Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
Rapamycin and Chloroquine Modulate Insulin Resistance and Hepatic Steatosis in a High-Fat/High-Cholesterol Diet-Induced Metabolic Dysfunction-Associated Steatotic Liver Disease Mouse Model
Authors Liu Q, Du Q, Yuan X, Zhang T, Li J, Xu J, Xie R ![]()
Received 10 May 2025
Accepted for publication 8 October 2025
Published 5 November 2025 Volume 2025:18 Pages 4059—4072
DOI https://doi.org/10.2147/DMSO.S539555
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Jae Woong Sull
Qi Liu,1,* Qian Du,2,* Xiaolu Yuan,3 Ting Zhang,3 Jiajing Li,3 Jingyu Xu,4 Rui Xie2
1Department of Gastroenterology, Zhejiang Provincial People’s Hospital Bijie Hospital, Bijie, Guizhou, People’s Republic of China; 2Department of Endoscopy and Digestive System, Guizhou Provincial People’s Hospital, Guiyang, Guizhou, People’s Republic of China; 3Department of Gastroenterology, Digestive Disease Hospital, Affiliated Hospital of Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China; 4School of Preclinical Medicine of Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingyu Xu, School of Preclinical Medicine of Zunyi Medical University, No. 6, Xuefu West Road, Xinpu New District, Zunyi, Guizhou, 563003, People’s Republic of China, Email [email protected] Rui Xie, Department of Endoscopy and Digestive System, Guizhou Provincial People’s Hospital, 83 Zhongshan East Road, Yunyan District, Guiyang, Guizhou, 550002, People’s Republic of China, Email [email protected]
Objective: To establish a short-term high-fat/high-cholesterol (HFHC) diet-induced Metabolic dysfunction-associated steatotic liver disease (MASLD) mouse model, and evaluate the effects of rapamycin (RaPa) and chloroquine (CQ) on this model to explore their therapeutic potential and side effects.
Methods: An early MASLD mouse model was constructed via short-term HFHC diet feeding. Model mice were intraperitoneally injected with RaPa or CQ. Drug effects were analyzed on body weight, liver weight, lipid metabolism-related genes (APOB, FASN, PLIN2), inflammatory factors (IL-6, IL-10), and fibrosis markers (LOX, Col-1α-1, CCL2, TGFβ 1, PDGFRβ, α-SMA) at mRNA and protein levels.
Results: RaPa ameliorated body weight and liver weight in early MASLD mice, downregulated FASN and PLIN2 expression, upregulated IL-10 mRNA levels, and alleviated hepatic steatosis, but induced metabolic disorders such as Insulin resistance and hyperlipidemia. In contrast, CQ promoted FASN and PLIN2 expression, exacerbated hepatic steatosis, reduced IL-10 mRNA levels, and upregulated fibrosis-related markers (LOX, TGFβ 1, PDGFRβ, α-SMA) at both mRNA and protein levels, thereby driving MASLD progression to liver fibrosis. Notably, CQ improved metabolic abnormalities in model mice, including obesity, hyperlipidemia, and Insulin resistance.
Conclusion: RaPa and CQ exhibit dual effects on early MASLD: RaPa alleviates hepatic steatosis but exacerbates metabolic disorders, whereas CQ improves metabolic abnormalities but accelerates liver fibrosis. This paradox highlights the need to balance metabolic regulation and liver injury prevention in MASLD treatment, providing critical experimental insights for targeted drug development.
Keywords: MASLD, MASH, autophagy modulation, rapamycin, chloroquine
Introduction
In recent years, metabolic dysfunction-associated steatotic liver disease (MASLD, formerly known as MAFLD/NAFLD) has emerged as a major global public health challenge. Epidemiological data indicate that MASLD currently affects over 25% of the global population, making it a significant chronic metabolic disorder that seriously threatens human health.1 MASLD exhibits a progressive pathological trajectory. Under sustained metabolic stress, hepatic steatosis can gradually develop into metabolic dysfunction-associated steatohepatitis (MASH), which subsequently triggers progressive liver fibrosis through pro-inflammatory cytokine cascades and extracellular matrix deposition. This ultimately leads to irreversible liver damage including end-stage cirrhosis and hepatocellular carcinoma (HCC), significantly increasing treatment difficulty and mortality rates.2 Simultaneously, individuals with Metabolic dysfunction-associated steatotic liver disease (MASLD) face an increased risk of developing type 2 diabetes mellitus (T2DM), cardiovascular disease, chronic kidney disease, and extrahepatic cancers, with the majority of MASLD-related mortality attributable to cardiac causes.3 MASLD exacerbates hepatic and peripheral insulin resistance, predisposes to atherogenic dyslipidemia, and promotes the systemic release of pro-inflammatory cytokines and hepatokines, thereby contributing to the development of T2DM.4 However, the molecular pathogenesis of MASLD remains largely elusive, so treatment options for this disease are still limited.
Abnormal lipid accumulation in hepatocytes is the initial stage of hepatic steatosis. Free fatty acids are taken up by hepatocytes and esterified into triglycerides (TG),5–7 for storage as lipid droplets (LDs).8 Under physiological conditions, LDs are degraded through hydrolysis mediated by the PLIN1-hormone-sensitive lipase axis and via lipophagy. Excessive free fatty acid intake leads to LD accumulation, triggering lipotoxicity and further promoting endoplasmic reticulum stress, apoptosis, inflammation, and metabolic abnormalities such as insulin resistance.9
Clearance of LDs is crucial for maintaining liver function. Autophagy acts as a cellular “recycling station” that maintains homeostasis by degrading abnormal cellular components. Autophagy is an evolutionarily conserved catabolic process characterized by the degradation of cellular proteins and damaged organelles to promote cell survival and maintain cellular homeostasis.10 Previous studies have suggested that autophagy can serve as an effective defense mechanism against various pathological damages, including alcoholic and non-alcoholic fatty liver disease.11 Fat autophagy can affect the severity of fat degeneration, which in turn affects liver damage. CQ can exacerbate liver injury in mice fed with HFD.12 3-methyladenine (3MA), an inhibitor of autophagy, can significantly increase TG levels in liver cells in the absence or presence of exogenous lipid supplements such as oleates, while treatment with rapamycin (an autophagy activator) significantly reduces LD numbers and TGs.13 However, its role in the pathogenesis of MASLD remains controversial.
In this study, we employed the autophagy agonist rapamycin and autophagy inhibitor chloroquine to intervene in the HFHC mouse model, as they target key nodes of the mTOR-autophagy pathway respectively. Although rapamycin inhibits mTORC1 to ameliorate lipid deposition, it disrupts mTORC2 and consequently induces hyperglycemia;14 Chloroquine blocks autophagic flux by inhibiting autophagosome-lysosome fusion, thereby suppressing lipophagy,15,16 and potentially exacerbating hepatic lipid accumulation. This study investigated the roles of autophagy activation and inhibition in MASLD and the advantages and disadvantages of autophagy pathway in regulating MASLD mouse models by intraperitoneal injection of autophagy agonist RaPa and autophagy inhibitor CQ.
In this study, we employed the autophagy agonist rapamycin and autophagy inhibitor chloroquine to intervene in the HFHC mouse model, as they target key nodes of the mTOR-autophagy pathway respectively. Although rapamycin inhibits mTORC1 to ameliorate lipid deposition, it disrupts mTORC2 and consequently induces hyperglycemia;14 Chloroquine blocks autophagic flux by inhibiting autophagosome-lysosome fusion, thereby suppressing lipophagy,15,16 and potentially exacerbating hepatic lipid accumulation. This study investigated the roles of autophagy activation and inhibition in MASLD and the advantages and disadvantages of autophagy pathway in regulating MASLD mouse models by intraperitoneal injection of autophagy agonist RaPa and autophagy inhibitor CQ. Previous studies have shown that a high-fat and high-cholesterol diet can lead to more severe hepatic steatosis, severe inflammation, and perisinusoidal fibrosis (fatty liver disease), but this usually occurs after 30 weeks. However, the HFHD diet model fed for less than 12 weeks does not form significant liver fibrosis.17 To facilitate the observation of the effect of autophagy on MASLD/MASH and liver fibrosis, we constructed a relatively early MASLD model. To avoid the influence of some imperceptible inflammation and fibrosis, we shortened the feeding time of the HFHD diet to 10 weeks to avoid uncontrollable inflammatory responses and fibrosis that might mask the direct effect of the drug on the early pathogenesis.
Materials and Methods
Construction of Animal Models
Eight male wild-type C57 mice were purchased and randomly divided into two groups (n = 4 per group). One group was fed a standard diet (SD), while the other group received a high-fat/high-cholesterol (HFHC) diet (42% fat and 0.2% cholesterol) provided by Nantong Troffey Feed Co., Ltd. After 10 weeks, liver tissue samples were collected for HE staining, Sirius red staining, and Masson staining to assess whether the model mice exhibit early-stage MASLD.
After verifying that high-fat and high-cholesterol diets for 10 weeks can form an early model of MASLD, we constructed interference models of rapamycin(RaPa) and chloroquine(CQ). Forty-eight 8-week-old mice were randomly divided into 4 groups with 12 mice in each group. The mice in the first group were fed the standard diet + intraperitoneal injection of normal saline (SD), the others in the second group were fed the HFHC diet + intraperitoneal injection of normal saline (HFHC), and the others in the third group were fed the HFHC diet + intraperitoneal injection of RaPa (2mg/kg/d)18(HFHC+RaPa). The mice in the fourth group were fed HFHC diet plus intraperitoneally injected CQ (60mg/kg/d)19(HFHC+CQ). GTT and ITT tests were performed before cervical dislocation in 10W mice, and blood from the eyes was collected for blood biochemical analysis. The liver weight and liver weight/body weight ratio of 10W euthanized mice were monitored, and the liver tissues were collected for paraffin embedding and frozen section. The remaining liver tissue is frozen at −80°C for future use.
The experimental mice were purchased from Beijing Huafukang Biotechnology Co., LTD. The ethical approval (No.: ZMU21-2203-551) was approved by the Experimental Animal Welfare Ethics Committee of Zunyi Medical University. Animal handling was performed in accordance with the Chinese “Guidelines for Ethical Review of Animal Welfare” (GB/T 35892–2018) and ARRIVE guidelines.
GTT and ITT Experiments
(1) GTT, mice were fasted (with free access to water) for 12 hours, their tail tips were cut off, and blood was quickly squeezed out from the tail tips. Fasting blood glucose was immediately measured using a blood glucose meter, and then glucose (2g/kg) was injected intraperitoneally. Blood glucose levels were monitored at 30, 60, 90, and 120 minutes after injection.
(2) ITT, fasting for 4 hours (with free access to water), with the same treatment method as above. After measuring fasting blood glucose, intraperitoneal injection of insulin (0.5 IU/kg) is administered; Monitor blood glucose levels at 30, 60, 90, and 120 minutes.
Serum Biochemical Index Detection
After fasting overnight and before euthanizing the mice, collect eye blood, centrifuge to obtain supernatant, and perform biochemical measurements according to standard procedures. Lipids were measured using electrochemiluminescence immunoassay with an automated analyzer (Roche Cobas 8000 System, CV 0.6–0.9%, Indianapolis, IN) for total cholesterol (CHOL), triglycerides (TRIG), low-density lipoprotein cholesterol (LDLc), and high-density lipoprotein cholesterol (HDLc). Measure liver enzymes [aspartate aminotransferase (AST), alanine aminotransferase (ALT)] using UV absorbance (Roche Cobas 8000 System, AST CV 0.5–3.2%, ALT CV 0.5–3.2%, ALT CV 0.5–3.1%).
Liver Histopathology
For histological analysis, liver tissue (0.1–0.2g) was fixed in 10% formalin solution, dehydrated with different concentrations of ethanol solution, and embedded in paraffin. Subsequently, the tissue embedded in paraffin was cut to a thickness of 4μm for staining with hematoxylin and eosin (H&E) as well as Sirius red and Masson. Use Olympus IX81S1F-3 (Olympus, Tokyo, Japan) to examine H&E, Sirius Red, and Masson liver sections at magnification of 40x and 20x, respectively. Use ImageJ software (version 1.37c) to analyze the images.
Hepatic steatosis was quantified through systematic examination of H&E-stained liver sections. For each specimen, 12–40 representative regions of interest (ROIs) were randomly selected to calculate lipid droplet occupancy. Lipid droplets were identified as hollow circular structures under brightfield microscopy, with their proportional area quantified using standardized image analysis software. Fibrotic progression was evaluated using Sirius Red and Masson’s trichrome-stained sections to visualize collagen deposition. A systematic sampling approach was implemented, with 8–16 ROIs analyzed per specimen. All digital images underwent uniform threshold adjustment prior to computational quantification of collagen-positive areas. Results were expressed as percentage of stained tissue relative to total ROI area, ensuring consistent inter-sample comparability.
Protein Imprinting Experiments
The protein lysates were obtained from each mouse liver sample (about 100mg) by adding grinding bead to a mixture of 1mL RIPA buffer and 10μL protease inhibitor. After grinding the resulting tissue homogenate in a grinder and centrifugation at 4°C and 12,000×g for 20 min, supernatant was taken to obtain the protein lysates. The protein concentration in the lysate was measured by spectrophotometer at a wavelength of 595nm using a protein determination dye reagent (Bio-Rad Laboratories, Inc). Thereafter, equal amounts of protein (15μg) were separated from all samples by electrophoresis in 12.5% and 10% sodium dodecyl sulfate polyacrylamide gels. The isolated proteins were transferred to the Immobilon-P membrane (Millipore, Billerica, MA, USA).
Western blot analysis was performed by first treating the membrane with Protein-Free Rapid Blocking Buffer (1×, Yatase PS108P) at room temperature for 15 minutes. Primary antibody incubation was conducted overnight at 4°C using antibodies against FASN, PLIN2, LC3, P62, and α-SMA (Proteintech, 1:5000 dilution), followed by 1-hour incubation with HRP-conjugated secondary antibody (1:5000 dilution) at room temperature (The specific genetic information can be found in Supplementary Information Tables 1 and 2). Protein bands were visualized using an Immobilon immunoblotting detection system (Millipore) combined with a ChemiDoc imaging system (Bio-Rad Laboratories, Inc)., with band intensity quantified via Image Lab 6.1 software and normalized to the housekeeping gene GAPDH (Proteintech 60004-1-Ig).
mRNA Experiments
Total RNA was isolated from liver tissues or cultured cells using TransZol Up Reagent (TransGen Biotech ET111-01), reverse-transcribed to cDNA using the EasyScript One-Step gDNA Removal Kit (TransGen Biotech AE311-04), and amplified with TransStart Top Green qPCR Super Mix (TransGen Biotech AQ131-04). Relative gene expression was calculated using the 2−ΔΔCt method with GAPDH as the internal reference gene. The specific details of the RNA primers can be found in Supplementary Information Table 3.
Statistical Analysis
Statistical analysis is mainly conducted using GraphPad Prism8.0 software. Non paired t-test was used between two samples, one-way analysis of variance was used for pairwise component comparison, LSD test was used for homogeneity of variance, and Dunnett T3 test was used for heterogeneity of variance. The significant results are represented by * P<0.05, * * P<0.01, and * * * P<0.001, respectively.
Result 1: Feeding Mice an HFHC Diet for 10 weeks Effectively Induced an Early-Stage MASLD Model
Eight 8-week-old C57 mice were randomly divided into two groups and fed standard diet (SD) and HFHC diet respectively. In order to avoid severe inflammatory response and fibrosis, the feeding time was shortened to 10W. After euthanizing the mice, liver tissue was collected for HE staining, Sirius red staining, and Masson staining. As shown in Figure 1A, after induction of 10W with HFHC diet, the liver became significantly yellow, indicating smooth and glossy appearance. Compared with the SD diet group, HE staining in the HFHC group mainly manifested as simple fat degeneration with a small amount of balloon like changes, and no inflammatory cell infiltration was observed (Figure 1B). Masson staining and Sirius red staining were used to observe the liver tissue. Red staining only observed a small amount of fibrosis around the sinus and portal vein (Figure 1C and D). We used the Non alcoholic Fatty Liver Disease Activity Score20 and found that all models had a NAS score<5, with no liver fibrosis or liver fibrosis in Stage 1. Therefore, we believe that short-term feeding with HFHC can form an early model of MASLD, without causing severe inflammation, liver fibrosis, or cirrhosis.
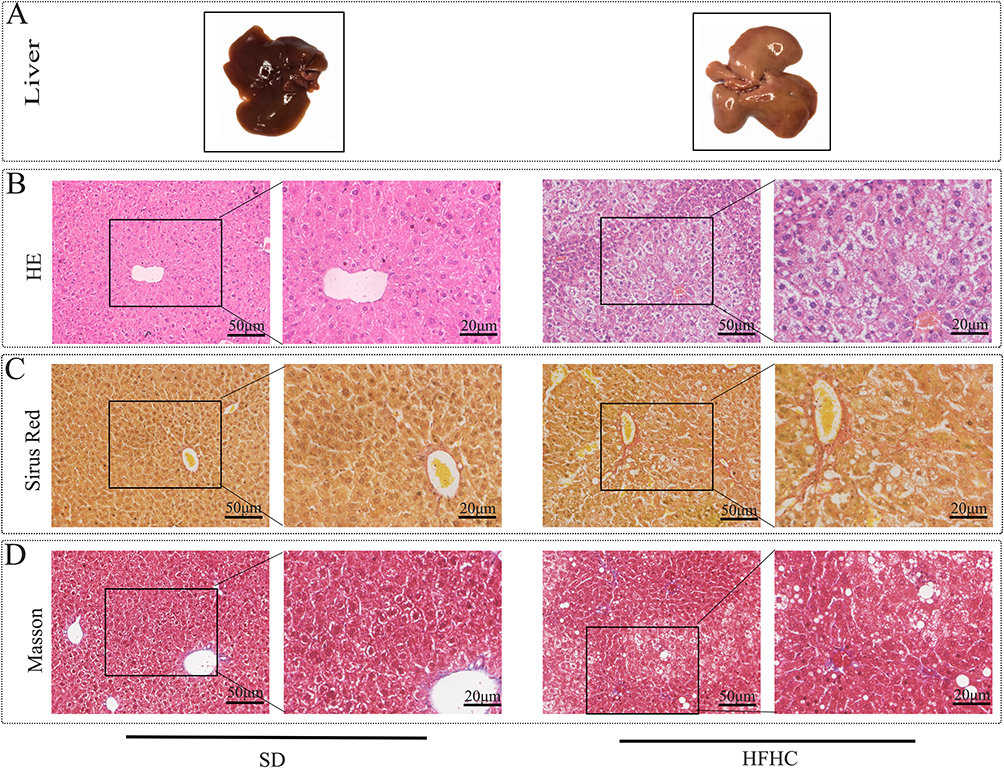 |
Figure 1 HFHC diet induced early NAFL/NASH model. (A) Compared with SD diet, representative images of liver appearance induced by HFHC for fatty liver formation. (B) HE staining was used to observe lipid degeneration and inflammatory infiltration. (C) Representative image of Sirius Red observing liver fibrosis. (D) Masson staining observed representative images of liver fibrosis. |
Result 2: Effects of Autophagy on Body Weight, Liver Weight, and Liver/Body Weight Ratio in MASLD Model
Forty-eight 7-week-old C57 male mice were randomly divided into 4 groups to establish the animal model. There was no significant difference in body weight between each group (Figure 2A). The control group was given SD diet +8 weeks of age at the beginning of intrabitoneal injection of normal saline, and the three model groups were given HFHC diet + intrabitoneal injection of normal saline /RaPa/CQ group after 1 week of excessive standard diet respectively (Figure 2B). Body weight was monitored weekly, and the HFHC-induced mouse model was found to lead to significant obesity, as shown in Figure 2A. Starting from the fourth week, the weight gain in HFHC-fed mice became statistically significant (p < 0.05) compared to the SD group, while the weight loss effect of RaPa began to manifest from the third week. Interestingly, the CQ group also showed statistically significant weight loss from week 5 onwards. After mice were euthanized at 10 weeks, both RaPa and CQ were found to reduce liver weight, with CQ showing a more significant effect (Figure 2C).
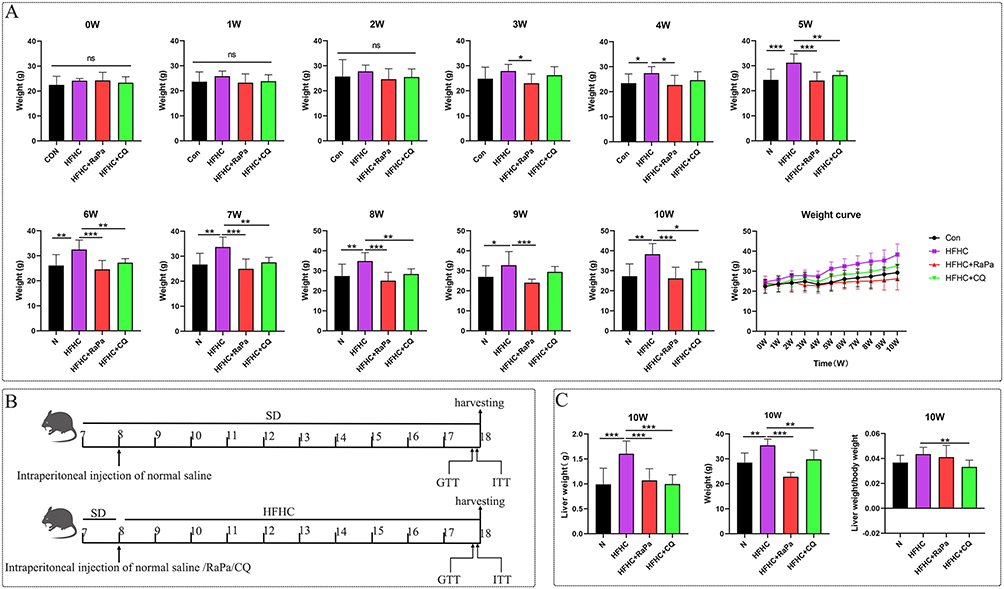 |
Figure 2 Effects of autophagy on mouse body weight, liver weight, and body weight/liver weight ratio. (A) Monitor weight changes weekly; (B) Schematic diagram of model construction and processing methods. (C) Liver weight, body weight, and liver weight to body weight ratio. |
Result 3: Regulation of Autophagy on Blood Glucose, Lipids, and Liver Function in Early MASLD Models
Before euthanizing the mice, insulin tolerance and glucose tolerance experiments were conducted. CQ significantly improved insulin resistance induced by the HFHC diet, while RaPa led to insulin resistance (Figure 3A). The results suggested that RaPa promoted adverse metabolic states such as hyperlipidemia and hyperglycemia, whereas CQ was found to play a protective role.
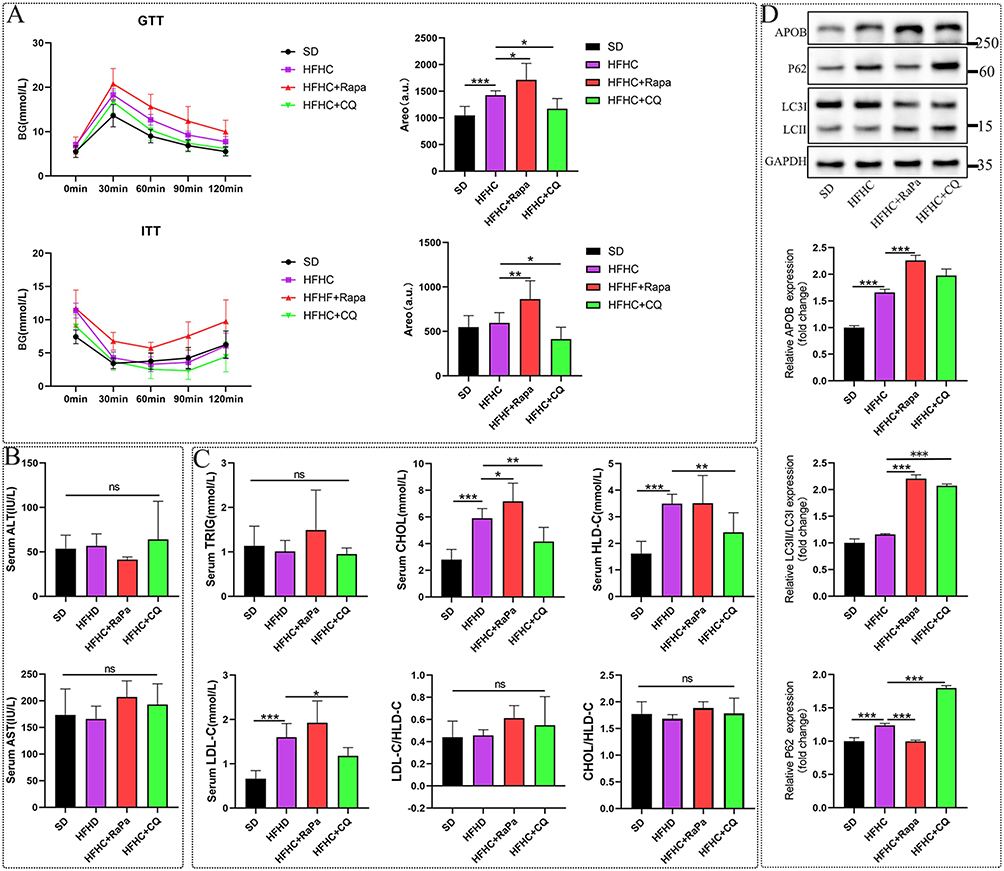 |
Figure 3 The regulatory effect of autophagy on blood glucose, liver function, and blood lipids in an early MASLD model. (A) Blood glucose change curves and statistical graphs of GTT and ITT tests before killing mice at 10W. (B) ALT and AST statistical charts. (C) TRIG, CHOL, HLD-C, LDL-C, LDL-C/HDL-C, CHOL/HLD-C statistical charts. (D) The impact of autophagy on APOB. |
After 10W, serum ALT, AST, cholesterol, triglycerides, high-density lipoprotein, and low-density lipoprotein were detected in the HFHC mouse model. Quantitative detection of serum ATL and AST showed no statistical significance. Compared with the normal diet control group, short-term feeding of HFHC did not cause liver function damage in mice, and that activating and inhibiting autophagy did not aggravate liver function damage (Figure 3B). HFHC can induce an increase in CHOL, HLD-C, and LDL-C, while no significant differences in serum TRIG levels were observed among the groups. It was also found that activating autophagy can further increase CHOL, HLD-C, and LDL-C, while inhibiting autophagy can reverse the upward trend of these indicators (Figure 3C). Therefore, we believe that RaPa poses a risk of causing hyperlipidemia, while chloroquine can lower blood lipids. APOB is a major component of very low-density lipoprotein and low-density lipoprotein,21 and in a study related to coronary heart disease, it was found that the clinical benefits of reducing triglyceride and LDL-C levels may be directly proportional to the absolute changes in ApoB.22 Therefore, our detection of changes in liver APOB may provide a meaningful clinical evaluation for predicting the association between autophagy and coronary heart disease. As shown in Figure 3D, rapamycin can lead to the upregulation of APOB expression. APOB-100 is the core structural protein for the synthesis of VLDL in the liver. VLDL is the primary carrier for transporting endogenous triglycerides from the liver to peripheral tissues. After VLDL is secreted, its triglycerides are taken up by peripheral tissues, and the remaining particles are gradually converted into LDL.23 Therefore, we believe that the increase in blood LDL levels induced by rapamycin may be related to the enhancement of hepatic triglyceride synthesis and secretion. And CQ cannot completely reverse the expression of APOB, which means that CQ’s lipid-lowering effect may involve other pathways.
Result 4: The Effect of Autophagy on Hepatic Steatosis
As shown in Figure 4A, after induction with the HFHC diet for 10 weeks, the liver exhibited significant yellowing and enlargement, consistent with pre-experiment observations. RaPa treatment significantly improved the liver appearance, restoring it to a color close to normal. HE staining revealed steatosis with a small amount of ballooning, and little inflammatory cell infiltration was observed. At the same time, it was found that the liver steatosis in the RaPa group was significantly reduced, while the steatosis in the CQ group was heavier than that in the HFHC group, and inflammatory cell infiltration and Mallory Denk bodies formation were observed (yellow circle in Figure 4B). Oil red staining revealed that HFHC diet can induce a large amount of lipid droplet aggregation, while RaPa can reduce lipid droplet aggregation, while CQ can increase lipid droplet aggregation (Figure 4C and D). Meanwhile, Western blot detection found that HFHC can induce upregulation of PLIN2 and FASN expression, RaPa can downregulate PLIN2 and FASN expression, while CQ can upregulate PLIN2 and FASN expression (Figure 4E). Therefore, we believe that the activation of autophagy may alleviate liver steatosis by inhibiting de novo synthesis of fat and reducing lipid droplet aggregation. After inhibiting autophagy, de novo synthesis of fat is enhanced, lipid droplet aggregation is obvious, leading to aggravated lipid degeneration.
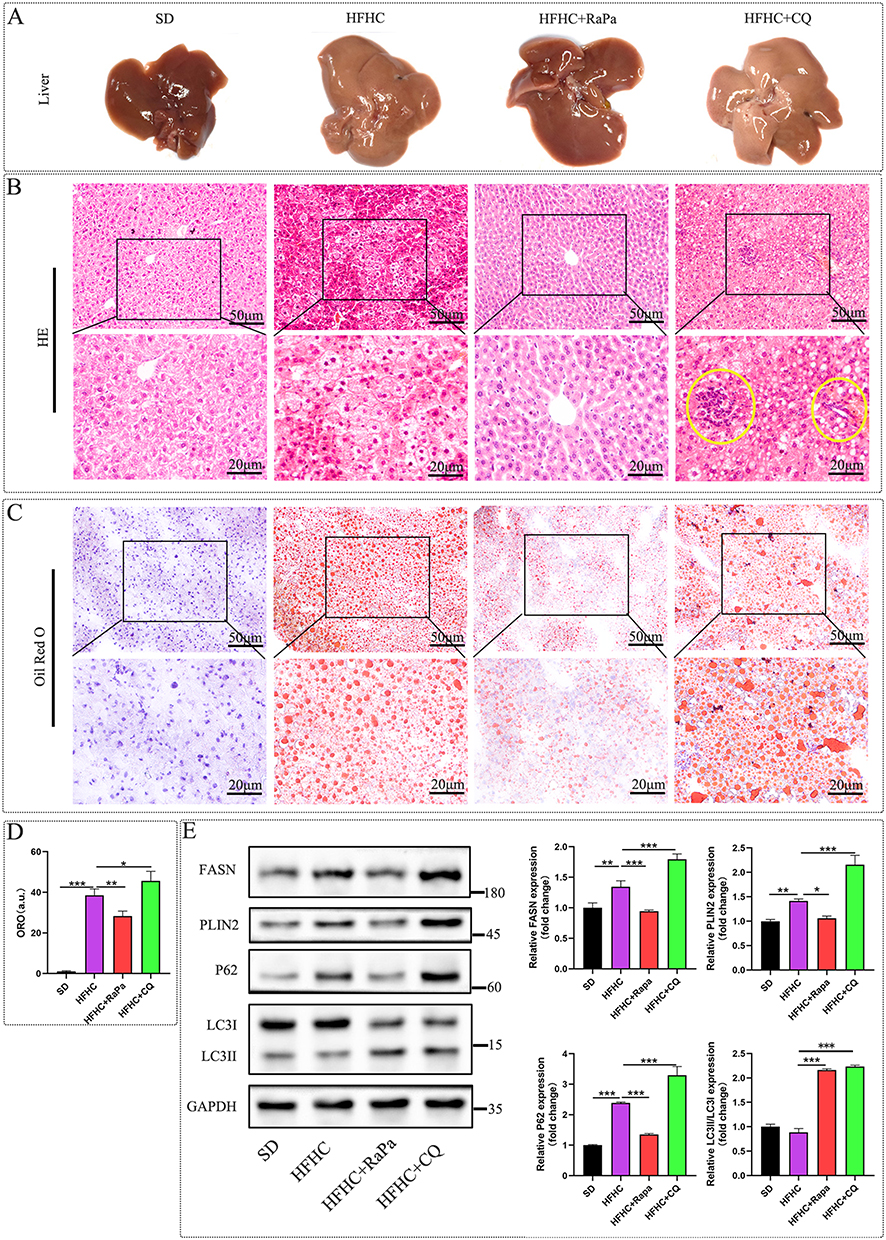 |
Figure 4 Effects of autophagy on lipid degeneration, droplet formation, and adipogenesis. (A) Representative images of the overall appearance of the liver. (B) Representative images of HE staining, inflammatory cell infiltration and Mallory Denk bodies formation were observed in yellow circle. (C) Representative images of oil red staining. (D) Oil red staining statistical chart. (E) Representative Western blot images of the effect of autophagy on FASN and PLIN2 expression. |
Result 5: Inhibition of Autophagy May Exacerbate Liver Fibrosis in MASLD/MASH Mouse Models
As shown in Figure 5A, we validated the expression of inflammation and fibrosis markers in the early MASLD/MASH model by detecting the relative mRNA expression levels of liver inflammation and fibrosis markers. Previous studies have suggested a positive correlation between IL6 and MASH inflammation,24,25 and IL-10 plays a protective role in liver inflammation;26,27 PDGFRβ, TGF β1, Col1-α1, LOX, CCL2, and α- SMA are marker factors for liver fibrosis formation.28–30 Among them, PDGFRβ levels gradually increase with the increase of fibrosis stage, and the largest difference is observed in patients with significant fibrosis compared to those without fibrosis or mild fibrosis.30 We detected the nucleic acid levels of IL-6, IL-10, PDGFRβ, TGFβ1, Col1-α1, LOX, CCL2, and α- SMA. IL-10 was upregulated after RaPa induction and downregulated after CQ treatment. Therefore, we believe that RaPa may alleviate inflammation induced by HFHC, while CQ may exacerbate liver inflammation. At the same time, it was found that compared with the SD diet, HFHC induced an increase in the expression level of CCL2, and there was no significant statistical difference in other fibrosis indicators. RaPa did not change the nucleic acid expression levels of TGF β1, LOX, CCL2, and α - SMA. However, after CQ treatment, the expression levels of TGF β1, LOX, CCL2, and α- SMA were significantly upregulated, and TGF β1, LOX, CCL2, and α- SMA were positively correlated with the degree of liver fibrosis. Furthermore, we observed fibrosis in each group by staining with Sirius Red and Masson. We found that after CQ inhibition of autophagy, the expression of collagen fibers in the liver increased, and there was significant fibrosis around the central vein and portal area, which spread to the liver parenchyma. Some areas around the sinusoids showed obvious fibrosis (Figure 5B–D). At the same time, we used Western blot analysis of liver tissue to detect the expression level of α- SMA (Figure 5E). We found that the expression level of α- SMA was significantly upregulated in the HFHC+CQ group. Therefore, we believe that CQ inhibition of autophagy may lead to the development of MASLD and MASH towards liver fibrosis. This phenomenon may be attributed to mechanisms such as autophagy blockade, subsequent p62 accumulation, activation of hepatic stellate cells, and increased exosome secretion by hepatocytes following chloroquine administration.31
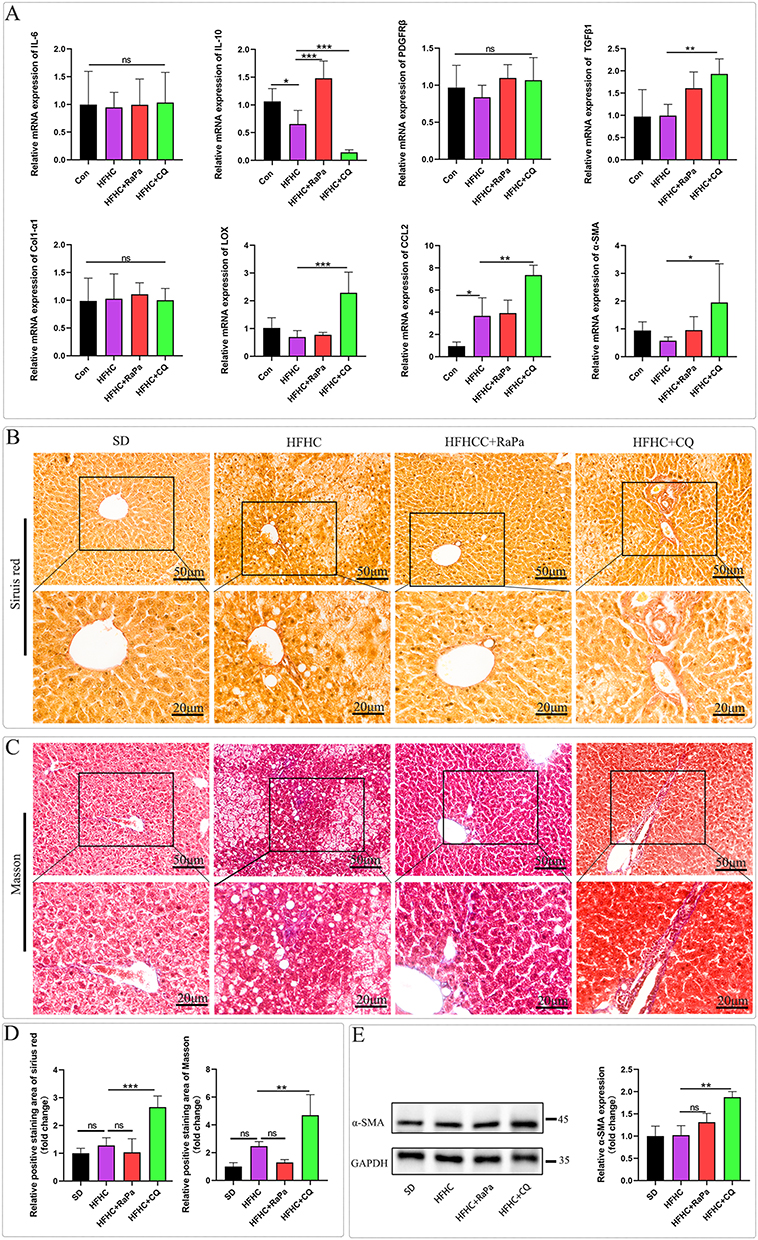 |
Figure 5 RaPa alleviates liver inflammation, while CQ leads to the progression of NAFL and NASH towards liver fibrosis. (A) The nucleic acid levels of IL-6, IL-10, PDGFRβ, TGFβ1, Col1-α1, LOX, CCL2, and α-SMA; (B) Representative image of Sirius red staining. (C) Representative image of Masson staining. (D) Statistical chart of positive areas stained with Sirius Red and Masson. (E) Western blot detection of representative images of α- SMA protein levels. |
Discussion
Early diagnosis and intervention are critical for effective disease management. Through the development of an early MASLD model and the exploration of autophagy’s role, this study aims to identify potential strategies to inhibit MASLD progression, thereby mitigating the risk of cirrhosis and liver cancer.
The autophagy agonist rapamycin is widely used as an immunosuppressant in clinical practice, and it has also been found that RaPa is beneficial in improving liver steatosis and body weight.11,18 Our study found that RaPa can downregulate the expression of hepatic FASN and PLIN2, which may lead to hepatic fat synthesis and lipid droplet reduction, thereby alleviating the causes of fatty liver. But interestingly, its risk of causing hyperlipidemia and glucose intolerance cannot be ignored.32 Rapamycin has been shown to induce glucose intolerance in mice.33 Clinical studies, particularly those involving long-term rapamycin use in kidney transplant patients, corroborate our findings by demonstrating that rapamycin can cause hyperlipidemia and reduce fat mass. The underlying mechanism may involve RaPa’s modulation of the insulin signaling pathway, leading to increased adipose tissue lipase activity and/or decreased lipoprotein lipase activity. This, in turn, promotes hepatic triglyceride synthesis, VLDL secretion, and hypertriglyceridemia.34 Our research confirms that RaPa can lead to upregulation of liver APOB expression, which is a major component of VLDL, and LDL. Therefore, upregulation of APOB expression may be one of the causes of hypercholesterolemia. The induction of gluconeogenesis in the liver by rapamycin is the basis for the development of severe glucose tolerance.32 Long term inhibition of mTORC1 by rapamycin can impair insulin secretion and cause insulin resistance.33 Rapamycin is not only an inhibitor of the mTORC1 target, but the destruction of mTORC2 in the liver damages liver insulin sensitivity, which may be one of the main cause of rapamycin induced glucose intolerance.35 Mechanistically, mTORC1 orchestrates lipogenic gene expression through multiple pathways. First, mTORC1 promotes the proteolytic activation and nuclear translocation of SREBPs, enabling the transcription of genes involved in fatty acid (eg, FASN, SCD) and cholesterol (eg, HMGCR) biosynthesis.36 Second, the mTORC1 downstream effector S6K1 mediates the activation of SREBP and its target genes37 Rapamycin-mediated mTORC1 inhibition likely attenuates this S6K1-dependent SREBP activation, potentially reducing the expression of lipogenic enzymes. However, the net effect on plasma lipids is complex due to compensatory mechanisms. For instance, chronic mTORC1 inhibition can relieve the negative feedback on insulin/PI3K/Akt signaling.36 Enhanced Akt activity may then paradoxically promote SREBP activity and hepatic lipogenesis through alternative pathways, potentially explaining the observed APOB upregulation and hypercholesterolemia in our model.
Autophagy inhibitors CQ/HCQ were originally developed to combat malaria and are used to treat rheumatic autoimmune diseases. They are currently being tested in clinical trials as therapeutic drugs for other diseases.38 Previous studies suggested that HCQ improved beta cell function and insulin sensitivity in non-diabetic patients and increased adiponectin levels, which may have had a beneficial impact on glucose metabolism.39 It was found that CQ improved HFHC-induced glucose intolerance. We also found that CQ significantly decreased serum total cholesterol and LDL-C levels. Other studies have also found that CQ alleviates insulin intolerance, hyperglycemia, hyperinsulinemia, hypertriglyceridemia, and hypercholesterolemia in mice induced by HFD intake.19 This is consistent with previous clinical studies on the lipid-lowering effects of hydroxychloroquine in patients with rheumatoid arthritis.40 At the same time, it can also reduce high-density lipoprotein. Simultaneously, this study discovered that chloroquine reduces the level of high-density lipoprotein (HDL). It is widely acknowledged that maintaining a certain level of HDL can decrease the incidence of coronary heart disease. Therefore, whether the observed reduction in HDL expression induced by chloroquine in this study could potentially increase the risk of coronary heart disease warrants further investigation.Previous studies have suggested that CQ can exacerbate the damage of fatty liver.11,41 Our research results indicate that CQ may promote the formation of liver inflammation, steatosis, and fibrosis, which may be related to the upregulation of LOX, TGFβ1, and α- SMA by CQ.
Inflammatory and fibrotic factors were detected under autophagy intervention conditions, and it was found that the inflammatory factor IL-6 did not show statistical significance with the hepatic fibrotic inflammatory factor PDGFRβ. Previous studies have suggested that the level of PDGFRβ gradually increases with the increase of fibrosis stage.30 The reason for the above phenomenon may be due to the short establishment time of the model, as the inflammatory or fibrotic factors mentioned above are not yet sufficient to produce statistically significant changes in nucleic acid levels. Under HFHC induction, the anti-inflammatory factor IL-10 was downregulated, while RaPa reversed this change. CQ further downregulated the mRNA expression of IL-6. At the same time, HE staining of liver slices showed no significant inflammatory cell infiltration in the RaPa intervention group, while inflammatory cell aggregation was observed in the liver lobules in the CQ intervention group, indicating that RaPa has anti-inflammatory effects in NAFL and MASH, while CQ has a certain pro-inflammatory effect in the liver. The role of CQ in promoting liver inflammation seems to contradict its role in the rheumatic immune system, and its specific mechanism needs further clarification.
Overall, consistent with previous studies, we conclude that RaPa ameliorates hepatic steatosis,10 but induces insulin resistance, whereas CQ improves adverse metabolic parameters yet increases the risk of hepatic fibrosis.19 Significantly, our study reveals that neither simple autophagy activation nor inhibition confers comprehensive therapeutic benefits. Instead, our data underscore the existence of a critical and subtle tissue-specific balance, urging future strategies for targeted autophagy modulation to disentangle the hepatic benefits from systemic or detrimental hepatic effects.
Conclusions
In an early metabolic dysfunction-associated steatotic liver disease (MASLD) mouse model induced by short-term HFHC diet, rapamycin alleviates hepatic steatosis through downregulating lipid synthesis markers (FASN, PLIN2) and enhancing anti-inflammatory IL-10, but exacerbates metabolic disorders including hyperglycemia and hyperlipidemia via APOB-mediated VLDL/LDL secretion. Conversely, chloroquine improves insulin sensitivity and dyslipidemia, yet paradoxically accelerates liver fibrosis by upregulating profibrotic factors (TGFβ1, α-SMA) and promoting collagen deposition. This dual-edged effect highlights the tissue-specific dilemma of autophagy modulation: rapamycin protects the liver at metabolic cost, while chloroquine benefits metabolism but provokes hepatic injury. Therapeutic strategies for MASLD must therefore balance hepatic protection with systemic metabolic safety, potentially requiring combination therapies to dissect these opposing effects.
Abbreviation
MASLD, metabolic dysfunction-associated steatotic liver disease; non-alcoholic fatty liver disease; NAFL, Non-alcohol fatty liver; MASH, non-alcoholic steatohepatitis; RaPa, rapamycin; CQ, chloroquine; HCQ, hydroxychloroquine; HFHC, High fat, high cholesterol diet; FASN, fatty acid synthase; APOB, Apolipoprotein B; α-SMA, α-smooth muscle actin; PLIN2, Perilipin 2; LC3, Microtubule-associated protein light chain 3; P62, Sequestosome 1; mTORC, rapamycin complex; HDL-C, high density lipoprotein cholesterol; LDL-C, low density lipoprotein cholesterin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; TRIG, triglycerides; CHOL, cholesterol; GTT, Glucose Tolerance Test; ITT, Insulin tolerance test.
Data Sharing Statement
The key datasets supporting the findings of this study, including all quantitative results presented in the graphs and statistical analyses, are included within the main manuscript and its figure panels. Additional detailed data, such as full-length blot images and primary numerical data for gene expression analyses, are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
All experiments were approved by the Institute of Animal Care and Use Committee of the Affiliated Hospital of Zunyi Medical University.
Consent for Publication
We have obtained consents to publish this paper from all the participants of this study.
Acknowledgments
We also thank Professor Biguang Tuo (Department of Gastroenterology, Affiliated Hospital to Zunyi Medical University) for highly professional services.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by research grants the National Natural Science Foundation of China (No.81660099, No.81770610) and the Science and Technology Plan Project of Guizhou Province (QIAN KE HE JI CHU-ZK(2023)YI BAN556).
Disclosure
The authors declare that they have no competing interests.
References
1. Rinella ME, Lazarus JV, Ratziu V, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;78:1966–1986. doi:10.1097/hep.0000000000000520
2. Pan C, Liu J, Gao Y, et al. Hepatocyte CHRNA4 mediates the MASH-promotive effects of immune cell-produced acetylcholine and smoking exposure in mice and humans. Cell Metab. 2023;35:2231–2249.e2237. doi:10.1016/j.cmet.2023.10.018
3. Stefan N, Yki-Järvinen H, Neuschwander-Tetri BA. Metabolic dysfunction-associated steatotic liver disease: heterogeneous pathomechanisms and effectiveness of metabolism-based treatment. Lancet Diabetes Endocrinol. 2025;13:134–148. doi:10.1016/s2213-8587(24)00318-8
4. Targher G, Corey KE, Byrne CD, Roden M. The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nat Rev Gastroenterol Hepatol. 2021;18:599–612. doi:10.1038/s41575-021-00448-y
5. Jenkins CM, Mancuso DJ, Yan W, et al. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968–48975. doi:10.1074/jbc.M407841200
6. Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem. 2004;279:47066–47075. doi:10.1074/jbc.M403855200
7. Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi:10.1126/science.1100747
8. Onal G, Kutlu O, Gozuacik D, Dokmeci Emre S. Lipid droplets in health and disease. Lipids Health Dis. 2017;16:128. doi:10.1186/s12944-017-0521-7
9. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–155. doi:10.1038/s41580-018-0085-z
10. Park HS, Song J-W, Park J-H, et al. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy. 2021;17:2549–2564. doi:10.1080/15548627.2020.1834711
11. Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi:10.1016/j.jhep.2013.01.011
12. Zhang H, Yan S, Khambu B, et al. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy. 2018;14:1779–1795. doi:10.1080/15548627.2018.1490850
13. Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi:10.1038/nature07976
14. Hughes KJ, Kennedy BK. Cell biology. Rapamycin paradox resolved. Science. 2012;335:1578–1579. doi:10.1126/science.1221365
15. Seo J, Kwon D, Kim SH, et al. Role of autophagy in betaine-promoted hepatoprotection against non-alcoholic fatty liver disease in mice. Curr Res Food Sci. 2024;8(100663):100663. doi:10.1016/j.crfs.2023.100663
16. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi:10.1080/15548627.2018.1474314
17. Savard C, Tartaglione EV, Kuver R, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology. 2013;57:81–92. doi:10.1002/hep.25789
18. Chang GR, Chiu Y-S, Wu -Y-Y, et al. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci. 2009;109:496–503. doi:10.1254/jphs.08215fp
19. Ortiz-Silva M, Leonardi BF, Castro É, et al. Chloroquine attenuates diet-induced obesity and glucose intolerance through a mechanism that might involve FGF-21, but not UCP-1-mediated thermogenesis and inhibition of adipocyte autophagy. Mol Cellular Endocrinol. 2023;578:112074. doi:10.1016/j.mce.2023.112074
20. Sheka AC, Adeyi O, Thompson J, et al. Nonalcoholic steatohepatitis: a review. JAMA. 2020;323:1175–1183. doi:10.1001/jama.2020.2298
21. Muscella A, Stefàno E, Marsigliante S. The effects of exercise training on lipid metabolism and coronary heart disease. Am J Physiol Heart Circ Physiol. 2020;319:H76–h88. doi:10.1152/ajpheart.00708.2019
22. Ference BA, Kastelein JJP, Ray KK, et al. Association of triglyceride-lowering LPL variants and LDL-C–lowering LDLR variants with risk of coronary heart disease. JAMA. 2019;321:364–373. doi:10.1001/jama.2018.20045
23. Olofsson SO, Borèn J. Apolipoprotein B: a clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Internal Med. 2005;258:395–410. doi:10.1111/j.1365-2796.2005.01556.x
24. Fontes-Cal TCM, Mattos RT, Medeiros NI, et al. Crosstalk between plasma cytokines, inflammation, and liver damage as a new strategy to monitoring NAFLD progression. Front Immunol. 2021;12:708959. doi:10.3389/fimmu.2021.708959
25. Duan Y, Pan X, Luo J, et al. Association of Inflammatory Cytokines With Non-Alcoholic Fatty Liver Disease. Front Immunol. 2022;13:880298. doi:10.3389/fimmu.2022.880298
26. Paredes-Turrubiarte G, González-Chávez A, Pérez-Tamayo R, et al. Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin Exp Med. 2016;16:193–202. doi:10.1007/s10238-015-0347-4
27. Miyamoto Y, Kikuta J, Matsui T, et al. Periportal macrophages protect against commensal-driven liver inflammation. Nature. 2024;629:901–909. doi:10.1038/s41586-024-07372-6
28. Chen W, Yang A, Jia J, et al. Lysyl oxidase (LOX) family members: rationale and their potential as therapeutic targets for liver fibrosis. Hepatology. 2020;72:729–741. doi:10.1002/hep.31236
29. Pedroza M, To S, Smith J, Agarwal SK. Cadherin-11 contributes to liver fibrosis induced by carbon tetrachloride. PLoS One. 2019;14:e0218971. doi:10.1371/journal.pone.0218971
30. Lambrecht J, Verhulst S, Mannaerts I, et al. A PDGFRβ-based score predicts significant liver fibrosis in patients with chronic alcohol abuse, NAFLD and viral liver disease. EBioMedicine. 2019;43:501–512. doi:10.1016/j.ebiom.2019.04.036
31. Zhang XW, Zhou J-C, Peng D, et al. Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis by restoring autophagy and suppressing exosome-mediated HSC activation. Autophagy. 2020;16:782–796. doi:10.1080/15548627.2019.1635383
32. Houde VP, Brûlé S, Festuccia WT, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi:10.2337/db09-1324
33. Yang SB, Lee HY, Young DM, et al. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J Mol Med. 2012;90:575–585. doi:10.1007/s00109-011-0834-3
34. Morrisett JD, Abdel-Fattah G, Hoogeveen R, et al. Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J Lipid Res. 2002;43:1170–1180. doi:10.1194/jlr.M100392-JLR200
35. Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi:10.1126/science.1215135
36. Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA. 2020;117:31189–31197. doi:10.1073/pnas.2017152117
37. Düvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi:10.1016/j.molcel.2010.06.022
38. Nirk EL, Reggiori F, Mauthe M. Hydroxychloroquine in rheumatic autoimmune disorders and beyond. EMBO Mol Med. 2020;12:e12476. doi:10.15252/emmm.202012476
39. Wasko MC, McClure CK, Kelsey SF, et al. Antidiabetogenic effects of hydroxychloroquine on insulin sensitivity and beta cell function: a randomised trial. Diabetologia. 2015;58:2336–2343. doi:10.1007/s00125-015-3689-2
40. Morris SJ, Wasko MCM, Antohe JL, et al. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res. 2011;63:530–534. doi:10.1002/acr.20393
41. Chen R, Wang Q, Song S, et al. Protective role of autophagy in methionine-choline deficient diet-induced advanced nonalcoholic steatohepatitis in mice. Eur J Pharmacol. 2016;770:126–133. doi:10.1016/j.ejphar.2015.11.012
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Statin Monotherapy Not Inferior to Aspirin or Combined Aspirin and Statins Reducing the Incidences of Cirrhosis, HCC, and Mortality in MAFLD/MASH Patients: A Population Cohort Study
Lee CH, Huang YH, Hsu TJ, Yen TH, Hsieh SY
International Journal of General Medicine 2024, 17:6495-6511
Published Date: 27 December 2024
A Review of the Therapeutic Potential of Ginseng and Its Bioactive Components in Nonalcoholic Fatty Liver Disease
Hao L, Li S, Li C, Zhang Z, Hu X, Yan H
Drug Design, Development and Therapy 2025, 19:83-96
Published Date: 8 January 2025
Exploring Mechanisms of Lang Qing Ata in Non-Alcoholic Steatohepatitis Based on Metabolomics, Network Pharmacological Analysis, and Experimental Validation

Li S, Zhu H, Zhai Q, Hou Y, Yang Y, Lan H, Jiang M, Xuan J
Drug Design, Development and Therapy 2025, 19:1681-1701
Published Date: 11 March 2025
Retrospective Study of Real-World Treatment Patterns of Subcutaneous Semaglutide Use Among Patients with Metabolic Dysfunction-Associated Steatohepatitis in the United States
Kim Y, Zeng N, Winer-Jones JP, Bonafede M, Lobo F, O’Donnell J, Ryan T
ClinicoEconomics and Outcomes Research 2025, 17:743-754
Published Date: 22 October 2025