")
Back to Journals » International Journal of General Medicine » Volume 14
Retarding Progression of Chronic Kidney Disease in Autosomal Dominant Polycystic Kidney Disease with Metformin and Other Therapies: An Update of New Insights
Authors Carullo N, Zicarelli MT, Casarella A , Nicotera R, Castagna A, Urso A, Presta P, Andreucci M, Russo E , Bolignano D, Coppolino G
Received 12 July 2021
Accepted for publication 13 September 2021
Published 22 September 2021 Volume 2021:14 Pages 5993—6000
DOI https://doi.org/10.2147/IJGM.S305491
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Nazareno Carullo,1 Maria Teresa Zicarelli,1 Alessandro Casarella,2 Ramona Nicotera,1 Alberto Castagna,2 Alessandra Urso,1 Pierangela Presta,1 Michele Andreucci,1 Emilio Russo,2 Davide Bolignano,1 Giuseppe Coppolino1
1Renal Unit, Department of Health Sciences, “Magna Graecia” University, Catanzaro, Italy; 2Department of Health Sciences, “Magna Graecia” University, Catanzaro, Italy
Correspondence: Giuseppe Coppolino
Renal Unit, Department of Health Sciences, “Magna Graecia” University, Viale Europa - Germaneto, Catanzaro, 88100, Italy
Tel +39 09613697170
Email [email protected]
Abstract: Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent single-gene disorder leading to renal failure. Current therapies are aimed to treat renal and extrarenal complications of ADPKD, but improved knowledge of the pathophysiological mechanisms leading to the generation and growth of cysts has permitted the identification of new drug candidates for clinical trials. Among these, in this review, we will examine above all the role of metformin, hypothesized to be able to activate the AMP-activated protein kinase (AMPK) pathway and potentially modulate some mechanisms implicated in the onset and the growth of the cysts.
Keywords: cystogenesis, renal cells, metformin, chronic renal disease, renal tubule
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent single-gene disorder leading to renal failure.1 It occurs in all races with a prevalence estimated to be from 1 case every 400 to 1 case every 1000 births.2 ADPKD essential feature is the formation of multiple and bilateral renal growing cysts determining kidney enlargement, progressive parenchyma damage and often leading finally to end-stage renal disease (ESRD) and renal replacement therapy in adulthood. It is considered a multisystem disorder as cysts appear also in other organs such as liver, pancreas, arachnoid membrane, etc., and this characteristic causes extrarenal complications like intracranial aneurysms and cardiac valvular disease.3,4 ADPKD is genetically heterogenous, but two commons genes are identified, PKD1 and PKD2, that respectively encode for polycystin-1 and -2 (PC1, PC2).1
Polycystins are transmembrane proteins mainly found in plasma membranes but also in the primary cilium localized in renal tubular epithelia, hepatic bile ductuli and pancreatic ducts, in which alteration cause uncontrollable cellular proliferation and abnormal fluid secretion.5–7 The main molecular pathogenetic mechanisms involve the pathways of cAMP, mTOR, MAPK/ERK and JAK/STAT.8
Current therapies are aimed to treat renal and extrarenal complications of ADPKD, but improved knowledge of the pathophysiological mechanisms leading to the generation and growth of cysts has permitted the identification of new drug candidates for clinical trials. Among these, in this review, we will examine above all the role of metformin, hypothesized to be able to activate the AMP-activated protein kinase (AMPK) pathway and potentially modulate some mechanisms implicated in the onset and the growth of the cysts.9 Despite evidence from numerous preclinical studies, there are still many questions about the clinical efficacy of metformin in ADPKD patients.
ADPKD: From Genetic to Impaired Signaling Pathways
PKD Gene Products
PC1 and PC2 are members of a subfamily of transient receptor potential channels controlling both intracellular calcium homeostasis and signaling. Polycystins are largely expressed in epithelial (among them renal tubules) and in many other tissues.8,10 They detect extracellular signals at primary cilia, cell–cell junctions and cell-matrix contacts and are essential to preserve the differentiated phenotype of the tubular epithelium. The reduction of one of the polycystins makes it impossible to maintain planar polarity and enhances cellular proliferation and apoptosis, expression of secretory phenotype.11 PC1 (~660 kDa) is a receptor-like protein; it consists of a large extracellular N region, 11 transmembrane regions and a brief intracellular C region. It is localized in primary cilium, plasma membrane, desmosome, adherens junctions and interacts with PC2 creating a complex that is believed to play a role in the regulation of intracellular Ca2+. Moreover, PC1 interacts with a wide network of proteins and has an essential role in ciliary mechanosensory functions.12 PC2 (~110 kDa) is Ca2+-responsive cation channel and consists of an N-terminal cytoplasmatic region, six transmembrane domains and short C-terminal region. It is found primarily in endoplasmatic endothelium but furthermore in plasma membrane, primary cilium, mitotic spindles, centrosome and is involved in PC1 regulation.11,13 The function of the PC complex on the cilium is a debated and unresolved question. One of the cilium functions could be as a flux detector, supporting calcium influx when flow is present and curtailing Ca2+ import in response to a lack of flow or loss of the PC complex.14,15
Via ryanodine receptors, this influx regulates further release of cation from subcellular compartments, modulating the downstream of calcium pathways. The alteration of intracellular calcium concentration mediates gene expression, apoptosis, differentiation, and cell division. The impaired calcium pathway causes the major pathophysiological features of ADKPD, resulting in cystogenesis.14
The Function of cAMP, mTOR, AMPK and JAK-STAT Pathway in the Cellular Proliferation and Fluid Secretion Regulation in Cystic Epithelial Cells in ADPKD
Cyclic adenosine monophosphate (cAMP) is involved in crucial cellular processes such as fluid and electrolyte regulation, differentiation, transcription and cell proliferation. Many evidence have shown that elevated intracellular cAMP concentration causes cyst growth, kidney expansion and progression of renal disease. Indeed, in animal models of PKD, high levels of cAMP were frequently found, not only in the kidney but also in vascular smooth muscle and liver.16 Intracellular cAMP accumulation and altered calcium levels could have an essential role in ADPKD pathogenesis and cystogenesis.17
Levels of intracellular cAMP are determined by the activities of membrane-bound and soluble adenylyl cyclase (ACs), which drive the formation of cAMP from ATP, and phosphodiesterases (PDEs) that transform cAMP to AMP.18 Reduced intracellular calcium levels (present in PKD) could be responsible for AC6 or AC5 activation, which directly inhibit calcium-/calmodulin-dependent PDE119 and inhibit (indirectly) cGMP inhibitable PDE3, consequently leading to accumulation of cAMP and activation of protein kinase A (PKA).20 This, in turn, stimulates cystic fibrosis transmembrane conductance regulator (CFTR)-driven chloride and fluid secretion and cell proliferation.21,22
In summary, in cystic epithelium, the reduced intracellular calcium concentration, due to PC complex alteration, causes activation of Ras/Raf/MEK/ERK pathway by cAMP, in contrast to normal kidney epithelia.23–25 The elevated cAMP levels activate CFTR and potassium channels, producing respectively outflow of chloride and sodium.8,26 This determines an increased osmotic pressure and consequent transfer of water by aquaporins, enlarging the cysts.
In addition, the functional disorder of PC1 leads to mTOR pathway activation, through unregulated phosphorylation of TSC2 (tuberous sclerosis complex-2) mediated by AKT. In ADPKD, AMPK down-regulates both CFTR channels and mTOR pathway that, as already mentioned, are partially responsible for cystogenesis.9,27,28
The JAK-STAT signalling system is probably also involved in the pathogenesis of ADPKD. STAT-6 is activated by the interleukin (IL) 4- and IL-13 and indeed the cyst fluid contains high concentrations of IL-13 and its receptors.29
Current Therapy for ADPKD
Treatment of ADPKD includes management of renal and extrarenal complications, chronic kidney disease (CKD), and specific therapies (disease-modifying therapies). Most patients with ADPKD could require renal replacement treatment after the fourth decade.30 In all patients with ADPKD, management of hypertension is critical. It is thought that it may be secondary to the expansion of renal cysts by causing areas of renal ischemia and leading to increased activity of the renin-angiotensin-aldosterone system.31,32 For this reason, the first-line treatment is ACE inhibitors or, in case of intolerance, an angiotensin receptor blocker.32,33 In addition, sodium restriction (maximum 2 grams of sodium per day corresponding to approximately 5 grams of salt) is recommended for all patients because increased sodium excretion has been shown to be associated with growth of renal volume and reduced estimated glomerular filtration rate (eGFR).34 To identify patients who might benefit from specific therapy (disease-modifying therapies), it is necessary to recognize those at high risk for CKD progression. The most widely used method is based on the Mayo classification.
Patients with ADPKD are divided into five classes (1A, 1B, 1C, 1D, 1F), from lowest to highest risk; classes 1C, 1D, and 1E are considered to be at high risk of progression to ESRD.35 Using this method, it is possible to predict the rate of eGFR reduction through total kidney volume (TKV).36 This can be calculated with the TKV calculator (available online) which is based on the length, width and depth of both kidneys obtained from computed tomography (CT) or magnetic resonance imaging (MRI) images, both without contrast medium [http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754]. All present therapies aim to treat renal and extrarenal complications of ADPKD such pain, urinary tract and cyst infection, nephrolithiasis, hypertension and obviously the CKD. Improved knowledge of the pathophysiological mechanisms leading to the generation and growth of cysts has permitted the identification of new drug candidates for clinical trials. These include vasopressin antagonists, somatostatin analogs, mammalian target of rapamycin (mTOR) inhibitors and so other investigational drugs. Of these, the only one to have entered in clinical practice is Tolvaptan, a short-acting vasopressin V2-receptor (V2R) antagonist.
Vasopressin Antagonist
It has been seen, in animal studies, that vasopressin acting on V2Rs, activates a cascade of intracellular signals leading to cell proliferation and fluid secretion, crucial for cystogenesis.37 Conversely, suppression of vasopressin release, action, or production through high water intake, antagonism on the V2R, or genetic elimination of vasopressin results in lesser cyst burden and, consequently, a slowing of kidney damage with increased animal survival.38,39 After these results, two large randomized clinical trials were conducted on the use of Tolvaptan in patients with ADPKD. In TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal dominant Polycystic Kidney Disease and Its Outcomes) were enrolled patients with eGFR >60 mL/min/1.73m2 and a TKV >750 mL. In the treatment group, Tolvaptan has been shown to reduce the annual increase of TKV and the decline in renal function, although there was a higher rate of aquaretic-related adverse events (thirst, polyuria, polydipsia, hypernatremia, nocturia) and a clinically significant increase in liver enzymes in tolvaptan-treated patients.40,41 In REPRISE (Replicating Evidence of Preserved Renal Function: An Investigation of Tolvaptan Safety and Efficacy in ADPKD) were enrolled patients with more advanced CKD (eGFR 25 to 65 mL/min/1.73m2).42 In this study the data showed, also, that Tolvaptan slows the decline in renal function, even in patients in whom the baseline eGFR is significantly reduced. It is thought that increased oral water intake, suppressing vasopressin release, may reduce cyst growth in patients with ADPKD. To answer this question, a pilot study was conducted to evaluate the effect of water loading (acute and chronic) on urinary osmolarity and cAMP levels. Chronic water loading (at least 3 L/day) has been seen to increase urinary volume and to reduce urinary osmolarity (mean value 270 mosmol/L). With chronic water loading, cAMP excretion did not change, whereas with acute loading it did.43,44 These data suggest that increased oral water intake of at least 3 L/day may, by partially suppressing vasopressin release, effectively reduce urinary osmolarity.
Somatostatin Analogues
Somatostatin is a hormone that inhibits intracellular cAMP production, but due to its short half-life, it has limited therapeutic potential.45 For this reason, more stable synthetic peptides have been tried; among them octreotide (long-acting somatostatin), lanreotide (somatostatin analogue) and pasireotide (pansomatostatin analogue) have the potential to reduce fluid accumulation in renal and hepatic cysts in patients with PKD.46–52 However, these drugs have not demonstrated to slow the decline in kidney function and are potentially responsible for serious adverse events;, for these reasons, they are not recommended.
Mammalian Target of Rapamycin (mTOR) Inhibitors
As previously described, mTOR is activated in animal models of PKD. Studies in rodent models of PKD have demonstrated that sirolimus and everolimus (mTOR inhibitors) are able to prevent cystic expansion and protect renal function.53–55 Two large clinical trials testing everolimus and sirolimus have shown that these agents are yes able to slow renal and hepatic volume growth but without preserving renal function, at least in the short term (18–24 months).56
Other Experimental Therapies
So many other medical treatments are currently under investigation in animal models of ADPKD: methylprednisolone, lovastatin, epidermal growth factor receptor tyrosine kinase inhibitors, cyclin-dependent kinase inhibitor (roscovitine), bardoxolone methyl, mitogen-activated protein kinase inhibitor.30,57–63 Pravastatin, an HMG-CoA reductase inhibitor, in a small randomized clinical trial that included children and young adults with ADPKD, has shown to slow the rate of increase in kidney volume.64 Bosutinib, a Src-inhibitor (overactivated in ADPKD), in a Phase II, double-blind randomized clinical trial, reduced the rate of kidney growth but with similar eGFR annual decline compared to placebo.65 In animal model of cystic disorder, amiloride and low caffeine use may reduce cyst enlargement but has not been shown to be as effective in humans with ADPKD.66–68 Another important agent that is emerging as a possible medical treatment in patients with ADPKD is metformin. Below we discuss its mechanism of action and preclinical and clinical studies demonstrating its potential efficacy.
Metformin’s Mechanism of Action
Metformin currently represents the treatment of first choice in most patients with first-diagnosis type 2 diabetes mellitus (T2DM), in the absence of contraindications.69,70 All the effects and benefits of metformin are numerous and are not fully known. Its main mechanism of action is to reduce hepatic glucose release through an inhibition of gluconeogenesis.71 In addition, it activates AMPK in hepatocytes leading to a reduction of lipid levels.72,73
The Role of Metformin in ADPKD
Preclinical Evidence About the Metformin Efficacy on ADPKD
A variety of preclinical studies have shown an effect of metformin on cystogenesis in ADPKD. Actually, it is believed that impaired glucose metabolism may play an important role in kidney cyst formation. It has been shown that, in a murine model of PKD1 and in human kidney cells with ADPKD, there is a shift in energy metabolism that causes an enhanced aerobic glycolysis. Because of this metabolic change, ADPKD cells has a broken proliferation/apoptosis equilibrium. In contrast, glucose deprivation causes an increase in apoptosis and a reduction in autophagy and proliferation in PKD1 mutant cells. The administration of a non-metabolized glucose analog (2-deoxyglucose, 2DG), modulating AMPK and m-TOR pathways, reduced proliferation rates, kidney volume and cystic index.74 Metformin, inhibiting both the m-TOR and CFTR pathways, can activate AMPK that leads to a reduction of renal cyst growth in mouse models affected by ADPKD. Takiar et al have noticed that in the renal parenchyma of mouses treated with metformin, cysts persist; this proves the inability of metformin to revert cystogenesis when the cysts have already been produced. Indeed, the action of metformin on cystogenesis before its occurrence was to reduce moderately the cyst growth,9 which suggests that it might significantly delay the progression of the cysts, retarding the onset of ESRD. Lian et al have investigated the effects of the combination of metformin and 2DG in pig model. In the control group, there was a pronounced enlargement of renal parenchyma with growth of large vesicles and cysts, while in the treatment group, there was a minimal presence of the same traits. The maximal difference was showed in the combination-therapy group. After 20 months, in the treatment group, was shown a significant reduction in creatinine serum concentration. The interventional group showed, a lower activity of several elements of mTOR, while AMPK was over expressed.75
Clinical Evidence About Metformin Efficacy in ADPKD
The clinical efficacy of metformin in ADPKD patients has been studied only in a few small clinical trials, while more extensive studies are currently in progress and results are awaited in 2022 (ClinicalTrials.gov). These studies will assess the feasibility of using metformin as a therapy for ADPKD; one of these will compare the efficacy of metformin versus tolvaptan.
The first literature data about the possible effectiveness of metformin in ADPKD patients are connected to the occasional observation that one of three sisters was diagnosed T2DM in 2016, so metformin (1500 mg/day) was prescribed. Two years later, through retrospective analysis, was seen an eGFR decline three times slower than before, while the eGFR decline of the other two sisters was unchanged.3,76–78 In another retrospective study, data from seven patients with CKD stage 3 and T2DM were analyzed. These subjects were in chronic treatment with at least 1000 mg of metformin. After the first year of monitoring, in the metformin group, eGFR decreased by only 2.5%, while in the control group, it was reduced by 16%. Between the second and third year, a similar trend to the previous one was registered: in the metformin group, eGFR was stable around the baseline (48.1 mL/min/1.73m2) while continuing to decrease in the control group. Of course, all these data must be interpreted within the limits of the type of study and the population under consideration. Major limitations include the small sample size, the retrospective design of the study and the absence of direct evidence of a cause-and-effect relationship between metformin administration and protection on renal tissue.79 METROP was the first prospective study to assess the potential of Metformin on renal function in ADPKD patients. This is a single-center pilot study in which were enrolled 34 non-diabetic ADPKD patients in a single-arm of treatment, that consisted of 500–1000 mg/day metformin. The follow-up time was 24 months and has been completed by only 16 of 34 patients (47%). Twelve of these dropped out of the study for unspecified reasons, affecting the reliability of the results. This study demonstrated the tolerability of metformin, with its limitations. Only a few adverse events occurred, which were mainly of a gastrointestinal character (63.6%). In synthesis, in concert with intention-to-treat population (ITT) analysis, eGFR had changed by –2.59% and by −7.2%, in line with per-protocol population (PP) analysis. The results of the study were affected by several limitations, among them, the small sample size, the chosen design and the large number of dropouts (54%). For that, obviously every conclusion should be made with caution.80
Finally, two randomized clinical trials (ClinicalTrials.gov NCT02656017 and NCT02903511) are in progress with the goal of verifying the potential metformin efficacy in ADPKD patients. A third study (ClinicalTrials.gov NCT03764605), still in the recruitment phase, will aim to evaluate metformin efficacy in slowing renal cystogenesis in ADPKD compared to the actual gold standard (tolvaptan).
Conclusions
The optimal management of ADPKD should consist not only in the treatment of its complications and the general measures used in CKD but also with drugs that can modify the course of the disease itself as is partially the case of tolvaptan (vasopressin antagonist). These agents may be able to slow, stop the growth of cysts or even prevent their onset, reducing the renal damage that unfortunately often leads to renal replacement treatment (Figure 1).
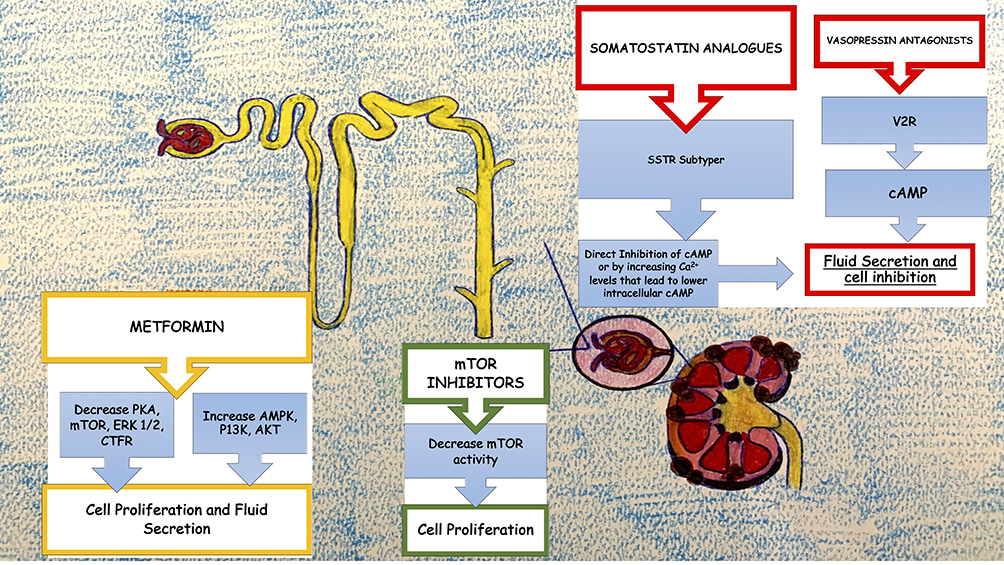 |
Figure 1 Graphical representation of mechanisms involving current new therapies to slow progression of CKD in ADPKD. |
Metformin may be the near-ideal agent to achieve these results, considering its well-known good tolerability already partially affirmed in ADPKD patients. Its potential use is sustained by one of its possible mechanisms of action in predicting efficacy by the involvement of AMPK in the pathophysiology of ADPKD. In addition, strong preclinical data suggested that metformin prevents the development of renal cysts by slowing the progression of the disease and subsequent kidney damage. This supports the testing of metformin in randomized clinical trials. The possibility to act therapeutically before the onset of renal cysts makes it crucial the early diagnosis of the disease, even before the appearance of clinical manifestations (often very late because of the important functional reserve of the kidney). Data to enable its use in clinical practice do not yet exist, but promising results from ongoing clinical trials are expected in the short term.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis. 2016;67:792–810. doi:10.1053/j.ajkd.2015.07.037
2. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi:10.1038/ki.2009.128
3. Cornec-le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019;393:919–935. doi:10.1016/S0140-6736(18)32782-X
4. Chapman AB, Devuyst O, Eckardt K-U, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int. 2015;88:17–27. doi:10.1038/ki.2015.59
5. Geng L, Segal Y, Peissel B, et al. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest. 1996;98:2674–2682. doi:10.1172/JCI119090
6. Ibraghimov-Beskrovnaya O, Dackowski WR, Foggensteiner L, et al. Polycystin: in vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc Natl Acad Sci U S A. 1997;94:6397–6402. doi:10.1073/pnas.94.12.6397
7. Huan Y, van Adelsberg J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest. 1999;104:1459–1468. doi:10.1172/JCI5111
8. Malekshahabi T, Khoshdel Rad N, Serra AL, Moghadasali R. Autosomal dominant polycystic kidney disease: disrupted pathways and potential therapeutic interventions. J Cell Physiol. 2019;234:12451–12470. doi:10.1002/jcp.28094
9. Takiar V, Nishio S, Seo-Mayer P, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A. 2011;108:2462–2467. doi:10.1073/pnas.1011498108
10. Su Q, Hu F, Ge X, et al. Structure of the human PKD1-PKD2 complex. Science. 2018;361:eaat9819. doi:10.1126/science.aat9819
11. Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124:2315–2324. doi:10.1172/JCI72272
12. Xu GM, Sikaneta T, Sullivan BM, et al. Polycystin-1 interacts with intermediate filaments. J Biol Chem. 2001;276:46544–46552. doi:10.1074/jbc.M107828200
13. Hildebrandt F, Benzing T, Katsanis N, Schwartz RS. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi:10.1056/NEJMra1010172
14. Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi:10.1038/ng1076
15. Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi:10.1007/s00232-001-0075-4
16. Wallace DP. Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta. 2011;1812:1291–1300. doi:10.1016/j.bbadis.2010.11.005
17. Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi:10.1681/ASN.2013040398
18. Houslay MD, Milligan G. Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem Sci. 1997;22:217–224. doi:10.1016/s0968-0004(97)01050-5
19. Defer N, Best-Belpomme M, Hanoune J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am J Physiol Renal Physiol. 2000;279:F400–F416. doi:10.1152/ajprenal.2000.279.3.F400
20. Wang X, Ward CJ, Harris PC, Torres VE. Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int. 2010;77:129–140. doi:10.1038/ki.2009.438
21. Hanaoka K, Devuyst O, Schwiebert EM, Wilson PD, Guggino WB. A role for CFTR in human autosomal dominant polycystic kidney disease. Am J Physiol. 1996;270:C389–C399. doi:10.1152/ajpcell.1996.270.1.C389
22. Wallace DP, Grantham JJ, Sullivan LP. Chloride and fluid secretion by cultured human polycystic kidney cells. Kidney Int. 1996;50:1327–1336. doi:10.1038/ki.1996.445
23. Yamaguchi T, Pelling JC, Ramaswamy NT, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000;57:1460–1471. doi:10.1046/j.1523-1755.2000.00991.x
24. Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17–31. doi:10.1111/j.1365-2796.2006.01743.x
25. Yamaguchi T, Wallace DP, Magenheimer BS, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi:10.1074/jbc.M405079200
26. Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. 2009;297:F849–F863. doi:10.1152/ajprenal.00181.2009
27. Dere R, Wilson PD, Sandford RN, Walker CL, Blagosklonny MV. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS One. 2010;5:e9239. doi:10.1371/journal.pone.0009239
28. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi:10.1016/s0092-8674(03)00929-2
29. Olsan EE, Mukherjee S, Wulkersdorfer B, et al. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A. 2011;108:18067–18072. doi:10.1073/pnas.1111966108
30. Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009;18:99–106. doi:10.1097/MNH.0b013e3283262ab0
31. Bell PE, Hossack KF, Gabow PA, et al. Hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1988;34:683–690. doi:10.1038/ki.1988.233
32. Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323:1091–1096. doi:10.1056/NEJM199010183231602
33. Ecder T, Edelstein CL, Chapman AB, et al. Reversal of left ventricular hypertrophy with angiotensin converting enzyme inhibition in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 1999;14:1113–1116. doi:10.1093/ndt/14.5.1113
34. Torres VE, Abebe KZ, Schrier RW, et al. Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney Int. 2017;91:493–500. doi:10.1016/j.kint.2016.10.018
35. Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26:160–172. doi:10.1681/ASN.2013101138
36. Bae KT, Sun H, Lee JG, et al. Novel methodology to evaluate renal cysts in polycystic kidney disease. Am J Nephrol. 2014;39:210–217. doi:10.1159/000358604
37. Torres VE. Pro: tolvaptan delays the progression of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2019;34:30–34. doi:10.1093/ndt/gfy297
38. Torres VE, Wang X, Qian Q, et al. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10:363–364. doi:10.1038/nm1004
39. Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:102–108. doi:10.1681/ASN.2007060688
40. Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol. 2011;6:2499–2507. doi:10.2215/CJN.03530411
41. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–2418. doi:10.1056/NEJMoa1205511
42. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377:1930–1942. doi:10.1056/NEJMoa1710030
43. Barash I, Ponda MP, Goldfarb DS, Skolnik EY. A pilot clinical study to evaluate changes in urine osmolality and urine cAMP in response to acute and chronic water loading in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2010;5:693–697. doi:10.2215/CJN.04180609
44. Torres VE, Bankir L, Grantham JJ. A case for water in the treatment of polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4:1140–1150. doi:10.2215/CJN.00790209
45. Messchendorp AL, Casteleijn NF, Meijer E, Gansevoort RT. Somatostatin in renal physiology and autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2020;35:1306–1316. doi:10.1093/ndt/gfz054
46. Meijer E, Visser FW, van Aerts RMM, et al. Effect of lanreotide on kidney function in patients with autosomal dominant polycystic kidney disease: the DIPAK 1 randomized clinical trial. JAMA. 2018;320:2010–2019. doi:10.1001/jama.2018.15870
47. Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:206–216. doi:10.1111/j.1523-1755.2005.00395.x
48. Caroli A, Antiga L, Cafaro M, et al. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5:783–789. doi:10.2215/CJN.05380709
49. Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–1061. doi:10.1681/ASN.2009121291
50. Hogan MC, Masyuk TV, Page L, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant. 2012;27:3532–3539. doi:10.1093/ndt/gfs152
51. Chrispijn M, Nevens F, Gevers TJG, et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther. 2012;35:266–274. doi:10.1111/j.1365-2036.2011.04923.x
52. Hogan MC, Chamberlin JA, Vaughan LE, et al. Pansomatostatin agonist pasireotide long-acting release for patients with autosomal dominant polycystic kidney or liver disease with severe liver involvement: a randomized clinical trial. Clin J Am Soc Nephrol. 2020;15:1267–1278. doi:10.2215/CJN.13661119
53. Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21:489–497. doi:10.1681/ASN.2009040421
54. Wu M, Wahl PR, Le Hir M, et al. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press Res. 2007;30:253–259. doi:10.1159/000104818
55. Zafar I, Ravichandran K, Belibi FA, Doctor RB, Edelstein CL. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010;78:754–761. doi:10.1038/ki.2010.250
56. Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–829. doi:10.1056/NEJMoa0907419
57. Gattone VH
58. Gile RD, Cowley BD, Gattone VH, et al. Effect of lovastatin on the development of polycystic kidney disease in the Han: SPRDrat. Am J Kidney Dis. 1995;26:501–507. doi:10.1016/0272-6386(95)90497-2
59. Sweeney WE
60. Muto S, Aiba A, Saito Y, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002;11:1731–1742. doi:10.1093/hmg/11.15.1731
61. Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. doi:10.1038/nature05348
62. Fujiki T, Ando F, Murakami K, et al. Tolvaptan activates the Nrf2/HO-1 antioxidant pathway through PERK phosphorylation. Sci Rep. 2019;9:9245. doi:10.1038/s41598-019-45539-8
63. Omori S, Hida M, Fujita H, et al. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol. 2006;17:1604–1614. doi:10.1681/ASN.2004090800
64. Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2014;9:889–896. doi:10.2215/CJN.08350813
65. Tesar V, Ciechanowski K, Pei Y, et al. Bosutinib versus placebo for autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2017;28:3404–3413. doi:10.1681/ASN.2016111232
66. Grantham JJ, Uchic M, Cragoe EJ, et al. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int. 1989;35:1379–1389. doi:10.1038/ki.1989.137
67. Gardner KD
68. Belibi FA, Wallace DP, Yamaguchi T, et al. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002;13:2723–2729. doi:10.1097/01.asn.0000025282.48298.7b
69. Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669–2701. doi:10.2337/dci18-0033
70. American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2019. Diabetes Care. 2019;42:S90–S102. doi:10.2337/dc19-S009
71. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi:10.1007/s00125-017-4342-z
72. Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi:10.2337/diabetes.51.8.2420
73. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi:10.1172/JCI13505
74. Rowe I, Chiaravalli M, Mannella V, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19:488–493. doi:10.1038/nm.3092
75. Lian X, Wu X, Li Z, et al. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br J Pharmacol. 2019;176:711–724. doi:10.1111/bph.14558
76. Capuano I, Riccio E, Caccavallo S, De Simone I, Pisani A. ADPKD and metformin: from bench to bedside. Clin Exp Nephrol. 2019;23:1341–1342. doi:10.1007/s10157-019-01770-1
77. Coppolino G, Bolignano D, Rivoli L, et al. Tumour markers and kidney function: a systematic review. Biomed Res Int. 2014;2014:647541. doi:10.1155/2014/647541
78. Coppolino G, Leporini C, Rivoli L, et al. Exploring the effects of DPP-4 inhibitors on the kidney from the bench to clinical trials. Pharmacol Res. 2018;129:274–294. doi:10.1016/j.phrs.2017.12.001
79. Pisani A, Riccio E, Bruzzese D, Sabbatini M. Metformin in autosomal dominant polycystic kidney disease: experimental hypothesis or clinical fact? BMC Nephrol. 2018;19:282. doi:10.1186/s12882-018-1090-3
80. Sorohan BM, Ismail G, Andronesi A, et al. A single-arm pilot study of metformin in patients with autosomal dominant polycystic kidney disease. BMC Nephrol. 2019;20:276. doi:10.1186/s12882-019-1463-2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.