Back to Archived Journals » Journal of Neurorestoratology » Volume 3
Research progress in animal models and stem cell therapy for Alzheimer’s disease
Received 11 September 2014
Accepted for publication 29 October 2014
Published 19 December 2014 Volume 2015:3 Pages 11—22
DOI https://doi.org/10.2147/JN.S74160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Hongyun Huang
Fabin Han,1,2 Wei Wang1, Chao Chen1
1Centre for Stem Cells and Regenerative Medicine, 2Department of Neurology, Liaocheng People’s Hospital/The Affiliated Liaocheng Hospital, Taishan Medical University, Shandong, People’s Republic of China
Abstract: Alzheimer’s disease (AD) causes degeneration of brain neurons and leads to memory loss and cognitive impairment. Since current therapeutic strategies cannot cure the disease, stem cell therapy represents a powerful tool for the treatment of AD. We first review the advances in molecular pathogenesis and animal models of AD and then discuss recent clinical studies using small molecules and immunoglobulins to target amyloid-beta plaques for AD therapy. Finally, we discuss stem cell therapy for AD using neural stem cells, olfactory ensheathing cells, embryonic stem cells, and mesenchymal stem cell from bone marrow, umbilical cord, and umbilical cord blood. In particular, patient-specific induced pluripotent stem cells are proposed as a future treatment for AD.
Keywords: amyloid-beta plaque, neurofibrillary tangle, neural stem cell, olfactory ensheathing cell, mesenchymal stem cell, induced pluripotent stem cell
Introduction
Dementia is mainly characterized by impairment of cognitive and memory functions and is involved in a series of neurological diseases. These diseases include Alzheimer’s disease (AD), frontotemporal dementia, stroke-induced vascular dementia, and mixed dementia. AD and stroke-induced vascular dementia are reported to account for 80%–90% of all dementia cases. AD is a neurodegenerative disease characterized by a gradual loss of memory, language, and cognitive function.
AD is estimated to affect over 5 million people in the USA and more than 30 million people worldwide. In the USA, more than 3%–5% of persons over the age of 65 years are reported to suffer from AD.1,2 In this review, we focus on animal models and stem cell treatment for AD.
Etiology and pathological mechanisms of AD
In the past two decades, the most important progress in AD research has been the identification of senile plaques composed of amyloid-beta (Aβ) peptide and tau protein-related neurofibrillary tangles (NFTs) as pathological hallmarks in the brains of patients with AD. Etiologically, AD is classified into two forms, ie, familial AD and sporadic AD. Clinically and pathologically, familial AD and sporadic AD are similar, with both types of patients exhibiting progressive cognitive impairment and pathological plaque deposits in their brains. Familial AD and sporadic AD also share other neuronal phenotypic characteristics, including axonal transport defects, synapse loss, and selective neuronal death.1,3 Most AD cases are sporadic, whereas familial AD accounts for only 5% of all AD cases. Molecular genetic studies have identified causative genes for familial AD, the most common being the amyloid precursor protein (APP) gene and two presenilin genes (PS1 and PS2). Although no mutations in the tau gene have been identified in AD, several studies have shown an association between Aβ and hyperphosphorylated tau. Direct interaction between tau and Aβ peptide was shown to induce tau aggregation and hyperphosphorylation.4
The synaptic plasticity impairments induced by Aβ peptide are associated with tau phosphorylation, indicating tau protein is required for the neurotoxic effects of Aβ deposits.5 In the pathogenesis of AD, aggregated Aβ plaque and tau-related NFTs deposit in the brain to induce neuronal cytotoxicity, extensive neuronal degeneration, and programmed death of nerve cells by the end stages of the disease (Figure 1A). The APP protein is highly expressed in the central nervous system, where it is thought to play important roles in synapse formation, neurogenesis, axonal transport, cell signaling, and plasticity. As a type I transmembrane protein, APP is normally cleaved by β-secretase or γ-secretase. When APP is sequentially cleaved, first by β-secretase and then by γ-secretase, Aβ peptide fragments of the 40 and 42 amino acids (Aβ40 and Aβ42) are produced. These Aβ peptides eventually form Aβ-oligomers or Aβ–polymers, which aggregate in the neural cells to form Aβ plaques (Figure 1B).6 PS1 and PS2 are transmembrane protein components of the γ-secretase enzyme, which cleaves multiple substrates, including APP. In addition to γ-secretase activity, PS1 and PS2 are also involved in regulation of the endosome/lysosome pathway and autophagy.7 NFTs are mainly composed of tau aggregates. In the process of formation of NFTs, tau protein is first phosphorylated, and phosphorylated tau proteins are then aggregated to form highly ordered fibrils and NFTs, as shown in Figure 1C.8,9 Because of the Aβ plaque deposits and NFTs in the brains of patients with AD, the neural cells shrink and induce apoptosis progressively. Such programmed cell death occurs first in the brain regions responsible for memory and language and ultimately spreads to the entire brain. In the apoptotic process for neuronal cells, cholinergic neurons and neuronal synapses are first affected and gradually degenerate to induce shrinkage of the brain. Many brain regions are then affected, and display amyloid plaques and NFTs.10–12
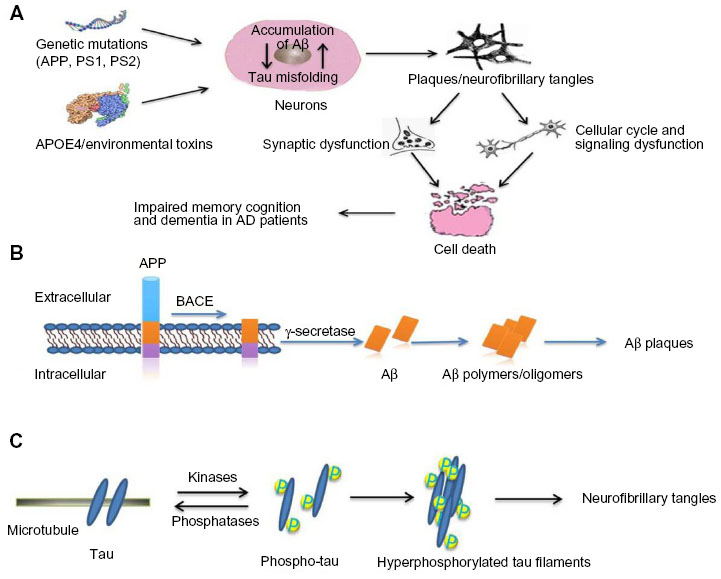 | Figure 1 Pathological molecular mechanism of AD. |
Animal models of AD
Since the etiology of AD is not completely known, a wide range of animal models have been developed to study the pathological process and screen therapeutic agents for the disease. Even though none of these animal models have fully recapitulated the phenotypes of AD, they have been proved to be valuable resources for studying the pathogenesis of AD. Current animal models of AD include natural models, lesioned models, and transgenic models.
Natural AD models
Aging dog, cats, and nonhuman primates (mouse lemurs, Rhesus monkeys, Octodon degus) develop some forms of neurological dysfunction, but most of these animals do not develop AD-related neuropathology similar to that occurring in humans. Aβ plaque deposition has been found in the brains of aging dogs with cognitive dysfunction, but no NFTs were detected. Like humans, dogs have a fully conserved Aβ amino acid sequence, so aged dogs have been used as a natural AD model to evaluate whether or not certain therapeutic drugs can reduce Aβ deposits and produce cognitive improvement.13 Mouse lemurs, being nonhuman primates, are potential animal models for AD because they live for 8–10 years and develop certain age-related changes similar to those that occur in aging humans. Aging mouse lemurs show amyloid plaque, NFTs, and other AD-related neuropathological changes such as neuron loss in their brains. It is reported that about 20% of mouse lemurs aged 5 years or older show some degree of significant brain atrophy, amyloid plaque deposits, and loss of cholinergic neurons similar to the pathological features found in patients with AD.14–16 Rhesus monkeys are the models most commonly used to develop effective treatments for human neurological disorders as they share many aging diseases with humans. Monkeys over 20 years of age develop significant amyloid plaque-like inclusions in regions of the brain responsible for cognitive and memory. However, most aged monkeys do not show the rapid cognitive decline commonly found in AD patients.17 The Octodon degu, a South American rodent, has recently been reported to spontaneously develop AD-related amyloid deposits and NFT-like intracellular inclusions in the cortex and hippocampus in older age. Pathological examination showed the presence of cholinergic neurons in the cortex of the adult Octodon degu similar to those found in humans. Some age-associated cognitive impairment and memory dysfunction could also be observed in the adult Octodon degu. The high degree of homology of amino acid sequences (97.5%) between the Octodon degus and humans might contribute to the appearance of AD plaques and NFT-like deposits, making the aged Octodon degu a natural rodent model for studying the neurodegenerative mechanisms associated with AD.18
Lesioned AD models
Lesioned AD models are produced by applying chemical substances or physical instruments to destroy specific brain regions such as the hippocampus and cortex to induce cognitive dysfunction. Some studies have introduced Aβ peptide into the brains of rats or rhesus monkeys to induce some of the characteristic features of AD. However, although these models replicated some features similar to the clinical signs of AD, they did not resemble the pathological process of AD.19,20 The advantages of these models are that they induce specific brain lesions for study of memory dysfunction, motor dysfunction, and the mechanism of neuronal cell death, allowing understanding of the inflammation, neurotoxicity, neurodegeneration, and synaptic function of lesioned brain regions. The disadvantages of the lesioned models include the nonspecificity of the lesions and their failure to capture disease progression and other aging aspects of AD.21,22
Transgenic animal models
The development of transgenic technology makes it possible to overexpress or silence genes in different animals, and thereby shed light on the molecular mechanisms involving APP, PS1, PS2, and other genes in AD. APP transgenic mice (a Tg2576 mouse line) were generated by overexpressing the APP gene and show marked Aβ deposition similar to those seen in human AD, but at an early age. By 9–12 months, these mice develop extracellular AD-like Aβ deposits in the cerebral cortex and hippocampus. Nevertheless, the biochemical composition of the Aβ deposits in the transgenic mouse is different from that in the brains of human AD patients. In addition, these transgenic mice do not develop tau-related NFTs, as seen in the brains of AD patients.23,24 Transgenic mice overexpressing PS1 were shown to have some related neuronal loss in the hippocampus and neocortex and elevated levels of Aβ42, but abnormal pathological AD plaques were not observed.25 Subsequently, a double transgenic mouse model was generated by crossing the transgenic APP line (Tg2576) and a mutant PS1 (with M146L mutation) line, which was shown to have large numbers of fibrillary Aβ deposits in the cerebral cortex and hippocampus far earlier than seen in single transgenic Tg2576 mice. The double transgenic mice also showed a selective increase in Aβ42/43 in their brains. The neuron loss was seen to be more severe in double transgenic mice with both APP and PS1 mutations.26 In order to study further the molecular pathological process induced by genetic mutations of APP, PS1, and PS2, a triple transgenic mouse model with APP, PS1, and PS2 was generated and shown to have AD-relevant pathological features in some regions of the brain. These mice developed extracellular Aβ deposits and NFTs, and showed impaired synaptic plasticity, including long-term potentiation and cognitive function. This transgenic mouse model also provided a valuable approach to the study of inflammation, oxidative stress, and mitochondrial dysfunction in the pathogenesis of AD.27,28 However, the progressive neuronal loss in the hippocampus and other specific neocortical regions of the human AD brain is not evident in most of the transgenic mouse models, and this is a major limitation of these murine systems. Currently, there is no mouse model that can fully reproduce the features of pathological progression found in the majority of sporadic or late-onset AD cases. In addition, any experiment results acquired from animal models need to be consolidated in clinical trials before they can be extrapolated to humans. Transgenic rat models of AD were also generated by expressing AD-related human genes for APP and PS1 a decade ago.20,29 However, few rat models have been reported to develop intracellular and extracellular Aβ accumulation and cognitive deficits.30,31 A recent study developed a transgenic rat line (McGill-R-Thy1-APP) expressing mutant human APP carrying double mutations. These rats were found to develop extracellular Aβ deposits by 6 months of age and impairment of cognition as early as 3 months of age.32
The fruit-fly Drosophila melanogaster is widely used to model neurodegenerative diseases, including AD.33 The advantages of the fruit-fly are as follows. First, its brain is similar to that in humans in terms of the fundamental structures and functions of gene expression, membrane trafficking, neuronal connectivity, cell signaling, and synaptogenesis, even though it has only a small fraction of the cells found in the human brain.34 Second, it has a short life cycle and its genome was able to be completely sequenced to develop the transgenic fruit-fly model of AD. Third, the learning and memory dysfunction in patients with AD can be measured in the fruit-fly model. The limitation of this model is that the impaired cognitive functions found in human AD cannot be detected as in rodent models.
The zebrafish was originally used as a model organism for the study of vertebrate development and has also been used to study causes and mechanisms of human disease, such as AD.35 Although zebrafish lack the advanced cognitive behaviors seen in rodent models, their rapid development and large reproductive capacity (more than 100 embryos per spawning) provide obvious advantages over mammalian models. As a vertebrate model, zebrafish has been used to reveal particular characteristics of the various genes implicated in AD, which have been difficult to observe in other animal models.36,37 A mutant zebrafish lacking PSEN1 has been used to study the histaminergic neural system of the brain, which is essential in mediating the cognitive functions affected in AD. Analysis of histamine neuron numbers in the PSEN1–/– zebrafish brain revealed that PSEN1 is a regulator of histaminergic neuronal development.38 Blocking of translation of the PSEN2 protein in zebrafish has major effects on Notch signaling in comparison with PSEN2-/- mice which show only a minor phenotype.39–41 Furthermore, loss of PSEN2 expression affects the production of dorsal longitudinal ascending interneurons in the developing spinal cord of zebrafish larvae.40 Translation-blocking morpholinos have also been used to investigate the function of Appa and Appb (orthologs of human APP protein).42 Inhibition of Appa had little effect on the developing embryo, while inhibition of translation of Appb resulted in defective convergent extension cellular movements and a reduced body length. Loss of Appb activity in zebrafish has been shown to cause defective neural development, including defective axonal outgrowth patterning and synapse formation.43 These neural defects could only be rescued by full-length human APP and not by truncated forms, indicating that both the intracellular and extracellular domains of human APP are required for normal function.44 These studies demonstrate that zebrafish embryos can be exploited for analysis of different mutant forms of human APP when studying the molecular mechanism of AD.
Emerging small-molecule and antibody treatments for AD in clinical trials
The drugs currently approved by the US Food and Drug Administration for AD include four cholinesterase inhibitors (tacrine, donepezil, rivastigmine, or galantamine) and an N-methyl-D-aspartate receptor antagonist (memantine). Development of new treatments for AD involves inhibition of neurodegeneration and activation of neural regeneration. Given that Aβ deposits are the key pathological hallmarks of AD, blocking the signaling pathway initiated by Aβ may represent a promising therapeutic strategy in the disease.45–48 Several ongoing clinical trials include chemical agents, small molecules immunomodulation and growth factors focused on removing Aβ plaques and decreasing the production and toxicity of Aβ. More therapies have been designed to target Aβ plaques. Some anti-Aβ monoclonal antibodies, immunoglobulins, Aβ Vaccines, and γ-secretase or β-secretase inhibitors to reduce the formation of Aβ plaques have also been in clinical trials. Developing neurotrophic factor-based compounds that can increase neurogenesis and rescue neuronal plasticity represents a promising approach. A ciliary neurotrophic factor peptidergic compound (peptide 6c) was found to increase neurogenesis and rescue neuronal/synaptic plasticity in aged mice. Certain tetrapeptides corresponding to the active regions of brain-derived neurotrophic factor (BDNF) were also found to induce expression of neuronal markers in primary hippocampal neuronal cultures of mouse embryonic day 18, suggesting that BDNF tetrapeptides could modulate BDNF signaling to improve neurogenesis.49,50 Most recently, a large study has examined a public website (http://www.clinicaltrials.gov) that records clinical trials from 2002 to 2012. In total, 413 AD trials were performed, including 124 Phase I trials, 206 Phase II trials, and 83 Phase III trials. Most of these trials involved agents intended to improve symptomatic cognition, but some trials using small molecules to modify the disease process and immunoglobulins to improve immunomodulation in AD were performed.51 Although some treatments cleared Aβ plaques from the brains of AD patients, the treated patients showed even worse performance than the placebo-treated controls. One Phase III clinical trial on immunoglobulin-based therapy reduced Aβ pathology but failed to improve cognitive deficits.52,53 No new treatments have been approved for AD since 2003. Most of the Aβ-based treatments failed to show efficacy or were toxic in human clinical trials, suggesting new therapeutic approaches need to be explored.54
Stem cell therapy for AD
Over the past two decades, stem cell-based therapies have been widely studied as a novel strategy for the treatment of AD. Stem cell studies have shown that neurogenesis occurs in the developing brain and that neural progenitor cells (NPCs) reside in the adult central nervous system. Thus, stem cells hold great potential for repair of the injured central nervous system and treatment of neurological diseases, particularly neurodegenerative conditions like AD. To this end, two therapeutic strategies are being considered, ie, stimulation of endogenous NPCs in the adult brain to regenerate neurons and transplantation of neural stem cells (NSCs) to repair degenerated or injured neural cells and tissues.55 Sources of stem cells for transplantation include human embryonic stem cells (ESCs), NSCs in the fetal brain, adult bone marrow-derived mesenchymal stem cells (MSCs), olfactory ensheathing cells, umbilical cord MSCs, and human induced pluripotent stem cells (iPSCs), as summarized in Figure 2.
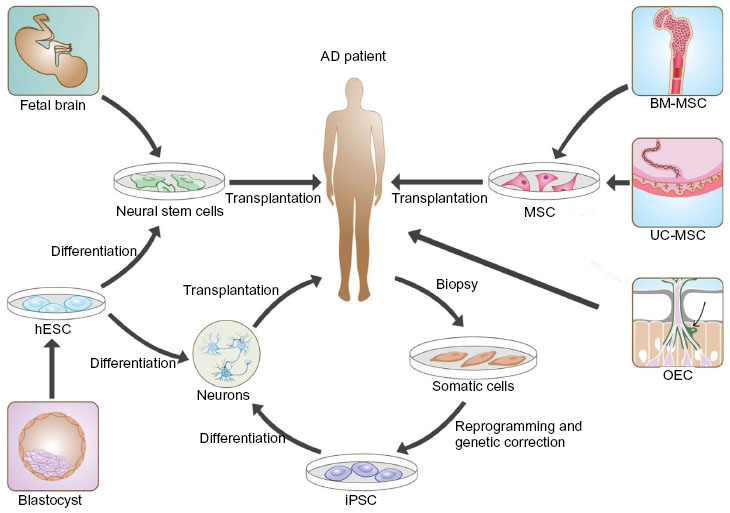 | Figure 2 Stem cell sources for AD therapy. |
Endogenous activation of adult NSCs
Some chemical compounds and cellular factors, such as allopregnanolone, fluoxetine, granulocyte colony-stimulating factor and stromal cell-derived factor-1α have been shown to be able to activate endogenous neurogenesis. Allopregnanolone has been shown to induce endogenous neurogenesis in triple transgenic AD mice and to induce activation of NPCs and the production of BrdU+ cells to improve learning and memory function. In addition, activation of microglia and increased neuroprotection were also found to be induced by allopregnanolone.56 Combined administration of granulocyte colony-stimulating factor, AMD3100, and stromal cell-derived factor-1α stimulated the migration of bone marrow-derived hematopoietic progenitor cells into different brain areas to produce a therapeutic effect in AD mice. A neuropathological study showed that hippocampal neurogenesis and memory function were increased even though Aβ deposition was not changed in the AD mouse brain.57 A recent study has shown that treatment with fluoxetine promoted neuronal differentiation and had increased protective effects against Aβ-induced cell death in AD.58 Because of aging and neurodegeneration in AD patients, activating endogenous NSCs to differentiate into neurons in the adult brain is limited. To increase differentiation of endogenous NSCs, some mechanisms and signal pathways underlying the specific neurogenic niches will need to be unraveled and modified to enhance the therapeutic potential of adult endogenous NSCs.59
NSC therapy
NSCs isolated from postnatal day 14 mice were transplanted to the hippocampus in the mouse AD model to explore the effectiveness of NSC transplantation. The transplanted mouse NSCs were able to survive, differentiate, and improve memory in the mouse brain.60 A mouse transgenic model of AD was generated using the tetracycline-inducible promoter system to temporally and spatially regulate expression of the neural toxin, diphtheria toxin A-chain and thereby selectively induce neural loss in the forebrain. Cognitive function in the transgenic AD mice was evaluated both before and after an inducible brain lesion.60 After confirmation of the cognitive and memory deficits, mouse NSCs were transplanted into the hippocampi of the transgenic AD mice. The transplanted NSCs were found to survive for at least 4.5 months, and to migrate and differentiate into neurons and glia. NSC transplantation significantly increased hippocampal-dependent memory, synaptic density, and neuronal numbers in the mouse brain, suggesting that NSCs have some therapeutic role in improving memory dysfunction in transgenic AD mice. A 2 year-study was performed by transplanting mouse NSCs into the brains of triple transgenic AD mice and showed that NSC transplantation improved memory and learning, even though the pathological tau tangles were not changed in this AD model.61 A recent study modified NSCs to express metalloproteinase 9 (MMP-9), a secreted protease that degraded aggregated Aβ peptides once NSCs were transplanted into APP/PS1 AD mice via direct injection into their brains. These engineered MMP-9 NSCs survived in the AD mouse brain, but were mainly distributed in the white matter tracts without migrating to amyloid plaques, indicating that the delivery procedure needs to be improved to enhance the therapeutic efficacy of NSC transplantation.62 In a recent study, NSCs were transplanted bilaterally into hippocampal regions in the APP/PS1 transgenic mouse. NSCs were found to improve spatial learning and memory function in these mice, but did not decrease Aβ deposits.63
Immunohistochemical analyses showed that NSCs proliferated, migrated, and differentiated into three neuronal cell types. The improvement in cognitive function was correlated with enhanced long-term potentiation and increased neuronal expression of proteins related to cognitive function, such as N-methyl-D-aspartate, synaptophysin, and BDNF.
Human NSCs (hNSCs) are derived from the fetal brain at 3–4 months’ gestation. hNSCs were shown to differentiate into neurons and glial cells, and have been used in the treatment of neurological diseases, including AD. In order to increase survival and neural differentiation, several studies have used genetic approaches to modify hNSCs to express growth factors such as nerve growth factor (NGF) or BDNF to make hNSC-NGF cells or hNSC-BDNF cells, respectively. NSCs expressing BDNF or NGF were shown to restore learning and memory function in AD animals.64 Intrahippocampal transplantation of hNSC-NGF cells was found to have increased survival and differentiation of hNSC-NGF cells into neurons and astrocytes in the rat brain. The transplanted hNSC-NGF cells were also found to express NGF and improve learning and memory function in chemically lesioned AD rats.65 Given that dysfunction of cholinergic neurons and decreased activity of choline acetyltransferase (ChAT) are the main causes of the cognitive deficits in AD, another study genetically engineered hNSCs to express the ChAT gene (hNSC-ChAT cell line) to improve cognitive function. After transplantation of hNSC-ChAT cells into rats with learning deficits induced by kainic acid, learning and memory function was improved, with elevation of acetylcholine levels seen in the cerebrospinal fluid.66
Olfactory ensheathing cells are isolated from adult or fetal olfactory bulb tissue, and are capable of enhancing neuronal transduction through the axons of the neurons to promote the function of neurons. Olfactory ensheathing cells were reported to continuously secrete neurotrophic factor, increasing the survival of transplanted cells. Expression of ChAT was significantly increased in rats cotransplanted with olfactory ensheathing cells and neural progenitor cells when compared with animals transplanted with either type of cells alone and cognitive dysfunction was improved in rats with co-transplantation of these two types of cells.67 In some studies, olfactory ensheathing cells have been cotransplanted with NSCs or other cell types to repair brain and spinal cord injuries.68–70 Cotransplantation of NPCs with olfactory ensheathing cells in the kainic acid-lesioned rat model was reported to improve functional recovery of learning and memory.67 Another study showed that learning and memory function recovered when hNSCs overexpressing the human ChAT gene were transplanted into the AD rat brain.71 Most recently, genetically modified human olfactory bulb NSCs have been shown to reverse the cognitive deficit in a rat model of AD. These cells were engineered to express human NGF by lentivirus-mediated infection and were transplanted into the hippocampi of rats with AD induced with ibotenic acid. Olfactory bulb NSC-human NGF cells were found to differentiate into mature neurons, oligodendrocytes, and astrocytes, and to improve memory and learning in AD rats.72
ESC therapy
Since NSCs derived from the fetal brain have limited ability to differentiate into neurons in vitro, some studies have investigated ESCs isolated from the inner cell mass of an embryonic blastocyst for the treatment of AD. Tang et al reported that transplantation of NSCs derived from mouse ESCs significantly improved memory 16 weeks after transplantation in AD rats.73 Moghadam et al investigated ESC-derived NPCs and cholinergic neurons of primed NPCs from mouse ESCs for treatment of AD in a rat model. ESCs were induced to primed NPCs by addition of Shh to the neural induction medium and were found to differentiate into cholinergic neurons better when NPCs were primed with Shh. Behavioral analysis revealed a significant improvement in memory deficits following transplantation with primed NPCs (cholinergic neurons), indicating that ESC-derived primed NPCs are more suitable for restoring memory deficits in AD rats.74
MSC therapy
Some studies have shown that memory and cognitive deficits can be alleviated by transplanting MSCs derived from adult bone marrow, the umbilical cord, and umbilical cord blood into the brain regions of animals with AD.75 Lee et al produced an AD model by injecting Aβ into the hippocampi of C57BL/6 mice and found that transplantation of bone marrow-derived MSCs into the mouse brain significantly reduced Aβ deposits. They also transplanted human bone marrow-derived MSCs into double-mutant APP and PS1 transgenic mice and found reduced Aβ deposition and improved restoration of defective microglial function by increasing Aβ-degrading factors and decreasing inflammatory responses. APP/PS1 transgenic mice treated with bone marrow-derived MSCs were also found to have improved cognitive function and decreased tau hyperphosphorylation. They found that reduction of Aβ deposits is accompanied with increasing activation of microglia in the mouse model of AD, suggesting bone marrow-derived MSC transplantation may promote the reduction of Aβ deposition through microglial activation.76,77 A recent study found that transplantation of bone marrow-derived MSCs significantly reduced cerebral Aβ deposition in young AD mice without cognitive features of the disease. Molecular analyses showed that the presynaptic proteins dynamin 1 and synapsin 1, which are typically decreased in the brains of AD patients, were upregulated in the brains of AD mice treated with bone marrow-derived MSCs.75 This study demonstrated that bone marrow-derived MSC transplantation has potential therapeutic effects in early AD. In another study, transplanted bone marrow-derived MSCs were distributed near Aβ deposits in the brains of transgenic AD mice and the distributions of the transplanted cells were associated with decreased Aβ deposits, indicating that human bone marrow-derived MSC transplantation is effective in AD therapy.78
Inflammation of the central nervous system has recently been suggested to play an important role in the pathogenesis of AD.79 Thus, transplantation of immune-regulating cells could improve the inflammation and have a therapeutic role in AD. Combined transplantation of T regulatory cells and MSCs from human umbilical cord tissue into AβPPswe/PS1dE9 double transgenic AD mice has been reported to ameliorate impaired cognition and reduced Aβ plaque deposition. In addition, there was an increase in soluble Aβ and a decrease in activated microglia and systemic inflammatory factors, indicating that cotransplantation of T regulatory cells and MSCs may have a synergistic effect in the treatment of AD.80
Molecular mechanisms of stem cell transplantation
The molecular mechanisms via which stem cell transplantation improves memory and cognition in animal models of AD are not clear, but possibly include cell replacement, release of neurotrophic and neuroprotective factors, endogenous activation of neurogenesis, anti-inflammatory activity, and stem cells as carriers to deliver therapeutic proteins to the degenerated regions of the brain and decrease Aβ deposits. NSCs can express many neurotrophins, including BDNF, GDNF and NGF.61 BDNF, GDNF and NGF are major regulators of synaptic plasticity. Rats with traumatic brain injury were intravenously treated with human MSCs, and tissue samples taken from the traumatized cerebral hemispheres with grafted MSCs 24 hours after injury showed significantly increased expression of NGF, BDNF, and neurotrophin-3.81
Stem cells can secrete anti-inflammatory factors, such as interleukin-10, an anti-inflammatory cytokine, and prostaglandin E2, to inhibit the inflammatory process in AD. Transplantation of NSCs can also influence endogenous neurogenesis in the host. Transplanted NSCs derived from human ESCs reduced infarct volume and improved behavioral outcome after distal middle cerebral artery occlusion in rats. In this study, transplantation was able to increase the number of neurons expressing doublecortin in the subventricular zone (SVZ), but not in the contralateral SVZ or dentate gyrus zone in rat brains 60 days post-transplantation.82 To determine if MSCs stimulate endogenous neurogenesis, MSCs were transplanted to the normally restrictive SVZ of mice and were found to increase proliferation and neuronal differentiation of neural progenitors within the SVZ. The proportion of newborn neurons was increased out of the total neurons.83 A recent study showed that adipose tissue-derived MSCs transplanted into the hippocampi of APP/PS1 transgenic AD mice significantly increased the number of BrdU/doublecortin-stained cells in the SVZ of the dentate gyrus in the hippocampus, suggesting that MSC transplantation improves memory and cognition by enhancing neurogenesis in APP/PS1 transgenic AD mice.84 Bone marrow-derived MSCs were shown to increase leukocyte activation and promote leukocyte-endothelial interactions, and were also found to significantly inhibit transcriptional activation of nuclear factor-kappa B (NF-κB) and inhibit DNA binding of NF-κB subunits p50 and p65 to putative NF-κB DNA binding sites by the direct effects of MSCs on interleukin-10 and NF-κB.85
In a study that focused on alleviating amyloid pathology by transplanting NSCs from postnatal mice, the NSCs was modified to express MMP-9, a protease that degrades aggregated Aβ peptides. Even though some endogenous MMP-9 was detected around amyloid plaques in the mouse model, stem cell-delivered MMP-9 was not found to have an impact on Aβ plaques. This study suggested that this delivery approach needs to be improved to enhance the therapeutic efficacy of the transplanted cells.62
iPSCs for modeling and treating AD
Our understanding of the pathogenesis of AD is currently limited by difficulties in obtaining viable neurons from patients and our inability to model the sporadic form of the disease. Current transgenic animal models have provided important insights into AD; however, these models often do not recapitulate all of the human AD phenotype and pathology by overexpressing mutant proteins or introducing genetic mutations into endogenous genes in transgenic models.86
Recent advances in somatic cell reprogramming have generated iPSCs by overexpressing four transcription factors, ie, OCT4, SOX2, c-MYC, and KLF4, in skin fibroblasts, blood cells, and urine cells. These iPSCs can be derived from autologous human cells and can be differentiated into any type of cell, including neurons and glial cells. Since iPSCs overcome the shortages of immune rejection and ethical issues related to other human stem cells, iPSCs will have great potential in modeling diseases and autologous transplantation therapy for neurodegenerative diseases, including AD.87–90
The first iPSC model of AD was generated from fibroblasts of AD patients with mutations in PS1 (A246E) and PS2 (N141I) and characterized by differentiating these iPSCs into neurons. These neurons derived from iPSCs of AD patients were shown to increase Aβ42 deposits induced by mutant PS1/PS2. Importantly, secretion of Aβ42 from these neurons was shown to be regulated by certain γ-secretase inhibitors and modulators, suggesting their potential use in identifying candidate drugs for AD.91 A recent study has generated iPSCs from AD patients with a duplication of the APP gene. The neurons differentiated from these iPSCs had significantly higher levels of Aβ, phospho-tau, and active glycogen synthase kinase-3β. In addition, treatment of the iPSC-derived neurons with β-secretase inhibitors caused significant reductions in phospho-tau and active glycogen synthase kinase-3β levels. These results indicate that iPSCs can be used to analyze disease phenotypes relevant to AD.92 Since these iPSCs were generated by retroviral or lentiviral vectors to express reprogramming factors of OCT4, SOX2, c-MYC, and KLF4, these exogenous genes and retroviral or lentiviral vectors may integrate into the iPSC genome to disrupt the genome and cause mutations or carcinogenesis. Thus, retroviral or lentiviral iPSCs may not be suitable for transplantation therapy. Most recently, there has been a study showing generation of virus-free human iPSCs from AD patients with a PS1 mutation (A246E) using nonintegrating episomal vectors. Neurons from mutant iPSC cell lines were found to express PS1-A246E mutations and to cause Aβ deposits, as seen in the brains of AD patients. These iPSCs harboring a PS1 gene mutation could be used as humanized models to explore the pathogenesis of AD and screen for therapeutic drugs.93 However, iPSC-derived neurons still carry APP, PS1 and PS2 gene mutations, and cannot be directly transplanted into AD patients for therapy. To overcome this problem, several genetic approaches have been developed to correct the mutations in mutant iPSCs by homologous recombination, as shown in other studies, and the mutation-corrected neurons can then be transplanted into patients for treatment of AD.94,95
Conclusion and perspectives
Over the past decade, treating AD with drugs targeting Aβ plaques has failed in almost all of the clinical trials, suggesting new approaches are needed to improve learning and cognitive function in patients with AD. Stem cells are able to differentiate into neurons and glial cells to replace damaged cells, secreting neurotrophic molecules or modulating inflammation to restore memory and cognitive function in AD patients. However, clinical application of stem cells to treat AD will require more basic research so that the mechanisms underlying the pathological process of AD and regulation of the proliferation, migration, differentiation, survival, and function of stem cells and their derivatives are better understood and can be effectively controlled. While some therapeutic effects have been observed following transplantation of stem cells in animal models of AD, to date few studies have been explored to transplant stem cells for clinical trials of AD. Some stem cell sources, such as MSCs derived from bone marrow, umbilical cord, and umbilical cord blood, are easily accessible; however, these cells have limited neural differentiation and may induce immune rejection because they are allogenic. Human ESCs and NSCs can be induced to generate a large amount of neurons for transplantation, but these cells come with ethical issues that have hindered their clinical application in AD therapy. iPSCs are derived from reprogrammed somatic cells such as fibroblast, blood or urine cells from patients themselves and overcome the immune rejection and ethical issues concerning stem cells. Thus, patient-specific iPSCs will have invaluable potential for treatment of AD. However, before iPSCs can be applied in AD patients, several aspects regarding mutation correction, neural differentiation, purification, and long-term survival of iPSCs following transplantation need to be optimized and standardized.
Acknowledgments
The authors are grateful to Shichao Wu and Junyu Gao for their technical assistance in preparation of the figures for the manuscript. This work was supported by the Developmental Fund of Shandong Science and Technology Department, Shandong, People’s Republic of China (2011YD18054).
Disclosure
The authors report no conflicts of interest in this work.
References
Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr1. | |
Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–828. | |
Goldstein LS. Axonal transport and neurodegenerative disease:can we see the elephant? Prog Neurobiol. 2012;99(3):186–190. | |
Rank KB, Pauley AM, Bhattacharya K, et al. Direct interaction of soluble human recombinant tau protein with Abeta 1-42 results in tau aggregation and hyperphosphorylation by tau protein kinase II. FEBS Lett. 2002;514(2–3):263–268. | |
Shipton OA, Leitz JR, Dworzak J, et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31(5):1688–1692. | |
Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615–29619. | |
Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. | |
Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2(7):a006247. | |
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau:sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33 Suppl 1:S123–S139. | |
Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32(3):389–401. | |
Brunholz S, Sisodia S, Lorenzo A, Deyts C, Kins S, Morfini G. Axonal transport of APP and the spatial regulation of APP cleavage and function in neuronal cells. Exp Brain Res. 2012;217(3–4):353–364. | |
Reitz C. Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis. 2012;2012:369808. | |
Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res. 1991;10(4):299–305. | |
Languille S, Blanc S, Blin O, et al. The grey mouse lemur: a non-human primate model for ageing studies. Ageing Res Rev. 2012;11(1):150–162. | |
Bons N, Rieger F, Prudhomme D, Fisher A, Krause KH. Microcebus murinus: a useful primate model for human cerebral aging and Alzheimer’s disease? Genes Brain Behav. 2006;5(2):120–130. | |
Kraska A, Dorieux O, Picq JL, et al. Age-associated cerebral atrophy in mouse lemur primates. Neurobiol Aging. 2011;32(5):894–906. | |
Buccafusco JJ. Estimation of working memory in macaques for studying drugs for the treatment of cognitive disorders. J Alzheimers Dis. 2008;15(4):709–720. | |
Inestrosa NC, Reyes AE, Chacon MA, et al. Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol Aging. 2005;26(7):1023–1028. | |
Castane A, Theobald DE, Robbins TW. Selective lesions of the dorsomedial striatum impair serial spatial reversal learning in rats. Behav Brain Res. 2010;210(1):74–83. | |
Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T. Progressive brain dysfunction following intracerebroventricular infusion of beta(1–42)-amyloid peptide. Brain Res. 2001;912(2):128–136. | |
Van Dam D, De Deyn PP. Animal models in the drug discovery pipeline for Alzheimer’s disease. Br J Pharmacol. 2011;164(4):1285–1300. | |
Li W, Wu Y, Min F, Li Z, Huang J, Huang R. A nonhuman primate model of Alzheimer’s disease generated by intracranial injection of amyloid-beta42 and thiorphan. Metab Brain Dis. 2010;25(3):277–284. | |
Kalback W, Watson MD, Kokjohn TA, et al. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry. 2002;41(3):922–928. | |
Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimers Dis. 2006;9(Suppl 3):195–207. | |
Chui DH, Tanahashi H, Ozawa K, et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5(5):560–564. | |
Rutten BP, Van der Kolk NM, Schafer S, et al. Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am J Pathol. 2005;167(1):161–173. | |
Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. | |
Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14(10):1097–1105. | |
Lopez EM, Bell KF, Ribeiro-da-Silva A, Cuello AC. Early changes in neurons of the hippocampus and neocortex in transgenic rats expressing intracellular human a-beta. J Alzheimers Dis. 2004;6(4):421–431. | |
Vercauteren FG, Clerens S, Roy L, et al. Early dysregulation of hippocampal proteins in transgenic rats with Alzheimer’s disease-linked mutations in amyloid precursor protein and presenilin 1. Brain Res Mol Brain Res. 2004;132(2):241–259. | |
Flood DG, Lin YG, Lang DM, et al. A transgenic rat model of Alzheimer’s disease with extracellular Abeta deposition. Neurobiol Aging. 2009;30(7):1078–1090. | |
Leon WC, Canneva F, Partridge V, et al. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-β-associated cognitive impairment. J Alzheimers Dis. 2010;20(1):113–126. | |
Iijima K, Iijima-Ando K. Drosophila models of Alzheimer’s amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-beta 42. J Alzheimers Dis. 2008;15(4):523–540. | |
Sang TK, Jackson GR. Drosophila models of neurodegenerative disease. NeuroRx. 2005;2(3):438–446. | |
Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8(5):353–367. | |
Liao HK, Wang Y, Noack Watt KE, et al. Tol2 gene trap integrations in the zebrafish amyloid precursor protein genes appa and aplp2 reveal accumulation of secreted APP at the embryonic veins. Dev Dyn. 2012;241(2):415–425. | |
Newman M, Nornes S, Martins RN, Lardelli MT. Robust homeostasis of presenilin1 protein levels by transcript regulation. Neurosci Lett. 2012;519(1):14–19. | |
Sundvik M, Chen YC, Panula P. Presenilin1 regulates histamine neuron development and behavior in zebrafish, Danio rerio. J Neurosci. 2013;33(4):1589–1597. | |
Nornes S, Newman M, Verdile G, et al. Interference with splicing of presenilin transcripts has potent dominant negative effects on presenilin activity. Hum Mol Genet. 2008;17(3):402–412. | |
Nornes S, Newman M, Wells S, Verdile G, Martins RN, Lardelli M. Independent and cooperative action of Psen2 with Psen1 in zebrafish embryos. Exp Cell Res. 2009;315(16):2791–2801. | |
Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A. 1999;96(21):11872–11877. | |
Joshi P, Liang JO, DiMonte K, Sullivan J, Pimplikar SW. Amyloid precursor protein is required for convergent-extension movements during zebrafish development. Dev Biol. 2009;335(1):1–11. | |
Abramsson A, Kettunen P, Banote RK, et al. The zebrafish amyloid precursor protein-b is required for motor neuron guidance and synapse formation. Dev Biol. 2013;381(2):377–388. | |
Song P, Pimplikar SW. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PLoS One. 2012;7(4):e34209. | |
Nygaard HB, Strittmatter SM. Cellular prion protein mediates the toxicity of beta-amyloid oligomers: implications for Alzheimer disease. Arch Neurol. 2009;66(11):1325–1328. | |
Dodel R, Rominger A, Bartenstein P, et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol. 2013;12(3):233–243. | |
Coric V, van Dyck CH, Salloway S, et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012;69(11):1430–1440. | |
Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. | |
Blanchard J, Chohan MO, Li B, Liu F, Iqbal K, Grundke-Iqbal I. Beneficial effect of a CNTF tetrapeptide on adult hippocampal neurogenesis, neuronal plasticity, and spatial memory in mice. J Alzheimers Dis. 2010;21(4):1185–1195. | |
Cardenas-Aguayo Mdel C, Kazim SF, Grundke-Iqbal I, Iqbal K. Neurogenic and neurotrophic effects of BDNF peptides in mouse hippocampal primary neuronal cell cultures. PLoS One. 2013;8(1):e53596. | |
Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. | |
Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562. | |
Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9(4):363–372. | |
Mullane K, Williams M. Alzheimer’s therapeutics: continued clinical failures question the validity of the amyloid hypothesis – but what lies beyond? Biochem Pharmacol. 2013;85(3):289–305. | |
Taupin P. The therapeutic potential of adult neural stem cells. Curr Opin Mol Ther. 2006;8(3):225–231. | |
Singh C, Liu L, Wang JM, et al. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol Aging. 2012;33(8):1493–1506. | |
Shin JW, Lee JK, Lee JE, et al. Combined effects of hematopoietic progenitor cell mobilization from bone marrow by granulocyte colony stimulating factor and AMD3100 and chemotaxis into the brain using stromal cell-derived factor-1alpha in an Alzheimer’s disease mouse model. Stem Cells. 2011;29(7):1075–1089. | |
Chang KA, Kim JA, Kim S, et al. Therapeutic potentials of neural stem cells treated with fluoxetine in Alzheimer’s disease. Neurochem Int. 2012;61(6):885–891. | |
Taupin P. Adult neural stem cells, neurogenic niches, and cellular therapy. Stem Cell Rev. 2006;2(3):213–219. | |
Yamasaki TR, Blurton-Jones M, Morrissette DA, Kitazawa M, Oddo S, LaFerla FM. Neural stem cells improve memory in an inducible mouse model of neuronal loss. J Neurosci. 2007;27(44):11925–11933. | |
Blurton-Jones M, Kitazawa M, Martinez-Coria H, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106(32):13594–13599. | |
Njie EG, Kantorovich S, Astary GW, et al. A preclinical assessment of neural stem cells as delivery vehicles for anti-amyloid therapeutics. PLoS One. 2012;7(4):e34097. | |
Zhang W, Wang PJ, Sha HY, Ni J, Li MH, Gu GJ. Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol Neurobiol. 2014;50(2):423–437. | |
Fan X, Sun D, Tang X, Cai Y, Yin ZQ, Xu H. Stem-cell challenges in the treatment of Alzheimer’s disease: a long way from bench to bedside. Med Res Rev. 2014;34(5):957–978. | |
Lee HJ, Lim IJ, Park SW, Kim YB, Ko Y, Kim SU. Human neural stem cells genetically modified to express human nerve growth factor(NGF) gene restore cognition in the mouse with ibotenic acid-induced cognitive dysfunction. Cell Transplant. 2012;21(11):2487–2496. | |
Park D, Joo SS, Kim TK, et al. Human neural stem cells overexpressing choline acetyltransferase restore cognitive function of kainic acid-induced learning and memory deficit animals. Cell Transplant. 2012;21(1):365–371. | |
Srivastava N, Seth K, Khanna VK, Ansari RW, Agrawal AK. Long-term functional restoration by neural progenitor cell transplantation in rat model of cognitive dysfunction: co-transplantation with olfactory ensheathing cells for neurotrophic factor support. Int J Dev Neurosci. 2009;27(1):103–110. | |
Wang G, Ao Q, Gong K, Zuo H, Gong Y, Zhang X. Synergistic effect of neural stem cells and olfactory ensheathing cells on repair of adult rat spinal cord injury. Cell Transplant. 2010;19(10):1325–1337. | |
Sun T, Ye C, Zhang Z, Wu J, Huang H. Cotransplantation of olfactory ensheathing cells and Schwann cells combined with treadmill training promotes functional recovery in rats with contused spinal cords. Cell Transplant. 2013;22 Suppl 1:S27–S38. | |
Huang H, Xi H, Chen L, Zhang F, Liu Y. Long-term outcome of olfactory ensheathing cell therapy for patients with complete chronic spinal cord injury. Cell Transplant. 2012;21 Suppl 1:S23–S31. | |
Park D, Lee HJ, Joo SS, et al. Human neural stem cells over-expressing choline acetyltransferase restore cognition in rat model of cognitive dysfunction. Exp Neurol. 2012;234(2):521–526. | |
Marei HE, Farag A, Althani A, et al. Human olfactory bulb neural stem cells expressing hNGF restore cognitive deficit in Alzheimer’s disease rat model. J Cell Physiol. 2015;230(1):116–130. | |
Tang J, Xu H, Fan X, et al. Embryonic stem cell-derived neural precursor cells improve memory dysfunction in Abeta(1–40) injured rats. Neurosci Res. 2008;62(2):86–96. | |
Moghadam FH, Alaie H, Karbalaie K, Tanhaei S, Nasr Esfahani MH, Baharvand H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation. 2009;78(2–3):59–68. | |
Bae JS, Jin HK, Lee JK, Richardson JC, Carter JE. Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-beta deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer’s disease. Curr Alzheimer Res. 2013;10(5):524–531. | |
Lee JK, Jin HK, Bae JS. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-beta deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci Lett. 2009;450(2):136–141. | |
Lee JK, Jin HK, Endo S, Schuchman EH, Carter JE, Bae JS. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells. 2010;28(2):329–343. | |
Malm TM, Koistinaho M, Parepalo M, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18(1):134–142. | |
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012;2012:756357. | |
Yang H, Xie Z, Wei L, Bi J. Systemic transplantation of human umbilical cord derived mesenchymal stem cells-educated T regulatory cells improved the impaired cognition in AbetaPPswe/PS1dE9 transgenic mice. PLoS One. 2013;8(7):e69129. | |
Kim HJ, Lee JH, Kim SH. Therapeutic effects of human mesenchymal stem cells on traumatic brain injury in rats: secretion of neurotrophic factors and inhibition of apoptosis. J Neurotrauma. 2010;27(1):131–138. | |
Jin K, Xie L, Mao X, et al. Effect of human neural precursor cell transplantation on endogenous neurogenesis after focal cerebral ischemia in the rat. Brain Res. 2011;1374:56–62. | |
Kan I, Barhum Y, Melamed E, Offen D. Mesenchymal stem cells stimulate endogenous neurogenesis in the subventricular zone of adult mice. Stem Cell Rev. 2011;7(2):404–412. | |
Yan Y, Ma T, Gong K, Ao Q, Zhang X, Gong Y. Adipose-derived mesenchymal stem cell transplantation promotes adult neurogenesis in the brains of Alzheimer’s disease mice. Neural Regen Res. 2014;9(8):798–805. | |
Letourneau PA, Menge TD, Wataha KA, et al. Human bone marrow derived mesenchymal stem cells regulate leukocyte-endothelial interactions and activation of transcription factor NF-kappa B. J Tissue Sci Eng. 2011;Suppl 3:001. | |
Duff K, Suleman F. Transgenic mouse models of Alzheimer’s disease: how useful have they been for therapeutic development? Brief Funct Genomic Proteomic. 2004;3(1):47–59. | |
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. | |
Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797–801. | |
Park I-H, Arora N, Huo H, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134(5):877–886. | |
Zhou T, Benda C, Dunzinger S, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7(12):2080–2089. | |
Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. | |
Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482(7384):216–220. | |
Machairaki V, Ryu J, Peters A, et al. Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev. August 21, 2014. [Epub ahead of print.] | |
Garate Z, Davis BR, Quintana-Bustamante O, Segovia JC. New frontier in regenerative medicine:site-specific gene correction in patient-specific induced pluripotent stem cells. Hum Gene Ther. 2013;24(6):571–583. | |
Hockemeyer D, Wang H, Kiani S, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29(8):731–734. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.