Back to Journals » International Journal of General Medicine » Volume 10
Recognition and management of platelet-refractory bleeding in patients with Glanzmann’s thrombasthenia and other severe platelet function disorders
Authors Chitlur M, Rajpurkar M ![]() , Recht M
, Recht M ![]() , Tarantino MD, Yee DL, Cooper DL
, Tarantino MD, Yee DL, Cooper DL ![]() , Gunawardena S
, Gunawardena S
Received 29 November 2016
Accepted for publication 20 February 2017
Published 3 April 2017 Volume 2017:10 Pages 95—99
DOI https://doi.org/10.2147/IJGM.S128953
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Meera Chitlur,1 Madhvi Rajpurkar,1 Michael Recht,2 Michael D Tarantino,3 Donald L Yee,4 David L Cooper,5 Sriya Gunawardena5
1Carman and Ann Adams Department of Pediatrics, Wayne State University and Children’s Hospital of Michigan, Detroit, MI, USA; 2Oregon Health and Science University, Portland, OR, USA; 3Bleeding and Clotting Disorders Institute, Peoria, IL, USA; 4Department of Pediatrics, Baylor College of Medicine, Houston, TX, USA; 5Clinical Development, Medical and Regulatory Affairs, Novo Nordisk Inc., Plainsboro, NJ, USA
Abstract: Patients with rare qualitative platelet disorders or platelet function disorders (PFDs) may present to the hospital physician with severe bleeding episodes or excessive surgical bleeding. Although standard treatment consists of platelet transfusions, repeated transfusions may result in the development of antiplatelet antibodies (APA) or clinical refractoriness, rendering further platelet therapy ineffective. In such settings, an approved treatment option for patients with Glanzmann’s thrombasthenia (GT), one of the well-known rare PFDs, is recombinant activated coagulation factor VII (rFVIIa). Data regarding the efficacy of rFVIIa in patients with GT and platelet refractoriness are available from a large patient registry, an international survey, and multiple case reports and demonstrate efficacy in patients with and without refractoriness or APA. This article reviews the rFVIIa clinical data in patients with GT and platelet refractoriness and discusses clinical implications relevant to the hospital-based physician. Because uncontrolled bleeding can be life-threatening, hospital physicians should be alert to the signs of platelet refractoriness, be able to recognize continued internal or external bleeding, and know how to adapt treatment regimens for the effective management of bleeding. The management of patients who receive rFVIIa should occur in consultation with a hematologist with experience in PFDs, and patients with suspected platelet refractoriness should be referred to such a hematologist as early as possible. A critical unmet need is the development of a definition of an adequate response to platelet transfusion, which would facilitate early recognition of platelet refractoriness in patients with PFDs who exhibit a normal platelet count.
Keywords: platelet function disorder, Glanzmann’s thrombasthenia, alloantibodies, antiplatelet antibodies, platelet refractoriness, bleeding management
Glanzmann’s thrombasthenia – clinical presentation and diagnosis
Inherited platelet function disorders (PFDs) are a group of rare bleeding disorders characterized by qualitative defects in platelet adhesion, aggregation, secretion, or hemostatic activity.1,2 Bleeding associated with PFDs varies in frequency and intensity, although typical manifestations include predominantly mucocutaneous symptoms, such as epistaxis, gingival hemorrhage, menorrhagia, and easy bruising. Bleeding in response to trauma can also be severe, and therefore, surgical procedures often require careful hemostatic management.
Glanzmann’s thrombasthenia (GT) is one of the well-known rare PFDs, occurring in an estimated 1 per 1 million individuals and is associated with abnormalities in glycoprotein IIb/IIIa (GPIIb/IIIa), the integrin glycoprotein complex responsible for platelet aggregation through binding to fibrinogen. Because of the autosomal recessive nature of GT, its prevalence is highest among ethnic groups in which consanguinity is common.3 Unlike other rare PFDs, literature on GT extends beyond case reports to include an international survey and a large patient registry, and therefore GT often serves as a prototype for broader discussions of PFD diagnosis and management.
Coagulation disorders may be considered in cases of unexplained bleeding or in patients with an individual or family history of severe bleeding or easy bruising. Diagnosing GT can be challenging because of the similarity in bleeding patterns across a variety of platelet disorders and the absence of diagnostic indicators revealed through initial laboratory assessments.4 Unlike quantitative platelet disorders (thrombocytopenias), patients with GT have normal platelet counts and a normal prothrombin time (PT) and partial thromboplastin time (PTT). In most institutions, assessment of closure time (a screening test performed via platelet function analyzer [PFA-100]) can be used to detect abnormalities in platelet function; however, this tool lacks disease specificity and may be most effectively used as a screening tool or to exclude severe platelet defects.5 Advanced hematologic assessments necessary to diagnose GT require consultation with a hematologist and include platelet aggregometry (the “gold standard” diagnostic tool) or flow cytometry for GPIIb/IIIa.
Bleeding management and platelet refractoriness
The standard treatment for severe bleeding and perioperative management in patients with GT is platelet transfusion,1,2,6 although less severe episodes may be controlled with local measures such as fibrin sealants and topical thrombin, or with antifibrinolytics or desmopressin.7,8An important limitation in the use of platelet transfusions is the potential for platelet refractoriness after repeated transfusions, which often involves development of antiplatelet alloantibodies (APA) targeting human leukocyte antigens (HLAs) or deficient glycoproteins (GPIIb/IIIa in patients with GT).2,7,9,10 Interestingly, APA have been reported in patients who have not received platelet transfusions, suggesting that alternative mechanisms for sensitization may occur, such as molecular mimicry, partial GP expression, or exposure to platelets/platelet microparticles via red blood cell transfusions.11 Because of the rarity of GT, consistent epidemiologic data regarding the incidence of refractoriness or typical number of exposures associated with the development of APA or refractoriness are lacking; however, the risk of developing refractoriness is thought to increase with repeated exposures.7,8
The potential for APA development may be reduced by administering platelets obtained from HLA-matched donors1,8 or by using leukocyte-depleted blood components;12 however, platelet availability may be limited because of short shelf life and variable blood bank capacity,13 leading to difficulties in obtaining HLA-matched platelets in a timely manner when faced with severe bleeding, including that occurring in the perioperative setting. An additional challenge in patients with GT is monitoring hemostatic response to platelet transfusions; unlike thrombocytopenias, which can be followed with platelet counts, response in patients with GT must be assessed functionally, either through clinical improvements in bleeding or through laboratory testing of platelet function.14 However, these assessments may be difficult to perform, may take several hours, and are not available at all hospitals.
An important alternative therapeutic option is recombinant activated coagulation factor VII (rFVIIa, NovoSeven® RT; Novo Nordisk A/S, Bagsvaerd, Denmark).7,8 rFVIIa was approved by the European Medicines Agency (EMA) in 2004 for the treatment of bleeding episodes and prevention of bleeding during surgery in patients with GT who are refractory to platelets and have APA15 and was approved by the Food and Drug Administration (FDA) in 2014 for the treatment of bleeding episodes and perioperative management in adults and children with GT with refractoriness to platelet transfusions, with or without APA.16 Here we review data regarding the clinical experience with rFVIIa in patients with GT and platelet refractoriness and discuss the importance of rapidly recognizing refractoriness and initiating alternative treatments.
Clinical experience with rFVIIa in patients with GT
Following early investigations into the use of rFVIIa in Von Willebrand’s disease and thrombocytopenia, the first reported use of rFVIIa for GT was in 1995 for refractory epistaxis in a 2-year-old child.17 Subsequent reports include case reports and series,18 an international survey regarding the use of rFVIIa in patients with GT,19 an open-label Iranian study including clinical responses to rFVIIa,20 and a multinational GT registry (GTR).21,22
The largest source of data regarding rFVIIa in patients with GT and APA or refractoriness is the GTR, a multinational registry study initiated as an EMA postmarketing surveillance commitment, which evaluated use of systemic hemostatic treatments in GT.21,22 At the request of the FDA, APA status, refractoriness, and rFVIIa efficacy data from the GTR were independently evaluated by a hematology expert panel, which reviewed data on a case-by-case basis using all available patient information. Of 266 rFVIIa-treated bleeding episodes and 160 rFVIIa-treated surgical procedures evaluated by this panel, most bleeding episodes were in children aged 16 years or younger (65%), and most surgical procedures were in adults more than 16 years of age (86%).16 The most common types of bleeding episodes were epistaxis (44%), gum bleeding (18%), menorrhagia (14%), tooth or dental extraction related (11%), and gastrointestinal (9%), and the most common types of surgical procedures were dental (66%), endoscopic (8%), nasal (5%), excision related (4%), gastrointestinal (4%), and orthopedic (4%). Overall, rFVIIa was considered effective in 251 (94.4%) bleeding episodes and 159 (99.4%) surgical procedures (Table 1). Adjudicated rFVIIa efficacy was also high across treatment categories (rFVIIa ± platelets ± other hemostatic agents) and across APA or refractory groups. Among patients with refractoriness, rFVIIa was considered effective in 94.9% of bleeding episodes and 98.6% of surgical procedures. The recommended dosing for severe bleeding episodes is 90 µg/kg every 2–6 hours and for surgery 90 µg/kg every 2 hours.16
 | Table 1 Adjudicator evaluation of GTR rFVIIa efficacy data Notes: aPatient numbers are not additive. Patients may have episodes with different treatment regimens and have more than one antibody/refractory status. bAll treatment regimens that included treatment with rFVIIa. cIncludes GPIIb/IIIa, HLA, and unspecified platelet-specific antibodies. dTreatment was rFVIIa only for 26/79 episodes with refractoriness with or without antibodies, 2/10 episodes with platelet-specific antibodies only, and 81/177 episodes with neither or unknown. The remainder received rFVIIa with platelets and/or antifibrinolytic agents. eAssumes no platelet-specific antibodies or refractoriness reported or antibody and refractory status unknown. fNo reports of failure or lack of consensus were reported. gTreatment was rFVIIa only for 22/70 episodes with refractoriness with or without antibodies, 13/24 episodes with platelet-specific antibodies only, and 31/66 episodes with neither or unknown. The remainder received rFVIIa with platelets and/or antifibrinolytic agents. Abbreviations: GP, glycoprotein; GTR, Glanzmann’s Thrombasthenia Registry; HLA, human leukocyte antigen; NA, not available; rFVIIa, recombinant activated coagulation factor VII. |
Additional clinical information was provided by an international survey including physician reports from 37 hospitals in 14 countries across Europe, North America, Australia, and Asia.19 Use of rFVIIa was reported in 142 events (108 bleeding episodes and 34 invasive procedures) which occurred in 59 patients; most of these patients were female (59%) and median age was 22 years (range, 1–72 years). Reasons for using rFVIIa included prevention of antiplatelet immunization (43%), history of APA (42%), prevention of blood-borne pathogen transmission (37%), and platelet inefficacy for previous bleeds (35%) or for the current bleed (14%). Among 103 evaluable bleeding episodes, rFVIIa was reported as effective (defined as the cessation of bleeding and a lack of rebleeding within 48 hours) in 69 episodes (67%); furthermore, investigators reported comparable efficacy between patients with and without APA or refractoriness. Among 31 evaluable surgical procedures, rFVIIa was considered effective in 29 procedures (94%); however, the number of surgeries in patients with APA and/or refractoriness was not reported. Two serious adverse events possibly related to rFVIIa were reported by the investigators (deep venous thrombosis with pulmonary embolism and clotting in one ureter).
Multiple case reports have also described rFVIIa use in patients with GT and were supportive of the FDA approval of rFVIIa in GT.18 Of 49 available reports of patients with GT who received rFVIIa, 36 bleeding episodes and 29 invasive, surgical, or dental procedures were documented. Patient ages ranged from 1 to 60 years, and 53% were female. Of reported bleeding events, rFVIIa was considered effective in 25 bleeding episodes (69%) and 28 procedures (97%). This rate of efficacy is comparable to that reported among the subset of patients with reported APA and/or refractoriness (n=15); within this population, an effective response was reported for 4 of 8 bleeding episodes (50%) and 9 of 10 procedures (90%). Overall, one serious adverse event (jugular vein thrombosis) and two nonserious adverse events (nausea and vomiting, experienced by a single patient) were reported as possibly or probably related to rFVIIa administration.
Discussion and clinical recommendations
Defining platelet refractoriness in patients with PFDs is a significant challenge, as standard assessments for effectiveness of platelet transfusions have been established only in the context of thrombocytopenia.23 Developing a standard measurement of platelet transfusion effectiveness independent of platelet count is therefore a critical unmet need. For hospital-based physicians, persistent external (visible) or internal bleeding (ie, a continuous drop in blood hemoglobin concentration or continuous drainage from surgical drain) after platelet transfusions should be viewed as a potential indicator of platelet refractoriness (Figure 1).
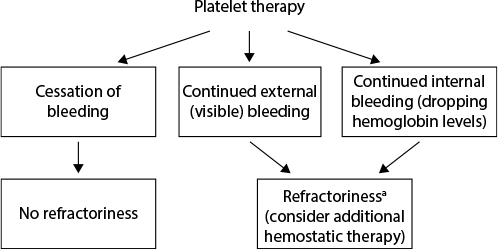 | Figure 1 Platelet refractoriness management algorithm in severe or postsurgical bleeding. aIn postsurgical bleeding, causes of bleeding unrelated to hemostasis should also be considered. |
Hospital physicians should be aware of alternative treatments such as rFVIIa for patients experiencing an inadequate response to platelet transfusions for severe bleeding episodes or for surgical management after failing to respond to local or topical (eg, gelatin sponges, packing) and antifibrinolytic (oral or intravenous) therapies. Bleeding in these patients should be managed preferably in consultation with a hematologist with expertise in coagulation disorders, and those with suspected platelet refractoriness should be referred to an experienced hematologist to ensure optimal long-term management.
Because of the risk of alloimmunization with repeated platelet transfusions, rFVIIa may be administered as a preemptive alternative to platelet transfusions, even in patients without evidence of refractoriness, especially when early treatment is important. Interesting data regarding real-world use of rFVIIa were obtained from the GTR, which showed that despite the labeled indication in Europe of GT with refractoriness and APA, 177 of 266 rFVIIa-treated bleeds (67%) and 66 of 160 rFVIIa-treated surgeries (41%) in this registry occurred in patients with neither refractoriness nor APA (or who had unknown status for both).
Because platelet transfusions are standard treatment for severe bleeding episodes and perioperative management in patients with GT, the potential development of refractoriness is an important clinical concern. Uncontrolled bleeding can be life threatening, and therefore hospital-based physicians should be on alert to identify patients with ongoing bleeding suggestive of platelet refractoriness and should be aware of the importance of adapting treatment regimens for the effective management of bleeding.
Acknowledgments
Writing assistance was provided by Anna Abt, PhD, ETHOS Health Communications, Yardley, Pennsylvania, with financial assistance from Novo Nordisk Inc., in compliance with international guidelines on Good Publication Practice.
Disclosure
M Chitlur is a paid consultant for Novo Nordisk Inc. and has received honoraria from Baxter, Bayer, Biogen Idec, and Pfizer. M Rajpurkar has received honoraria from Novo Nordisk Inc. M Recht has received grant/research support from Baxter, Biogen Idec, Novo Nordisk Inc., and Pfizer and served as a consultant to Kedrion and Novo Nordisk Inc. MD Tarantino has received grant/research support from Novo Nordisk Inc. and has been a consultant to Baxalta, Biogen, Bioproducts Laboratory, Grifols, Novo Nordisk Inc., Octapharma, and Pfizer. DL Yee is a local principal investigator on clinical trials funded by Novo Nordisk Inc. and served as a consultant to Octapharma. DL Cooper is an employee of Novo Nordisk Inc. S Gunawardena is an employee of Novo Nordisk Inc. The authors report no other conflicts of interest in this work.
References
Alamelu J, Liesner R. Modern management of severe platelet function disorders. Br J Haematol. 2010;149(6):813–823. | ||
Sharathkumar AA, Shapiro A. Platelet Function Disorders. 2nd ed. Montréal, QC: World Federation of Hemophilia (WFH); 2008. | ||
Nurden AT. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;1:10. | ||
Israels SJ, El-Ekiaby M, Quiroga T, Mezzano D. Inherited disorders of platelet function and challenges to diagnosis of mucocutaneous bleeding. Haemophilia. 2010;16 (Suppl 5):152–159. | ||
Harrison P. The role of PFA-100 testing in the investigation and management of haemostatic defects in children and adults. Br J Haematol. 2005;130(1):3–10. | ||
DiMichele DM, Hathaway WE. Use of DDAVP in inherited and acquired platelet dysfunction. Am J Hematol. 1990;33(1):39–45. | ||
Di Minno G, Coppola A, Di Minno MN, Poon MC. Glanzmann’s thrombasthenia (defective platelet integrin alphaIIb-beta3): proposals for management between evidence and open issues. Thromb Haemost. 2009;102(6):1157–1164. | ||
Bolton-Maggs PH, Chalmers EA, Collins PW, et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol. 2006;135(5):603–633. | ||
Bellucci S, Caen J. Molecular basis of Glanzmann’s thrombasthenia and current strategies in treatment. Blood Rev. 2002;16(3):193–202. | ||
George JN, Caen JP, Nurden AT. Glanzmann’s thrombasthenia: the spectrum of clinical disease. Blood. 1990;75(7):1383–1395. | ||
Ghosh K, Kulkarni B, Shetty S, Nair S. Antiplatelet antibodies in cases of Glanzmann’s thrombasthenia with and without a history of multiple platelet transfusion. Indian J Hum Genet. 2009;15(1):23–27. | ||
Seligsohn U. Treatment of inherited platelet disorders. Haemophilia. 2012;18 (Suppl 4):161–165. | ||
Haijema R, van der Wal J, van Dijk NM. Blood platelet production: optimization by dynamic programming and simulation. Compt Oper Res. 2007;34(3):760–779. | ||
Male C, Koren D, Eichelberger B, Kaufmann K, Panzer S. Monitoring survival and function of transfused platelets in Glanzmann thrombasthenia by flow cytometry and thrombelastography. Vox Sang. 2006;91(2):174–177. | ||
NovoSeven [summary of product characteristics]. Bagsvaerd, Denmark: Novo Nordisk A/S; 2013. | ||
NovoSeven RT Coagulation Factor VIIa (Recombinant) room temperature stable [prescribing information]. Plainsboro, NJ: Novo Nordisk; 2015. | ||
Tengborn L, Petruson B. A patient with Glanzmann thrombasthenia and epistaxis successfully treated with recombinant factor VIIa. Thromb Haemost. 1996;75(6):981–982. | ||
Rajpurkar M, Chitlur M, Recht M, Cooper DL. Use of recombinant activated factor VII in patients with Glanzmann’s thrombasthenia: a review of the literature. Haemophilia. 2014;20(4):464–471. | ||
Poon MC, D’Oiron R, Von Depka M, et al. Prophylactic and therapeutic recombinant factor VIIa administration to patients with Glanzmann’s thrombasthenia: results of an international survey. J Thromb Haemost. 2004;2(7):1096–1103. | ||
Lak M, Scharling B, Blemings A, et al. Evaluation of rFVIIa (NovoSeven) in Glanzmann patients with thromboelastogram. Haemophilia. 2008;14(1):103–110. | ||
Poon MC, d’Oiron R, Zotz RB, Bindslev N, Di Minno MN, Di Minno G. The international prospective Glanzmann Thrombasthenia Registry: treatment and outcomes in surgical intervention. Haematologica. 2015;100(8):1038–1044. | ||
Di Minno G, Zotz RB, d’Oiron R, Bindslev N, Di Minno MN, Poon MC. The international prospective Glanzmann Thrombasthenia Registry: treatment modalities and outcomes in non-surgical bleeding episodes in Glanzmann thrombasthenia patients. Haematologica. 2015;100(8):1031–1037. | ||
Stanworth SJ, Navarrete C, Estcourt L, Marsh J. Platelet refractoriness – practical approaches and ongoing dilemmas in patient management. Br J Haematol. 2015;171(3):297–305. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.