Back to Journals » Journal of Inflammation Research » Volume 17
Pyroptosis in Osteoarthritis: Molecular Mechanisms and Therapeutic Implications
Authors Chen Y ![]() , Zeng D, Wei G
, Zeng D, Wei G ![]() , Liao Z, Liang R, Huang X, Lu WW
, Liao Z, Liang R, Huang X, Lu WW ![]() , Chen Y
, Chen Y ![]()
Received 6 November 2023
Accepted for publication 20 January 2024
Published 8 February 2024 Volume 2024:17 Pages 791—803
DOI https://doi.org/10.2147/JIR.S445573
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Yeping Chen,1,* Daofu Zeng,1,* Guizheng Wei,1,* Zhidong Liao,1,2 Rongyuan Liang,1 Xiajie Huang,1,2 William W Lu,3 Yan Chen1,2
1Department of Bone and Joint Surgery, the First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 2Collaborative Innovation Centre of Regenerative Medicine and Medical BioResource Development and Application Co-Constructed by the Province and Ministry, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 3Department of Orthopedics and Traumatology, the University of Hong Kong, Hong Kong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Chen, Department of Bone and Joint Surgery, the First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, People’s Republic of China, Tel +86-18077790338, Email [email protected]
Abstract: Osteoarthritis (OA) is a chronic disease that causes pain and functional impairment by affecting joint tissue. Its global impact is noteworthy, causing significant economic losses and property damage. Despite extensive research, the underlying pathogenesis of OA remain an area of ongoing investigation. It has recently been discovered that the OA progression is significantly influenced by pyroptosis. Pyroptosis is a complex process that involves three pathways culminating in the assembly of Gasdermin-D (GSDMD)-N-terminal (GSDMD-NT) into pores through aggregation on the plasma membrane. The aggregation of GSDMD-NT proteins stimulates the release of inflammatory mediators, such as Interleukin-1β (IL-1β), Interleukin-18 (IL-18), and Matrix Metallopeptidase 13 (MMP13), ultimately leading to cellular lysis. The pyroptosis process in specific cells, including synovial macrophages, fibroblast-like synoviocytes (FLS), chondrocytes, and subchondral osteoblasts, contributs factor to the development of OA. Currently, the specific cells that undergo pyroptosis first are not yet fully understood, and it remains unknown whether pyroptosis in one cell can trigger the same process in other cells. Therefore, targeting pyroptosis could potentially offer a novel treatment approach for OA patients. We present a comprehensive analysis of the molecular mechanisms and key features of pyroptosis. We also outline the current research progress on various aspects, including synovial tissue, articular cartilage, extracellular matrix (ECM), and subchondral bone, with a focus on pyroptosis. The aim is to provide theoretical references for the effective management of OA.
Keywords: pyroptosis, osteoarthritis, caspases-1/4/5/11, GSDMD, synovium, chondrocytes
Introduction
OA is a type of joint ailment affecting both large and small joints, such as the hip, knee, and hand. The disease causes structural changes in the articular cartilage, ligaments, subchondral bone, joint capsule, synovial tissue, and periarticular muscles.1,2 The primary symptoms of OA are joint pain and loss of function,3,4 which may lead to disability and the need for joint replacement.5–7 It is pertinent to note that OA is more prevalent in the elderly population, and its prevalence increases with age.8 The condition causes significant economic and property losses globally,6,7 particularly for individuals over 65 years old.9,10 OA results from a combination of individual and joint factors, including age, gender, obesity, genetics, diet, injury, joint malalignment, and abnormal joint loading.5,11,12
Previous studies have demonstrated that the programmed death of multiple cells in the bone and joint, which is regulated by various death modalities, is closely associated with the occurrence and progression of OA. The mechanisms involved in this process include Apoptosis, autophagy, necrotizing Apoptosis, ferroptosis, and pyroptosis.13–15 There has been a notable surge in the study of pyroptosis,14–17 a recently discovered type of programmed cell death that is closely linked with OA.18 Pyroptosis is induced by the activation of cysteinyl aspartate-specific proteinase-1 (caspase-1),18,19 resulting in the formation of inflammatory bodies, cell membrane perforation, and release of inflammatory cells and factors.18,20–22 It affects various tissues and cells, such as osteoporotic cartilage, ECM, subchondral bone, synovium, and joint fluid, in different ways. While several studies have explored pyroptosis-related molecules in OA, there is still no consensus on which cells undergo pyroptosis or which cells are affected first. Additionally, the relationship between pyroptosis and subchondral bone remains largely unexplored. Our aim is to investigate the mechanisms of pyroptosis in OA and its impact on the synovial membrane, articular cartilage, ECM, and subchondral bone. Through our research, we hope to reveal the pathological mechanisms of OA and provide valuable insights for further research and clinical treatment.
Molecular Mechanisms of Pyroptosis
Pyroptosis is a type of inflammatory cell death that differs from other forms, such as apoptosis.17,23 Initially, it was thought to be a type of apoptosis, but it was later defined as pyroptosis in 2001.23 This type of cell death is caused by caspase-1 in macrophages infected with Salmonella. Pyroptosis is morphologically and biochemically different from other forms, as it leads to cell infiltration, swelling, rupture, and death.16,20,24 Pyroptosis causes cell membranes to form ruptured stomata with an inner diameter of 14–20 nm called GSDMD pores through a series of reactions mediated by inflammatory caspase-1.22 As a result, it leads to cell infiltration, swelling, rupture, and death; inflammatory factors are released into the tissue, and spherical vesicles form around the nucleus.25,26 The nucleus coagulates and solidifies, and DNA fragmentation occurs.20 However, unlike apoptosis, nuclear DNA is not degraded in pyroptosis.18
Pyroptosis is a form of cell death that is more intricate than apoptosis and employs diverse primary mediators in contrast to apoptosis. Pyroptosis utilizes caspase-1/4/5/11 instead of caspase-2, caspase-9, and caspase-10.16,21,27 These caspases aid in the fabrication of inflammatory vesicles and the liberation of IL-1β, IL-18, and heat shock proteins (HSPs) from the stomata.28
Three Different Pathways of Pyroptosis
Canonical Pathway
Canonical pyroptosis is a complex process that involves the assembly of inflammatory complexes. Pathogenic microbes and injury factors trigger this process through molecular patterns.23,29 Toll-like receptors (TLRs) play a vital role in the initiation phase by binding to molecular patterns, which activate the nuclear transcription factor-kappa B (NF-κB) and mitogen-activated protein kinases (MAPK) pathways. This results in increased expression of pro-interleukin-1β (pro-IL-1β) and nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3). Although the mechanism through which NLRP3 activates canonical pyroptosis is still unclear, it is understood that during the activation phase, the assembly of inflammatory vesicles takes center stage, ultimately leading to the activation of caspase-1.30–33 Canonical pyroptosis is triggered by the activation of mitochondria through exposure to NLRP3 activators, including microparticles, toxins, adenosine triphosphate (ATP), pathogens, crystals, or aggregates.30,34–38 Following tissue injury, advanced oxidative protein product (AOPP) increases and causes a reduction in mitochondrial membrane potential.39,40 This decrease in mitochondrial membrane potential leads to an increase in mitochondrial reactive oxygen species (mtROS), resulting in the formation and fragmentation of oxidized mitochondrial DNA (Ox-mtDNA), which is released into the cytosol.41–44 The Ox-mtDNA can activate and assemble NLRP3 inflammatory vesicles. Inflammatory vesicles,45 formed by oligomeric receptor proteins such as NLRP1, NLRP3, and NLRP4, and absent in melanoma 2 (AIM2), are involved in intrinsic immunity. These multi-protein complexes include apoptosis-associated speck-like proteins containing a caspase-associated recruitment domain (CARD) (ASC) and pro-caspase-1.46–49 The NLRP3 inflammasome is formed when aggregated ASC recruit pro-caspase-1 through homotypic CARD and CARD interactions. The oligomeric NLRP3 protein is capable of recruiting ASC through its homotypic pyrin domain (PYD) and PYD interactions, ultimately leading to the formation of an ASC patch.28 Once assembled, the NLRP3 inflammasome forms by the interaction of homotypic Nacht structural domains, resulting in a complex with a high molecular weight that triggers the self-activation of caspase-1. This activation subsequently modulates the activation of pro-IL-1β and pro Interleukin-18 (pro-IL-18) through protein hydrolysis and releases active IL-1β and IL-18, inducing pyroptosis.28,30
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) activate pyroptosis through complex mechanisms. PAMPs encompass viral RNA, toxins, and bacterial surface components, while DAMPs comprise uric acid crystals, ATP, aluminum adjuvants, and β-amyloid peptides. PAMPs and DAMPs bind to evolutionarily conserved pattern recognition receptors (PRRs).28,30,50 The synthesis of mitochondrial DNA (mtDNA) is essential for NLRP3 signaling, which is triggered by one of the three principal PRR families - TLRs stimulated through myeloid differentiation factor 88 (MyD88) and Toll/IL-1R domain-containing adaptor-inducing interferon-beta (TRIF) dapters in the NF-κB pathway or mitogen-activated protein kinase (MAPK) pathway.46,51 Interferon regulatory factor 1 (IRF1) leads to increased cytidine/uridine monophosphate kinase 2 (CMPK2) and TRIF adapter-dependent pro-IL-1β expression. CMPK2 is an enzyme that facilitates mtDNA synthesis, which leads to Ox-mtDNA fragments.33 The NLRP3 inflammasome necessitates Ox-mtDNA from the cytosol, which relies on CMPK2 activity. It helps regulate inflammatory vesicle-associated diseases.30,33 The CMPK2 protein’s IRF1-dependent catalytic activity regulates mtDNA replication and NLRP3 activation. However, it does not affect pro-IL-1β and NLRP3 or tumor necrosis factor-alpha (TNF-α) secretion. Deficiency does not affect caspase-11-mediated activation of the non-canonical inflammasome, NLRP3 activation-induced mitochondrial damage, or mtROS production. IRF1 can influence Ox-mtDNA and NLRP3 inflammasome activation by influencing mtDNA replication. However, it does not explain why the morphological characteristics of nuclear DNA occur.33 Recent research has uncovered that the never-in mitosis gene A (NIMA)-related kinase 7 (NEK7) plays a crucial role in activating the NLRP3 inflammasome complex in canonical and non-canonical pyroptosis. NEK7 also plays a role in cellular potassium efflux by interacting with the inflammatory complex. Without NEK7, the inflammatory complex cannot aggregate, which is necessary to activate the NLRP3 inflammasome complex and caspase 1.52,53
It is imperative to establish a consensus on the precise mechanisms that activate NLRP3 inflammatory vesicles. Such mechanisms encompass ion fluxes, including K+, Cl−, Na+, and Ca2+, mitochondrial dysfunction, ROS, and mtDNA release, lysosomal disruption, trans-Golgi catabolism, calcium changes, formation of non-specific membrane pores, and potassium efflux.28,54 Clarifying these mechanisms and their interactions is crucial to enhance our understanding of the inflammatory response and develop effective treatment strategies.
Non-Canonical Pathway
Non-canonical pyroptosis is a less complex way of pyroptosis that is initiated by caspase-11 in mice or caspase-4/5 in humans. This process is triggered by the detection of lipopolysaccharide (LPS) directly within the cytoplasm, which induces the activation of caspase-11 or caspase-4/5 independently of TLR4.55,56 This leads to the expression and activation of these caspases,48,55 which can then cleave pro-IL-1b, inducing its activation and release into the extracellular space.57
Caspase-1 plays a vital role in the non-canonical inflammasome pathway by inducing pyroptosis and releasing IL-1β and IL-18, while caspase-11 does not require these functions. It is pertinent to note that caspase-1 is a direct substrate of pro-IL-1β, not caspase-11, although these caspases may have reciprocal regulation. For caspase-11 induced pyroptosis, GSDMD is essential, as it facilitates the maturation of IL-1β, TLR initiation, and processing of pro-caspase-1 and pro-IL-1β.57 ATP triggers the canonical pyroptosis pathway in tissues through the recombinant purinergic receptor (P2X7R). The functional ion channel of the P2X7R subunit is a stable plasma membrane trimer.58 Upon ATP binding to the P2X7R gate, Na+ and Ca2+ flow in, while K+ flows out rapidly, leading to fast depolarization. P2X7R plays a crucial role in inflammation by activating the inflammasome, including the NLRP3, ASC, and pro-caspase-1. NLRP3 initiates and cleaves pro-caspase-1, producing biologically active IL-1β and triggering an inflammatory response59 (Figure 1).
 |
Figure 1 Mechanisms of three types of pyroptosis pathway. In the canonical pathway, PRRs detect signals both inside and outside cells and trigger the NF-κB signaling pathway, which boosts the expression of multiple genes. Other factors, such as ion flow, mitochondrial damage, and reactive molecules, can also activate NLRP3 inflammatory vesicles. These vesicles then change GSDMD into GSDMD-NT, leading to the formation of membrane pores as they aggregate and insert into the plasma membrane. Additionally, Caspase-1 helps create functional IL-1β and IL-18, which exit through these pores. In the non-canonical pathway, intracellular LPS triggers the activation of pro-caspase-4/-5/-11 via GBP and then promotes the production of active IL-1β and IL-18. LPS activates the MAPK and NF-κB signaling pathways and activates GSDMD-NT by binding to the receptor in the alternative pathway. It leads to the forming of the GSDMD pore and NLRP3 inflammatory vesicle through caspase-8. Then IL-1β is activated and released extracellularly through the GSDMD pore. The process can be inhibited by cFLIPL. It’s worth noting that there is mutual regulation between the canonical and non-canonical pathways. |
Alternative Pathway
The administration of LPS leads to the activation of apoptosis and regulation of cellular FLICE-like inhibitory protein (cFLIP) expression, thereby controlling pyroptosis. Moreover, increasing mtDNA stimulates pyroptosis by activating PRRs and their signaling pathways. LPS also influences the release of IL-1β through alternative pyroptosis via TRIF adapter,30 receptor-interacting protein 1 (RIP1) activity, and caspase-8.28 The production of pro-IL-1β requires TRIF, caspase-8, and RIP1. In addition, the spot-forming effect of ASC is caspase-8 dependent.46 It is noteworthy that the NLRP3 inflammasome components are responsible for mediating macrophage IL-1β secretion, not pyroptosis.57 Recent studies have identified the caspase-8 and FADD-like apoptosis regulator (cFLAR) gene, which encodes the caspase-8 enzyme inactive homolog cellular FLIP long isoform protein (cFLIPL), as a crucial component in regulating pyroptosis.46 Cells use cFLIP to control caspase-8 activation, with the short isoform (cFLIPR) completely blocking it and the cFLIPL partially blocking it. The quantity of cFLIPs in cells affects pyroptosis. The cFLIP protein obstructs the activation caspase-8 and minimizes pyroptosis triggered by LPS.46 Furthermore, cFLIPL regulates caspase-8 activation and complex II formations, protecting macrophages from LPS-induced pyroptosis. Caspase-8 initiates two fundamental processes: first, it activates GSDMD, leading to pyroptosis; second, it activates the NLRP3 inflammasome, which, in turn, drives the maturation and release of IL-1β. It is necessary to understand the exact mechanism by which NLRP3 and caspase-8 promote alternative pyroptosis via NLRP-3 inflammasomes.28
The activation of caspase-1 has been found to stimulate the production of IL-1β and IL-18, while the activation of caspase-8, caspase-11, or caspase-4/5 leads to pyroptosis. The cleavage of GSDMD in its central linker domain by caspase-1 in humans and caspase-4/5 in mice results in the creation of GSDMD-C-terminal and GSDMD-NT fragments.60 These fragments accumulate on lipid membranes, creating pores.31 Notably, the full-length GSDMD or C-terminal cleavage fragments can only lead to pyroptosis after being cleaved and released. GSDMD-NT interacts with phosphatidylinositol and cardiolipin on the inner leaflet membrane, forming a 16-monomer pore of undefined size on the plasma membrane.28,61 The process leads to cell depolarization and the release of inflammatory factors such as IL-1β, IL-18, and TNF-β. While it remains unclear how the GSDMD-NT fragment enters endothelial cell mitochondria,56 the pores release inflammatory substances, causing cell inflammation and death by allowing molecules to move in and out of the cell.28,33 The endosomal sorting complex required for transport (ESCRT) complex has been found to repair the cell membrane integrity and reduce the cell volume when there are only a few GSDMD pores in the cell membrane.28 Drugs can prevent pore formation by inhibiting GSDMD, which can help control the release of inflammatory factors during GSDMD activation.56 There are also interconnections between family members,28 and it is expected that GSDMD, its family members, and interventions for disease control will be the next primary research focus.
Pyroptosis and Synovium in OA
Synovial inflammation plays a pivotal role in the development of OA. It is an indicator of progression and is closely associated with cartilage loss, changes in synovial fluid, pain, and the formation of bone fragments.62,63 Our study aims to examine the correlation between synovial cells, inflammation in OA, and coping mechanisms for pyroptosis. The synovium is primarily composed of FLS and macrophages.64
Fibroblast-Like Synoviocytes in OA Synovium
OA leads to synovial lesions resulting from the growth of synovial tissue cells, causing hyperplasia and fibrosis. Specialized mesenchymal cells, known as fibroblast-like synoviocytes (FLS), make up most synovial tissue cells and are responsible for producing synovial fluid rich in lubricants and hyaluronic acid, which lubricate cartilage. FLS are the primary effector cells of synovial fibrosis in knee osteoarthritis (KOA).65 However, altered synovial permeability and decreased fat and hyaluronic acid concentrations in OA may result in pain.62 Pyroptosis, a type of cell death, is associated with OA. Pyroptosis-related molecules, such as NLRP3, caspase-1, and GSDMD, have been found in human OA synovial.66–68 LPS,66 collagenase, and both LPS and ATP together67,68 lead to upregulating of NLRP1 and NLRP3 inflammasome-associated proteins in FLS cells. Increased inflammasome expression can cause pyroptosis.67 In an animal model of synovial tissue inflammation, NLRP3 inflammatory vesicle-mediated pyroptosis was positively associated with temporomandibular joint osteoarthritis (TMJOA) synovitis.64 FLS cells from synovial tissue of OA patients treated with LPS/ATP led to an elevation of related factors in pyroptosis and inflammatory factors in the model.65–68 However, it was found that stimulating FLS with LPS or ATP alone does not induce pyroptosis. In vitro, induction of FLS resulted in changes in NLRP3 and caspase-1 mRNA, but only the GSDMD protein had corresponding changes. Detectable changes in both mRNA and protein of pyroptosis related-molecules occurred when co-culturing FLS with THP-1 macrophages, indicating that FLS cell-mediated synovial fibrosis may depend on macrophage pyroptosis.64 While Ac-YVAD-CMK (AYC), a caspase-1 inhibitor, did not affect NLRP3, it downregulated IL-1β, GSDMD, caspase-1, and ASC. A selective inhibitor of NLRP3 downregulated the expression of IL-1β, GSDMD, and caspase-1 in synovial tissues, suggesting that both inhibitors affect pyroptosis related-factors. Rat OA models showed elevated hypoxia-inducible factor-1 (HIF-1) levels. SiRNA of HIF-1α treatment decreased dead FLS numbers and downregulated pyroptosis related-protein expression.64 Pyroptosis of FLS exacerbates synovial inflammation and fibrosis.65 In rat models, inhibiting pyroptosis through activating adenosine monophosphate-activated protein kinase (AMPK) with stromal cell-derived factor-1 (SDF-1) improved the severity of OA.66
Macrophage in OA Synovium
OA is a condition that affects the joints of individuals. The synovium in the affected joints contains various macrophages that possess pro-inflammatory and anti-inflammatory traits. Some of these macrophages have heightened phagocytic and immunosuppressive activity.69 Past studies have cited that pro-inflammatory macrophages cause OA development by infiltrating and activating synovial tissue related to synovial fibrosis in KOA.70 Reducing synovial macrophages with drugs can help alleviate OA symptoms such as pain and stiffness.71 However, some studies have demonstrated that lowering synovial macrophage numbers can result in an increase in synovial inflammation.72 Further research is necessary to understand the role and mechanism of different phenotypes of macrophage pyroptosis in OA and its potential for treating OA. The previous studies did not clarify the macrophage phenotype and its varying effects on inflammation. Synovial macrophages are mainly M2 and control inflammation when exposed to stimuli. M2 macrophages can turn into M1 macrophages, which cause inflammation.73 In individuals with OA, macrophages deposit crystals in the synovium. Dead cells release ATP, which activates the NLRP3 inflammasome to produce IL-1β and IL-18. Sodium urate crystals in joints are linked to OA severity and bone flab formation; its decrease can protect joint function.62 Caspase-1 causes GSDMD pores to form, prolonging inflammation in the synovium.21 Zhang et al observed increased NLRP3 inflammasome components in synovial tissues of rats with KOA. Treatment with caspase-1 inhibitors improved tissue organization and reduced inflammation, fibrosis, and expression of pro-inflammatory factors. However, caspase-1 knockout reduced pyroptosis induced by LPS, but their macrophages were not directly from the diseased synovium.74 In agreement with the previous note,64 after the co-culture of macrophages undergoing pyroptosis and FLS, FLS cells also exhibited characteristics of pyroptosis and elevated expression of fibrogenic factors.74 Thus, Macrophage pyroptosis plays a role in the development of OA synovial. Further research is necessary to explore the connection between macrophages and FLS and their role in OA.
Pyroptosis and Cartilage in OA
The articular cartilage protects the bone plate by absorbing loads and strains while minimizing strain transfer to the bone.75 Chondrocytes, the specialized cells, respond to various stimuli such as mechanical loading and growth factors to maintain cartilage homeostasis. The primary component of cartilage is the ECM, which is primarily composed of type II collagen.76,77
OA is a debilitating medical condition that arises due to degeneration of cartilage, chondrocyte death, and ECM degradation. Activation of the MAPK/NF-κB pathway can trigger inflammation in chondrocytes, leading to a range of conditions, including OA.78 In an OA model, the expression of NLRP3 was found to be higher in medial cartilage than in lateral cartilage, and inhibition of the MAPK/NF-κB/NLRP3 can alleviate chondrocyte pyroptosis, proteoglycan loss, collagen degradation, articular cartilage degeneration, and subchondral bone destruction.79 Pyroptosis, induced by activators that promote chondrocyte NLRP3 inflammasome activation via the MAPK/NF-κB pathway, can exacerbate these conditions.37,38,79 Bai et al conducted a study that revealed the potential role of pyroptosis in OA. Their findings showed that selective inhibition of NLRP3 and caspase-1 led to a reduction in the expression of pyroptosis-related molecules such as NLRP3, ASC, caspase-1, and GSDMD in both an OA animal model and rat chondrocytes stimulated with hydrogen peroxide (H₂O₂). Additionally, the authors noted that H2O2 stimulation resulted in the elevation of ROS and Ca2+ but a decrease in K+ in chondrocytes. Interestingly, the use of an A3 adenosine receptor (A3AR) activator to inhibit the pyroptosis pathway led to a decrease in ROS and related pyroptosis molecules, along with an increase in Ca2+. The A3AR activator did not affect intracellular K+ and Ca2+.80 It is important to note that pyroptosis pathways can be triggered via three distinct pathways, with NLRP3 and caspase-1 playing a prominent role in the canonical pathway, which is dependent on K+ efflux.81 However, non-canonical pyroptosis cannot reveal two contradictory phenomena in chondrocyte pyroptosis stimulated by H2O2.80 In post-traumatic osteoarthritis (PTOA) models and OA patients, RNA levels of GSDMD, IL-1β, and IL-18 were found to be higher in cartilage tissue. GSDMD knockout mice in the PTOA model experienced less inflammation, cartilage loss, and osteosclerosis, indicating that inhibiting GSDMD may be a new approach to treating OA.81 Although chondrocytes have been found to induce pyroptosis in vitro, recent research suggests that chondrocyte pyroptosis is likely induced by inflammatory factors such as IL-1β, IL-18, and TNF-α, which are released during synovial pyroptosis rupture.80,81 In an animal model undergoing surgical destabilization of the medial meniscus (DMM) under multiple fatty acid treatments, higher expression of TLR4, MyD88, phosphor-NF-κB (p-NF-κB), NLRP3, caspase-1, and IL-1β was detected in the articular cartilage tissue than in the normal group. Similar results were observed in chondrocytes treated with subsequent LPS, where TLR4, p-NF-κB, NLRP3, Caspase-1, GSDMD, and IL-1β protein expression decreased or increased accordingly after TLR4 inhibition or overexpression, highlighting the involvement of the TLR4 and NF-κB pathway in the process of chondrocytes pyroptosis in OA. However, the level of IL-1β was detected, but not IL-18.82 Furthermore, in an OA rat model, a decrease in phosphorylated AMPK and an increase in Caspase-1 were observed. Treatment with the AMPK activator AICAR significantly reduced the expression of inflammasome and IL-1β in LPS-induced chondrocyte inflammation.83,84 The study revealed that the AMPK pathway leads to pyroptosis and contributes to OA development, though it also did not mention changes in IL-18.85,86
The article discusses various pathways that can affect chondrocyte pyroptosis and ECM degradation.85 Upregulating TGF-β1, activating Smad2/3, and inhibiting the NF-κB pathway may potentially reduce oxidative stress and protect chondrocytes from pyroptosis, ultimately mitigating OA development. Additionally, moderate stimulation of articular cartilage may potentially suppress chondrocyte pyroptosis and protect joints in OA patients by inhibiting the phosphoinositide 3‑kinase (PI3K)/protein kinase B (Akt)/NF-κB signaling pathway.86 However, inappropriate mechanical stress can harm chondrocytes and activate the NF-κB pathway, leading to increased oxidative stress, pyroptosis, and ECM degradation during OA development.85 Moreover, in rat chondrocytes treated with H2O2, the expression of USP7, ROS, NLRP3, GSDMD-NT, active caspase-1, procaspase-1, MMP1, and MMP13 increased. Silencing, inhibition, or overexpression of the ubiquitin-specific protease 7 (USP7) gene accordingly affected the expression of those factors, whereas NLRP1, NLRP6, NLR-family CARD-containing protein 4 (NLRC4), AIM2, and ASC remained unchanged. USP7 interacts with nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4), leading to chondrocyte pyroptosis. This finding was confirmed in OA animal models and human OA cartilage.87 Furthermore, increased ATP in rats with OA was found to be induced by monosodium iodoacetate (MIA) injection. It also detected upregulation of inflammatory switch purinergic receptors P2X7, ROS, MMP13, NF-κB, p65, NLRP3, caspase-1, and IL-1β, ultimately resulting in worse ECM degradation, cartilage loss, and bone resorption, thereby accelerating OA progression.88,89 Conversely, P2X7R suppression led to the opposite effect.59
When chondrocytes or synovial tissue undergo pyroptosis, they release inflammatory factors such as IL-1β, IL-18, TNF-α and MMPs, particularly MMP1 and MMP13,18,87,90–92 which increase the levels of inflammation in the joint fluid and surrounding tissues. This leads to the release of more catabolic enzymes and pro-inflammatory factors, resulting in the degradation of cartilage and a more severe inflammatory response. This process contributes to the development of OA.18,22,84. Licochalcone A is a natural compound that can attenuate the effects of LPS on chondrocytes. It works by promoting the production of proteoglycans, aggregating collagen type II, and remodeling the ECM through the nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1)/NF-κB axis. Additionally, it inhibits pyroptosis, which can increase cartilage ECM degradation.84
Pyroptosis and Subchondral Bone in OA
Subchondral bone, a thin layer of dense bone beneath calcified cartilage, consists of subchondral cortical bone, subchondral trabecular bone, and epiphyseal trabecular bone. It is a crucial component of the knee joint structure and function, containing 48% minerals, 31% organic material, and 21% water in healthy samples.84 However, in cases of OA, the thickness of the subchondral cortical bone is affected, leading to subchondral sclerotic consistency, bone redundancy, osteophytes, and bone cysts in the advanced stages.93,94 Our research findings have revealed the consistent subchondral bone foundation supporting the articular cartilage.7 However, in cases of OA, the trabecular rods are missing, and the trabecular plates become thick and fragile, disrupting the cartilage’s typical support system. Additionally, injuries and abnormal mechanical stress exacerbate the irregular dispersion of microstructures like trabecular rods and plates within the bone, leading to localized hardening of the subchondral bone.7 While our study primarily focuses on the mechanical and morphological aspects of the subchondral bone, we have overlooked the specific cells involved in this process.7,95–97 Furthermore, we have neglected to consider the chemical reactions within these living cells, including iron death, apoptosis, and pyroptosis. Pyroptosis-associated molecules are elevated in cases of destabilization of the medial meniscus (DMM) model, resulting in significantly higher OA Research Society International (OARSI) scores.84 Yan et al discovered the effect of pyroptosis on an OA model and identified increased expression of pyroptosis factors, such as NLRP3, Caspase-1, GSDMD, and IL-1β, in chondrocytes, along with elevated OARIS scores. In the initial stage of the study, they observed a decrease in subchondral bone mass, an increase in trabecular separation (Tb. Sp), an elevation in osteoclasts, and a decrease in osteoprotegerin (OPG). However, in the later stages of OA, they noticed an increase in subchondral bone volume, a decrease in Tb. Sp and osteoclasts, and an increase in OPG.98 Ting et al conducted similar experiments that yielded comparable outcomes.79 These findings demonstrate a significant correlation between pyroptosis and subchondral bone changes during OA development. The severity of OA is directly proportional to the duration and intensity of pyroptosis effects, resulting in more prolonged and intense pyroptosis effects in severe cases.98 (Figure 2).
 |
Figure 2 Mechanism of pyroptosis-mediated osteoarthritis. When certain signals, either inside or outside the cell, increase the expression of the NLRP3 inflammatory body, it triggers the cleavage of GSDMD and pro-IL-1β by caspase-1. This process produces GSDMD-NT which creates membrane pores that induce cytotoxicity and release inflammatory factors. In a prolonged chronic inflammatory environment, this can lead to synovial inflammation, osteophyte formation, cartilage degradation, and ultimately result in osteoarthritis. |
Interconnection of Tissues and Cells of Pyroptosis
OA affects the entire joint, including the synovial membrane, articular cartilage, ECM, joint fluid, subchondral bone, and accessory structures. Pyroptosis causes lesions that affect multiple structures in the joint, involving synovial macrophages, FLS, chondrocytes, and subchondral bone. The aim of the current study is to determine which cell undergoes pyroptosis first and leads to the development of OA. The study found upregulated pyroptosis-related molecules in knee synovial tissue and chondrocytes in human and animal models of OA. Pyroptosis was induced in vitro in different cells, and while some showed pyroptosis responses, it was not observed in FLS. However, co-cultures of activator-stimulated macrophages and FLS did show pyroptosis. Therefore, it is suggested that macrophages may preferentially undergo pyroptosis when induced by stimulants.65,67,68
It has been discovered through research that the inflammatory extracellular vesicles from macrophages can trigger both canonical and non-canonical pyroptosis in chondrocytes, leading to cartilage breakdown.73,80,81 The synovium plays a crucial role in providing essential nutrition to the avascular cartilage tissue, and synovitis, the inflammation of the synovial tissue, is a significant factor in the development of OA. When the synovial tissue undergoes pyroptosis, it releases pro-inflammatory cartilage degradation products into the synovial fluid, which causes inflammation in the surrounding synovium, leading to further degradation of chondrocytes.84 The degradation of cartilage leads to a decrease in lubricin and hyaluronic acid (HA) and an increase in matrix-degrading enzymes in the body that help protect the joint.18 During the early stages of OA, synovial macrophages are stimulated by activators from both in vivo and ex vivo sources, leading to the initiation of pyroptosis through MAPK or NF-κB pathways. GSDMD-NT, which forms intracellular aggregated empty pores, releases inflammatory factors, leading to macrophages undergoing lysis. The intracellular stimulants released into the tissue by macrophages further stimulate remaining macrophages, synovial fibroblasts, and chondrocytes, leading to more severe OA. This causes synovial fibrosis, chondrocyte death, and the release of TNF-α, MMPs, and other inflammatory factors, leading to advanced OA lesions. (Figure 3).
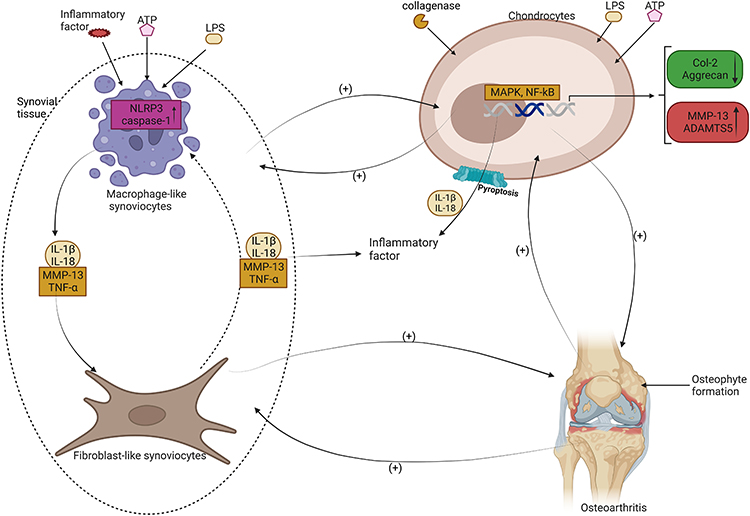 |
Figure 3 OA is a condition that involves the breakdown of cartilage in the joints. The role of pyroptosis, which is the rupture of cells, in the progression of OA has been studied. MLS and chondrocytes, which are cells in the joints, can detect foreign substances and activate an inflammatory response through the assembly of NLRP3 inflammasome. This can lead to the rupture of synoviocytes and the release of inflammatory factors that cause synovial fibroblasts to be affected. In addition, chondrocytes can activate MAPK and NF-κB signaling pathways, leading to the expression of MMP13 and ADAMTS5 while reducing the synthesis of Col-2 and Aggrecan, which are important components of cartilage. Pyroptosis of chondrocytes can also result in the release of inflammatory factors such as IL-1β and IL-18, which fuel each other in a vicious cycle, leading to the formation of osteophytes and worsening of OA. |
Summary and Perspective
OA is a prevalent and complex health issue that can result in severe, persistent disability. Although several treatments exist for OA, more targeted and beneficial therapies are needed. The condition is influenced by various factors, including pyroptosis, a type of inflammation that plays a significant role in OA.5,6,13–15,17,23 Pyroptosis involves three distinct pathways: canonical, non-canonical, and alternative, each with unique molecular characteristics. The process consists of two phases: initiation and activation. During the initiation phase of the canonical pathway, PAMPs and DAMPs bind with TLRs, leading to the synthesis of NLRP3 inflammasomes in the cytoplasm via the MAPK and NF-κB pathways triggered by mtDNA and ROS changes. In the activation phase, the NLRP3 inflammasome activates caspase-1.30–33,45 Intracellular LPS can directly induce the formation of caspase-4/5 or caspase-11, which further form IL-1β55–57 and NLRP-3 inflammasomes and also form IL-1β via PAMPs and DAMPs dependent on caspase-8. GSDMD can be converted into GSDMD-NT by caspase-1, caspase-4/5, or both caspase-11 and caspase-8.46 These can aggregate at the plasma membrane to form pores that lead to the release of inflammatory factors and cause death.28,61 Several molecules associated with pyroptosis, such as caspase-1, caspase-4/5, caspase-11, and caspase-8, of the caspase family, as well as NLRP-3 and GSDMD and its isoforms, and various inflammatory factors, have been observed to change in animal models of OA and the joints of OA patients. These changes were also detected in macrophages induced with LPS in vitro. Inhibition of these molecules or pyroptosis-associated factors resulted in a decrease in the expression of pyroptosis-associated molecules and alleviation of OA. However, it’s worth noting that these studies have some controversial points and limitations. Despite these limitations, pyroptosis remains a crucial area of investigation in the search for more targeted and effective treatments for OA.
Numerous studies have delved into the role of NLRP3 inflammasome in canonical pyroptosis, while only a few have explored non-canonical or alternative pyroptosis.73 The current limitations make it difficult to create animal models of non-canonical or alternative pyroptosis and identify the activators that cause pyroptosis in vitro. Although pyroptosis of synovial macrophages, FLS, and chondrocytes is believed to play a role in OA development, the exact function of pyroptosis remains contested due to variations in animal models and stimuli.30,56 Numerous studies have failed to establish a definitive causal relationship between pyroptosis and OA during different stages. Some studies have focused solely on IL-1β while overlooking the crucial role of IL-18 in activating the knee’s inflammatory and immune response.17,67,82,99 Furthermore, the specific cell type that undergoes pyroptosis in OA and its potential effects on other cells remain unresolved. We can potentially regulate pyroptosis, which is a form of programmed cell death, in the future by targeting specific components of inflammatory vesicles, such as NLRP3, caspase-1, GSDMD, and ASC, or activation pathways, such as NF-κB, MAPK, P2X7, Nrf2, ROS, USP7, and HIF-1α, in chondrocytes, synoviocytes, and synovial cells. The use of macrophages and synovial cells in the joint, a multi-tissue motor system, is a vital area of research for the prevention and treatment of OA. We hope to identify particular factors that can be treated with medication or other methods to impede the pyroptosis process in OA patients, with the ultimate aim of reducing inflammation, alleviating pain, and potentially curing the condition for those afflicted
Abbreviations
PRRs, pattern recognition receptors; LPS, Lipopolysaccharides; NF-κB, nuclear factor-k-gene binding; NLRP3, NOD-like receptor thermal protein domain associated protein 3; IRF1, Interferon regulatory factor 1; pro-caspase-1, pro-cysteinyl aspartate specific proteinase-1; pro-IL-1β, pro-interleukin-1β; pro-IL-18, pro-interleukin-18; ROS, reactive oxygen species; Ox-mtDNA, oxidized mitochondrial DNA; GSDMD, Gasdermin D; caspase-1, cysteinyl aspartate specific proteinase-1; GSDMD-NT, Gasdermin D- N terminal; IL-1β, interleukin-1β; IL-18, interleukin-18; pro-caspase-4/-5/-11, pro-cysteinyl aspartate specific proteinase-4/-5/-11; caspase-1, cysteinyl aspartate specific proteinase −1;caspase-4/-5/-11, cysteinyl aspartate specific proteinase-4/-5/-11; GBP, guanylate binding protein; MAPK: mitogen-activated protein kinases; caspase-8, cysteinyl aspartate specific proteinase −8; P2X7R, P2X7 receptor; cLFLIPL, cellular FLIP long isoform protein; Nur77, nerve growth factor-induced gene B; PI4P, phosphatidylinositol-4-phosphate; ATP, adenosine triphosphate; MyD88, myeloid differentiation primary response 88; TRIF, Toll/IL-1R domain-containing adaptor-inducing IFN-β; CMPK2, cytidine/uridine monophosphate kinase 2; MMP13, matrix metalloproteinases 13; ADAMTS5, a disintegrin and metalloproteinase protein 5; Col-2, Collagen-2.
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (82060406), Natural Science Foundation of Guangxi (2022JJA141126), Advanced Innovation Teams and Xinghu Scholars Program of Guangxi Medical University, China Postdoctoral Science Foundation (2019M650235), Key R&D Project of Qingxiu District, Nanning, Guangxi (2021003).
Author Contributions
Yan Chen serves as the corresponding author for this paper. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of figures, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that the authors involved in the research had absolutely no commercial or financial relationships that could in any way be seen as a potential conflict of interest.
References
1. Martel-Pelletier J, Barr AJ, Cicuttini FM, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072.
2. Glyn-Jones S, Palmer AJ, Agricola R, et al. Osteoarthritis. Lancet. 2015;386(9991):376–387.
3. Ondrésik M, Azevedo Maia FR, da Silva Morais A, et al. Management of knee osteoarthritis. Current status and future trends. Biotechnol Bioeng. 2017;114(4):717–739.
4. Wallace IJ, Worthington S, Felson DT, et al. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc Natl Acad Sci U S A. 2017;114(35):9332–9336.
5. Palazzo C, Nguyen C, Lefevre-Colau -M-M, Rannou F, Poiraudeau S. Risk factors and burden of osteoarthritis. Ann Phys Rehabil Med. 2016;59(3):134–138.
6. Abramoff B, Caldera FE. Osteoarthritis: pathology, Diagnosis, and Treatment Options. Med Clin North Am. 2020;104(2):293–311.
7. Chen Y, Hu Y, Yu YE, et al. Subchondral Trabecular Rod Loss and Plate Thickening in the Development of Osteoarthritis. J Bone Miner Res. 2018;33(2):316–327.
8. Fan Y, Li Z, Zhang H, et al. Valgus knee bracing may have no long-term effect on pain improvement and functional activity in patients with knee osteoarthritis: a meta-analysis of randomized trials. J Orthop Surg Res. 2020;15(1):373.
9. Boer CG, Radjabzadeh D, Medina-Gomez C, et al. Intestinal microbiome composition and its relation to joint pain and inflammation. Nat Commun. 2019;10(1):4881.
10. Ma Z, Wang D, Weng J, Zhang S, Zhang Y. BNIP3 decreases the LPS-induced inflammation and apoptosis of chondrocytes by promoting the development of autophagy. J Orthop Surg Res. 2020;15(1):284.
11. Zeng D, Chen Y, Liao Z, et al. Cartilage organoids and osteoarthritis research: a narrative review. Front Bioeng Biotechnol. 2023;11:1278692.
12. Wei G, et al. Risk of metabolic abnormalities in osteoarthritis: a new perspective to understand its pathological mechanisms. Bone Res. 2023;11(1):63.
13. Zhang S, Xu J, Si H, Wu Y, Zhou S, Shen B. The Role Played by Ferroptosis in Osteoarthritis: evidence Based on Iron Dyshomeostasis and Lipid Peroxidation. Antioxidants (Basel). 2022;11:9.
14. Hwang HS, Kim HA. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int J Mol Sci. 2015;16(11):26035–26054.
15. Castrogiovanni P, Ravalli S, Musumeci G. Apoptosis and Autophagy in the Pathogenesis of Osteoarthritis. J Invest Surg. 2020;33(9):874–875.
16. Hsu SK, Li CY, Lin IL, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. 2021;11(18):8813–8835.
17. Yang J, Hu S, Bian Y, et al. Targeting Cell Death: pyroptosis, Ferroptosis, Apoptosis and Necroptosis in Osteoarthritis. Front Cell Dev Biol. 2021;9:789948.
18. An S, Hu H, Li Y, Hu Y. Pyroptosis Plays a Role in Osteoarthritis. Aging Dis. 2020;11(5):1146–1157.
19. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
20. Lu F, Lan Z, Xin Z, et al. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J Cell Physiol. 2020;235(4):3207–3221.
21. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42(4):245–254.
22. Paludan SR, Reinert LS, Hornung V. DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat Rev Immunol. 2019;19(3):141–153.
23. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114.
24. Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12(5):259–268.
25. Chao L, Li Z, Zhou J, et al. Shen-Ling-Bai-Zhu-San Improves Dextran Sodium Sulfate-Induced Colitis by Inhibiting Caspase-1/Caspase-11-Mediated Pyroptosis. Front Pharmacol. 2020;11:814.
26. Jiao Y, Zhao H, Chen G, et al. Pyroptosis of MCF7 Cells Induced by the Secreted Factors of hUCMSCs. Stem Cells Int. 2018;2018:5912194.
27. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019;52(2):e12563.
28. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–2127.
29. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166(1):1–15.
30. Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140(6):821–832.
31. Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243(1):136–151.
32. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, Inflammation, and Immunity: a Troika Governing Cancer and Its Treatment. Cell. 2016;166(2):288–298.
33. Zhong Z, Liang S, Sanchez-Lopez E, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203.
34. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230.
35. Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164(5):896–910.
36. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225.
37. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022.
38. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420.
39. Liu Z, Yao X, Jiang W, et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation. 2020;17(1):90.
40. Barnett KC, Ting JP. Mitochondrial GSDMD Pores DAMPen Pyroptosis. Immunity. 2020;52(3):424–426.
41. Keller MD, Torres VJ, Cadwell K. Autophagy and microbial pathogenesis. Cell Death Differ. 2020;27(3):872–886.
42. Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza A Virus Infection Triggers Pyroptosis and Apoptosis of Respiratory Epithelial Cells through the Type I Interferon Signaling Pathway in a Mutually Exclusive Manner. J Virol. 2018;92:14.
43. Yao Y, Zang Y, Qu J, Tang M, Zhang T. The Toxicity Of Metallic Nanoparticles On Liver: the Subcellular Damages, Mechanisms, And Outcomes. Int J Nanomed. 2019;14:8787–8804.
44. Donado CA, Cao AB, Simmons DP, Croker BA, Brennan PJ, Brenner MB. A Two-Cell Model for IL-1β Release Mediated by Death-Receptor Signaling. Cell Rep. 2020;31(1):107466.
45. Tumurkhuu G, Shimada K, Dagvadorj J, et al. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ Res. 2016;119(6):e76–90.
46. Muendlein HI, Jetton D, Connolly WM, et al. cFLIP(L) protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science. 2020;367(6484):1379–1384.
47. Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193–1206.
48. Hu Z, Chai J. Structural Mechanisms in NLR Inflammasome Assembly and Signaling. Curr Top Microbiol Immunol. 2016;397:23–42.
49. Wang L, Sharif H, Vora SM, Zheng Y, Wu H. Structures and functions of the inflammasome engine. J Allergy Clin Immunol. 2021;147(6):2021–2029.
50. Ambrogini E, Que X, Wang S, et al. Oxidation-specific epitopes restrain bone formation. Nat Commun. 2018;9(1):2193.
51. Hari P, Millar FR, Tarrats N, et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci Adv. 2019;5(6):eaaw0254.
52. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357.
53. Sharif H, Wang L, Wang WL, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570(7761):338–343.
54. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: an Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20:13.
55. Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671.
56. Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745.
57. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121.
58. Linden J, Koch-Nolte F, Dahl G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu Rev Immunol. 2019;37:325–347.
59. Li Z, Huang Z, Zhang H, et al. P2X7 Receptor Induces Pyroptotic Inflammation and Cartilage Degradation in Osteoarthritis via NF-κB/NLRP3 Crosstalk. Oxid Med Cell Longev. 2021;2021:8868361.
60. Ma C, Yang D, Wang B, et al. Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci Adv. 2020;6(21):eaaz6717.
61. Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116.
62. Sanchez-Lopez E, Coras R, Torres A, Lane NE, Guma M. Synovial inflammation in osteoarthritis progression. Nat Rev Rheumatol. 2022;18(5):258–275.
63. Liao Z, et al. ‘Transient receptor potential vanilloid 1: a potential therapeutic target for the treatment of osteoarthritis and rheumatoid arthritis’ Cell Prolif. 2023;e13569.
64. Xin Y, Wang W, Mao E, Yang H, Li S. Targeting NLRP3 Inflammasome Alleviates Synovitis by Reducing Pyroptosis in Rats with Experimental Temporomandibular Joint Osteoarthritis. Mediators Inflamm. 2022;2022:2581151.
65. Zhang L, Zhang L, Huang Z, et al. Increased HIF-1α in Knee Osteoarthritis Aggravate Synovial Fibrosis via Fibroblast-Like Synoviocyte Pyroptosis. Oxid Med Cell Longev. 2019;2019:6326517.
66. Wang S, Mobasheri A, Zhang Y, Wang Y, Dai T, Zhang Z. Exogenous stromal cell-derived factor-1 (SDF-1) suppresses the NLRP3 inflammasome and inhibits pyroptosis in synoviocytes from osteoarthritic joints via activation of the AMPK signaling pathway. Inflammopharmacology. 2021;29(3):695–704.
67. Zhao LR, Xing RL, Wang PM, et al. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP‑induced pyroptosis in knee osteoarthritis. Mol Med Rep. 2018;17(4):5463–5469.
68. Xiao Y, Ding L, Yin S, et al. Relationship between the pyroptosis of fibroblast‑like synoviocytes and HMGB1 secretion in knee osteoarthritis. Mol Med Rep. 2021;23:2.
69. Chou CH, Jain V, Gibson J, et al. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep. 2020;10(1):10868.
70. Lopes EBP, Filiberti A, Husain SA, Humphrey MB. Immune Contributions to Osteoarthritis. Curr Osteoporos Rep. 2017;15(6):593–600.
71. Kraus VB, McDaniel G, Huebner JL, et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage. 2016;24(9):1613–1621.
72. Wu CL, McNeill J, Goon K, et al. Conditional Macrophage Depletion Increases Inflammation and Does Not Inhibit the Development of Osteoarthritis in Obese Macrophage Fas-Induced Apoptosis-Transgenic Mice. Arthritis Rheumatol. 2017;69(9):1772–1783.
73. Ebata T, Terkawi MA, Kitahara K, et al. Noncanonical Pyroptosis Triggered by Macrophage-Derived Extracellular Vesicles in Chondrocytes Leading to Cartilage Catabolism in Osteoarthritis. Arthritis Rheumatol. 2023;75(8):1358–1369.
74. Zhang L, Xing R, Huang Z, et al. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediators Inflamm. 2019;2019:2165918.
75. Aziz AH, Eckstein K, Ferguson VL, Bryant SJ. The effects of dynamic compressive loading on human mesenchymal stem cell osteogenesis in the stiff layer of a bilayer hydrogel. J Tissue Eng Regen Med. 2019;13(6):946–959.
76. Ramaswamy G, Sohn P, Eberhardt A, Serra R. Altered responsiveness to TGF-β results in reduced Papss2 expression and alterations in the biomechanical properties of mouse articular cartilage. Arthritis Res Ther. 2012;14(2):R49.
77. Guo T, Noshin M, Baker HB, et al. 3D printed biofunctionalized scaffolds for microfracture repair of cartilage defects. Biomaterials. 2018;185:219–231.
78. Li Z, Dai A, Yang M, Chen S, Deng Z, Li L. p38MAPK Signaling Pathway in Osteoarthritis: pathological and Therapeutic Aspects. J Inflamm Res. 2022;15:723–734.
79. Jiang T, Gong Y, Zhang W, et al. PD0325901, an ERK inhibitor, attenuates RANKL-induced osteoclast formation and mitigates cartilage inflammation by inhibiting the NF-κB and MAPK pathways. Bioorg Chem. 2023;132:106321.
80. Bai H, Zhang Z, Liu L, Wang X, Song X, Gao L. Activation of adenosine A3 receptor attenuates progression of osteoarthritis through inhibiting the NLRP3/caspase-1/GSDMD induced signalling. J Cell Mol Med. 2022;26(15):4230–4243.
81. Yang T, Sun K, Wang C, et al. Gasdermin D deficiency attenuates arthritis induced by traumatic injury but not autoantibody-assembled immune complexes. Arthritis Res Ther. 2021;23(1):286.
82. Jin X, Dong X, Sun Y, Liu Z, Liu L, Gu H. Dietary Fatty Acid Regulation of the NLRP3 Inflammasome via the TLR4/NF-κB Signaling Pathway Affects Chondrocyte Pyroptosis. Oxid Med Cell Longev. 2022;2022:3711371.
83. Chen Y, Liu Y, Jiang K, Wen Z, Cao X, Wu S. Linear ubiquitination of LKB1 activates AMPK pathway to inhibit NLRP3 inflammasome response and reduce chondrocyte pyroptosis in osteoarthritis. J Orthop Translat. 2023;39:1–11.
84. Yan Z, Qi W, Zhan J, et al. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J Cell Mol Med. 2020;24(22):13046–13057.
85. Wang Y, Jin Z, Jia S, Shen P, Yang Y, Huang Y. Mechanical stress protects against chondrocyte pyroptosis through TGF-β1-mediated activation of Smad2/3 and inhibition of the NF-κB signaling pathway in an osteoarthritis model. Biomed Pharmacother. 2023;159:114216.
86. Jia S, Yang Y, Bai Y, et al. Mechanical Stimulation Protects Against Chondrocyte Pyroptosis Through Irisin-Induced Suppression of PI3K/Akt/NF-κB Signal Pathway in Osteoarthritis. Front Cell Dev Biol. 2022;10:797855.
87. Liu G, Liu Q, Yan B, Zhu Z, Xu Y. USP7 Inhibition Alleviates H(2)O(2)-Induced Injury in Chondrocytes via Inhibiting NOX4/NLRP3 Pathway. Front Pharmacol. 2020;11:617270.
88. Zeng D, Yao P, Zhao H. P2X7, a critical regulator and potential target for bone and joint diseases. J Cell Physiol. 2019;234(3):2095–2103.
89. McCarthy AE, Yoshioka C, Mansoor SE. Full-Length P2X(7) Structures Reveal How Palmitoylation Prevents Channel Desensitization. Cell. 2019;179(3):659–670.e613.
90. Hu J, Zhou J, Wu J, et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-κB activity and pyroptosis in chondrocytes. J Ethnopharmacol. 2020;247:112261.
91. Ortega N, Behonick DJ, Werb Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004;14(2):86–93.
92. Hu Q, Ecker M. Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. Int J Mol Sci. 2021;22(4):56.
93. Mukherjee S, Nazemi M, Jonkers I, Geris L. Use of Computational Modeling to Study Joint Degeneration: a Review. Front Bioeng Biotechnol. 2020;8:93.
94. Hirvasniemi J, Thevenot J, Kokkonen HT, et al. Correlation of Subchondral Bone Density and Structure from Plain Radiographs with Micro Computed Tomography Ex Vivo. Ann Biomed Eng. 2016;44(5):1698–1709.
95. Chen Y, et al. ‘Abnormal subchondral bone remodeling and its association with articular cartilage degradation in knees of type 2 diabetes patients’. Bone Res. 2017;5:17034.
96. Chen Y, et al. ‘Bone turnover and articular cartilage differences localized to subchondral cysts in knees with advanced osteoarthritis’. Osteoarthritis Cartilage. 2015;23(12):2174–2183.
97. Wen CY, et al. ‘Bone loss at subchondral plate in knee osteoarthritis patients with hypertension and type 2 diabetes mellitus’. Osteoarthritis Cartilage. 2013;21(11): 1716–1723.
98. Yan J, Feng G, Yang Y, et al. Autophagy attenuates osteoarthritis in mice by inhibiting chondrocyte pyroptosis and improving subchondral bone remodeling. Biomol Biomed. 2023;23(1):77–88.
99. Matsui K, Tsutsui H, Nakanishi K. Pathophysiological roles for IL-18 in inflammatory arthritis. Expert Opin Ther Targets. 2003;7(6):701–724.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.