote IL-1β and IL-18 secretion, leading to pyroptosis cone cell death.149 P2RX7 receptors respond to local extracellular ATP levels, and it has been reported that P23H retina tissue promotes ATP levels.150,151 Author and colleagues proposed that rod photoreceptors undergoing necroptotic cell death release their cellular components including ATP and HMGB1, which lead to inflammasome activation in cones.148 Caspase-1-mediated pyroptosis triggers photoreceptor cell death in photo-oxidative damage-produced retinal degeneration.152 As a consequence, these outcomes report that P2RX7 plays a significant role in DN progression. Zha et al have documented that down-regulation of METTL3 promotes HG-treated RPE cell pyroptosis by regulating the miR-25-3p/PTEN/Akt signaling axis through DGCR8.153 Therefore, METTL3 regulates the pathogenesis of DR.153 Extensive reports have documented a potential role of pro-inflammatory and pro-apoptotic TXNIP in developing both T1 and T2D and its vascular complications.154,155 TXNIP is highly upregulated in diabetic retinal tissues and HG-treated retinal cell culture. Multiple non-mutually unique mechanisms of inflammasome initiation in DR have been projected.156,157 Hyperglycemia-mediated ROS synthesis promotes NLRP3 inflammasome activation through dissociation of TRX1-TXNIP and aggregation of AGEs and ALEs. Previous studies have also reported that theNLRP3 inflammasome activation via TXNIP overexpression promotes DR progression.156,158 Perrone et al showed that overactivation of TXNIP leads to DR progression.159 TXNIP-induced inflammasome activation promotes diabetic ocular complications.159 In a previous study, authors and colleagues have reported that ROS-TXNIP pathway mediates NLRP3 inflammasome activation promoting HRMECs death.160 Decreased miR-590-3p activates pyroptosis by enhancing the NLRP1 inflammasome and inducing the NOX4/ROS/TXNIP/NLRP3 signaling pathway. These notable findings indicate that inflammasome-induced pyroptosis contributes to the pathogenesis and progression of DR.
Diabetic Peripheral Neuropathy (DPN)
Diabetic peripheral neuropathy (DPN) is a common and severe complication of DM. The early changes in patients with DPN include accumulation of the extracellular matrix proteins, chronic inflammation, axonal degeneration, and loss of the unmyelinated fibers, leading to sensorimotor conduction delays and irreversible nerve damage. Current research has documented that pyroptosis and inflammasome play significant roles in DPN progression.161 However, Sun et al have reported pyroptosis-mediated proteins, including GSDMD, NLRP3, pro-caspase-1, cleaved-caspase-1, and IL-1β expression levels, are substantially higher in STZ-induced diabetic rat peripheral tissues.161 ELISA analysis reported that serum IL-1β and IL-18 levels are tremendously augmented in STZ-treated DPN injured experimental rats.162 Excessive ROS generation and NLRP3 inflammasome activation promote inflammation and pyroptosis in DPN.156,163 Author and colleagues have evaluated the potential effects of mediating TXNIP on pre-diabetic neuropathy and its actual molecular mechanism in HFD-treated diabetic mice and palmitate-stimulated neurons.164 TXNIP activation is commonly associated with pre-diabetic neuropathy in the HFD-fed mice model. Zhou et al demonstrated that TXNIP/NLRP3 inflammasome activation could promote the pathogenesis of diabetes and regulate miR-23a mediated neuropathic pain in spinal glial cells.165 Previous research has also reported that NLRP3 inflammasome activation plays a significant role in DPN progression.164 Cheng et al have reported that Schwann cell pyroptosis further promotes DPN progression.166 The authors evaluated pyroptosis-signaling pathways in HG-treated rat Schwann cell line RSC96. Importantly, they found higher cleaved-GSDMD, NLRP3, P2X7, TXNIP, ASC, caspase-1, IL-1β, and IL-18 expression levels in HG-treated RSC96 cells. Immunofluorescence staining and quantitative RT-PCR confirmed that the pyroptosis-associated proteins are significantly enhanced in HG-stimulated RSC96 cells.166 Furthermore, increased ROS generation and NF-κB nuclear translocation are notably observed in HG-stimulated RSC96 cells. These findings strongly indicate that pyroptosis plays a significant role in DPN development.
Targeting Inflammasome and Pyroptosis for the Therapeutic Implications of Diabetic Complications
Metformin (MET) is an antidiabetic drug widely used for treating T2D. Current studies have documented that MET exerts powerful anti-aging, anti-tumor, and cardioprotective effects.167,168 However, the actual mechanism of MET in regulating the NLRP3 inflammasome and pyroptosis in DCM remains elusive. Yang et al have demonstrated that MET administration can reduce mTOR, NLRP3, caspase-1, GSDMD-N, and IL-1β protein expression levels in STZ-induced diabetic C57BL/6 mice models and HG-treated primary cardiomyocytes from neonatal mice.169 Immunohistochemistry staining shows the NLRP3, caspase-1, and IL-1β protein expression levels are considerably lower in the MET-treated DCM mice model.169 In addition, immunofluorescence staining reports that MET treatment could hinder GSDMD- mediated pyroptosis in HG-treated primary cardiomyocytes. MET treatment could hinder the NLRP3 inflammasome activation via promoting the AMPK/mTOR signaling pathway in diabetic mice model. Besides, MET treatment can induce AMPK, thus promoting autophagy via hindering the mTOR signaling pathway and suppressing pyroptosis in DCM. A previous study has suggested that Ac-YVAD-CMK, BAY11-7082, or NAC attenuate HG- and H/R-stimulated H9C2 cell injury and suppress NLRP3 inflammasome-dependent pyroptosis.170 Suppression of the NLRP3 inflammasome and ROS synthesis attenuates cell injury and pyroptosis in LPS-stimulated H9C2 cells under HG and H/R conditions.170 BAY11-7082 and NAC treatment can hinder NLRP3 inflammasome by decreasing the NLRP3, ASC, and NF-κB p65 protein expression levels in LPS and HG+H/R-stimulated H9C2 cells. In addition, Ac-YVAD-CMK and BAY11-7082 treatment can promote cell viability. These findings suggest that Ac-YVAD-CMK and BAY11-7082 can suppress pyroptosis for the novel therapeutic approach in treating DCM. Jeyabal et al have reported that treatment with miR-9 mimics can hinder hyperglycemia-stimulated ELAVL1 and suppress pyroptosis in cardiomyocytes.171 MicroRNA-9 suppresses HG-stimulated pyroptosis by targeting ELAVL1 in human ventricular cardiomyocytes.171 ELAVL1 knockdown also suppresses the canonical inflammasome pathway and pyroptotic cell death in mouse macrophage RAW 264.7 cells.171 MiRNAs are small endogenous non-coding RNAs that contribute to DCM progression. The knockdown of miR-30d can significantly suppress cardiomyocyte pyroptosis in DCM.114 The author and colleagues suggest that mir-30d may be a potential therapeutic target in treating DCM. RNA Kcnq1ot1 promotes several pathophysiological mechanisms of multiple diseases, including arrhythmia and acute myocardial damage. The potential role of Kcnq1ot1 regulating pyroptosis in HG-induced cardiac fibroblasts remains unclear. Kcnq1ot1 silencing by a lentivirus-shRNA can promote cardiac function and inhibit pyroptosis-signaling pathways in the STZ-induced C57BL/6 mice models.106 Growing evidence suggests that NLRP3-gene silencing ameliorates cardiac inflammation and pyroptosis in the STZ-induced T2D rat model.172 Furthermore, silencing of the NLRP3 inflammasome suppresses cardiomyocyte pyroptosis in HG-treated H9c2. Interestingly, ROS inhibition can reduce NF-kB phosphorylation, TXNIP, NLRP3 inflammasome, and mature IL-1β in HG-stimulated H9c2 cells. Importantly, NF-kB inhibition attenuates the NLRP3 inflammasome activation. TXNIP-siRNA can significantly reduce the stimulation of caspase-1 and IL-1β.172 Wei et al have reported that the TXNIP-gene silencing reduces the NLRP3 inflammasome expression and inhibits caspase-1 activation and IL-1β secretion in HG-stimulated cardiomyocytes.112 Therefore, inhibiting the TXNIP-induced NLRP3 inflammasome could be a novel therapeutic target in treating DCM in the future. AIM2-gene silencing alleviates cardiac dysfunctions, promoting a metabolic disorder and ventricular remodeling in the STZ-induced diabetic rat model.119 AIM2-siRNA suppresses GSDMD-N-associated pyroptosis in H9c2 cardiomyoblasts.119 Xie et al have reported that the silencing of CMKLR1 in DCM rats attenuates the NLRP3 and caspase-1 and IL-1β protein expression levels.107 The authors have demonstrated that the silencing of either CMKLR1 or NLRP3 can significantly suppress caspase-1 and IL-1β expression levels. These findings show that CMKLR1-gene silencing can have a protective effect against DCM. The emerging study also reported that knockdown of the CACR in cardiomyocytes opposed HG-stimulated caspase-1 activation.173 Intriguingly, miR-214-3p knockdown could partly abolish the favorable outcomes of CACR silencing on pyroptosis in cardiomyocytes.173 These important findings suggest that CACR could be a promising therapeutic avenue via CACR/miR-214-3p/caspase-1 pathways in DCM. Exendin-4 is considered a GLP analog and has a prolonged half-life than GLP-1 by shirking removal through DPP-4. Exendin-4 is a potent pyroptotic blocker preserving against hyperglycemia-stimulated cardiomyocyte pyroptosis via regulating the AMPK-TXNIP signaling axis.112 Ye et al reported that inhibition of the SGLT-2 with dapagliflozin could reduce the NLRP3/ASC inflammasome and attenuate DCM progression in the T2D diabetic mice model (Table 1).174 The authors also reported that inhibition of the SGLT-2 with dapagliflozin could suppress the NLRP3, caspase-1, ASC, IL-1β, IL-18, and TNF-α mRNA expression levels in cardiac tissues of the T2D mice model. Li et al have shown that Klotho can improve DCM by inhibiting the NLRP3 inflammasome.175 The authors documented that Klotho’s protective effect on diabetes-mediated cardiac injury is intensively associated with the suppression of NLRP3 inflammasome, recommending its potential therapeutic effects in treating DCM. Zhang et al have suggested that H3 relaxin treatment for two weeks can protect against myocardial injury in experimental DCM by suppressing the NLRP3 inflammasome, IL-1β, and IL-18 expression levels.176 Thus, this study recommends that H3 relaxin could be a novel therapeutic agent for DCM treatment. Growing research shows that vaspin ameliorates myocardial injury in rats’ DCM model by suppressing the NLRP3 inflammasome and reducing the cleavage of caspase-1 and maturation of IL-1β in STZ-induced diabetic cardiac tissues and HG-stimulated H9C2cells.177 Gypenosides are the major ingredients of Gynostemma pentaphyllam that have exerted significant anti-hyperglycemia and anti-inflammation effects. Zhang et al have reported that Gypenosides treatment can improve DCM by hindering ROS-mediated NLRP3 inflammasome activation.178 Previous studies have documented that MSCs exert substantial cardioprotective effects in CVB3-stimulated myocarditis. MSCs can significantly hinder NLRP3 inflammasome in CVB3-triggered inflammatory DCM.179 Rosuvastatin alleviates DCM by attenuating the NLRP3 inflammasome and MAPK pathway in a T2D rat model.108 These findings suggest that inhibiting the pyroptosis and inflammasome factors might be a promising therapeutic avenue in treating DCM.
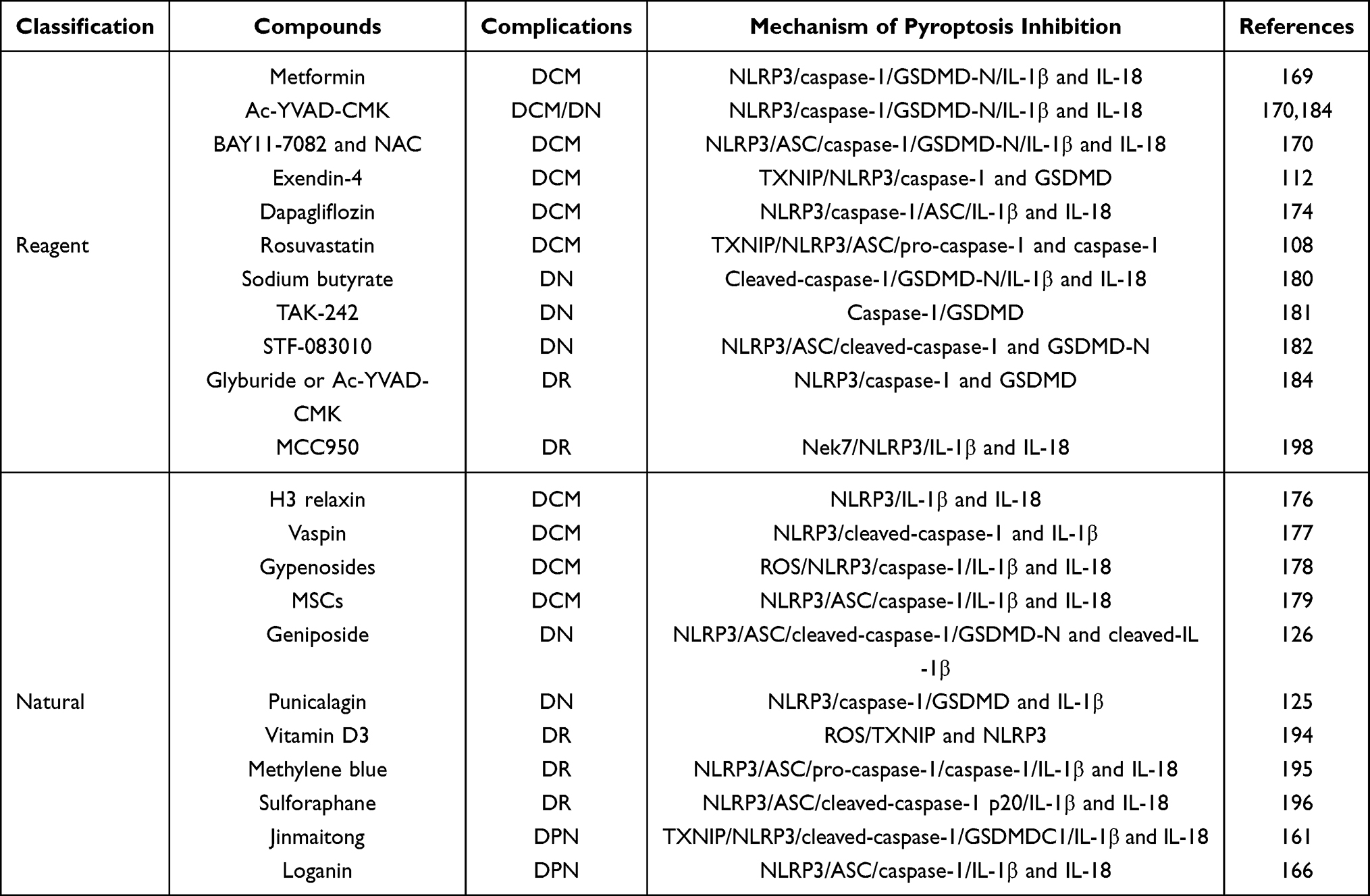 |
Table 1 Compounds Inhibiting Pyroptosis-Signal Pathway to Treat and Manage Diabetic Complications |
Sodium butyrate (NaB) has potential anti-inflammatory activities in the DN mice model and HG-stimulated mouse glomerular mesangial cells. Gu et al performed an investigation to explore the possible therapeutic effects of NaB regulating pyroptosis in HG-treated renal glomerular endothelial cells (GECs).180 Exogenous NaB administration can suppress the pyroptosis-associated proteins, including GSDMD-N, caspase-1, and IL-18 expression levels in HG-induced GECs. Furthermore, caspase-1 blocker Ac-YVAD-CMK or GSDMD-gene silencing could significantly attenuate DN progression. The authors also indicated that PI-positive cells were further promoted by hindering the caspase-1 and GSDMD pathway. Therefore, suppression of the pyroptosis-regulated cell death attenuates DN progression. Blocking the TLR4 prevents tubular cell injury by attenuating GSDMD-mediated pyroptosis in db/db mice model.181 TLR4 or NF-κB inhibition reduces GSDMD-NT, p-p65, and caspase-1 p20 expression levels in HG-treated HK-2 cells.181 This study suggests that suppressing the pyroptosis-mediated cell death could be a novel therapeutic strategy for DN treatment. Accumulating results report that Geniposide (GE) can effectively suppress DN progression. Li et al have demonstrated that GE can significantly alleviate DN by suppressing pyroptosis-corrected proteins, such as p-NF-κB, NLRP3, and ASC cleaved-caspase-1, GSDMD-N, and cleaved-IL-1β expression levels in STZ-treated DN mice and HG-induced podocyte injury models.126 GE alleviates renal dysfunctions by attenuating BUN, Scr, TNF-α, IL-1β, and IL-6 expression levels. Histological findings indicate GE administration efficaciously attenuates renal damages, including glomerular basement membrane thickening and inflammatory cell accumulation. These remarkable findings recommend that GE could be a prospective therapeutic molecule targeting pyroptosis-signaling pathways for DN treatment. TUNEL staining indicates that administration of the TLR4 blocker TAK-242 can significantly suppress tubular cell death in the kidneys of diabetic mice model.181 These crucial findings suggest that the TLR4/NF-κB signaling-pathway inhibition could be a promising therapeutic avenue to prevent and treat DN. A recent study has also reported that inhibiting TXNIP can attenuate NLRP3, ASC, cleaved-caspase-1, and GSDMD-N expression levels in DN rats.182 TUNNEL staining indicates that the ER-stress induced TXNIP molecule inhibition can tremendously reduce NRK-52E cell death.182 The current study also suggests that IRE1-α inhibitor STF-083010 treatment can inhibit pyroptosis-mediated renal tubular epithelial cell death. These findings suggest that inhibiting the ER-mediated pyroptosis could be another novel therapeutic strategy for managing and treating DN in the near future. Punicalagin (PU) has substantial anti-inflammatory properties and can alleviate DN progression. The Western blotting analysis has confirmed that PU administration can tremendously inhibit NLRP3, GSDMD, and caspase-1 expression levels in diabetic mice models.125 PU administration can also inhibit the IL-1β inflammatory factor secretion in the kidney tissues of HFD/STZ-treated diabetic animals. Suppressing the pyroptosis-mediated inflammatory response plays a vital role in the protective effects of PU. These findings strongly conclude that the TXNIP/NLRP3 signaling axis is a crucial player promoting DN progression, and PU can be a potential therapeutic agent in treating DN. Previous studies have shown that caspase-4/11 or GSDMD knockdown by siRNA can considerably attenuate DN progression.122 lncRNAs improve DN by changing miRNAs expression. Xie et al demonstrated that GAS5 overexpression could considerably attenuate GSDMD-N, NLRP3, caspase-1, and IL-1β expression levels.140 Immunofluorescence also verified GAS5 overexpression could significantly decrease that pyroptosis-related protein expression levels. Wen et al have reported that circACTR2 knockdown inhibits pyroptosis and reduces IL-1β secretion in HG-stimulated fibrosis in renal tubular cells.183 The authors have detected a new circRNA, circACTR2, that modulates HG-stimulated fibrosis, inflammation, and pyroptosis in proximal tubular cells. Therefore, targeting the circRNAs can serve unique insight regulating pyroptosis into DN’s pathogenesis and create a novel therapeutic strategy in the future.
Recent research has reported that caspase-1 blocker YVAD or NLRP3 blocker glyburide can significantly suppress GSDMD, NLRP3, caspase-1 protein expression levels in retinal pericytes.184 Notably, the silencing of GSDMD can also inhibit HG-induced retinal pericytes. Studies have documented that miRNAs play a significant role in the pathogenesis of DR development by regulating cell proliferation, migration, and death.185–187 Gu et al have established miR-590-3 to suppress pyroptosis in DR by targeting the NLRP1 inflammasome and inhibiting the NOX4 signaling pathway in HRMECs.188 The author and colleagues have demonstrated that the miR-590-3p overexpression or NLRP1 and NOX4 knockdown can promote cell activity and suppress HRMECs pyroptosis. Immunofluorescence staining and cell viability assay confirm that miR-590-3p overexpression can significantly suppress caspase-1-dependent pyroptosis in HRMECs. The authors claimed that targeting the miR-590-3 and NOX4/ROS/TXNIP/NLRP3 signaling axis could be a novel therapeutic avenue in treating DR. Previous reports indicate that METTL3 regulates multiple cell functions and diseases by modulating N6-methyladenosine (m6A) modifications.189–191 Upregulation of METTL3 and miR-25-3p can alleviate the cytotoxic effects of HG on RPE cells, and knockdown of METTL3 and miR-25-3p had a contrary impact.153 Furthermore, METTL3 overexpression can promote miR-25-3p expression in RPE cells in a microprocessor protein DGCR8-dependent manner, and miR-25-3p removal abolishes the effects of overexpressed METTL3 on cell functions in HG-induced RPE cells.153 More importantly, METTL3 overexpression can attenuate GSDMD, caspase-1, NLRP3, IL-1β, and IL-18 pyroptosis-related proteins and mRNA expression levels in HG-stimulated RPE cells.153 Flow cytometry and CCK-8 tests confirm that METTL3 overexpression levels can significantly suppress pyroptotic RPE cells in HG conditions. Therefore, METTL3 may help to discover a potential therapeutic gateway for DR treatment in the clinic. Zhang et. reported that the upregulation of miR-214 could remarkably attenuate pyroptosis. KCNQ1OT1 knockdown using a small interfering RNA can reduce pyroptosis-related markers expression.192 The authors suggest that the KCNQ1OT1/miR-214/caspase-1 signaling pathway shows a novel diabetic corneal endothelial keratopathy progression and can be a promising therapeutic target in the future.192 In addition, increasing evidence suggests that vitamin D3 has substantial therapeutic effects in treating DR.193 Vitamin D3 administration can significantly protect against DR by inhibiting the HG-induced ROS/TXNIP/NLRP3 inflammasome signaling pathway.194 Methylene blue (MB) administration can tremendously attenuate DR by impeding the NLRP3 inflammasome activation in STZ-treated diabetic rats.195 Thus, MB treatment can be a potential therapeutic molecule in treating DR. Sulforaphane (SFN) is abundant in cruciferous plants, serving substantial protective effects against multiple chronic diseases. Previous research demonstrated that NLRP3, ASC, cleaved-caspase-1 p20, and IL-1β expression levels were tremendously reduced in SFN-treated diabetic mice retinal tissues.196 MCC950 is considered an effective and definite blocker of the NLRP3 inflammasome.197 Zhang et al have investigated the substantial anti-inflammatory activities of MCC950 on HG-treated HRECs.198 The authors have demonstrated that MCC950 can significantly inhibit Nek7, NLRP3 expression, and IL-1β secretion in HG-induced HRECs. This study suggested that MCC950 could suppress HRECs dysfunction under HG conditions. This research can provide new insights for a future pharmacological therapeutic approach in treating DR in the future. PKR may regulate the NLRP3 inflammasome, and PKR deficiency can downregulate NLRP3, IL-1β, and HMGB1 expression levels in macrophages.199 Jiang et al have documented that Epac1 attenuates PKR to downregulate the NLRP3 inflammasome in HG-induced RECs.200 These shreds of evidence suggest that suppressing the NLRP3 inflammasome could be a promising therapeutic target in treating DR in the near future.
Jinmaitong (JMT) is regarded as a traditional Chinese medicine used in treating multiple diseases.201,202 Extensive studies have reported that JMT has significant clinical efficacy to prevent and treat DPN.202,203 However, Sun et al have documented that JMT treatment can significantly downregulate the GSDMDC1, NLRP3, TXNIP, cleaved-caspase-1, IL-1β, and IL-18 expression levels in the STZ-treated rat model.161 Furthermore, immunohistochemical assay indicates that JMT treatment can also hinder TXNIP-induced NLRP3 inflammasome in SNs of diabetic rats. The authors suggest that JMT can be a potential therapeutic candidate targeting pyroptosis in treating and managing DPN. Loganin (LOG) is an iridoid glycoside extracted from the fruit Cornus officinalis. Previous investigations have reported that LOG has substantial antioxidant, anti-inflammatory, neuroprotective effects.204–206 Cheng et al documented that LOG treatment could significantly suppress pyroptosis-related proteins and mRNA expression levels in HG-induced rat Schwann cell line RSC96.166 The authors demonstrated that LOG treatment could suppress Schwann cell pyroptosis by attenuating ROS synthesis and NLRP3 inflammasome. These findings suggest that LOG could be a novel therapeutic agent suppressing pyroptosis and inflammasome to treat DPN.
Conclusion and Future Prospectives
Pyroptosis is considered a pro-inflammatory programmed cell death mechanism. Studies have implicated that pyroptosis plays a crucial role in multiple diseases, such as liver disease, cardiovascular, cancer metastasis, immunological diseases, and CNS disorder. However, the activation of pyroptosis and its actual mechanism remains elusive in diabetes. Extensive reports have documented that pyroptosis plays a critical role in the development of diabetes and its complications. Targeting pyroptosis and inflammasome components has been exceptional attention to the researchers and clinicians. Recent research strongly suggests that some potential molecules can be applied to suppress the pyroptosis and inflammasome signaling pathways for a novel therapeutic target to treat diabetes in the near future. Our narrative review highlighted the significant role of pyroptosis and inflammasome in diabetic complications (Figure 3). Moreover, our article illustrated some potential agents that could be a possible therapeutic target for the management and treatment of diabetes and its complications. The studies on the fundamental role of pyroptosis-mediated cell death in diabetes and its complications are still limited and challenging to experimental and clinical investigations. Further studies are highly essential to investigate and clarify the exact molecular mechanism and the fundamental role of pyroptosis in diabetic complications. Clinical studies of those potential agents targeting the pyroptosis-mediated cell death and inflammasome may create a new window to achieve a possible therapeutic avenue for managing and treating diabetes and its complications in the future.
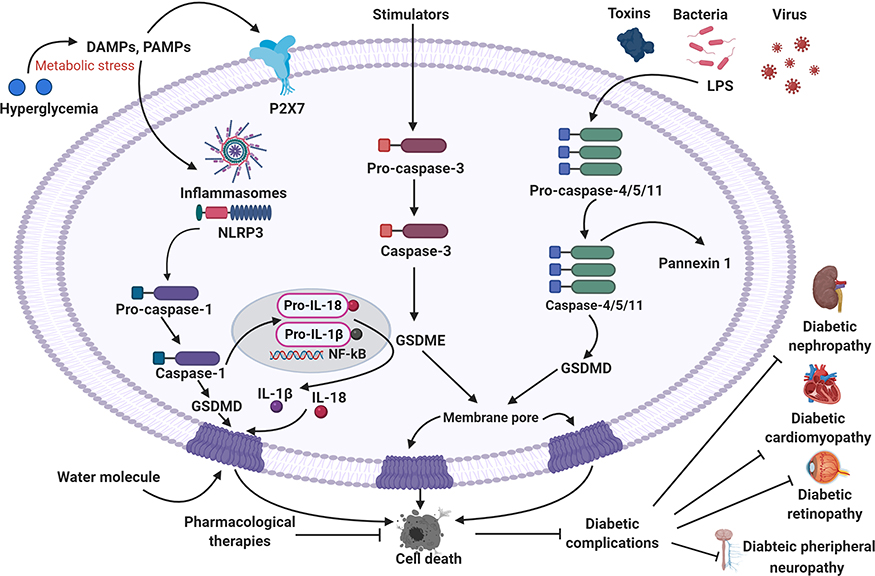 |
Figure 3 Summary of the pyroptosis in the development of diabetic complications and its therapeutic implications. |
Abbreviations
ROS, Reactive oxygen species; AGEs, Advanced glycation end products; ER, Endoplasmic reticulum; PRRs, Pattern recognition receptors; GSDMD, Gasdermin D; PAMPs, Pathogen-associated molecular patterns; DAMPs, Damage-associated molecular patterns; TLRs, Toll-like receptors; NLRs, NOD-like receptors; AIMs, Absent in melanomas; NLRP1, NOD-like receptor protein 1; NLRP3, NOD-like receptor protein 3; ASC, apoptosis-associated speck-like protein; miRNAs, MicroRNAs; HFD, High-fat diet; STZ, Streptozotocin; TXNIP, Thioredoxin-interacting protein; NF-κB, Nuclear factor-kappa B; EVs, Extracellular vesicles; siRNA, Small interfering RNA; MALAT1, Metastasis associated lung adenocarcinoma transcript 1; HRPCs, human retinal progenitor cells; CMKLR1, Chemokine-Like Receptor 1; GLP, Glucagon-like peptide; DPP-4, Dipeptidyl peptidase-4; SGLT-2, Sodium-glucose co-transporter-2; CCK-8, Cell counting kit-8; MCMV, Murine cytomegalovirus; CVB3, coxsackievirus B3; IRE1α, Inositol-requiring enzyme 1 α; MSCs, Mesenchymal stem cells; PKR, Protein kinase R.
Funding
This work was supported by the Natural Science Foundation of China (81972150, 81722028, 81801233); Natural Science Foundation of Zhejiang Province (LR18H50001), Research Unit of Research and Clinical Translation of Cell Growth Factors and Diseases of Chinese Academy of Medical Science (2019RU010).
Disclosure
The authors reported no conflicts of interest for this work.
References
1. de Candia P, Prattichizzo F, Garavelli S, et al. Type 2 diabetes: how much of an autoimmune disease? Front Endocrinol (Lausanne). 2019;10:451. doi:10.3389/fendo.2019.00451
2. Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015;5(1):194–222. doi:10.3390/biom5010194
3. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9(2):119.
4. Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des. 2013;19(32):5695–5703.
5. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–164.
6. Labbé K, Saleh M. Cell death in the host response to infection. Cell Death Differ. 2008;15(9):1339–1349.
7. Demarco B, Chen KW, Broz P. Cross talk between intracellular pathogens and cell death. 2020;297(1):174–193.
8. Coll NS, Epple P, Dangl JL. Programmed cell death in the plant immune system. Cell Death Differ. 2011;18(8):1247–1256.
9. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516.
10. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541.
11. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254.
12. Cheng S-B, Nakashima A, Huber WJ, et al. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019;10(12):927.
13. Jung S, Jeong H, Yu S-W. Autophagy as a decisive process for cell death. Exp Mol Med. 2020;52(6):921–930.
14. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21(7):398–414.
15. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–592.
16. Chen Q, Kang J, Fu C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduction Targeted Ther. 2018;3(1):18.
17. Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157(5):1415–1430.
18. Lall S. Activating apoptosis. Nat Struct Mol Biol. 2009;16(6):614.
19. Ditzel M, Meier P. Apoptosis: more than meets the eye. Cell. 2002;111(4):465–467.
20. Zampieri C, Ganini C, Melino G. The biochemistry of cell death. Cell Death Dis. 2020;11(4):259.
21. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352(6281):aaf2154.
22. Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146.
23. Cai Z, Jitkaew S, Zhao J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65.
24. Chen X, Li W, Ren J, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–121.
25. Dondelinger Y, Declercq W, Montessuit S, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–981.
26. Robinson N, Ganesan R, Hegedűs C, Kovács K, Kufer TA, Virág L. Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019;26:101239.
27. Minton K. Pyroptosis heats tumour immunity. Nat Rev Immunol. 2020;20(5):274–275.
28. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–142.
29. Storek KM, Monack DM. Bacterial recognition pathways that lead to inflammasome activation. Immunol Rev. 2015;265(1):112–129.
30. Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–1142.
31. Jorgensen I, Zhang Y, Krantz BA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. 2016;213(10):2113–2128.
32. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
33. Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature. 1992;356(6372):768–774.
34. Bolívar BE, Vogel TP, Bouchier-Hayes L. Inflammatory caspase regulation: maintaining balance between inflammation and cell death in health and disease. FEBS J. 2019;286(14):2628–2644.
35. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–247.
36. Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012;3(7):e338–e338.
37. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis. Inflammatory Caspases and Inflammasomes in Infectious Diseases. 2017;277(1):61–75.
38. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16(3):319–326.
39. Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem. 1986;261(16):7123–7126.
40. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–169.
41. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114.
42. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206–214.
43. Lee BL, Stowe IB, Gupta A, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. 2018;215(9):2279–2288.
44. Huang X, Feng Y, Xiong G, et al. Caspase-11, a specific sensor for intracellular lipopolysaccharide recognition, mediates the non-canonical inflammatory pathway of pyroptosis. Cell Biosci. 2019;9(1):31.
45. Xia X, Wang X, Cheng Z, et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 2019;10(9):650.
46. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364.
47. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–73, Table of Contents.
48. Suresh R, Mosser DM. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv Physiol Educ. 2013;37(4):284–291.
49. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4(3).
50. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018;18(4):e27.
51. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411.
52. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery. 2020;6(1):36.
53. Kanneganti TD, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–559.
54. Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16(1):7–21.
55. Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. 2017;29(5):201–210.
56. Sharma D, Kanneganti T-D. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213(6):617–629.
57. Lieberman J, Wu H. Gasdermin D Activity in Inflammation and Host Defense. 2019;4(39).
58. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein Gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48(1):35–44.e6.
59. Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. 2017;130(23):3955–3963.
60. Dorfleutner A, Chu L, Stehlik C. Inhibiting the inflammasome: one domain at a time. Immunol Rev. 2015;265(1):205–216.
61. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022.
62. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25(5):308–315.
63. Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213(6):617–629.
64. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):128.
65. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489.
66. Zhou Y, Tong Z, Jiang S, Zheng W, Zhao J, Zhou X. The roles of endoplasmic reticulum in NLRP3 inflammasome activation. 2020;9(5).
67. Rajamäki K, Lappalainen J, Oörni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;e11765.
68. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018;39(5):393–406.
69. Zamani P, Oskuee RK, Atkin SL, Navashenaq JG, Sahebkar A. MicroRNAs as important regulators of the NLRP3 inflammasome. Prog Biophys Mol Biol. 2020;150:50–61.
70. Li XF, Shen WW, Sun YY, et al. MicroRNA-20a negatively regulates expression of NLRP3-inflammasome by targeting TXNIP in adjuvant-induced arthritis fibroblast-like synoviocytes. Joint Bone Spine. 2016;83(6):695–700.
71. Felekkis K, Touvana E, Stefanou C, Deltas C. microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia. 2010;14(4):236–240.
72. Ying SY, Chang DC, Lin SL. The microRNA (miRNA): overview of the RNA genes that modulate gene function. Mol Biotechnol. 2008;38(3):257–268.
73. Jiang Z, Yao L, Ma H, et al. miRNA-214 inhibits cellular proliferation and migration in glioma cells targeting Caspase 1 involved in pyroptosis. Oncol Res. 2017;25(6):1009–1019.
74. Diamond CE, Khameneh HJ, Brough D, Mortellaro A. Novel perspectives on non-canonical inflammasome activation. ImmunoTargets Ther. 2015;4:131–141.
75. Lee KH, Kang TB. The molecular links between cell death and inflammasome. 2019;8(9).
76. Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671.
77. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology. 2017;152(2):207–217.
78. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83.
79. Russo AJ, Behl B, Banerjee I, Rathinam VAK. Emerging insights into noncanonical inflammasome recognition of microbes. J Mol Biol. 2018;430(2):207–216.
80. Lupfer CR, Anand PK, Liu Z, et al. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog. 2014;10(9):e1004410.
81. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8(1):14128.
82. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689.
83. Jiang S, Gu H, Zhao Y, Sun L. Teleost Gasdermin E is cleaved by Caspase 1, 3, and 7 and induces pyroptosis. J Immunol. 2019;203(5):1369–1382.
84. Orning P, Weng D, Starheim K, Ratner D. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. 2018;362(6418):1064–1069.
85. Mandal P, Feng Y, Lyons JD, et al. Caspase-8 collaborates with Caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity. 2018;49(1):42–55.e6.
86. Chen C, Ma X, Yang C, et al. Hypoxia potentiates LPS-induced inflammatory response and increases cell death by promoting NLRP3 inflammasome activation in pancreatic β cells. Biochem Biophys Res Commun. 2018;495(4):2512–2518.
87. Lin CF, Kuo YT, Chen TY, Chien CT. Quercetin-Rich Guava (Psidium guajava) juice in combination with trehalose reduces autophagy, apoptosis and pyroptosis formation in the kidney and pancreas of Type II diabetic rats. Molecules. 2016;21(3):334.
88. <20170406.pdf>.
89. Lin Y, Hu Y, Hu X, et al. Ginsenoside Rb2 improves insulin resistance by inhibiting adipocyte pyroptosis. Adipocyte. 2020;9(1):302–312.
90. Ma H, Jeppesen JF, Jaenisch R. Human T cells expressing a CD19 CAR-T receptor provide insights into mechanisms of human CD19-Positive β cell destruction. Cell Reports Med. 2020;1(6):100097.
91. Carlos D, Costa FRC, Pereira CA, et al. Mitochondrial DNA activates the NLRP3 inflammasome and predisposes to Type 1 diabetes in murine model. Front Immunol. 2017;164.
92. Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–415.
93. Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188.
94. Kursawe R, Dixit VD, Scherer PE, et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes. 2016;65(3):610–618.
95. Chiazza F, Couturier-Maillard A, Benetti E, et al. Targeting the NLRP3 inflammasome to reduce diet-induced metabolic abnormalities in mice. Mol Med. 2016;21(1):1025–1037.
96. Chiazza F, Couturier-Maillard A, Benetti E, et al. Targeting the NLRP3 inflammasome to reduce diet-induced metabolic abnormalities in mice. Mol Med. 2015;21(1):1025–1037.
97. Kim Y, Wang W, Okla M, Kang I, Moreau R, Chung S. Suppression of NLRP3 inflammasome by γ-tocotrienol ameliorates type 2 diabetes. J Lipid Res. 2016;57(1):66–76.
98. Esser N, L’Homme L, De Roover A, et al. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia. 2013;56(11):2487–2497.
99. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204.
100. de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6(12):a016287.
101. Kanter JE, Hsu CC, Bornfeldt KE. Monocytes and macrophages as protagonists in vascular complications of diabetes. Front Cardiovasc Med. 2020;7:10.
102. Mohr S, Yego EK, Vincent J, et al. Hyperglycemia leads to müller cell death through induction of pyroptosis-like mechanisms. Invest Ophthalmol Vis Sci. 2009;50(13):15.
103. Boudina S, Abel ED. Diabetic cardiomyopathy. Causes and Effects, Rev Endocrine Metabol Disord. 2010;11(1):31–39.
104. Yang F, Li A, Qin Y, et al. RNA mediates pyroptosis of diabetic cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther NuclAcids. 2019;17:636–643.
105. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. 2020;42.
106. Yang F, Qin Y, Lv J, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018;9(10):1000.
107. Xie Y, Huang Y, Ling X, Qin H, Wang M, Luo B. Chemerin/CMKLR1 axis promotes inflammation and pyroptosis by activating NLRP3 inflammasome in diabetic cardiomyopathy rat. Front Physiol. 2020;11:381.
108. Luo B, Li B, Wang W, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovas Drugs Ther. 2014;28(1):33–43.
109. Zhang W, Xu W, Feng Y, Zhou X. Non-coding RNA involvement in the pathogenesis of diabetic cardiomyopathy. 2019;23(9):5859–5867.
110. He D, Zheng J, Hu J, Chen J, Wei X. Long non-coding RNAs and pyroptosis. Clin Chim Acta. 2020;504:201–208.
111. Yang F, Qin Y, Wang Y, et al. LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem. 2018;50(4):1230–1244.
112. Wei H, Bu R, Yang Q, et al. Exendin-4 Protects against Hyperglycemia-Induced Cardiomyocyte Pyroptosis via the AMPK-TXNIP Pathway. 2019;2019:8905917.
113. Wei H, Bu R, Yang Q, et al. Exendin-4 protects against hyperglycemia-induced cardiomyocyte pyroptosis via the AMPK-TXNIP pathway. J Diabetes Res. 2019;(2019):8905917.
114. Li X, Du N, Zhang Q, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5(10):e1479.
115. Cao R, Fang D, Wang J, Yu Y. ALDH2 overexpression alleviates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation. 2019;2019:4857921.
116. Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome treatment enhances anti-inflammatory M2 macrophages and reduces inflammation-induced pyroptosis in doxorubicin-induced cardiomyopathy. Cells. 2019;8(10).
117. Li S, Liu R, Xue M, et al. Spleen tyrosine kinase‑induced JNK‑dependent NLRP3 activation is involved in diabetic cardiomyopathy. Int J Mol Med. 2019;43(6):2481–2490.
118. Tavakoli Dargani Z, Singla DK. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am J Physiol Circulatory Physiol. 2019;317(2):H460–h471.
119. Wang X, Pan J, Liu H, et al. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019;221:249–258.
120. Kar S, Shahshahan HR, Hackfort BT, et al. Exercise training promotes cardiac hydrogen sulfide biosynthesis and mitigates pyroptosis to prevent high-fat diet-induced diabetic cardiomyopathy. Antioxidants. 2019;8(12).
121. Lin J, Cheng A, Cheng K, et al. New insights into the mechanisms of pyroptosis and implications for diabetic kidney disease. Int J Mol Sci. 2020;21(19).
122. Cheng Q, Pan J, Zhou ZL, et al. Caspase-11/4 and gasdermin D-mediated pyroptosis contributes to podocyte injury in mouse diabetic nephropathy. Acta Pharmacol Sin. 2020.
123. Zhan JF, Huang HW, Huang C, Hu LL, Xu WW. Long non-coding RNA NEAT1 regulates pyroptosis in diabetic nephropathy via mediating the miR-34c/NLRP3 axis. Kidney Blood Press Res. 2020;45(4):589–602.
124. Shahzad K, Bock F, Dong W, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87(1):74–84.
125. An X, Zhang Y, Cao Y, Chen J. Punicalagin protects diabetic nephropathy by inhibiting pyroptosis based on TXNIP/NLRP3 pathway. 12. 2020;5.
126. Li F, Chen Y, Li Y, Huang M, Zhao W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-κB pathway. Eur J Pharmacol. 2020;886:173449.
127. Liapis H, Romagnani P, Anders HJ. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol. 2013;183(5):1364–1374.
128. Dai H, Liu Q. Research progress on mechanism of podocyte depletion in diabetic nephropathy. 2017;2017:2615286.
129. Ding X, Jing N, Shen A, et al. MiR-21-5p in macrophage-derived extracellular vesicles affects podocyte pyroptosis in diabetic nephropathy by regulating A20. J Endocrinol Invest. 2020.
130. Huang F, Wang Q, Guo F, et al. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. EBioMedicine. 2019;48:491–504.
131. Xu W, Wang L, Li J, Cai Y, Xue Y. TXNIP mediated the oxidative stress response in glomerular mesangial cells partially through AMPK pathway. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie. 2018;107:785–792.
132. Abais JM, Xia M, Li G, et al. Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J Biol Chem. 2014;289(39):27159–27168.
133. Shah A, Xia L, Masson EA, et al. Thioredoxin-interacting protein deficiency protects against diabetic nephropathy. J Am Soc Nephrol. 2015;26(12):2963–2977.
134. Ignarski M, Islam R, Müller RU. Long Non-Coding RNAs in Kidney Disease. Int J Mol Sci. 2019;20(13).
135. Li X, Zeng L, Cao C, et al. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res. 2017;350(2):327–335.
136. Liu C, Zhuo H, Ye MY, Huang GX, Fan M, Huang XZ. LncRNA MALAT1 promoted high glucose-induced pyroptosis of renal tubular epithelial cell by sponging miR-30c targeting for NLRP3. 2020;36(9):682–691.
137. Wang K, Sun Q, Zhong X, et al. Structural Mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell. 2020;180(5):941–955.e20.
138. Song Z, Zhang Y, Gong B, Xu H, Hao Z, Liang C. Long noncoding RNA LINC00339 promotes renal tubular epithelial pyroptosis by regulating the miR-22-3p/NLRP3 axis in calcium oxalate-induced kidney stone. 2019;120(6):10452–10462.
139. Qiao Y, Tian X, Men L, et al. Spleen tyrosine kinase promotes NLR family pyrin domain containing 3 inflammasome‑mediated IL‑1β secretion via c‑Jun N‑terminal kinase activation and cell apoptosis during diabetic nephropathy. Mol Med Rep. 2018;18(2):1995–2008.
140. Xie C, Wu W, Tang A, Luo N, Tan Y. lncRNA GAS5/miR-452-5p reduces oxidative stress and pyroptosis of high-glucose-stimulated renal tubular cells. Diabetes Metabol Syndr Obesity. 2019;12:2609–2617.
141. Zhao W, He C, Wang F. [Screening potential Chinese materia medica and their monomers for treatment diabetic nephropathy based on caspase-1-mediated pyroptosis]. Nan Fang Yi Ke Da xue Xue Bao = Journal of Southern Medical University. 2020;40(9):1280–1287.
142. Yu X, Ma X, Lin W, Xu Q, Zhou H, Kuang H. Long noncoding RNA MIAT regulates primary human retinal pericyte pyroptosis by modulating miR-342-3p targeting of CASP1 in diabetic retinopathy. Exp Eye Res. 2020;108300.
143. Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017;95(8):803–808.
144. Yin Y, Chen F, Wang W, Wang H, Zhang X. Resolvin D1 inhibits inflammatory response in STZ-induced diabetic retinopathy rats: possible involvement of NLRP3 inflammasome and NF-κB signaling pathway. Mol Vis. 2017;23:242–250.
145. Gan J, Huang M. High glucose induces the loss of retinal pericytes partly via NLRP3-Caspase-1-GSDMD-mediated pyroptosis. 2020;(2020):4510628.
146. Kim LA, Amarnani D, Gnanaguru G, Tseng WA, Vavvas DG, D’Amore PA. Tamoxifen toxicity in cultured retinal pigment epithelial cells is mediated by concurrent regulated cell death mechanisms. Invest Ophthalmol Vis Sci. 2014;55(8):4747–4758.
147. Chien H, Dix RD. Evidence for multiple cell death pathways during development of experimental cytomegalovirus retinitis in mice with retrovirus-induced immunosuppression: apoptosis, necroptosis, and pyroptosis. J Virol. 2012;86(20):10961–10978.
148. Viringipurampeer IA, Metcalfe AL, Bashar AE, et al. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum Mol Genet. 2016;25(8):1501–1516.
149. Kerur N, Hirano Y, Tarallo V, et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54(12):7395–7401.
150. Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442(7102):527–532.
151. Acosta ML, Shin Y-S, Ready S, Fletcher EL, Christie DL, Kalloniatis M. Retinal metabolic state of the proline-23-histidine rat model of retinitis pigmentosa. Am J Physiol Cell Physiol. 2010;298(3):C764–C774.
152. Wooff Y, Fernando N, Wong JHC, et al. Caspase-1-dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration. Sci Rep. 2020;10(1):2263.
153. Zha X, Xi X, Fan X, Ma M, Zhang Y, Yang Y. Overexpression of METTL3 attenuates high-glucose induced RPE cell pyroptosis by regulating miR-25-3p/PTEN/Akt signaling cascade through DGCR8. Aging. 2020;12(9):8137–8150.
154. Chen J, Hui ST, Couto FM, et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB j. 2008;22(10):3581–3594.
155. Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes. 2008;57(4):938–944.
156. Chen W, Zhao M, Zhao S, et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflammation Res. 2017;66(2):157–166.
157. Devi TS, Lee I, Hüttemann M, Kumar A, Nantwi KD, Singh LP. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res. 2012;(2012):438238.
158. Singh LP, Devi TS, Yumnamcha T. The role of txnip in mitophagy dysregulation and inflammasome activation in diabetic retinopathy: a new perspective. JOJ Ophthalmol. 2017;4(4).
159. Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010;1(8):e65–e65.
160. Singh LP. Thioredoxin Interacting Protein (TXNIP) and pathogenesis of diabetic retinopathy. J Clin Exp Ophthalmol. 2013;4.
161. Sun Q, Wang C, Yan B, Shi X. Jinmaitong ameliorates diabetic peripheral neuropathy through suppressing TXNIP/NLRP3 inflammasome activation in the streptozotocin-induced diabetic rat model. 2019;12:2145–2155.
162. Sun Q, Wang C, Yan B, et al. [Expressions of thioredoxin interacting protein/nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome in the sciatic nerve of streptozotocin-induced diabetic rats]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2019;41(6):799–805.
163. Hayden MR, Salam M, Sowers JR. Reactive oxygen species and diabetic peripheral neuropathy – a closer look. In: Laher I, editor. Systems Biology of Free Radicals and Antioxidants. Berlin, Heidelberg: Springer Berlin Heidelberg; 2014:3375–3403.
164. Xu L, Lin X, Guan M. Verapamil attenuated prediabetic neuropathy in high-fat diet-fed mice through inhibiting TXNIP-mediated apoptosis and inflammation. 2019;2019:1896041.
165. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140.
166. Cheng YC, Chu LW, Chen JY, et al. Loganin attenuates high glucose-induced schwann cells pyroptosis by inhibiting ROS generation and NLRP3 inflammasome activation. 2020;9(9).
167. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71(9):3196–3201.
168. Driver C, Bamitale KDS, Kazi A, Olla M, Nyane NA, Owira PMO. Cardioprotective Effects of Metformin. J Cardiovasc Pharmacol. 2018;72(2):121–127.
169. Yang F, Qin Y, Wang Y, et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15(5):1010–1019.
170. Qiu Z, He Y, Ming H, Lei S. Lipopolysaccharide (LPS) aggravates high glucose- and hypoxia/reoxygenation-induced injury through activating ROS-dependent NLRP3 inflammasome-mediated pyroptosis in H9C2 Cardiomyocytes. 2019;2019:8151836.
171. Jeyabal P, Thandavarayan RA, Joladarashi D, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. 2016;471(4):423–429.
172. Luo B, Li B, Wang W, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9(8):e104771.
173. Yang F, Li A, Qin Y, et al. RNA mediates pyroptosis of diabetic cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther Nucleic Acids. 2019;17:636–643.
174. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. 2017;31(2):119–132.
175. Li X, Li Z, Li B, Zhu X, Lai X. Klotho improves diabetic cardiomyopathy by suppressing the NLRP3 inflammasome pathway. Life Sci. 2019;234:116773.
176. Zhang X, Pan L, Yang K, et al. H3 relaxin protects against myocardial injury in experimental diabetic cardiomyopathy by inhibiting myocardial apoptosis. Fibrosis and Inflammation, Cell Physiol Biochem. 2017;43(4):1311–1324.
177. Li X, Ke X, Li Z, Li B. Vaspin prevents myocardial injury in rats model of diabetic cardiomyopathy by enhancing autophagy and inhibiting inflammation. Biochem Biophys Res Commun. 2019;514(1):1–8.
178. Zhang H, Chen X, Zong B, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med. 2018;22(9):4437–4448.
179. Miteva K, Pappritz K, Sosnowski M, et al. Mesenchymal stromal cells inhibit NLRP3 inflammasome activation in a model of Coxsackievirus B3-induced inflammatory cardiomyopathy. Sci Rep. 2018;8(1):2820.
180. Gu J, Huang W, Zhang W, et al. Sodium butyrate alleviates high-glucose-induced renal glomerular endothelial cells damage via inhibiting pyroptosis. Int Immunopharmacol. 2019;75:105832.
181. Wang Y, Zhu X, Yuan S, et al. TLR4/NF-κB signaling induces GSDMD-related pyroptosis in tubular cells in diabetic kidney disease. Front Endocrinol (Lausanne). 10(603):2019.
182. Ke R, Wang Y, Hong S, Xiao L. Endoplasmic reticulum stress related factor IRE1α regulates TXNIP/NLRP3-mediated pyroptosis in diabetic nephropathy. Exp Cell Res. 2020;396(2):112293.
183. Wen S, Li S, Li L, Fan Q. circACTR2: a novel mechanism regulating high glucose-induced fibrosis in renal tubular cells via pyroptosis. Biol Pharm Bull. 2020;43(3):558–564.
184. Gan J, Huang M, Lan G, Liu L, Xu F. High glucose induces the loss of retinal pericytes partly via NLRP3-Caspase-1-GSDMD-mediated pyroptosis. Biomed Res Int. 2020;(2020):4510628.
185. Shafabakhsh R, Aghadavod E, Mobini M, Heidari-Soureshjani R, Asemi Z. Association between microRNAs expression and signaling pathways of inflammatory markers in diabetic retinopathy. 2019;234(6):7781–7787.
186. Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94(6):776–780.
187. Wang JY, Chen LJ. The role of miRNAs in the invasion and metastasis of cervical cancer. 2019;39(3).
188. Gu C, Draga D, Zhou C, et al. miR-590-3p inhibits pyroptosis in diabetic retinopathy by targeting NLRP1 and inactivating the NOX4 signaling pathway. Invest Ophthalmol Vis Sci. 2019;60(13):4215–4223.
189. Yang C, Hu Y, Zhou B, et al. The role of m6A modification in physiology and disease. Cell Death Dis. 2020;11(11):960.
190. Chen M, Wong CM. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol Cancer. 2020;19(1):44.
191. Zhang Y, Geng X, Li Q, et al. m6A modification in RNA: biogenesis, functions and roles in gliomas. J Exp Clin Cancer Res. 2020;CR 39(1):192.
192. Zhang Y, Song Z, Li X, et al. Long noncoding RNA KCNQ1OT1 induces pyroptosis in diabetic corneal endothelial keratopathy. Am J Physiol Cell Physiol. 2020;318(2):C346–c359.
193. Lu L, Lu Q, Chen W, Li J, Li C, Zheng Z. Vitamin D3 protects against diabetic retinopathy by inhibiting high-glucose-induced activation of the ROS/TXNIP/NLRP3 inflammasome pathway. J Diabetes Res. 2018;(2018):8193523.
194. Lu L, Lu Q. Vitamin D(3) protects against diabetic retinopathy by inhibiting high-glucose-induced activation of the ROS/TXNIP/NLRP3 inflammasome pathway. 2018;2018:8193523.
195. Hao J, Zhang H, Yu J, Chen X, Yang L. Methylene blue attenuates diabetic retinopathy by inhibiting NLRP3 inflammasome activation in STZ-induced diabetic rats. Ocul Immunol Inflamm. 2019;27(5):836–843.
196. Li S, Yang H, Chen X. Protective effects of sulforaphane on diabetic retinopathy: activation of the Nrf2 pathway and inhibition of NLRP3 inflammasome formation. Exp Animals. 2019;68(2):221–231.
197. Jiao J, Zhao G, Wang Y, Ren P, Wu M. MCC950, a selective inhibitor of NLRP3 inflammasome, reduces the inflammatory response and improves neurological outcomes in mice model of spinal cord injury. Front Mol Biosci. 2020;7:37.
198. Zhang Y, Lv X, Hu Z, et al. Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis. 2017;8(7):e2941.
199. Abdanipour A, Schluesener HJ, Tiraihi T. Effects of valproic acid, a histone deacetylase inhibitor, on improvement of locomotor function in rat spinal cord injury based on epigenetic science. Iran Biomed J. 2012;16(2):90–100.
200. Jiang Y, Steinle JJ. Epac1 inhibits PKR to reduce NLRP3 inflammasome proteins in retinal endothelial cells. J Inflamm Res. 2019;12:153–159.
201. Yin DH, Liang XC, Zhao LI, et al. Jinmaitong decreases sciatic nerve DNA oxidative damage and apoptosis in a streptozotocin-induced diabetic rat model. Exp Ther Med. 2015;10(2):778–786.
202. Song W, Jiang W, Wang C, et al. Jinmaitong, a traditional chinese compound prescription, ameliorates the streptozocin-induced diabetic peripheral neuropathy rats by increasing sciatic nerve IGF-1 and IGF-1R expression. Front Pharmacol. 2019;10:255.
203. Xie J, Song W, Liang X, et al. Jinmaitong ameliorates diabetic peripheral neuropathy in streptozotocin-induced diabetic rats by modulating gut microbiota and neuregulin 1. Aging. 2020;12(17):17436–17458.
204. Oszmiański J, Wojdyło A. Roots and leaf extracts of dipsacus fullonum L. and their biological activities. 2020;9(1).
205. Carrillo-Ocampo D, Bazaldúa-Gómez S, Bonilla-Barbosa JR, Aburto-Amar R, Rodríguez-López V. Anti-inflammatory activity of iridoids and verbascoside isolated from Castilleja tenuiflora. Molecules. 2013;18(10):12109–12118.
206. Kim H, Youn K, Ahn M-R, et al. Neuroprotective effect of loganin against Aβ25-35-induced injury via the NF-κB-dependent signaling pathway in PC12 cells. Food Funct. 2015;6(4):1108–1116.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.