Back to Journals » ImmunoTargets and Therapy » Volume 14
Regulation of Histone Acetylation During Inflammation Resolution
Authors Gong L, Lei J, Zhou Y, Zhang J, Wu L, Chen Y, Liu X, Li Y ![]()
Received 28 April 2025
Accepted for publication 11 October 2025
Published 22 October 2025 Volume 2025:14 Pages 1145—1158
DOI https://doi.org/10.2147/ITT.S537242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Li Gong,1,* Juan Lei,2,* Yu Zhou,2 Jiangang Zhang,2 Lei Wu,2 Yu Chen,2 Xudong Liu,2 Yongsheng Li1,2
1Department of Phase I Clinical Trial Ward, Chongqing Key Laboratory of Translational Research for Cancer Metastasis and Individualized Treatment, Chongqing University Cancer Hospital, Chongqing, People’s Republic of China; 2Department of Medical Oncology, Chongqing Key Laboratory of Translational Research for Cancer Metastasis and Individualized Treatment, Chongqing University Cancer Hospital, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongsheng Li, Email [email protected]
Background: Inflammatory resolution is an active and coordinated process. Histone acetylation represents the primary epigenetic alteration associated with inflammatory diseases. However, the precise role of histone acetylation in the resolution of inflammation remains poorly understood.
Methods: Lipopolysaccharide (1 μg/mL, 10 ng/mL) and Escherichia coli (105 c.f.u. 106 c.f.u.) were employed to establish inflammatory models both in vitro and in vivo. UPLC-MS/MS was utilized to quantify acetyl CoA and lipid mediators. qPCR was conducted to assess the expression of specific genes. Flow cytometry analysis was performed to enumerate polymorphonuclear neutrophils and monocytes/macrophages. Histone acetylation was evaluated using Western blotting.
Results: Our analysis reveals that histone acetylation and acetyl CoA are temporally regulated during the inflammatory response. A low-dose challenge results in heightened histone acetylation and reduced acetyl CoA. Metabolic repatterning during the inflammatory response promotes the generation of acetyl CoA and histone acetylation. Furthermore, the overexpression of histone acetylation enhances the production of anti-inflammatory lipid mediators, particularly the specialized pro-resolving lipid mediators (SPMs).
Conclusion: These findings illustrate that histone acetylation is not only temporally and differentially regulated during inflammatory responses but also interacts with metabolic reprogramming to promote the production of SPMs, thereby facilitating inflammation resolution.
Keywords: histone acetylation, inflammation resolution, lipid mediators, SPMs
Introduction
Increasing evidence indicates that dysfunction in inflammatory reactions can lead to a variety of diseases.1 Inflammatory resolution is an active and coordinated process.2,3 The ideal outcome of this resolution is a timely return to homeostasis.2,4 The initiation and resolution of inflammation are tightly regulated by lipid mediators (LMs), including specialized pro-resolving lipid mediators (SPMs).5 SPMs, such as lipoxins, resolvins, protectins, and maresins, have been shown to facilitate the resolution of inflammation by limiting polymorphonuclear neutrophil (PMN) infiltration and enhancing macrophage (MФ) activity.6,7 However, the underlying mechanisms governing the generation of SPMs and their regulatory functions remain to be fully elucidated.
Histone modifications, including histone methylation, acetylation, and deacetylation, are closely associated with inflammatory diseases.8,9 Histone acetylation, a significant epigenetic modification, plays critical roles in regulating the expression of pro-inflammatory and anti-inflammatory genes, serving as a mechanism for modulating inflammatory responses.10 For example, lactic acid induces transcriptional repression of macrophage pro-inflammatory functions through histone acetylation;11 the increased levels of histone acetylation triggered by inflammatory stimuli result in a more relaxed chromatin structure, thereby promoting the transcription and expression of pro-inflammatory genes such as TNF-α, IL-6, and IL-1β.12 Recent research has revealed that epigenetic modifications play a central role in the regulation of LMs.13,14 However, the role of histone acetylation in inflammatory resolution and the production of SPMs remains to be comprehensively investigated.
Therefore, this study aims to elucidate the mechanisms by which histone acetylation contributes to the resolution of inflammation, particularly its regulatory effects on the production and function of SPMs, with the goal of providing new theoretical insights and potential therapeutic targets for the treatment of inflammatory diseases.
Materials and Methods
Cell Lines
The immortalized bone marrow-derived macrophages (iBMDM) were generously provided by Professor Feng Shao from the National Institute of Biological Sciences in Beijing, China. These cells were cultured in RPMI-1640 medium (Gibco, Cat#C11875500BT), supplemented with 10% Fetal Bovine Serum (FBS) sourced from South America (ZETA, Cat#Z7185FBS), and maintained without penicillin-streptomycin (P/S) under standard culture conditions. To generate self-limited and delayed-resolving inflammatory models, iBMDM cells were induced with lipopolysaccharide (LPS) at concentrations of 10 ng/mL and 1 µg/mL, respectively.15,16 In certain experiments, a 10 µM histone acetyltransferase inhibitor (HATi) (WM-1119 Selleck S8776), a 10nM RvD2 (Obtained from Cayman Chemical) and a 1 µM ACSS2 inhibitor (VY-3-135, MCE) were administered.
Mice and Murine Peritonitis
Eight-week-old male C57BL/6Jnifdc mice were obtained from Vital River Laboratory (Beijing, China) and maintained under controlled, specific pathogen-free conditions with a 12-hour light cycle. The mice were fed a standard chow diet at Chongqing University Cancer Hospital. Self-limited and delayed-resolution murine peritonitis models were established as previously described.3 Mice were administered intraperitoneal injections of Escherichia coli (E. coli, serotype O6:K2:H1) at concentrations of 105 and 106 colony-forming units (c.f.u.) to induce self-limited and delayed-resolving inflammatory responses, respectively.17,18 E. coli was cultured in LB broth and harvested during the mid-log phase (OD450 nm ≈ 1.0; 1 × 109 c.f.u. mL−1). Peritoneal lavage was performed at 0, 4, and 24 hours post-injection, and the total number of infiltrated cells was assessed using trypan blue. PMNs and Mono/MФ were quantified using light microscopy and immunofluorescent staining techniques. In the specified experiments, HATi (WM-1119, Selleck S8776) was administered at a dosage of 10 mg/kg.
All animal experiments were approved by the Ethics Committee of Chongqing University Cancer Hospital (Chongqing, China, CZLS202107-A). These experiments adhered to national and international guidelines for the care and use of laboratory animals, and were conducted in compliance with the Declaration of Helsinki as approved by the Animal Care and Use Committee of Chongqing University Cancer Hospital.
Seahorse Experiment
This experiment was detailed in our previous research.19 Macrophages were seeded at a density of 1×104 cells per well in 96-well plates and allowed to adhere for 3 to 4 hours. Subsequently, they were treated with LPS at a concentration of 10 ng/mL for either 4 hours or 24 hours. Following treatment, the cells were transferred to unbuffered assay media and incubated in a non-CO2 incubator at 37°C for 1 hour. Oxygen consumption rates (OCR) were measured using an Agilent Seahorse XFp analyzer, following the sequential addition of oligomycin (1.5 µM, diluted in Seahorse XFp Base media, 495455, Sigma), FCCP (1.0 µM, diluted in Seahorse XFp Base media, C2920, Sigma), and Rotenone/Antimycin A (Rot/AA) (0.5 µM, diluted in Seahorse XFp Base media, 35410/R8875, Sigma). Additionally, the Extracellular Acidification Rate (ECR) was assessed using the same Agilent Seahorse XFp analyzer after the addition of Rot/AA (0.5 mmol/L, diluted in Seahorse XFp Base media, 35410/R8875, Sigma).
Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Total RNA was extracted from the cells using the RNA-easy Isolation Reagent (Vazyme, R701-02), and the RNA concentration was quantified with the NanoDrop 2000 (Thermo Scientific). One microgram of total RNA was converted to complementary DNA (cDNA) utilizing the PrimeScript RT-PCR Kit (Takara, RR014A) in accordance with the manufacturer’s protocol. qPCR was conducted with 2x SYBR Green qPCR Master Mix (Selleck, B21203) on a CFX384 system (BIO-RAD), and the expression levels of target genes were assessed using the relative quantification (2−ddCt) method. The expression of β-actin mRNA served as a reference for mRNA quantification. All qPCR experiments were performed in triplicate. Primer sequences were sourced from Primer Bank (https://pga.mgh.harvard.edu/primerbank/).
Flow Cytometry
Peritoneal exudates, along with cultured PMNs and Mono/MФ, were collected for flow cytometry analysis. The cells were incubated with the specified antibodies for 30 minutes at 4°C. The antibodies utilized included CD11b (clone M1/70, catalog no. 101206, BioLegend, San Diego, CA), F4/80 (clone BM8, catalog nos. 123110 and 123116, BioLegend), and Ly6G (clone 1A8, catalog no. 127618, BioLegend). Flow cytometry was conducted using CytoFLEX LX platforms, and the results were analyzed with FlowJo Software version 10.8.1.
Western Blot (WB) Analysis
Histone proteins were extracted from iBMDM cells and murine ascites using the EpiQuik™ Total Histone Extraction Kit (Base Catalog no. OP-0006). Total proteins were extracted from iBMDM cells using RIPA Lysis Buffer (Beyotime no. P0013B) and PMSF (Beyotime no. ST506). According to the previously established protocols and manufacturer’s instructions, Western blot analysis was subsequently performed to evaluate the levels of histone acetylation, histone acetyltransferase (HAT), 15-Lox and 5-Lox.20,21 The primary antibodies employed in this analysis included Histone H3 (D1H2 XP® Rabbit mAb no. 4499, Cell Signaling Technology), Pan-kac (Anti-Acetyllysine Mouse mAb no. PTM-101, Jingjie), HAT (Rabbit polyclonal antibody no. 11432-1-AP, Proteintech), 5-Lox (Rabbit mAb no. CY8019, Abways), 15-Lox (Rabbit polyclonal antibody no. H00000246-D01P, Abnova). Band quantification was carried out using densitometry with ImageJ software.
Detection of ATP
Intracellular ATP levels were quantified using a commercial ATP assay kit (catalog no. S0026, Beyotime). Following the administration of E. coli (serotype O6: K2: H1; 105 c.f.u.) to mice and the induction of iBMDM with LPS (10 ng/mL) for 24 hours, ATP was measured in accordance with the manufacturer’s protocol.
Determination of d‐glucose‐13C6 Incorporation into Acetyl-CoA
Following the established protocols,22 iBMDM cells were washed twice with PBS and incubated in 10 mL of culture medium containing 10% FBS, 90% glucose-free DMEM, and 25 mM d-glucose-13C6 (MCE, HY-B0389A) for 24 hours. After the culture medium was removed, the cells were washed twice with pre-cooled PBS, mixed with 1 mL of pre-cooled methanol, and subsequently collected by scraping. The cell extracts were then analyzed using ultra-high-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) to assess metabolites, including pyruvate, acetyl-CoA, and lactic acid.
UPLC-MS/MS Analysis
The levels of various lipids, acetyl-CoA, pyruvate and lactic acid were analyzed by UPLC-MS/MS. This process employed a UPLC I-Class system (Waters, Milford, Massachusetts, USA) coupled with an AB SCIEX Instruments 6500 Q-TRAP mass spectrometer (Applied Biosystems, Foster City, California, USA) operating in negative ionization mode. The mobile phase was composed of a mixture of methanol, water, and acetic acid at a ratio of 60:40:0.01 (v/v/v), which was progressively modified to 85:15:0.01 over 30 minutes, and subsequently to 100:0:0.01 over the following 5 minutes, maintaining a flow rate of 200 mL/min. Instrument control and data acquisition were performed using Analyst V.1.6 software (Applied Biosystems). For lipid identification, a minimum of six diagnostic ions along with retention times were utilized. Quantification was achieved through the peak area of multiple reaction monitoring transitions, and a linear calibration curve was established for each compound.
Statistical Analysis
All results were validated through a minimum of three independent experiments and are reported as mean ± SEM. To compare two groups, we employed unpaired two-tailed Student’s t-test or one-way or two-way analysis of variance (ANOVA) followed by Sidak’s multiple comparisons test, utilizing GraphPad Prism software (version 8.0) to determine statistical significance. A P-value of < 0.05 was deemed statistically significant. In the data presented, *denotes P < 0.05, **signifies P < 0.01, ***indicates P < 0.001, and ****means P < 0.0001.
Results
Temporal Regulation of Histone Acetylation During Inflammation Resolution
To investigate the relationship between histone acetylation and the resolution of inflammation, we employed well-established models of self-limited inflammation, both in vivo and in vitro. In the in vivo model, mice were intraperitoneally inoculated with E. coli at a concentration of 105 c.f.u. In the in vitro model, iBMDM were induced with LPS at 10 ng/mL. In the self-limited murine peritonitis model, PMN infiltration peaked at 12 hours, followed by a decline (Figure 1a). Conversely, the population of monocytes/macrophages (Mono/MФ) rebounded at 4 hours and gradually increased from 4 to 24 hours (Figure 1a). These findings are consistent with the anti-inflammatory actions of MФ.17
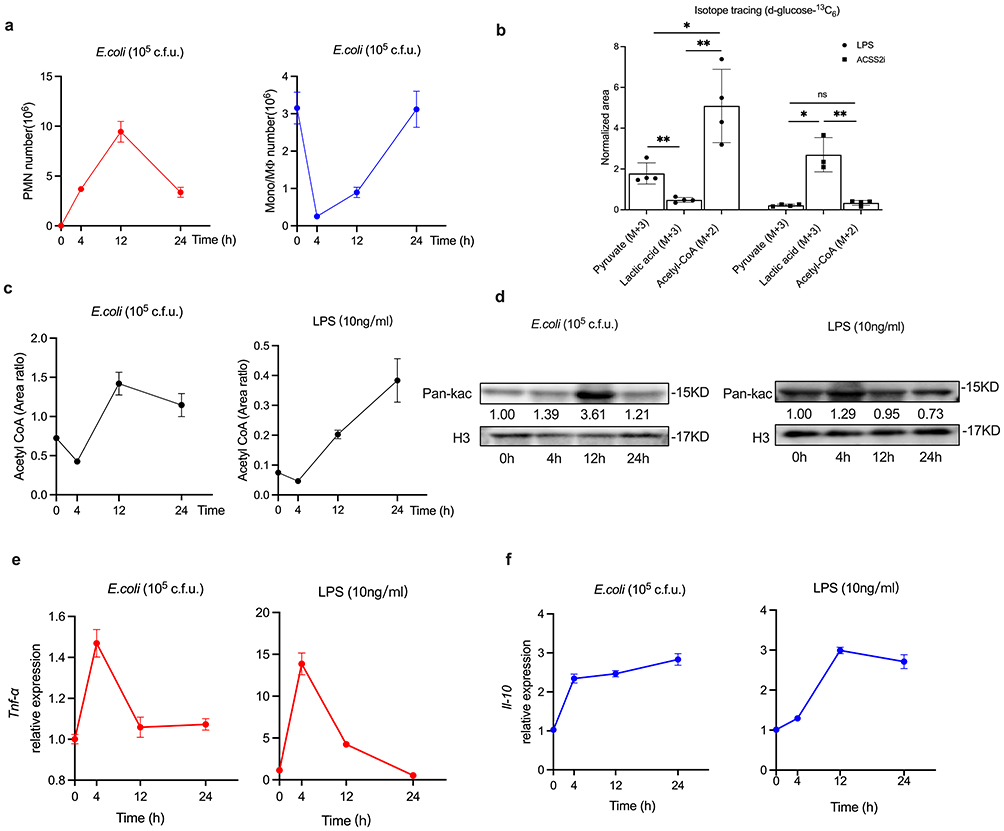 |
Figure 1 Temporal and differential levels of histone acetylation and inflammatory cytokines during self-limited inflammation resolution. (a) C57BL/6 mice (n=5 per time point) were injected intraperitoneally with E. coli at a concentration of 105 c.f.u. The exudate PMN and Mono/MФ were enumerated. (b) D-glucose-13C6 labeled pyruvate, lactate and acetyl-CoA in iBMDM cells after treatment with LPS (10ng/mL) and ACSS2i (1uM) for 24 hours. (c) Acetyl CoA levels in vivo and in vitro experiments were assessed using UPLC-MS/MS. (d) The levels of histone acetylation in vivo and in vitro experiments were analyzed by Western blotting using antibodies against Pan-Kac and Histone H3. (e and f) The expression levels of Tnf-α and Il10 were assessed by qPCR. Results are presented as mean ± SEM (n =3 or 4). *P < 0.05, **P < 0.01. Abbreviation: ns, not significant. |
Acetyl-CoA acts as the exclusive donor of the acetyl group for histone acetylation.23–25 We conducted a stable isotope tracing assay by treating iBMDM cells with d-glucose-13C6 (25 mM) and LPS (10 ng/mL) for 24 hours, subsequently measuring the levels of key intermediate metabolites, including pyruvate, lactate, and acetyl-CoA. The results indicated that the levels of pyruvate (M+3) and acetyl-CoA (M+2) were significantly elevated compared to lactate (M+3) (Figure 1b). Conversely, treatment of iBMDM with the acetyl-CoA synthetase inhibitor ACSS2i (1 µM) resulted in a significant reduction in both pyruvate (M+3) and acetyl-CoA (M+2) levels, along with a notable increase in lactate (M+3) (Figure 1b). These findings confirm the carbon flux into the acetyl-CoA pool during the inflammatory response. In vivo, acetyl-CoA decreased to a minimum at 4 hours and peaked at 12 hours (Figure 1c); but in vitro, the level of acetyl-CoA decreased to its lowest level at 4 hours and subsequently increased (Figure 1c). For histone acetylation, we observed an increase following the onset of inflammation, peaking at 12 hours in vivo (Figure 1d). In vitro, however, histone acetylation reached its maximum at 4 hours (Figure 1d), which was earlier than that observed in vivo.
Histone acetylation mediates the transcription of inflammation-related genes.11 We assessed the mRNA expressions of cytokines using quantitative real-time polymerase chain reaction (qPCR). From both in vivo and in vitro inflammatory experiments, we observed that tumor necrosis factor-α (Tnf-α), a pro-inflammatory cytokine, exhibited an increasing trend at 4 hours, followed by a decline at 12 and 24 hours (Figure 1e). In contrast, and as expected, the anti-inflammatory interleukin-10 (Il-10) significantly increased over the 24-hour period (Figure 1f). These results suggest that histone acetylation is involved in inflammation resolution and is temporally and differentially regulated.
Differential Histone Acetylation Patterns in Self-Limited versus Delayed-Resolving Inflammation
Our previous study demonstrated that self-limited inflammation exhibits distinct characteristics compared to delayed-resolving inflammation.3 To investigate the differences in histone acetylation between these two types of inflammation, we utilized murine models of peritonitis induced by administering 105 c.f.u. and 106 c.f.u. of E. coli intraperitoneally. In vitro, we induced iBMDM using LPS at concentrations of 10 ng/mL and 1 µg/mL, corresponding to the self-limited and delayed-resolving inflammation models, respectively.
In the murine peritonitis model, PMNs peaked at 12 hours before declining, while PMN levels continued to rise over 24 hours in the delayed-resolution group (Figure 2a). The levels of Mono/MФ decreased following the onset of inflammation but exhibited an increase at 12 hours, occurring later than in the self-limited inflammation model (Figure 2b). Histone acetylation levels in the self-limited group were observed to be higher than those in the delayed-resolving group at both the 4 and 12-hour time points; however, at the 24-hour mark, histone acetylation levels decreased but remained significantly higher in the delayed-resolving group (Figure 2c). Additionally, acetyl-CoA levels demonstrated a gradual increase over the 24-hour period, with significantly higher levels recorded in the delayed-resolving group (Figure 2d). In vitro analyses corroborated that the levels of histone acetylation (Figure 2e) and acetyl-CoA (Figure 2f) were consistent with the in vivo findings.
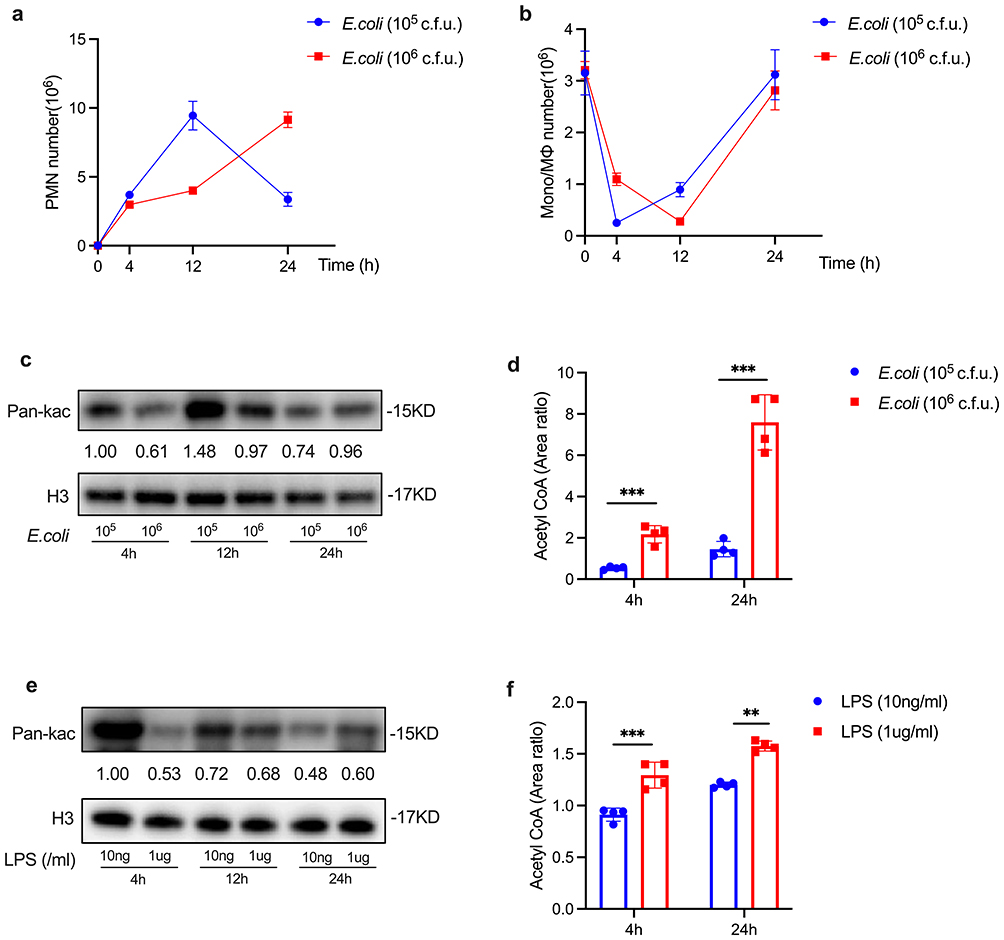 |
Figure 2 The differential histone acetylation and acetyl CoA levels in self-limited versus delayed-resolving inflammation. C57BL/6 mice were subjected to intraperitoneal injection of E. coli to induce peritonitis, with two groups established: self-limited (105 c.f.u.) and delayed-resolving (106 c.f.u.) (n=5 per time point). Additionally, LPS was cultured with iBMDM at concentrations of 10 ng/mL for the self-limited group and 1 µg/mL for the delayed-resolving group. Exudates were collected at specified intervals for analysis. PMN (a) and Mono/MФ (b) were quantified. The expression levels of histone acetylation (c and e) were assessed using Western blotting. The levels of acetyl CoA (d and f) were analyzed via UPLC-MS/MS. Results are presented as as mean ± SEM (n = 3 to 4). **P < 0.01, and ***P < 0.001. |
Regarding inflammatory cytokines, a high-dose LPS challenge significantly elevated the levels of pro-inflammatory cytokines, including Tnf-α, Il-1β, and Il-6 (Supplemental Figure S1). In contrast, a low-dose LPS challenge resulted in increased production of anti-inflammatory cytokines, such as Il-10, Cd206, and transforming growth factor-beta (Tgf-β) (Supplemental Figure S1). The inflammatory cytokines associated with self-limited and delayed-resolving inflammation in vitro are detailed in Supplemental Figure S1. Collectively, these findings suggest that the delayed increase in histone acetylation is associated with the prolonged resolution of inflammation.
Metabolic Reprogramming Modulates Histone Acetylation During Inflammation Resolution
Given that inflammation can modulate energy metabolism in both physiological and pathological contexts,26,27 we analyzed the characteristics of metabolic reprogramming during the resolution of inflammation. We observed that glycolytic genes, specifically Hif1a and Glut1, increased and peaked at 4 hours, subsequently declining to near baseline levels at 24 hours in the exudates of murine peritonitis. Additionally, lipid metabolism genes, including Pparg and Fasn, showed significant increases at 4 hours (Figure 3a and Supplemental Figure S2a). In vitro analyses revealed that the mRNA expressions of Hif1a, Glut1, Pparg, and Fasn paralleled those observed in vivo (Figure 3b and Supplemental Figure S2b). Notably, ATP levels exhibited a gradual increase over the 0–24 hour period (Figure 3c). These findings demonstrate that metabolic reprogramming of glucose and lipid metabolism occurs during the resolution of inflammation.
 |
Figure 3 Metabolic reprogramming during inflammation resolution. (a) C57BL/6Jnifdc mice (n=5 per time point) were inoculated with E. coli at a concentration of 105 c.f.u., and exudates were collected at 0, 4, and 24 hours post-inoculation; the expressions of Hif1a, Glut1, Pparg, and Fasn were analyzed using quantitative real-time PCR (qPCR). (b) iBMDM were cultured with LPS at a concentration of 10 ng/mL, and the expressions of Hif1a, Glut1, Pparg, and Fasn were quantified. (c) ATP levels in exudate cells were measured using an ATP Assay Kit. (d) The Seahorse experiments were conducted to evaluate OCR and ECAR using the Seahorse XFp analyzer. Results are presented as mean ± SEM (n = 3).*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviation: ns, not significant. |
We subsequently evaluated the extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR) using Seahorse experiments. Notably, during the onset of inflammation, the ECAR was significantly higher than during the resolution phase, indicating that energy metabolism is predominantly driven by glycolysis (Figure 3d). In contrast, the OCR was elevated at 24 hours compared to 4 hours, suggesting an increase in oxidative phosphorylation during the resolution of inflammation (Figure 3d). Glycolysis is the primary source of acetyl-CoA.28,29 Acetyl-CoA serves as a substrate for histone acetylation. Collectively, these findings suggest that metabolic reprogramming during the inflammatory response enhances histone acetylation.
Histone Acetylation Mediates Production of Anti-Inflammatory LMs
As noted above, histone acetylation is closely linked to metabolic reprogramming. We investigated whether histone acetylation regulates lipid metabolism, thereby promoting the production of anti-inflammatory lipid mediators, particularly SPMs. The process of histone acetylation is regulated by histone acetyltransferases (HAT).30 We employed a HAT inhibitor (HATi) in inflammatory models both in vivo and in vitro, identifying and profiling the characteristics of PMNs, Mono/MФ, histone acetylation, and acetyl-CoA. Our findings revealed that the count of PMNs increased at 4 hours post-inoculation with HATi, while the count of Mono/MФ rose at 24 hours following HATi treatment (Figure 4a). Histone acetylation levels decreased at both 4 and 24 hours post-inoculation with HATi (Figures 4b and c). Additionally, acetyl-CoA levels were elevated after HATi treatment (Figures 4d and e).
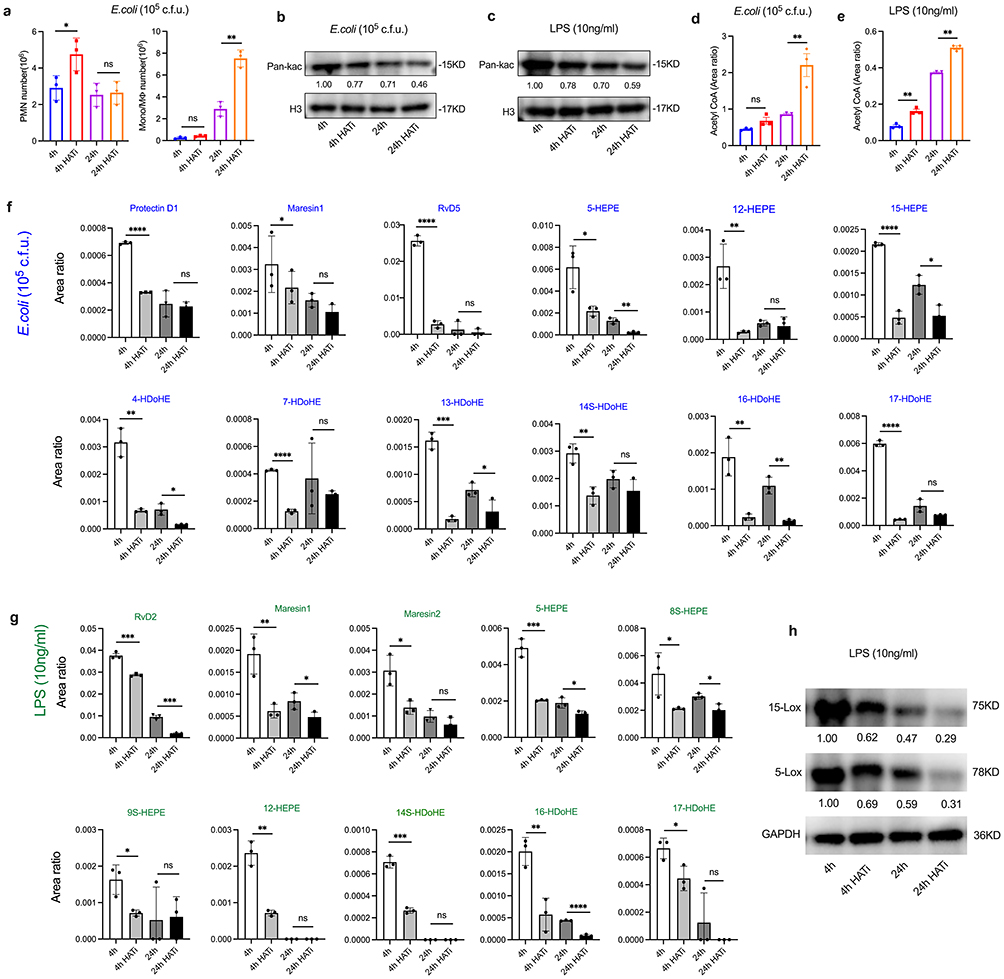 |
Figure 4 LMs-SPMs profiles are regulated by histone acetylation. Male mice (n=5 per time point)) and iBMDM cells were challenged with E. coli (105 c.f.u.) and LPS (10 ng/mL), respectively, to induce inflammatory responses, followed by treatment with HAT inhibitors (HATi) for 4 and 24 hours. (a) The enumeration of PMN and Mono/MФ. Histone acetylation in murine ascites (b) and in iBMDM cells (c) were assessed through WB experiments. The levels of acetyl CoA in murine ascites (d) and in iBMDM cells (e) were quantified using UPLC-MS/MS. (f and g) Lipid mediators (including SPMs) derived from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) were analyzed by UPLC-MS/MS. (h) The protein levels of 15-Lox and 5-Lox in HATi-treated and control groups were determined by WB. Results are presented as mean ± SEM (n = 3).*P < 0.05, **P < 0.01, ***P < 0.001,****P < 0.0001. Abbreviation: ns, not significant. |
Using UPLC-MS/MS, we assessed the expression levels of LMs in peritoneal exudates, including SPMs and other anti-inflammatory lipid mediators. Our results demonstrated that SPMs, specifically Protectin D1, Maresin1, and resolvin D5 (RvD5), along with anti-inflammatory LMs such as 5-HEPE, 12-HEPE, 15-HEPE, 4-HDoHE, 7-HDoHE, 13-HDoHE, 14S-HDoHE, 16-HDoHE, and 17-HDoHE, were significantly decreased at 4 hours post-HATi treatment (Figure 4f). In vitro experiments corroborated these findings, revealing that SPMs (RvD2, Maresin1, Maresin2) and anti-inflammatory LMs (5-HEPE, 8S-HEPE, 9S-HEPE, 12-HEPE, 14S-HDoHE, 16-HDoHE, and 17-HDoHE) also exhibited significant reductions at 4 hours after HATi treatment (Figure 4g). To further elucidate the mechanistic link between histone acetylation and SPM biosynthesis, we assessed the protein levels of key enzymes involved in SPM synthesis, specifically 15-LOX and 5-LOX, at 4 and 24 hours post-HATi treatment using Western blot analysis (Figure 4h). The results demonstrated a significant reduction in the levels of both enzymes in the HATi treatment group compared to the control group. Collectively, these findings suggest that histone acetylation could facilitate the production of anti-inflammatory LMs and promote the resolution of inflammation.
Discussion
The inflammatory response represents a common pathophysiological condition. Despite significant research efforts, the mechanisms governing its resolution remain poorly understood. In this study, we offer novel insights into the relationship between histone acetylation and the resolution of inflammation. Our findings suggest that histone acetylation is crucial for regulating the resolution of inflammation, in conjunction with metabolic reprogramming and the production of anti-inflammatory LMs, including SPMs.
Histone acetylation is a chemical modification catalyzed by histone acetyltransferases, all of which require acetyl-CoA as the essential donor of the acetyl group in eukaryotic cells.23–25 In our study, we conducted a d-glucose-13C6 tracing assay to measure the levels of pyruvate (M+3), lactate (M+3), and acetyl-CoA (M+2). The results indicated a carbon flux into the acetyl-CoA pool, thereby validating this pathway of acetyl-CoA synthesis during the inflammatory response. Furthermore, our findings demonstrated a strong positive correlation between elevated acetyl-CoA levels and increased histone acetylation. Due to limitations in detection technology, we were unable to observe d-glucose-13C6 labeled histone acetylation. Nevertheless, multiple lines of evidence support the close relationship between histone acetylation and acetyl-CoA, with acetyl-CoA serving as the exclusive donor of the acetyl group for histone acetylation.23–25
Histone acetylation plays a critical role in epigenetic modifications that influence various biological processes by altering chromatin conformation and regulating gene expression. This includes chromatin remodeling and inflammatory responses.30 Previous research has demonstrated that histone acetylation modulates inflammatory responses by upregulating the expression of both pro-inflammatory and anti-inflammatory genes.10,31 In our research, we investigated the relationship between histone acetylation and the inflammatory response through a series of inflammatory experiments. We observed that histone acetylation and inflammatory cytokines are temporally and differentially regulated during the resolution of inflammation. The pro-inflammatory factor, such as TNF-α, was upregulated during the initial phase of inflammation. This increase in pro-inflammatory mediators was accompanied by elevated levels of histone acetylation. As inflammation gradually resolved, the expression of pro-inflammatory factors declined, while anti-inflammatory factors, such as IL-10, remained relatively high, thereby contributing to the resolution of inflammation. Furthermore, the inflammatory response induced histone acetylation, which facilitated the production of SPMs, further promoting the resolution of inflammation.
Based on the results of in vitro experiments presented in Figures 1c and d, which illustrate an inverse relationship between histone acetylation and acetyl-CoA levels at 12 and 24 hours, we hypothesized that SPMs might inhibit HAT, thereby contributing to this observed phenomenon. To investigate this hypothesis, we treated iBMDM cells with RvD2 (10 nM)—a representative SPM—for 24 hours, following established experimental protocols designed to evaluate its potential inhibitory effect on HAT.3 Western blot analysis revealed a significant reduction in HAT protein levels after RvD2 treatment (Supplemental Figure S3), indicating that RvD2 mediates a negative feedback inhibition of HAT activity.
To model physiological and pathological inflammation, Tang et al tested varying concentrations of E. coli solution (104, 105, 106, 107, and 108 c.f.u.) in mice and monitored clinical symptoms.18 Their findings revealed that mice exposed to higher bacterial loads (106, 107, and 108 c.f.u.) developed severe inflammatory conditions, whereas those treated with lower doses (104 and 105 c.f.u.) exhibited milder symptoms.18 In a similar vein, Chiang et al utilized E. coli at concentrations of 105 and 107 c.f.u. to induce inflammatory responses in studies focused on self-limited and delayed-resolving inflammation.17 However, our preliminary experiments indicated that E. coli at a concentration of 107 c.f.u. resulted in unacceptably high mortality rates. Consequently, we opted for E. coli doses of 105 and 106 c.f.u. for our models of self-limited and delayed-resolving inflammation. In the context of in vitro inflammation modeling, Augello and Shen et al demonstrated that LPS at concentrations of 10 ng/mL and 1 μg/mL effectively simulates physiological and pathological inflammatory stimuli, respectively.15,16 Based on these findings, we adopted these LPS concentrations for our in vitro experiments.
In the context of self-limited versus delayed-resolving inflammation, a high-dose challenge resulted in a greater accumulation of acetyl-CoA during both the initiation and resolution phases compared to self-limited inflammation. Stronger stimuli were observed to elevate acetyl-CoA levels. Notably, a lower-dose challenge produced higher levels of histone acetylation during both the initiation and resolution phases, whereas a higher-dose challenge specifically increased histone acetylation levels during the resolution phase. These findings suggest that the delayed increase in histone acetylation is linked to the prolonged resolution of inflammation.
Inflammatory resolution is an active process in which cells undergo metabolic reprogramming to facilitate the resolution of inflammation and promote tissue repair.32 We examined the metabolic profile during inflammatory response and observed a significant upregulation of glycolysis-related genes (Hif1a and Glut1) and lipid metabolism genes (Pparg and Fasn) at the 4-hour time point. Glycolysis serves as the primary source of acetyl-CoA.28,29 The production of acetyl-CoA promotes histone acetylation.33,34 Furthermore, the reprogramming of lipid metabolism can also modulate the inflammatory response and aid in the repair of inflammatory damage.35 In our investigation of oxidative phosphorylation genes, we measured only Atp5α1, which exhibited a significant increase at the 4-hour mark. Notably, key oxidative phosphorylation genes, including those encoding cytochrome c oxidase (Cox), were not detected. Additionally, the results from the Seahorse experiment indicated a higher OCR at 24 hours compared to 4 hours, suggesting a marked transition in energy metabolism from glycolysis to oxidative phosphorylation. This metabolic reprogramming enhances energy efficiency and supports the repair process.36
Histone acetylation is tightly regulated by the enzymes of histone acetyltransferases and deacetylases.37 Previous studies have shown that inflammatory stimuli initially trigger the infiltration of PMNs, which subsequently recruit Mono/MФ to facilitate the anti-inflammatory response.2,3 SPMs, derived from polyunsaturated fatty acids, have been demonstrated to promote the resolution of inflammation and restore homeostasis.4,38,39 We analyzed the expressions of LMs before and after HATi treatment in both in vivo and in vitro inflammatory experiments. The results suggested that SPMs (Protectin D1, Maresin 1, RvD2, RvD5) and other anti-inflammatory LMs (5-HEPE, 12-HEPE, 14S-HDoHE, 16-HDoHE, 17-HDoHE, among others) significantly decreased following inoculation with HATi. These findings indicate that histone acetylation regulates SPMs production. The inhibitor of histone acetyltransferases presents a potential therapeutic agent for inflammatory conditions.40 However, in the present study, we did not validate the specificity of HATi (WM-1119), which represents a limitation of our research. However, numerous previous studies have established that WM-1119 is a potent and selective inhibitor of histone lysine acetyltransferases.41,42 Specifically, WM-1119 binds directly to the acetyl-CoA binding site of histone lysine acetyltransferases with high specificity.41,42 These established findings provide strong support for our selection of WM-1119 and enhance the reliability of our experimental results.
Our findings indicate that HATi and SPM analogs hold promise as potential interventions for alleviating inflammation. Currently, an SPM analog, Resolvin E1 (RvE1, TP-317), has successfully completed a Phase 1a clinical trial, with results indicating its potential as a breakthrough therapy for inflammatory diseases.43 Furthermore, several SPM analogs are undergoing clinical trials for various inflammatory conditions, including resolvins for inflammatory back pain (NCT06356844) and an SPM-enriched supplement targeting inflammation-induced vascular aging (NCT07038252). Presently, clinical investigations of HATi have primarily focused on oncology, particularly on malignancies characterized by dysregulation of the p300/CBP pathway.44,45 Notably, two HATi compounds, A-485 and CCS1477, have progressed to clinical testing for these specific indications.44,45
HATi may exhibit synergistic effects when combined with anti-inflammatory therapies, such as NSAIDs and biologics. For instance, HATi can suppress COX-2 expression by inhibiting NF-κB activity,46 suggesting that the combination of HATi with NSAIDs may provide a dual blockade of the NF-κB-driven inflammatory cascade. Additionally, HATi have been shown to inhibit p300-mediated acetylation of the TNF-α and IL-6 promoters, thereby reducing the production of these pro-inflammatory cytokines.47 This mechanism implies that combining HATi with biologics, such as infliximab (anti-TNF-α) or tocilizumab (anti-IL-6 receptor), could potentially enhance anti-inflammatory efficacy. However, since HAT regulates not only histones but also a variety of non-histone substrates, including transcription factors and signaling proteins, modulating HAT activity may lead to unpredictable alterations in gene expression profiles.48 Furthermore, the toxicity and pleiotropic effects of HATi pose significant challenges for their clinical translation.
In summary, this study utilized both in vivo and in vitro inflammatory experiments to demonstrate that histone acetylation is temporally and differentially regulated in association with metabolic reprogramming during the inflammatory response. The overexpression of histone acetylation was shown to enhance the production of anti-inflammatory lipid mediators (including SPMs), which possess pro-resolution properties. However, our investigation examined the relationship between histone acetylation and inflammation resolution at the whole-genome level. Future research focusing on histone acetylation at specific gene loci will yield a more precise understanding of its role in the inflammation resolution process.
Declaration
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Data Sharing Statement
Data relevant to our study are available upon reasonable request from the corresponding author.
Ethics Approval
This study was approved by Institutional Animal Care and Use Committee (ACUP 72209) at the Chongqing University Cancer Hospital.
Acknowledgments
Li Gong and Juan Lei are co-first authors for this study. We are grateful to Professor Feng Shao, National Institute of Biological Sciences in Beijing, for providing the iBMDM cells.
Funding
This work was supported by Funding for Chongqing Young and Middle-Aged Medical Excellence Team, National Outstanding Youth Reserve Talent Training Project, and Research Capacity Enhancement Project of Chongqing University Cancer Hospital (No. Y2024003).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Rajendran P, Chen YF, Chen YF, et al. The multifaceted link between inflammation and human diseases. J Cell Physiol. 2018;233(9):6458–6471. doi:10.1002/jcp.26479
2. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6(12):1191–1197. doi:10.1038/ni1276
3. Li Y, Dalli J, Chiang N, Baron RM, Quintana C, Serhan CN. Plasticity of leukocytic exudates in resolving acute inflammation is regulated by MicroRNA and proresolving mediators. Immunity. 2013;39(5):885–898. doi:10.1016/j.immuni.2013.10.011
4. Sugimoto MA, Vago JP, Perretti M, Teixeira MM. Mediators of the resolution of the inflammatory response. Trends Immunol. 2019;40(3):212–227. doi:10.1016/j.it.2019.01.007
5. Werz O, Gerstmeier J, Libreros S. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat Commun. 2018;9(1):59. doi:10.1038/s41467-017-02538-5
6. Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. 2014;7(2):a016311. doi:10.1101/cshperspect.a016311
7. Zhou Y, Lei J, Xie Q, et al. Fibrinogen-like protein 2 controls sepsis catabasis by interacting with resolvin Dp5. Sci Adv. 2019;5(11):eaax0629. doi:10.1126/sciadv.aax0629
8. Lin Y, Qiu T, Wei G, et al. Role of histone post-translational modifications in inflammatory diseases. Front Immunol. 2022;13:852272. doi:10.3389/fimmu.2022.852272
9. Clayton N, Pellei D, Lin Z. Histone acetylation, BET proteins, and periodontal inflammation. Mol Oral Microbiol. 2024;39(4):180–189. doi:10.1111/omi.12438
10. Daskalaki MG, Tsatsanis C, Kampranis SC. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J Cell Physiol. 2018;233(9):6495–6507. doi:10.1002/jcp.26497
11. Shi W, Cassmann TJ, Bhagwate AV, Hitosugi T, Ip WKE. Lactic acid induces transcriptional repression of macrophage inflammatory response via histone acetylation. Cell Rep. 2024;43(2):113746. doi:10.1016/j.celrep.2024.113746
12. Wu C, Li A, Hu J, Kang J. Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol Cell Biol. 2019;97(1):72–84. doi:10.1111/imcb.12203
13. Ferrari A, Fiorino E, Giudici M, et al. Linking epigenetics to lipid metabolism: focus on histone deacetylases. Mol Membr Biol. 2012;29(7):257–266. doi:10.3109/09687688.2012.729094
14. Schiattarella GG, Hill JA. Epigenetic control of lipid metabolism: implications for lifespan and healthspan. Cardiovasc Res. 2018;114(6):e33–e35. doi:10.1093/cvr/cvy048
15. Augello CJ. Investigating Gene Expression Effects of Neuregulin-1 (NRG-1) and Its Influence on Response to Inflammatory Stimulation with LPS in Microglial Cells in Vitro, in Bioengineering. University of California; 2022.
16. Shen X, He L, Cai W. Role of lipopolysaccharides in the inflammation and pyroptosis of alveolar epithelial cells in acute lung injury and acute respiratory distress syndrome. J Inflamm Res. 2024;17:5855–5869. doi:10.2147/JIR.S479051
17. Chiang N, Fredman G, Backhed F, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484(7395):524–528. doi:10.1038/nature11042
18. Tang W, Zhang Z, Nie D, Li Y, Liu S, Li Y. Protective effect of Citrus Medica Limonum essential oil against escherichia coli K99-Induced intestinal barrier injury in mice. Nutrients. 2023;15(12):2697. doi:10.3390/nu15122697
19. Wu L, Zhang X, Zheng L, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res. 2020;8(5):710–721. doi:10.1158/2326-6066.CIR-19-0261
20. Wang J, Zhao H, Zheng L, et al. FGF19/SOCE/NFATc2 signaling circuit facilitates the self-renewal of liver cancer stem cells. Theranostics. 2021;11(10):5045–5060. doi:10.7150/thno.56369
21. Sadler AJ, Suliman BA, Yu L, et al. The acetyltransferase HAT1 moderates the NF-κB response by regulating the transcription factor PLZF. Nat Commun. 2015;6(1):6795. doi:10.1038/ncomms7795
22. Peng JY, Cai DK, Zeng RL, et al. Upregulation of superenhancer-driven LncRNA FASRL by USF1 promotes de novo fatty acid biosynthesis to exacerbate hepatocellular carcinoma. Adv Sci. 2022;10(1):e2204711. doi:10.1002/advs.202204711
23. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13(12):877–919. doi:10.1007/s13238-021-00846-7
24. Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42(4):426–437. doi:10.1016/j.molcel.2011.05.004
25. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15(8):536–550. doi:10.1038/nrm3841
26. Soto‐Heredero G, Gómez de Las Heras MM, Gabandé‐Rodríguez E, Oller J, Mittelbrunn M. Glycolysis - a key player in the inflammatory response. FEBS J. 2020;287(16):3350–3369. doi:10.1111/febs.15327
27. Wang H, Ye J. Regulation of energy balance by inflammation: common theme in physiology and pathology. Rev Endocr Metab Disord. 2015;16(1):47–54. doi:10.1007/s11154-014-9306-8
28. Stacpoole PW, Dirain CO. The pyruvate dehydrogenase complex at the epigenetic crossroads of acetylation and lactylation. Mol Genet Metab. 2024;143(1–2):108540. doi:10.1016/j.ymgme.2024.108540
29. Jahng JWS, Zhang M, Wu JC. The role of metabolism in directed differentiation versus trans-differentiation of cardiomyocytes. Semin Cell Dev Biol. 2022;122:56–65. doi:10.1016/j.semcdb.2021.05.018
30. Patel AB, He Y, Radhakrishnan I. Histone acetylation and deacetylation - mechanistic insights from structural biology. Gene. 2024;890:147798. doi:10.1016/j.gene.2023.147798
31. Gao S, Xiao H, Luo H, Tang X, Zeng Y. Histone acetylation: a key regulator of inflammatory responses. Life Sci. 2025;380:123936. doi:10.1016/j.lfs.2025.123936
32. Jia N, Gao Y, Li M, et al. Metabolic reprogramming of proinflammatory macrophages by target delivered roburic acid effectively ameliorates rheumatoid arthritis symptoms. Signal Transduct Target Ther. 2023;8(1):280. doi:10.1038/s41392-023-01499-0
33. Moussaieff A, Rouleau M, Kitsberg D, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21(3):392–402. doi:10.1016/j.cmet.2015.02.002
34. Liu X, Cooper DE, Cluntun AA, et al. Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell. 2018;175(2):502–513e13. doi:10.1016/j.cell.2018.08.040
35. Batista-Gonzalez A, Vidal R, Criollo A, Carreno LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. 2019;10:2993. doi:10.3389/fimmu.2019.02993
36. Cortés M, Brischetto A, Martinez-Campanario MC, et al. Inflammatory macrophages reprogram to immunosuppression by reducing mitochondrial translation. Nat Commun. 2023;14(1):7471. doi:10.1038/s41467-023-42277-4
37. Ito K, P JB, I MA. Histone acetylation and deacetylation. Methods Mol Med. 2000;44:309–319. doi:10.1385/1-59259-072-1:309
38. Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim Biophys Acta. 2010;1801(12):1260–1273. doi:10.1016/j.bbalip.2010.08.002
39. Julliard WA, Myo YPA, Perelas A, Jackson PD, Thatcher TH, Sime PJ. Specialized pro-resolving mediators as modulators of immune responses. Semin Immunol. 2022;59:101605. doi:10.1016/j.smim.2022.101605
40. Dekker FJ, van den Bosch T, Martin NI. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today. 2014;19(5):654–660. doi:10.1016/j.drudis.2013.11.012
41. Baell JB, Leaver DJ, Hermans SJ, et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature. 2018;560(7717):253–257. doi:10.1038/s41586-018-0387-5
42. Huang F. New KAT6 inhibitors induce senescence and arrest cancer growth. Synth Syst Biotechnol. 2018;3(4):244–245. doi:10.1016/j.synbio.2018.10.006
43. Danese S, Siegmund B, Peyrin-Biroulet L, et al. P0627 pharmacokinetics, pharmacodynamics, safety, and efficacy of oral resolvin E1-based therapy in inflammatory bowel disease (IBD): translating resolvin E1 activation of BLT1 in experimental models to healthy volunteers. J Crohn’s Colitis. 2025;19(Supplement_1):i1238–i1238. doi:10.1093/ecco-jcc/jjae190.0801
44. Bishop TR, Subramanian C, Bilotta EM, et al. Acetyl-CoA biosynthesis drives resistance to histone acetyltransferase inhibition. Nat Chem Biol. 2023;19(10):1215–1222. doi:10.1038/s41589-023-01320-7
45. Welti J, Sharp A, Brooks N, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021;11(5):1118–1137. doi:10.1158/2159-8290.CD-20-0751
46. Kang H, Kim B. Bioactive compounds as inhibitors of inflammation, oxidative stress and metabolic dysfunctions via regulation of cellular redox balance and histone acetylation state. Foods. 2023;12(5):925. doi:10.3390/foods12050925
47. Yun JM, Jialal I, Devaraj S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem. 2011;22(5):450–458. doi:10.1016/j.jnutbio.2010.03.014
48. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–5318. doi:10.1038/sj.onc.1210599
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.