Back to Journals » Journal of Inflammation Research » Volume 16
Rapamycin Exacerbates Staphylococcus aureus Pneumonia by Inhibiting mTOR-RPS6 in Macrophages
Authors Yu FY, Zheng K, Wu YF, Gao SW, Weng QY, Zhu C, Wu YP, Li M, Qin ZN, Lou JF, Chen ZH ![]() , Ying SM, Shen HH, Li W
, Ying SM, Shen HH, Li W ![]()
Received 8 August 2023
Accepted for publication 17 November 2023
Published 30 November 2023 Volume 2023:16 Pages 5715—5728
DOI https://doi.org/10.2147/JIR.S434483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Adam Bachstetter
Fang-Yi Yu,* Kua Zheng,* Yin-Fang Wu,* Shen-Wei Gao, Qing-Yu Weng, Chen Zhu, Yan-Ping Wu, Miao Li, Zhong-Nan Qin, Jia-Fei Lou, Zhi-Hua Chen, Song-Min Ying, Hua-Hao Shen, Wen Li
Key Laboratory of Respiratory Disease of Zhejiang Province, Department of Respiratory and Critical Care Medicine, Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wen Li, Tel +86-571-87783729, Fax +86-571-87068001, Email [email protected]
Purpose: This study aimed to explore the effect of Rapamycin (Rapa) in Staphylococcus aureus (S. aureus) pneumonia and clarify its possible mechanism.
Methods: We investigated the effects of Rapa on S. aureus pneumonia in mouse models and in macrophages cultured in vitro. Two possible mechanisms were investigated: the mTOR-RPS6 pathway phosphorylation and phagocytosis. Furthermore, for the mechanism verification in vivo, mice with specific Mtor knockout in myeloid cells were constructed for pneumonia models.
Results: Rapa exacerbated S. aureus pneumonia in mouse models, promoting chemokines secretion and inflammatory cells infiltration in lung. In vitro, Rapa upregulated the secretion of chemokines and cytokines in macrophages induced by S. aureus. Mechanistically, the mTOR-ribosomal protein S6 (RPS6) pathway in macrophages was phosphorylated in response to S. aureus infection, and the inhibition of RPS6 phosphorylation upregulated the inflammation level. However, Rapa did not increase the phagocytic activity. Accordingly, mice with specific Mtor knockout in myeloid cells experienced more severe S. aureus pneumonia.
Conclusion: Rapa exacerbates S. aureus pneumonia by increasing the inflammatory levels of macrophages. Inhibition of mTOR-RPS6 pathway upregulates the expression of cytokines and chemokines in macrophages, thus increases inflammatory cells infiltration and exacerbates tissue damage.
Keywords: Staphylococcus aureus, pneumonia, macrophage, rapamycin, mTOR- ribosomal protein S6 signaling pathway
Introduction
Staphylococcus aureus (S. aureus), one of the most infamous and widespread bacterial pathogens in hospitals and communities, causes millions of severe invasive infections each year.1–3 Approximately 20–40% of the population asymptomatically harbor S. aureus colonization in the nasal squamous epithelium.3,4 As an opportunistic pathogen, immunocompromised patients, such as those using immunosuppressants, are at a greater risk of infection.5,6 Macrophages are important innate immune cells and serve as the first line of defense against pathogenic infections through phagocytosis and chemokine secretion, in addition to the role played by skin barrier.7 However, bacterial lipoproteins (LPPs) in Gram-positive bacteria, particularly S. aureus, can induce multiple distinct cellular states in macrophages, and these inflammatory trajectories change as the infection persists.6,8 These characteristics bring about significant complexities in researches and treatments.
Rapamycin (Rapa) is an immunosuppressive and antitumor drug.1,9 It is now commonly used in the clinic to combat rejection after organ transplantation and to resist tumor progression.10–14 Despite its beneficial effects, Rapa is reported to cause pneumonitis, such as non-infectious alveolitis and interstitial pneumonitis with inflammatory cells infiltration.15 In addition to non-infectious pneumonia,16 opportunistic infectious pneumonia is also well-reported in immunosuppressed state. In patients undergoing hematopoietic stem cell transplant (HSCT), who are in typical immunosuppressed states, pneumonia is reported to be the second most common nosocomial infection.17 Bacterial is the leading cause of microorganism-caused pneumonia in HSCT patients and accounts for 44.4%.18 Among them, S. aureus is one of the most common pathogens and is usually associated with multidrug-resistant (MDR) characteristics.19 It has also been pointed out that immunosuppression can worsen mortality in cases of ventilator-associated pneumonia (VAP).20 These facts prompt us to be vigilant about the impact of immunosuppressant use on S. aureus pneumonia.
Rapa inhibits its target, mTOR, a protein complex composed of three components. The catalytic subunit of mTOR is a serine/threonine protein kinase that can be activated through phosphorylation.21–24 mTOR is a crucial receptor in cell growth and metabolism, playing essential roles in numerous physiological and pathological processes. These include obesity, type 2 diabetes, cancer progression and aging.24 Due to the diverse roles of mTOR and its position as a central molecule in various signaling pathways, it is crucial to investigate mTOR and its downstream mechanisms. The main downstream protein of mTOR is ribosomal protein S6 kinase (S6K), which increase the phosphorylation of ribosomal protein S6 (RPS6). Phosphorylated RPS6(p-RPS6) regulates 5’ cap-dependent protein translation.24 For the pathogenesis of pneumonia, cytokines and chemokines are important proteins that can be synthesized and released by immune cells to regulate the inflammatory state. In other inflammatory diseases such as colitis, mTOR and one of its downstream proteins, RPS6, have been reported to be associated with inflammation levels.25 Therefore, we suppose that understanding this pathway can provide valuable insights into the regulation of cellular processes and potentially lead to the development of targeted therapies for pneumonia.
In this study, we attempted to elucidate the effect of Rapa on the severity of S. aureus infection. Our study showed that Rapa exacerbated S. aureus pneumonia in vivo and increased the inflammatory levels of macrophages in vitro. Inhibition of mTOR-RPS6 pathway upregulates the expression of cytokines and chemokines in macrophages and increases inflammatory cells infiltration and exacerbates tissue damage. Additionally, the drug does not enhance the clearance capacity of macrophages. This suggests that patients using Rapa in clinical settings should remain cautious about opportunistic infections.
Materials and Methods
Mice
The Mtorflox/flox mice (C57BL/6, The Jackson Laboratory, 011009) and LysMCre transgenic mice (C57BL/6, The Jackson Laboratory, 004781) were crossed to generate myeloid cell-specific Mtor conditional knockout mice, LysMCre-Mtorflox/floxmice.26,27 Transgene-negative littermates were used as the control (Mtorflox/flox) mice in the experiments. C57BL/6 mice (Shanghai SLAC laboratory animal Co. Ltd) were used as wild-type (WT) mice. Mice that were 4–6 weeks old were used for the in vitro bone marrow-derived macrophages (BMDMs) experiments. For in vitro peritoneal macrophages (PMs) experiments and in vivo S. aureus exposure models, mice that were 6–8 weeks old were used. All mice used in this study were housed under a constant 12-hour light/12-hour dark cycle in a specific pathogen-free facility, and all experimental protocols were approved by the Ethical Committee for Animal Studies at Zhejiang University. The primer sequences for gene identification were ordered from Hangzhou Youkang Biotechnology Co., Ltd and were shown in Table 1.
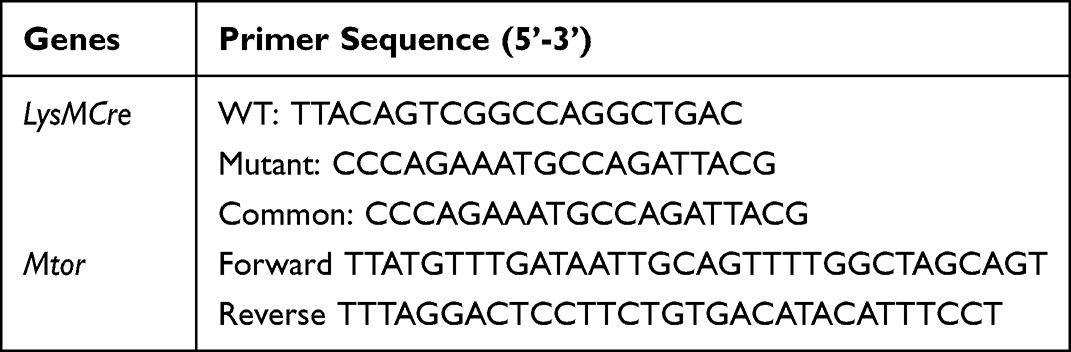 |
Table 1 The Primer Sequences for Gene Identification |
In vivo and in vitro S. aureus Exposure
In vivo, mice were infected by S. aureus by intratracheal injection of 2×107 S. aureus in 50 μL saline (control mice injected pure saline) for 1 day. In vitro, BMDMs and PMs were treated with S. aureus at 10 MOI or various concentrations noted in figure legend. The S. aureus strain was a clinical isolate (multilocus sequence type ST15 and agr type II) with Hld, PSMα, Hla, and PVL+++-.
Histological Analyses
After infection with S. aureus, the left lungs of experimental mice were fixed in 4% formaldehyde for 24 h. The lung tissues were dehydrated and embedded in paraffin, then sectioned into 4–5 μm thick slices. Hematoxylin and Eosin (H&E) staining was carried out following standard procedures. The degree of peribronchial and perivascular inflammation was assessed on a subjective scale of 0–3, and this quantification was measured by observers who were blinded to the group and treatment.
Bronchoalveolar Lavage Fluid Collection and Analysis
After 24 h of S. aureus infection, bronchoalveolar lavage fluid (BALF) was collected from the left lungs of experimental mice by three rounds of instillation and extraction of 0.4 mL PBS. After counting the number of total cells, the remaining BALF was centrifuged at 3000 g for 10 min at 4 °C to collect cells for Wright-Giemsa stain (Baso, BA4017) in order to obtain differential cell counts. The supernatant was stored at −80 °C for cytokine tests.
Cell Culture
BMDMs: Bone marrow cells were flushed out from the tibia and femur of 4-6-week-old C57BL/6 mice, and erythrocytes were eliminated using RBC Lysing Buffer (Sigma-Aldrich, USA). The remaining cells were seeded in plates and cultured in RPMI-1640 medium (biochannel, CNA) with 15% heat-inactivated fetal bovine serum (DCELL, CNA) and 1% penicillin-streptomycin (Beyotime, CNA). Macrophage differentiation was induced by 20 ng/mL of M-CSF (Novoprotein, CNA) added to the medium. Cells were incubated at 37 °C with 5% CO2, and on day 3 the medium needed to be replaced with fresh medium. By day 5, BMDMs would be mature.
PMs: Mouse peritoneal macrophages were collected from the peritoneal exudates of 6–8-week-old mice that received 2 mL of fluid thioglycolate medium through intraperitoneal injection for three times. The peritoneal exudate cells were cultured in RPMI-1640 medium (biochannel, CNA) containing 15% heat-inactivated fetal bovine serum (DCELL, CNA) for 3–4 h at 37 °C and 5% CO2. The non-adherent cells were then removed.
Real-Time Quantitative PCR(Q-PCR)
Total RNA was extracted from lung tissue or cells using the Trizol reagent (Vazyme, CNA) following the instructions of manufacturer. The RNA was then reverse transcribed using the HiScript II Q Select RT SuperMix for qPCR (Vazyme, CNA). Then the resulting cDNA was used for Real-Time Quantitative PCR with SYBR Green Master Mix (Vazyme, CNA) on a StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA). The method of presenting quantitative gene expression was relative quantification using comparative CT method. β-actin was selected as internal control gene.28 The primer sequences were ordered from Hangzhou Youkang Biotechnology Co., Ltd and are shown in Table 2.
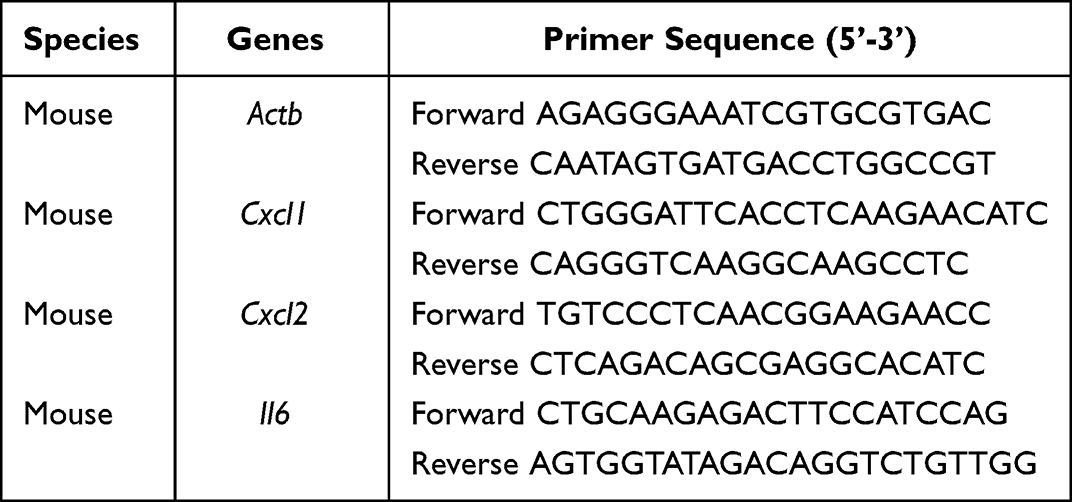 |
Table 2 The Primer Sequences for RT-PCR |
Western Blot (WB)
Mixed 1mL 5×Loading buffer (Beyotime, CNA), 4 mL RIPA (Beyotime, CNA), 200 μL PMSF (Beyotime, CNA) and 36 μL β-mercaptoethanol (sangon, CNA) to obtain the 1× Loading buffer. The cell lysate of BMDMs and PMs was collected using the 1× Loading buffer. After ultraphonic cell lysis and protein degeneration, Western blot was performed using 6% and 10% SDS-PAGE gels (Beyotime, CNA) and transferred to PVDF membranes (Phamtacia Corporation). The membranes were then blocked with 5% skim milk (sangon, CNA) in TBST buffer at room temperature for 1 h. Next, they were incubated overnight at 4 °C with primary antibodies diluted in 5% skim milk, followed by incubation with fluorescent secondary antibodies (EarthOx, USA) for 1 h at room temperature. The antibodies used were anti-β-actin (abclonal, CNA), anti-phosphorylated-mTOR (p-mTOR) (Cell Signaling Technology, USA) and anti-p-RPS6(Cell Signaling Technology, USA). The blots were imaged using the Odyssey two-color infrared laser imaging system (LI-COR, USA).
Immunofluorescence Analysis (IF)
The cells were fixated with 4% formaldehyde (Sinopharm, CNA) at room temperature for 20 min. After being washed with PBS, the cells were treated with 0.1% Triton-X-100 (Sigma-Aldrich, USA) for 10 min to permeabilize them, and then blocked with 5% BSA (sangon, CNA) at room temperature for 30 min. After washed with PBS, the cells were incubated overnight at 4 °C with primary antibodies, including anti-mouse-F4/80 and anti-rabbit-p-RPS6 (Cell Signaling Technology, USA), diluted at 1:200 in 5% BSA. This was followed by incubation for 30min at room temperature with fluorescent secondary antibody, Alexa Fluor® 488 goat anti-mouse IgG (HCL) antibody (Invitrogen, CNA) and Alexa Fluor® 555 goat anti-rabbit IgG (Invitrogen, CNA) diluted at 1:1000 in 5% BSA. Subsequently, the cells were incubated with DAPI (Invitrogen, CNA), diluted at 1:1000 in 5% BSA, for 20min at room temperature. Image capture was conducted using a fluorescence microscope (OLYMPUS IX83-FV3000, JPN), and the relative mean fluorescence intensities were normalized to the levels of the control.
Enzyme Linked Immunosorbent Assay (EILSA)
The BMDMs cell culture supernatant and BALF supernatant of animal model were collected. The concentrations of CXCL1, CXCL2 and IL6 were determined with ELISA kits (RD Systems, USA) following the protocol of manufacturer.
Phagocytic Activity
The phagocytic activity of BMDMs is assessed using fluorescenct beads (Sigma-Aldrich, USA) following the instructions of manufacturer. Flow Cytometry (CytoFLEX S, USA) was administered for phagocytose analysis.
Database Preparation
The 3D crystal structure of mTOR was obtained from the RCSB protein databank (www.rcsb.org, PDB ID: 5FLC).29 The structure of Rapa as the original ligands was separated from the active site of this complex, and the central coordinates of the removed original ligands were defined as the central coordinates for docking. The 3D structure of Pam2csk4 was downloaded from the Drugbank database (https://www.drugbank.com/). The ligands were prepared according to the default parameters (implemented with the MMFF94 force field) of RDKit. Ten most favored poses were then predicted, scored, and presented in kcal/mol using Ledock.30 The root-mean-square deviation (RMSD) of 1.0 Å was used for pose categorization, and a value of 20 was used for binding poses, as recommended. The results were visualized using Schrödinger 2023–1. The primer sequences for gene identification and Q-PCR were obtained from the National Library of Medicine (www.ncbi.nlm.nih.gov/gene/).
Statistical Analysis
The results are presented as mean ± SD. The statistical significance of differences was assessed using one-way ANOVA with Tukey’s multiple comparison test, performed with GraphPad Prism 8.4.3 software (GraphPad Software, San Diego, CA, USA). The level of significance (* p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001) was used for all tests.
Results
Rapa Exacerbates S. aureus Pneumonia in Mouse Models
The effect of Rapa on S. aureus pneumonia was investigated. After 3 days of Rapa treatment, mouse models of S. aureus pneumonia were constructed (Figure 1A). The representative histological pictures of H&E staining of mouse lungs and histological scores showed that the inflammation induced by S. aureus was exacerbated after the treatment of Rapa, as more inflammatory cells around the airways could be observed. And intraperitoneal injection of Rapa alone did not cause significant inflammatory cell infiltration in the lungs (Figure 1B and C). Accordingly, total cells and neutrophils in BALF were increased in Rapa treated mice with S. aureus pneumonia (Figure 1D–F). These results suggest that Rapa exacerbates S. aureus pneumonia in mouse models.
 |
Figure 1 Rapa exacerbates S. aureus pneumonia in mouse models.C57BL/6 mice were treated with Rapa 2 mg/kg within day 0–2. The control group was treated with DMSO. On day 3, the mice were exposed to S. aureus to establish the pneumonia model; (A) Experimental scheme; (B and C) Representative histological pictures (B) and semi-quantified score (C) of H&E staining of mouse lungs, Scale bar: 200 µm; (D) Amounts of total cells; (E and F) Proportion (E) and amounts (F) of neutrophils in BALF. Data represents 3–5 mice in each group and were replicated in three independent experiments. Data expressed as mean ± SD in each group. i.p., intraperitoneal injection; i.t., intratracheal instillation; Rapa, Rapamycin; DMSO, Dimethyl sulfoxide; S. aureus, Staphylococcus aureus; BALF, bronchoalveolar lavage fluid; + or -, with or without S. aureus infection; *p < 0.05, **p < 0.01. |
Rapa Upregulates S. aureus-Induced Inflammation in Macrophages
The previous results suggested that Rapa may exacerbate lung inflammation by promoting recruitment of neutrophils. Macrophages are the major source of neutrophil chemokines, CXCL1 and CXCL2,31 and play vital roles in immunomodulation, phagocytosis and antigen presentation. Therefore, BMDMs were cultured in vitro to investigate the effects. The results showed that inflammation in macrophages was significantly induced by S. aureus, and Rapa increased the mRNA levels of neutrophil-associated chemokines, Cxcl1, Cxcl2 (Figure 2A and B) and inflammatory cytokines Il6 (Figure 2C). Furthermore, Rapa increased the protein secretion of CXCL1, CXCL2 and IL6 (Figure 2D–F). These results suggest that Rapa upregulates the levels of S. aureus-induced neutrophil-related inflammatory cytokines and chemokines in macrophages.
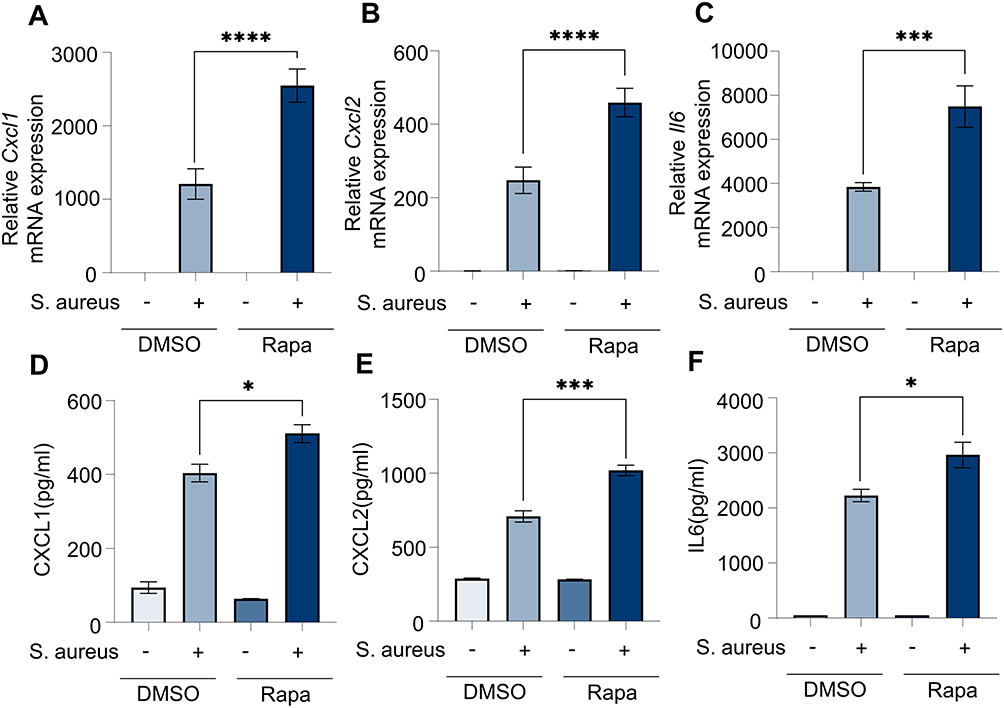 |
Figure 2 Rapa upregulates inflammation levels in BMDMs induced by S. aureus. BMDMs and culture supernatant were collected for Q-PCR and ELISA after incubation with Rapa (20 nM) for 3 hours and S. aureus (10 MOI) for 6 hours. (A) Cxcl1 mRNA; (B) Cxcl2 mRNA; (C) Il6 mRNA; (D) CXCL1 protein; (E) CXCL 2 protein; (F) IL6 protein. Data in panels (A–F) represent 3 samples for each group and were replicated in 3 independent experiments. Data expressed as mean ± SD in each group. Rapa, Rapamycin; DMSO, Dimethyl sulfoxide; S. aureus, Staphylococcus aureus; CXCL1, C-X-C motif chemokine ligand 1; CXCL2, C-X-C motif chemokine ligand 2; IL6, interleukin 6; + or -, with or without S. aureus infection; * p < 0.05, *** p < 0.001, ****p < 0.0001. |
S. aureus Upregulates mTOR Phosphorylation in Macrophages
Previous results (Figure 2) suggested that Rapa exacerbated the S. aureus-induced inflammatory response in macrophages. Rapa has been reported to inhibit the phosphorylation of mTOR. To investigate the effects of mTOR on macrophages infected by S. aureus, BMDMs and PMs were cultured in vitro and infected with S. aureus. Consistent with our expectations, levels of p-mTOR protein increased (Figure 3A–C), indicating the potential importance of mTOR in defense against S. aureus infection. Subsequently, the potential mechanism was explored. LPPs, a group of membrane proteins of S. aureus, are a major regulator of macrophage immune responses and metabolism, with di-acylated lipopeptides in LPPs showing the most significant effect.7 Molecular docking was performed to investigate the possible interaction of mTOR and Pam2csk4, a synthetic analogue of di-acylated lipopeptides. The dimerized mTOR and the most favored binding sites were shown (Figure 3D). The binding energy of the optimal binding conformation between Pam2csk4 and mTOR was −6.89 kcal/mol (Figure 3F), compared to −5.24 kcal/mol between Rapa and mTOR (Figure 3E), which indicated a possible strong affinity between Pam2csk4 and mTOR. The binding contribution between mTOR and Rapa (Figure 3G) or Pam2csk4 (Figure 3H) was visualized using 2D maps. For Rapa, hydrophobic interactions provided the most powerful free energy for conformational homeostasis. Moreover, Glu2032 and His2028 played the roles of hydrogen acceptor and donor, forming hydrogen bonds with the hydroxyl group of Rapa, respectively. For Pam2csk4, more electrostatic interactions were involved in the combination of them. Glu2032, Arg2036, Lys2166 were key residues. Therefore, we believe that di-acylated lipopeptides serve as potential ligands of mTOR.
 |
Figure 3 S. aureus induces mTOR activation in macrophages. Cells were infected with S. aureus for various concentrations (MOI) and timepoints (hours); (A and B) WB for p-mTOR protein in BMDMs; (C) WB for p-mTOR protein in PMs; (D) 3D structure of dimerized mTOR and the most favored binding sites; (E and F) 3D interaction structure between mTOR and Rapa (E) or Pam2csk4 (F); (G and H) 2D interaction structure between mTOR and Rapa (G) or Pam2csk4 (H). Data in panels (A–C) were replicated in 3 independent experiments. p-mTOR, phosphorylated mechanistic target of rapamycin; ACTB, β-actin; S. aureus, Staphylococcus aureus; MOI, multiplicity of infection; GLU, Glutamic Acid; HIS, Histidine; SER, Serine. |
Rapa Exacerbates S. aureus-Induced Inflammation in Macrophages via mTOR-RPS6 Pathway
RPS6, one of the downstream proteins of mTOR, can be phosphorylated and activated upon mTOR activation. In vitro, WB and IF results showed that p-RPS6 protein levels were increased in macrophages infected with S. aureus (Figure 4A–E). This effect was also observed in vivo, as demonstrated by the IF results of BALF cells (Figure 4F and G). These results suggest the activation of the mTOR-RPS6 pathway during S. aureus infection in macrophages. Subsequently, we investigated whether Rapa regulates inflammation by affecting this pathway. In BMDMs, Rapa inhibited the phosphorylation of RPS6 (Figure 4H). When utilizing PF4708671 to inhibit RPS6 phosphorylation, the levels of Cxcl1 and Cxcl2 induced by S. aureus significantly increased (Figure 4I and J). These results suggest that Rapa may upregulate the level of S. aureus-induced inflammation by inhibiting the activation of the mTOR-RPS6 pathway.
 |
Figure 4 Rapa exacerbates the S. aureus-induced inflammation via mTOR-RPS6 pathway in macrophages. Cells were infected with S. aureus for varying concentrations (MOI) and times (hours); (A and B) WB for p-RPS6 protein in BMDMs; (C) WB for p-RPS6 protein in PMs; (D and E) IF for p-RPS6 protein in BMDMs. C57BL/6 mice were exposed to S. aureus i.t. to establish a pneumonia model, and the control group was treated with saline; (F and G) IF for p-RPS6 protein in macrophages in BALF. Cells were incubated with Rapa (20 nM) or PF4708671 (10 μM) for 3 hours, and infected by S. aureus (10 MOI) for 6 hours. (H) WB for p-RPS6 protein in BMDMs; (I and J) Q-PCR for Cxcl1 and Cxcl2 mRNA in BMDMs. Data in panels (A–J) represent 3 samples for each group and were replicated in 3 independent experiments. Data expressed as mean ± SD in each group. Rapa, Rapamycin; DMSO, Dimethyl sulfoxide; S. aureus, Staphylococcus aureus; p-RPS6, phosphorylated Ribosomal protein S6; ACTB, β-actin; DAPI, 4’,6-diamidino-2-phenylindole; F4/80, mouse EGF-like module-containing mucin-like hormone receptor-like 1; Cxcl1, C-X-C motif chemokine ligand 1; Cxcl2, C-X-C motif chemokine ligand 2; MOI, multiplicity of infection; + or -, with or without S. aureus infection; *p < 0.05, **p < 0.01, ****p < 0.0001. |
Rapa Does Not Affect the Phagocytosis of Macrophages
To investigate whether the increased release of inflammatory cytokines and chemokines from macrophages is also affected by phagocytosis, we used PE-fluorescent beads to observe macrophage phagocytosis. The proportion of macrophages with PE fluorescence detected by flow cytometry (Figure 5A and B) can reflect the phagocytic activity of macrophages. Our results suggest that Rapa does not significantly increase the phagocytic activity of macrophages (Figure 5C). Furthermore, the S. aureus CFU from BMDMs culture supernatant appeared to be the same in two groups (Figure 5D), indicating that the phagocytosis of S. aureus was also not affected by Rapa. These results suggest that the upregulation of inflammation in macrophages is not due to an increased phagocytic capacity.
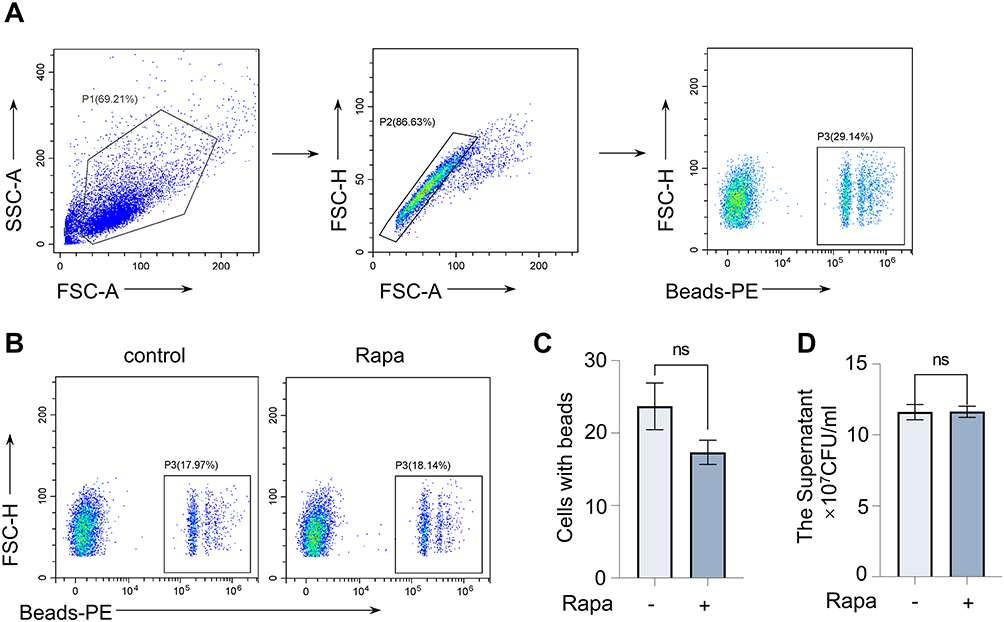 |
Figure 5 Rapa does not affect the phagocytosis of BMDMs. Cells were collected for FC after incubation with Rapa (20 nM) for 9 hours and fluorescent beads (3 μL/mL) for 0.5 hours. (A) The FC gating strategy; (B and C) The percentage of cells with beads. Cells were incubated with Rapa (20 nM) for 3 hours and infected with S. aureus (10 MOI) for 6 hours; (D) The culture supernatant concentration of S. aureus. Data in panels (A–D) represented 3 samples for each group and were replicated in 3 independent experiments. Data expressed as mean ± SD in each group. Rapa, Rapamycin; FSC-A, forward scatter- area; FSC-H, forward scatter- height; SSC-A, side scatter-area; PE, Phycoerythrin; + or -, with or without Rapa treatment; ns, no significance. |
Specific Knockout of Mtor in Mice Myeloid Cells Exacerbates S. aureus Pneumonia
To clarify whether mTOR can regulate the inflammation induced by S. aureus in vivo, Mtor-specific knockout in myeloid cells mice models with S. aureus pneumonia were constructed. The representative histological pictures of H&E-stained mouse lungs showed that the inflammation was significantly induced by S. aureus. Furthermore, an even greater increase in inflammatory cells around the airways could be observed in myeloid cells-specific Mtor knockout mice (Figure 6A and B). Accordingly, the total cell counts and the number of neutrophils in the BALF were increased (Figure 6C and D), and levels of neutrophil-related chemokines, CXCL1 and CXCL2, were also found to be increased in the supernatant of BALF (Figure 6E and F). These results suggest that specific knockout of Mtor in myeloid cells exacerbated S. aureus pneumonia in vivo.
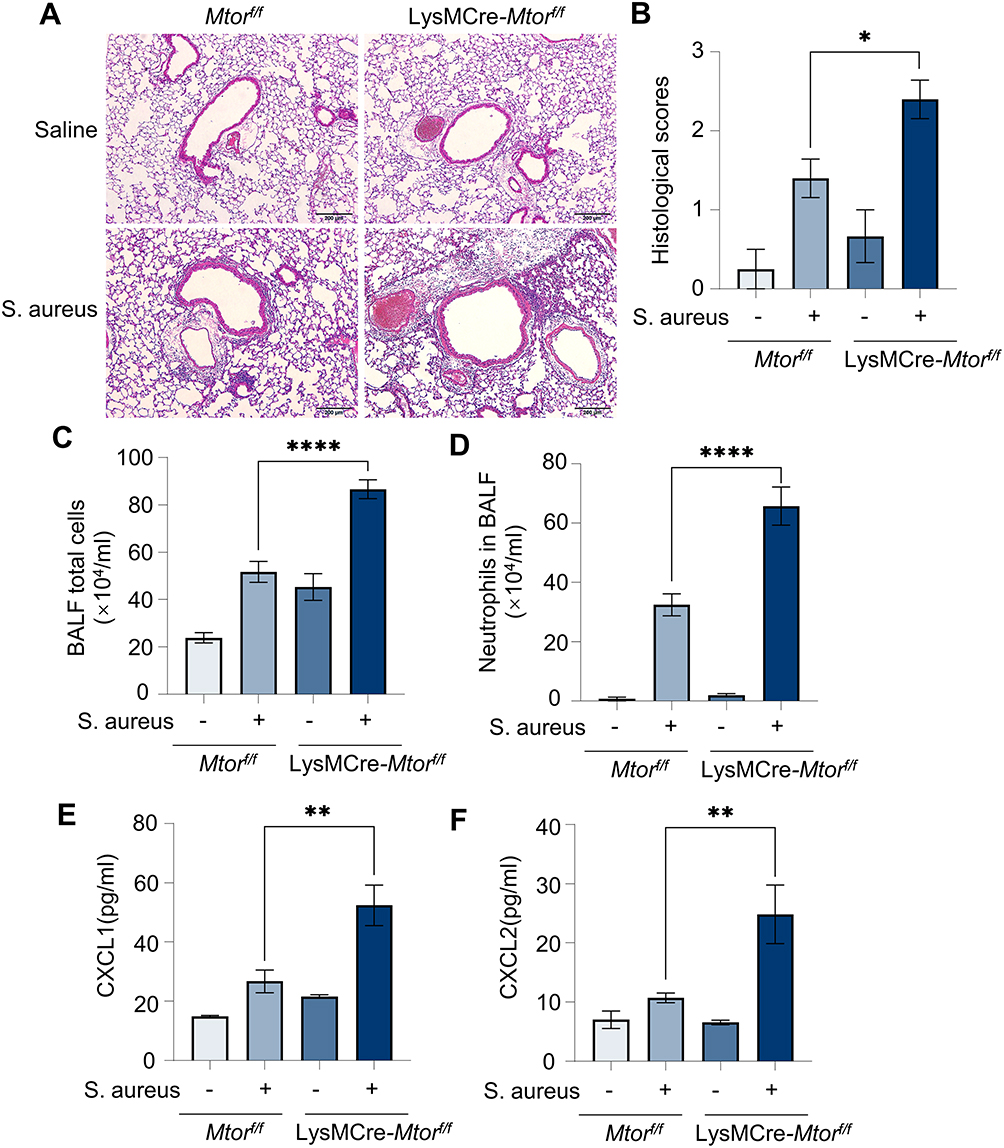 |
Figure 6 Specific knockout of Mtor in mouse myeloid cells exacerbates S. aureus pneumonia. Mtorf/f and LysMCre-Mtorf/f mice were exposed to S. aureus i.t. to establish a pneumonia model; The control group was treated with saline. (A and B) Representative histological pictures (A) and semi-quantified score (B) of H&E staining of mouse lungs, Scale bar: 200 µm; (C and D) Amounts of total cells (C) and neutrophils (D) in BALF; (E and F) chemokines in BALF, (E) CXCL1 protein; (F) CXCL2 protein. Data in panels represented 3–5 mice for each group and were replicated in three independent experiments. Data expressed as mean ± SD in each group. Mtorf/f, Mtorflox/flox; LysMCre, lysozyme M-Cre; S. aureus, Staphylococcus aureus; BALF, bronchoalveolar lavage fluid; CXCL1, C-X-C motif chemokine ligand 1; CXCL2, C-X-C motif chemokine ligand 2; + or -, with or without S. aureus infection; * p < 0.05, ** p < 0.01, ****p < 0.0001. |
Discussion
Although Rapa is a promising drug due to its significant immunosuppressive, antitumor and anti-aging effects, its inflammatory side effects are not negligible and should not be overlooked. Our study demonstrates that Rapa exacerbates inflammation in S. aureus pneumonia by upregulating the release of cytokines and chemokines from macrophages. Consistent with our results, a multicenter clinical study involving 30 renal transplant patients suggests that after the use of Rapa, two-thirds of patients exhibit at least one symptom of inflammation. The most common symptom reported is impaired skin mucosa, often accompanied by increased levels of IL6 and TNFα.32 A meta-analysis of 10 clinical trials, which includes 779 participants, has found an increasing risk of rash and stomatitis in cancer patients treated with Rapa. This risk is independent of the type of cancer being treated.33 And this inflammatory effect is also observed in healthy older adults. A randomized clinical study reveals a 264% increase in IL6 levels and a 16% increase in TNFα levels in the serum after six weeks of Rapa treatment at a dose of 1 mg/day.34 In addition to skin and mucosa inflammation, cases of Rapa-associated arthritis, colitis, and pneumonia have also been reported.15,35,36 Our study suggests that Rapa exacerbates the inflammation caused by S. aureus infection, which emphasizes the importance of monitoring Rapa’s side effects on opportunistic infections, in addition to non-infectious inflammation. There are also some studies suggesting the effect of Rapa on other bacterial pneumonia. An article shows that Rapa can alleviate LPS-induced pneumonia.37 Additionally, it has been shown that Rapa enhances resistance to Streptococcus pneumoniae pneumonia in aged mice by reducing cellular aging.38 These effects differ from our findings, indicating that Rapa may have varying effects against different bacteria.
mTOR is the target of Rapa and is commonly reported to be involved in cellular anabolic metabolism.24 Our results demonstrate an increase in mTOR phosphorylation when macrophages are infected with S. aureus. Additionally, inhibiting or knocking down mTOR can enhance the release of inflammatory cytokines induced by S. aureus. These findings suggest that mTOR may play a crucial role in regulating S. aureus infection and the levels of chemokines. Consistent with our study, enhancing the mTOR phosphorylation capacity in NK cells improved immune defense against opportunistic infections.39 In Epilepsy patients, the inhibition of mTOR increased the production of inflammatory cytokines in their peripheral mononuclear blood cells.40 However, when macrophages infected with Trueperella pyogenes were treated with mTOR siRNA, the relative levels of IL1β, IL6 and TNFα were observed to be decreased.41 These results suggest that the effects of mTOR on immune cells and cytokine levels may be convincible but heterogeneous.
The main functions of mTOR activation are the promotion of anabolism and the inhibition of catabolism. Its downstream effects include the activation of S6K to increase the phosphorylation of RPS6, as well as the inhibition of autophagy, lysosome biogenesis, and proteasome assembly.24 Autophagy is considered to be induced by S. aureus infection, facilitating its clearance.42 However, in some conditions, S. aureus can block autophagy flux, for example, by inhibiting the conversion of LC3-I to LC3-II and increasing the accumulation of p62. It can also increase the pH in autolysosomes, allowing it to evade autophagic degradation.43 Through this mechanism, S. aureus acquires an intracellular survival niche, consequently facilitating its dissemination within the host.44 Our study demonstrates that p-RPS6, another downstream molecule of mTOR that regulates 5’ cap-dependent protein translation, is increased in macrophages infected with S. aureus.24 These finding indicates that protein synthesis may play a role in regulating cytokine levels. The inhibition of mTOR-RPS6 leads to the expansion of the free amino acid pool, which may provide flexible possibilities for regulating chemokine and cytokine levels in the various conditions mentioned above.
Macrophages can phagocytose antigens and digest them into peptide fragments.45 There were still not enough convincing studies showing that mTOR regulates the bacterial phagocytosis process. Our results indicate that Rapa does not significantly increase the phagocytic activity of macrophages. Most of the current studies on the anti-bacterial effects of mTOR are focused on the digestion process following phagocytosis.46,47 It has been shown that inhibition of mTOR in dendritic cells induces autophagy and enhances cellular response to antigen peptide presentation.46 In macrophages infected by Trueperella pyogenes, mTOR functions as a potent regulator in autophagy-mediated bacteria elimination.41 These results indicate that mTOR in immune cells participants in the digestion of antigens after phagocytosis, and further research is needed to investigate the phagocytic process of bacteria in order to enhance bacterial clearance.
Our study still has some limitations, we did not collect data from patients with S. aureus pneumonia. Furthermore, additional investigation is needed to determine the detailed mechanisms of Rapa and the mTOR-RPS6 pathway in regulating S. aureus-induced macrophage inflammation and pneumonia.
Conclusion
Our study shows that Rapa exacerbates S. aureus pneumonia by increasing the inflammatory levels of macrophages. Inhibition of mTOR-RPS6 pathway upregulates the expression of cytokines and chemokines in macrophages, thus increases inflammatory cells infiltration and exacerbates tissue damage. However, phagocytosis is not affected. This suggests that patients using Rapa in clinic need to be alert to opportunistic infections, and that the drug does not increase the clearance capacity of macrophages. Instead, it may induce macrophages to release more chemokines, exacerbating tissue damage.
Data Sharing Statement
The data of this study are available from the corresponding author.
Ethics Approval and Informed Consent
This study was performed in line with the Ethical Committee for Animal Studies at Zhejiang University and followed the procedures and principles of the experimental animal welfare and ethical review of Zhejiang University for the care of laboratory animals.
Consent for Publication
This manuscript has never been published or accepted elsewhere. The manuscript is not currently under consideration for publication elsewhere. All authors of this paper have read and approved the publication of this manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (U22A20265, 82270023 and 82100001) and the Zhejiang Provincial Natural Science Foundation of China (LQ22H010006, LD21H010001).
Disclosure
The authors disclosed no relevant financial or non-financial interests in this work.
References
1. Eng CP, Sehgal SN, Vézina C. Activity of rapamycin (AY-22,989) against transplanted tumors. J Antibiot. 1984;37(10):1231–1237. doi:10.7164/antibiotics.37.1231
2. Rasigade JP, Dumitrescu O, Lina G. New epidemiology of Staphylococcus aureus infections. Clin Microbiol Infect. 2014;20(7):587–588. doi:10.1111/1469-0691.12718
3. van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. Predictors of mortality in staphylococcus aureus bacteremia. Clin Microbiol Rev. 2012;25(2):362–386. doi:10.1128/CMR.05022-11
4. Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532. doi:10.1056/NEJM199808203390806
5. von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N Engl J Med. 2001;344(1):11–16. doi:10.1056/NEJM200101043440102
6. Li M, Yu J, Guo G, Shen H. Interactions between macrophages and biofilm during Staphylococcus aureus-associated implant infection: difficulties and solutions. J Innate Immun. 2023. doi:10.1159/000530385
7. Nguyen M-T, Hu Z, Mohammad M, et al. Bacterial lipoproteins shift cellular metabolism to glycolysis in macrophages causing bone erosion. Microbiol Spectr. 2023;11:e0429322. doi:10.1128/spectrum.04293-22
8. Nguyen MT, Götz F. Lipoproteins of gram-positive bacteria: key players in the immune response and virulence. Microbiol Mol Biol Rev. 2016;80(3):891–903. doi:10.1128/MMBR.00028-16
9. Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 1977;55(1):48–51. doi:10.1139/y77-007
10. Andrassy J, Graeb C, Rentsch M, Jauch K-W, Guba M. mTOR inhibition and its effect on cancer in transplantation. Transplantation. 2005;80(1 Suppl):S171–S174. doi:10.1097/01.tp.0000186912.23630.85
11. Schnitzbauer AA, Filmann N, Adam R, et al. mTOR inhibition is most beneficial after liver transplantation for hepatocellular carcinoma in patients with active tumors. Ann Surg. 2020;272(5):855–862. doi:10.1097/SLA.0000000000004280
12. Verna EC, Patel YA, Aggarwal A, et al. Liver transplantation for hepatocellular carcinoma: management after the transplant. Am J Transplant. 2020;20(2):333–347. doi:10.1111/ajt.15697
13. de Fijter JW. Cancer and mTOR inhibitors in transplant recipients. Transplantation. 2017;101(1):45–55. doi:10.1097/TP.0000000000001447
14. Champion L, Stern M, Israël-Biet D, et al. Brief communication: sirolimus-associated pneumonitis: 24 cases in renal transplant recipients. Ann Intern Med. 2006;144(7):505–509. doi:10.7326/0003-4819-144-7-200604040-00009
15. Pham P-T-T, Pham P-CT, Danovitch GM, et al. Sirolimus-associated pulmonary toxicity. Transplantation. 2004;77(8):1215–1220. doi:10.1097/01.TP.0000118413.92211.B6
16. Buron F, Malvezzi P, Villar E, et al. Profiling sirolimus-induced inflammatory syndrome: a prospective tricentric observational study. PLoS One. 2013;8(1):e53078. doi:10.1371/journal.pone.0053078
17. Gudiol C, Sabé N, Carratalà J. Is hospital-acquired pneumonia different in transplant recipients? Clin Microbiol Infect. 2019;25(10):1186–1194. doi:10.1016/j.cmi.2019.04.003
18. Aguilar-Guisado M, Jiménez-Jambrina M, Espigado I, et al. Pneumonia in allogeneic stem cell transplantation recipients: a multicenter prospective study. Clin Transplant. 2011;25(6):E629–E638. doi:10.1111/j.1399-0012.2011.01495.x
19. Hoyo I, Linares L, Cervera C, et al. Epidemiology of pneumonia in kidney transplantation. Transplant Proc. 2010;42(8):2938–2940. doi:10.1016/j.transproceed.2010.07.082
20. Kreitmann L, Gaudet A, Nseir S. Ventilator-associated pneumonia in immunosuppressed patients. Antibiotics. 2023;12:2 doi:10.3390/antibiotics12020413.
21. Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. doi:10.1016/S0092-8674(02)00833-4
22. Kim D-H, Sarbassov DD, Ali SM, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular Cell. 2003;11(4):895–904. doi:10.1016/S1097-2765(03)00114-X
23. Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6(11):1122–1128. doi:10.1038/ncb1183
24. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
25. Kim H, Banerjee N, Barnes RC, et al. Mango polyphenolics reduce inflammation in intestinal colitis-involvement of the miR-126/PI3K/AKT/mTOR axis in vitro and in vivo. Mol Carcinog. 2017;56(1):197–207. doi:10.1002/mc.22484
26. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi:10.1023/A:1008942828960
27. Risson V, Mazelin L, Roceri M, et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol. 2009;187(6):859–874. doi:10.1083/jcb.200903131
28. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi:10.1038/nprot.2008.73
29. Aylett CHS, Sauer E, Imseng S, et al. Architecture of human mTOR complex 1. Science. 2016;351(6268):48–52. doi:10.1126/science.aaa3870
30. Zhang N, Zhao H. Enriching screening libraries with bioactive fragment space. Bioorg Med Chem Lett. 2016;26(15):3594–3597. doi:10.1016/j.bmcl.2016.06.013
31. De Filippo K, Henderson RB, Laschinger M, Hogg N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol. 2008;180(6):4308–4315. doi:10.4049/jimmunol.180.6.4308
32. Kraig E, Linehan LA, Liang H, et al. A randomized control trial to establish the feasibility and safety of rapamycin treatment in an older human cohort: immunological, physical performance, and cognitive effects. Exp Gerontol. 2018;105:53–69. doi:10.1016/j.exger.2017.12.026
33. Rana JS, Sheikh J. Serum sickness-like reactions after placement of sirolimus-eluting stents. Ann Allergy Asthma Immunol. 2007;98(2):201–202. doi:10.1016/S1081-1206(10)60699-0
34. Gomez-Fernandez C, Garden BC, Wu S, Feldman DR, Lacouture ME. The risk of skin rash and stomatitis with the mammalian target of rapamycin inhibitor temsirolimus: a systematic review of the literature and meta-analysis. Eur J Cancer. 2012;48(3):340–346. doi:10.1016/j.ejca.2011.11.028
35. Molinari M, Al-Saif F, Ryan EA, et al. Sirolimus-induced ulceration of the small bowel in islet transplant recipients: report of two cases. Am J Transplant. 2005;5(11):2799–2804. doi:10.1111/j.1600-6143.2005.01082.x
36. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. doi:10.1146/annurev-immunol-020711-075024
37. Nakajima T, Lin K-W, Li J, et al. T cells and lung injury. Impact of rapamycin. Am J Respir Cell Mol Biol. 2014;51(2):294–299. doi:10.1165/rcmb.2013-0171OC
38. Hinojosa CA, Mgbemena V, Van Roekel S, et al. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012;47(12):958–965. doi:10.1016/j.exger.2012.08.013
39. Bösken B, Hepner-Schefczyk M, Vonderhagen S, Dudda M, Flohé SB. An inverse relationship between c-Kit/CD117 and mTOR confers NK cell dysregulation late after severe injury. Front Immunol. 2020;11:1200. doi:10.3389/fimmu.2020.01200
40. Vieira ÉLM, Martins FMA, Bellozi PMQ, et al. PI3K, mTOR and GSK3 modulate cytokines’ production in peripheral leukocyte in temporal lobe epilepsy. Neurosci Lett. 2021;756:135948. doi:10.1016/j.neulet.2021.135948
41. Huang T, Cui K, Song X, et al. MTOR involved in bacterial elimination against Trueperella pyogenes infection based on mice model by transcriptome and biochemical analysis. Vet Microbiol. 2019;235:199–208. doi:10.1016/j.vetmic.2019.06.021
42. Lv Y, Fang L, Ding P, Liu R. PI3K/Akt-Beclin1 signaling pathway positively regulates phagocytosis and negatively mediates NF-κB-dependent inflammation in Staphylococcus aureus-infected macrophages. Biochem Biophys Res Commun. 2019;510(2):284–289. doi:10.1016/j.bbrc.2019.01.091
43. Cai J, Li J, Zhou Y, et al. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J Cell Mol Med. 2020;24(6):3460–3468. doi:10.1111/jcmm.15027
44. Mulcahy ME, O’Brien EC, O’Keeffe KM, Vozza EG, Leddy N, McLoughlin RM. Manipulation of autophagy and apoptosis facilitates intracellular survival of staphylococcus aureus in human neutrophils. Front Immunol. 2020;11:565545. doi:10.3389/fimmu.2020.565545
45. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi:10.1038/nature12034
46. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15(3):267–276. doi:10.1038/nm.1928
47. Fabri M, Realegeno SE, Jo E-K, Modlin RL. Role of autophagy in the host response to microbial infection and potential for therapy. Curr Opin Immunol. 2011;23(1):65–70. doi:10.1016/j.coi.2010.10.010
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.