Back to Journals » Cancer Management and Research » Volume 11
PTPN9 induces cell apoptosis by mitigating the activation of Stat3 and acts as a tumor suppressor in colorectal cancer
Authors Wang D, Cheng Z, Zhao M, Jiao C, Meng Q, Pan H, Xie Y, Li L, Zhu Y, Wang W, Qu C, Liang D
Received 10 September 2018
Accepted for publication 8 January 2019
Published 8 February 2019 Volume 2019:11 Pages 1309—1319
DOI https://doi.org/10.2147/CMAR.S187001
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Harikrishna Nakshatri
Dawei Wang,1,* Zhuoxin Cheng,2,3,* Ming Zhao,1,* Chengbin Jiao,2 Qinghui Meng,1 Huayang Pan,1 Yu Xie,1 Long Li,1 Yexing Zhu,1 Wei Wang,1 Chunlei Qu,1 Deshen Liang1
1Department of General Surgery, The First Affiliated Hospital of Harbin Medical University, Harbin 150001, People’s Republic of China; 2Department of General Surgery, The First Affiliated Hospital of Jiamusi University, Jiamusi 154002, People’s Republic of China; 3Heilongjiang Provincial Key Laboratory of Metabolic Disease, Jiamusi 154002, People’s Republic of China
*These authors contributed equally to this work
Background: Accumulating evidence has shown that protein tyrosine phosphatases (PTPs) are involved in regulating the transduction of many signaling pathways and play important roles in modulating the progression of some cancers, but the functions of PTPs in cancers have not been well elucidated until now. Here, we aimed to identify the roles of protein tyrosine phosphatase nonreceptor type 9 (PTPN9), a cytoplasmic PTP, in the development of colorectal cancer and elucidate the regulatory mechanism involved.
Materials and methods: Cell viability assessment, colony formation assay, caspase-3 and caspase-9 activity assay, real-time PCR, and Western blot analysis were applied.
Results: Our results showed that PTPN9 expression was frequently downregulated in colorectal cancer tissues compared with adjacent normal tissues. Overexpression of PTPN9 mitigated cell growth and colony formation and induced cell apoptosis in colorectal cancer. Conversely, PTPN9 knockdown promoted cell growth and survival. Moreover, PTPN9 negatively regulated the activation of Stat3 and depressed its nuclear translocation in colorectal cancer. The effects of PTPN9 knockdown on cell apoptosis were attenuated by inhibition of the Stat3 pathway.
Conclusion: These results indicate that PTPN9 inhibits cell growth and survival by repressing the activation of Stat3 in colorectal cancer, which suggests an important underlying mechanism of regulating cell growth and provides a novel candidate therapeutic target for colorectal cancer.
Keywords: PTPN9, apoptosis, colorectal cancer, Stat3, cell survival, PTPMeg2
Background
Colorectal cancer, whose morbidity has been increasing in recent years, is one of the most common malignancies in the digestive system.1 Surgical resection combined with chemotherapy-based comprehensive therapy is the primary and standard therapy for colorectal cancer. However, the 5-year survival rate of patients with colorectal cancer remains poor at <30%.2 An important reason is that the pathogenesis is still largely unknown, and there have been no effective treatment targets for colorectal cancer until now. Therefore, it is necessary to identify important factors involved in regulating the progression of colorectal cancer.
Protein tyrosine phosphatases (PTPs) are a class of enzymes that catalyze the dephosphorylation of phosphotyrosyl residues of proteins.3 Previous studies have shown that PTPs participate in regulating the transduction of signaling pathways and are involved in mediating some cellular processes (including cell apoptosis and proliferation), and the roles of PTPs in cancer progression have been widely studied.4,5 Inhibition of PTPN1B prevents ErbB2-induced mammary tumorigenesis and lung metastasis, and PTPN12 protects against cell growth and transformation in triple-negative breast cancer.6,7 Overexpression of PTPN11 facilitated hepatocellular carcinoma growth and metastasis.8 Protein tyrosine phosphatase nonreceptor type 9 (PTPN9), also known as PTPMeg2 or Meg2, is a cytoplasmic PTPs that is widely expressed in different tissues, such as brain tissue, leukocytes, and endocrine cells.9,10 PTPN9 has been shown to be involved in regulating several signaling pathways and physiological processes. It has been reported that PTPN9 can dephosphorylate and deactivate the nerve growth factor receptor TrkA, the insulin receptors in hepatocytes, vascular endothelial growth factor receptor 2, and Stat3.11–14 PTPN9 regulates the growth and proliferation of erythroid cells and embryonic development.15,16 Moreover, in some types of human cancers, the expression of PTPN9 is reportedly dysregulated, while the effects of PTPN9 on different cancers are not always consistent. Previous studies have shown that PTPN9 inhibited cell proliferation and mitigated the progression of cancer as a tumor suppressor in breast cancer and hepatocellular carcinoma.17,18 Decreased expression of PTPN9 can enhance cell proliferation and migration in cervical cancer and gastric cancer.19,20 Conversely, PTPN9, whose expression is upregulated in tumor tissues, facilitated cell growth and metastasis in esophageal squamous cell carcinoma.21 Nevertheless, the roles of PTPN9 in colorectal cancer and the corresponding molecular mechanism are still unknown and need to be elucidated.
We carried out this study to explore the effects of PTPN9 on cell growth and survival in colorectal cancer. Our results demonstrated the inhibitory effects of PTPN9 on cell survival and elucidated the molecular mechanism involved, revealing an important mechanism that regulates the survival of colorectal cancer cells.
Materials and methods
Cell cultures
Human colorectal cancer cell lines (HCT116, HT-29, SW480, SW620, and T84) were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were routinely cultured in a 5% CO2 and 37°C incubator and were grown in DMEM (Sigma-Aldrich Co., St Louis, MO, USA) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), 2.5 mg/L amphotericin B (Thermo Fisher Scientific), and 105 U/L penicillin–streptomycin (Thermo Fisher Scientific). H2O2 (Sigma-Aldrich Co.) was applied to induce an apoptotic model, and Stattic (Selleck Chemicals, Houston, TX, USA) was used as an inhibitor of Stat3.
Tumor specimens
Tumor specimens used in the study (including 33 cases of colorectal cancer tissues and paired adjacent normal tissues) were acquired from 77 patients who underwent curative surgery in the First Affiliated Hospital of Harbin Medical University. Patients were not treated with any preoperative anticancer therapy. Ethical approval was obtained from the Ethics Committee of Harbin Medical University and written informed consent was obtained from each patient. Tumor specimens were collected in accordance with the Declaration of Helsinki.
Generation of PTPN9 knockdown and overexpression lentiviruses
The lentiviruses containing a human-specific short hairpin RNA (shRNA)-targeted human-specific short hairpin RNA (shRNA)-targeted (shPTPN9) sequence and a negative control shRNA (shControl) were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, People’s Republic of China). The PTPN9-specific shRNA sequence (forward 5′-GATCCGAAGGAAGGAAGGCATTGTACTTCCTGTCAGATACAATGCCTTCCTTCCTTCTTTTTG-3′; reverse 5′-AATTCAAAAAGAAGGAAGGAAGGCATTGTATCTGACAGGAAGTACAATGCCTTCCTTCCTTCGG-3′) or the negative control sequence (forward, 5′-GATCCGTCCGGATACGCATGAATCCCTTCCTGTCAGAGGATTCATGCGTATCCGGACTTTTTG-3′; reverse, 5′-AATTCAAAAAGTCCGGATACGCATGAATCCTCTGACAGGAAGGGATTCATGCGTATCCGGACGG-3′) were utilized. The PTPN9 overexpression lentivirus (containing the whole coding sequence; NM_002833) was also obtained from Shanghai GenePharma Co., Ltd. The empty lentivirus (GFP) was used as a negative control. A total of 1×105 cells (HCT116 and SW480 cells) were infected with 1×106 recombinant lentivirus-transducing units in the presence of 6 µg/mL polybrene (Sigma-Aldrich Co.).
Cell viability assessment
The CCK-8 assay was utilized to determine cell viability. Briefly, cells were split into a 96-well plate (~8×103 cells per well), where they remained for 24–72 hours. Then, 10 µL of CCK-8 reagent (CCK-8; Dojindo Laboratories, Kumamoto, Japan) was added to each well, and the cells were incubated for 2 hours at 37°C. Finally, the absorbance was read at 450 nm using a microplate reader (BioTek, Winooski, VT, USA) according to the manufacturer’s instructions.
Colony formation assay
The plate colony formation assay was used to detect cell growth. Cells were seeded at 1×103 cells/well in 6-well plates. Approximately 15 days later, the cells were washed twice with PBS and then stained with a crystal violet solution (0.5% crystal violet, 20% methanol) for 10 minutes. After removal of the crystal violet solution, the plates were washed three times with PBS. Then, colonies could be observed directly, and the images were photographed by a camera (Nikon Corporation, Tokyo, Japan).
Real-time PCR
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) and suspended in 20 µL of RNase-free water according to the manufacturer’s protocol. Next, we determined the RNA concentration with a NanoDrop 1000 spectrophotometer (Thermo, Wilmington, DE, USA). The amplification was performed by a fast real-time PCR system (7900HT, ABI, Foster City, CA, USA) using the SYBR Premix Ex Taq Kit (TaKaRa, Dalian, People’s Republic of China). A universal two-step RT-PCR cycling condition was used as follows: 95°C for 30 seconds followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. The primer sequences used in this study were as follows: PTPN9 forward 5′-GATGTGCTCCGTGCCATAGA-3′, reverse 5′-CCTGGCAGTAAAGAGGGCAA-3′; and β-actin forward 5′-TTGTTACAGGAAGTCCCTTGCC-3′, reverse 5′-ATGCTATCACCTCCCCTGTGTG-3′. The mRNAs were normalized to the endogenous control β-actin in triplicate, and expression levels were calculated by the 2−ΔΔCt method.
Western blot analysis
Cell lysates in RIPA buffer containing protein (40 µg) were loaded onto a 12% or 15% resolving gel for electrophoresis. Proteins were transblotted onto a nitrocellulose membrane (Whatman Inc., Florham Park, NJ, USA). After the membrane was blocked with 5% nonfat milk at 25°C for 1 hour, the membrane was incubated with the primary antibodies (Stat3, Cell Signaling Technology, cat. no. 9139, 1:500; Phospho-Stat3 [Tyr705], Cell Signaling Technology, cat. no. 9145, 1:500; Bcl-xL, Cell Signaling Technology, cat. no. 2764, 1:500; cytochrome c, Cell Signaling Technology, cat. no. 4280, 1:200; anti-β-actin, Cell Signaling Technology, cat. no. 3700, 1:2,000; anti-histone H3, Cell Signaling Technology, cat. no. 4499, 1:1,000) overnight. Subsequently, the blots were incubated with HRP-conjugated secondary antibody (Santa Cruz Biotechnology Inc., Dallas, TX, USA) and then reacted with an enhanced chemiluminescence substrate (Pierce, Rockford, IL, USA). Bands were detected by a Bio-Rad ChemiDoc XRS system, and the densitometric analysis of protein bands were analyzed by Image Lab software 4.1 (Bio-Rad Laboratories Inc., Hercules, CA, USA). The protein levels were normalized to their internal controls (β-actin or histone H3), and then the data were expressed as relative changes compared with the control group (GFP group or shControl group), respectively.
Nuclear fraction isolation
Cell nuclear proteins were separated with the NE-PER Nuclear and Cytoplasmic Extraction Reagents (CERs, cat. no.78835, Thermo Fisher Scientific, Rockford, IL, USA). Briefly, cells were collected and washed with ice-cold PBS for three times. Then, cells were centrifuged at 500× g for 3 minutes, and CER I was added to the pellet followed by vigorous vortex for 15 seconds and incubation on ice for 10 minutes. Next, the cells were treated with CER II and the tube was shaken vigorously for 10 seconds and incubated on ice for 1 minute. After centrifugation at 16,000× g for 5 minutes, the pellet was resuspended with nuclear extraction reagent and incubated for 40 minutes on ice with occasional vortex. The nuclear fraction was collected after centrifugation at 16,000× g for 10 minutes.
Caspase-3 activity and caspase-9 activity assays
Caspase-3 activity and caspase-9 activity were measured using the luminescent substrates Z-DEVD-aminoluciferin (Caspase-Glo 3/7 assay; Promega Corporation, Fitchburg, WI, USA) and Z-LEHD-aminoluciferin (Caspase-Glo 9 assay; Promega Corporation), respectively. Then, the experiment was conducted according to the manufacturer’s instructions. Briefly, cells were cultured in DMEM containing 10% FBS. Then, cells were harvested and lysed in the Caspase-Glo reagent provided in the kit, and the luminescence (substrate turnover) of triplicate samples was determined at times 0 and 1 hour using a Victor2 luminometer (PerkinElmer Inc., Waltham, MA, USA). Protein quantification was carried out using the Bradford method. Caspase activities were calculated as Δ luminescence units per microgram protein.
Statistical analyses
Data were represented as mean ± SEM. Statistical analysis was carried out on a GraphPad Prism version 5.0 for Windows (GraphPad Software Inc., San Diego, CA, USA). A one-way ANOVA followed by Tukey’s multiple comparison test or an unpaired Student’s t-test was used to evaluate statistical significance. P<0.05 was considered statistically significant.
Results
Expression of PTPN9 is downregulated in colorectal cancer
To determine whether PTPN9 is involved in regulating the progression of colorectal cancer, a public data set (GSE44861, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44861) was utilized to validate PTPN9 expression in colorectal cancer. As shown in Figure 1A, B, the expression of PTPN9 was significantly downregulated in cancer tissues (n=47) compared with the paired adjacent normal tissues. Moreover, we examined the expression of PTPN9 in 33 colorectal cancer tissues and their paired adjacent normal tissues. Similar results were obtained; PTPN9 expression was frequently repressed in colorectal cancer tissues compared with adjacent normal tissues (Figure 1C, D). In addition, we examined the relationship between PTPN9 expression and the overall survival rate in 77 cases of colorectal cancer tissues using Kaplan–Meier survival probability estimates. As shown in Figure 1E, the overall survival rate of patients with low expression of PTPN9 was significantly lower than that of patients with high expression of PTPN9. These results show that PTPN9, a potential molecular marker for predicting the prognosis of colorectal cancer, might participate in regulating the development of colorectal cancer as a tumor suppressor.
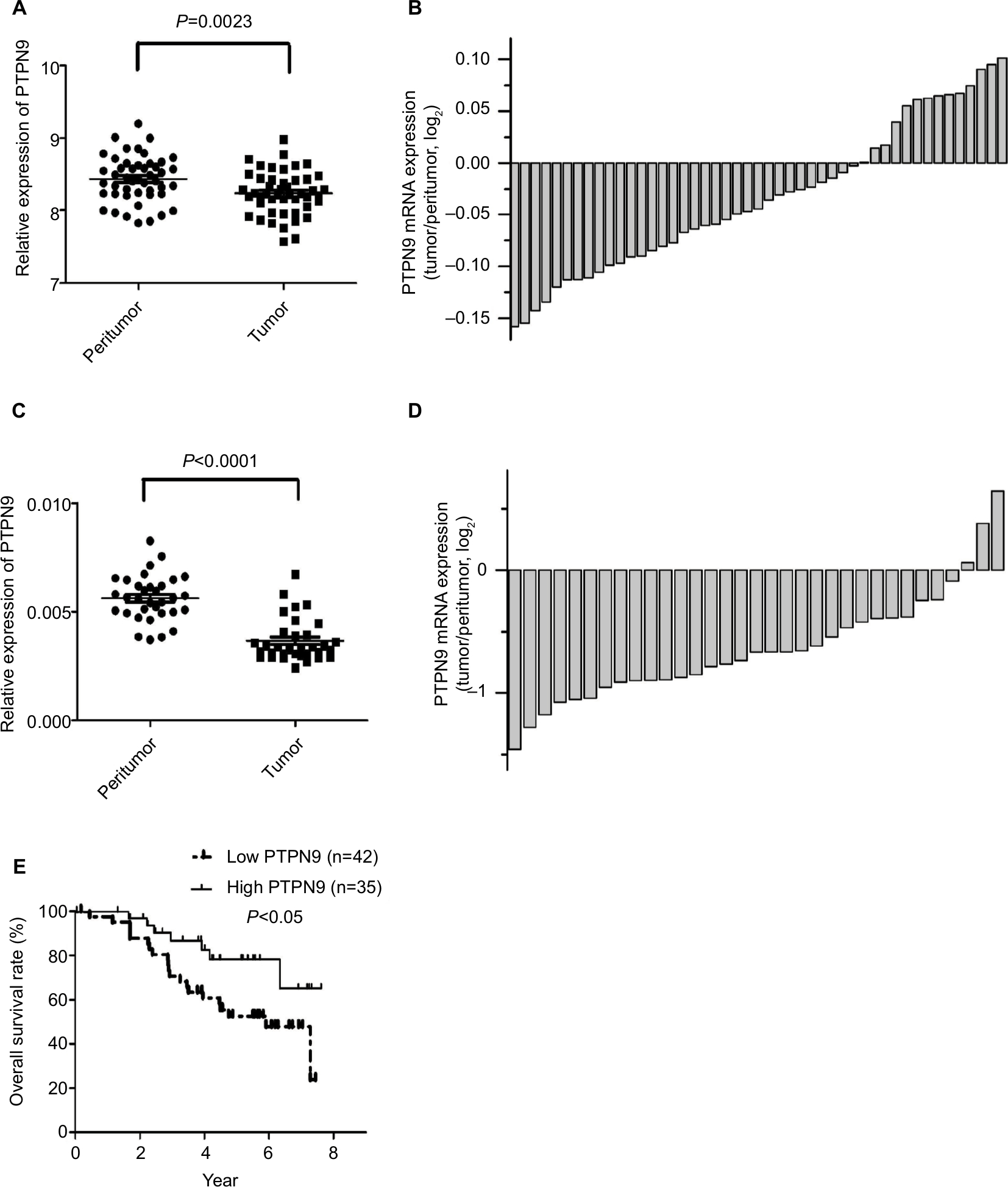 | Figure 1 PTPN9 expression is frequently downregulated in colorectal cancer tissues. Notes: (A and B) Expression of PTPN9 was downregulated in 47 samples of colorectal cancer tissues compared with the paired adjacent normal tissues. (C and D) Expression of PTPN9 in the colorectal cancer tissues was significantly downregulated compared with the paired adjacent normal tissues (n=33). (E) The overall survival rate of patients with low expression of PTPN9 was significantly lower than that of patients with high expression of PTPN9 in 77 samples of colorectal cancer tissues. A paired Student’s t-test (for comparisons of two groups) was used to evaluate statistical significance and Kaplan–Meier analysis was used for survival analysis. Abbreviation: PTPN9, protein tyrosine phosphatase nonreceptor type 9. |
Overexpression of PTPN9 inhibits cell growth and survival in colorectal cancer
To verify the precise role of PTPN9 in the survival of colorectal cancer cells, we established stable PTPN9-overexpressing cells. Endogenous expression of PTPN9 in five colorectal cancer cell lines was examined by real-time PCR (Figure 2A). The results showed that PTPN9 expression was relatively low in HCT116 cells, while endogenous expression of PTPN9 was high in SW480 cells. Overexpression efficiency was ascertained in HCT116 cells by real-time PCR (Figure 2B). As shown in Figure 2C, cell growth was significantly depressed by PTPN9 overexpression. The overexpression of PTPN9 mitigated the colony formation of cancer cells (Figure 2D). We then detected the effects of PTPN9 overexpression on cell apoptosis, a negative regulator of cell growth. The results showed that overexpression of PTPN9 led to increased caspase-9 activity and promoted the activation of caspase-3, and the release of cytochrome c into the cytoplasm was enhanced by PTPN9 overexpression (Figure 2E–G). These results imply that overexpression of PTPN9 mitigates cell growth and induces cell apoptosis in colorectal cancer.
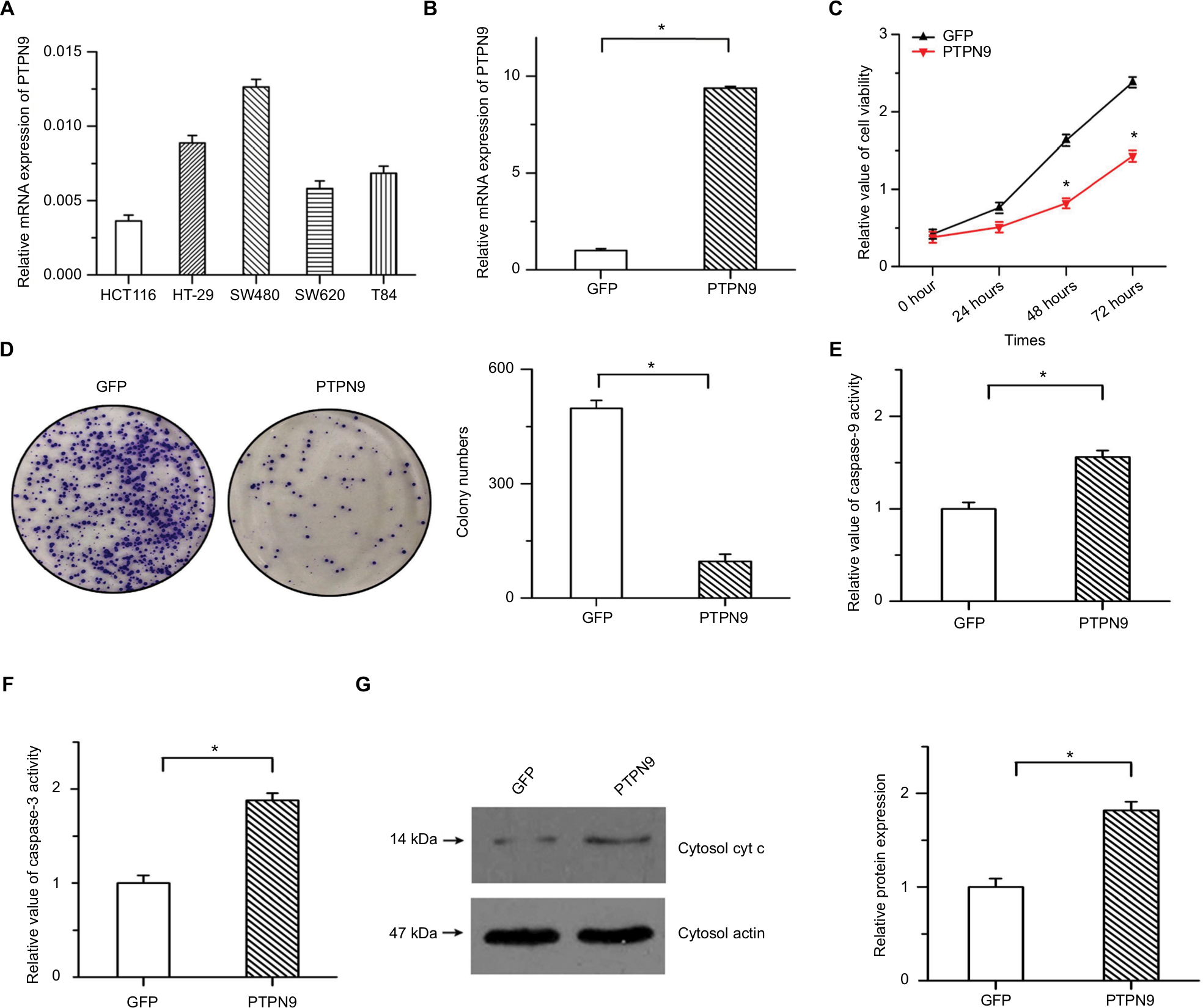 | Figure 2 Overexpression of PTPN9 inhibited cell growth and induced cell apoptosis in colorectal cancer cells. Notes: (A) Endogenous expression of PTPN9 was examined in five colorectal cancer cell lines. (B) Real-time PCR was utilized to validate the overexpression efficiency in HCT116. (C) Cell viability was repressed by overexpression of PTPN9 in HCT116 cells. (D) Overexpression of PTPN9 mitigated the colony formation of colorectal cancer cells. (E and F) Activities of caspase-9 and caspase-3 were increased by PTPN9 overexpression in HCT116 cells. (G) The release of cytochrome c into the cytoplasm was enhanced by PTPN9 overexpression. An unpaired Student’s t-test (for comparisons of two groups) was used to evaluate statistical significance (*P<0.05). Abbreviations: cyt c, cytochrome c; GFP, green fluorescent protein; PTPN9, protein tyrosine phosphatase nonreceptor type 9. |
PTPN9 knockdown represses cell apoptosis and promotes cell survival
We also knocked down the expression of PTPN9 in SW480 cells to explore its physiological function in cell survival. Real-time PCR was used to validate the knockdown efficiency (Figure 3A). As we all know the apoptotic rate of cancer cell is relatively low under normal condition. To examine the inhibitory roles of PTPN9 knockdown in cell apoptosis, we applied 100 µM H2O2 to induce an apoptotic model.22 The results of CCK-8 showed that the decreased cell viability and inhibition of colony formation induced by H2O2 were attenuated by PTPN9 knockdown (Figure 3B, C). Moreover, H2O2-induced increased caspase-9 and caspase-3 activities were reversed by PTPN9 knockdown (Figure 3D, E). PTPN9 knockdown protected against the release of cytochrome c into the cytoplasm under conditions of H2O2 treatment (Figure 3F). These results imply that knockdown of PTPN9 inhibits cell apoptosis in colorectal cancer.
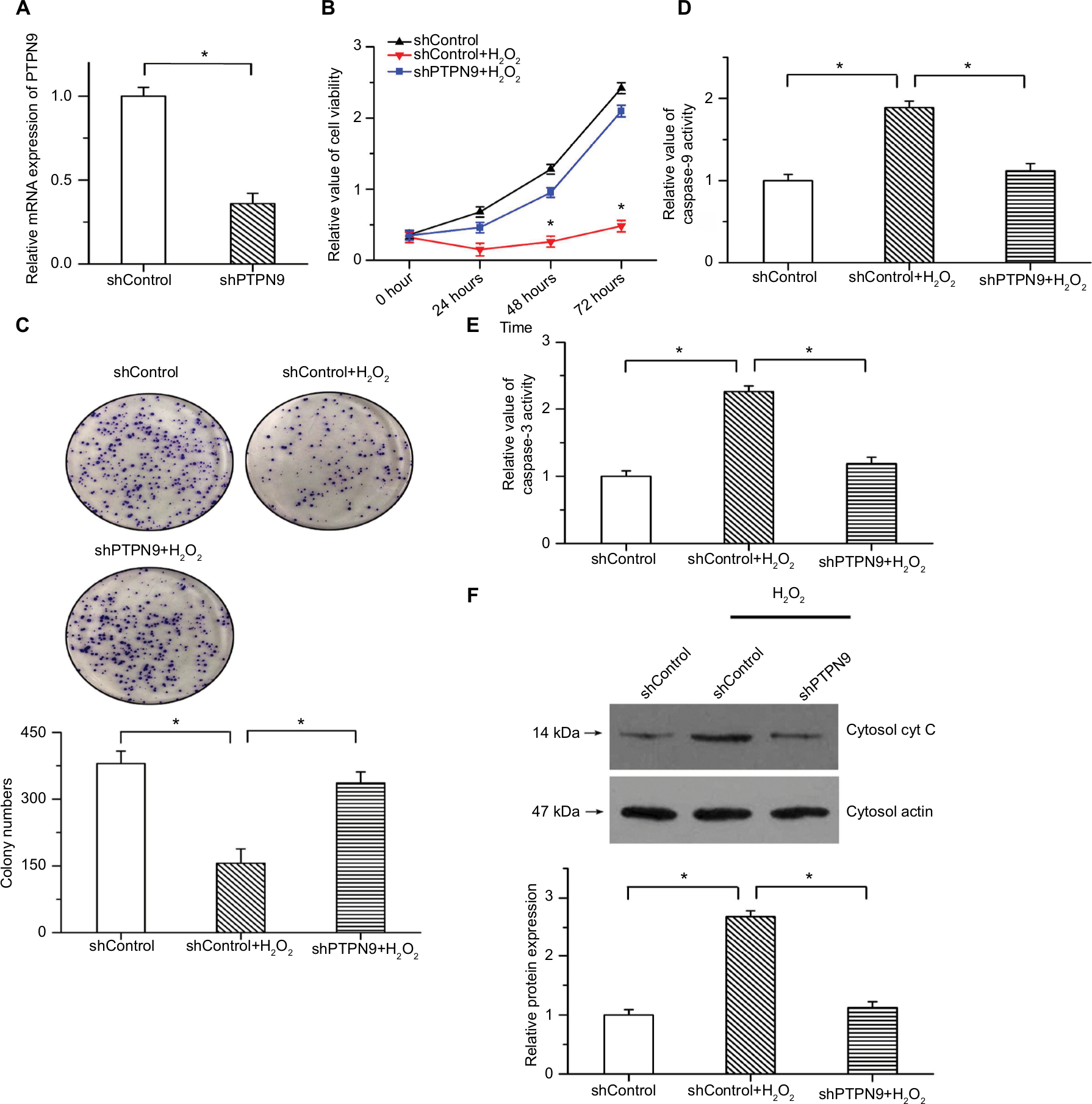 | Figure 3 PTPN9 knockdown repressed cell apoptosis and promoted cell survival. Notes: (A) Expression of PTPN9 was significantly depressed by PTPN9 shRNA in SW480 cells. (B) The decreased cell viability induced by H2O2 treatment was reversed by PTPN9 shRNA in SW480 cells. (C) H2O2-induced inhibition of colony formation was attenuated by PTPN9 knockdown. (D and E) Treatment with H2O2 led to the activation of caspase-9 and caspase-3, which was repressed by knockdown of PTPN9. (F) PTPN9 knockdown protected against the release of cytochrome c into the cytoplasm in SW480 cells. An unpaired Student’s t-test (for comparisons of two groups) or one-way ANOVA followed by Tukey’s multiple comparison test was used to evaluate statistical significance (*P<0.05). Abbreviations: cyt c, cytochrome c; PTPN9, protein tyrosine phosphatase nonreceptor type 9; sh, short hairpin. |
Activation of Stat3 is negatively regulated by PTPN9 in colorectal cancer cells
Previous studies have shown that Stat3 plays important roles in regulating cell survival and participates in mediating the development of colorectal cancer.23 PTPN9, as a potential regulator of Stat3, regulates the erythroid cell development of zebrafish and suppresses tumor growth in breast cancer by targeting Stat3.14,24 We then determined whether the regulatory roles of PTPN9 in cell apoptosis were mediated by Stat3 in colorectal cancer. Our results showed that PTPN9 overexpression repressed the phosphorylation of Stat3 but had no obvious effect on the expression of total Stat3 (Figure 4A). Conversely, the phosphorylation of Stat3 was significantly increased by knockdown of PTPN9 (Figure 4B). Moreover, translocation of Stat3 into the cellular nucleus was repressed by PTPN9 overexpression, while knockdown of PTPN9 had the opposite effect (Figure 4C, D).
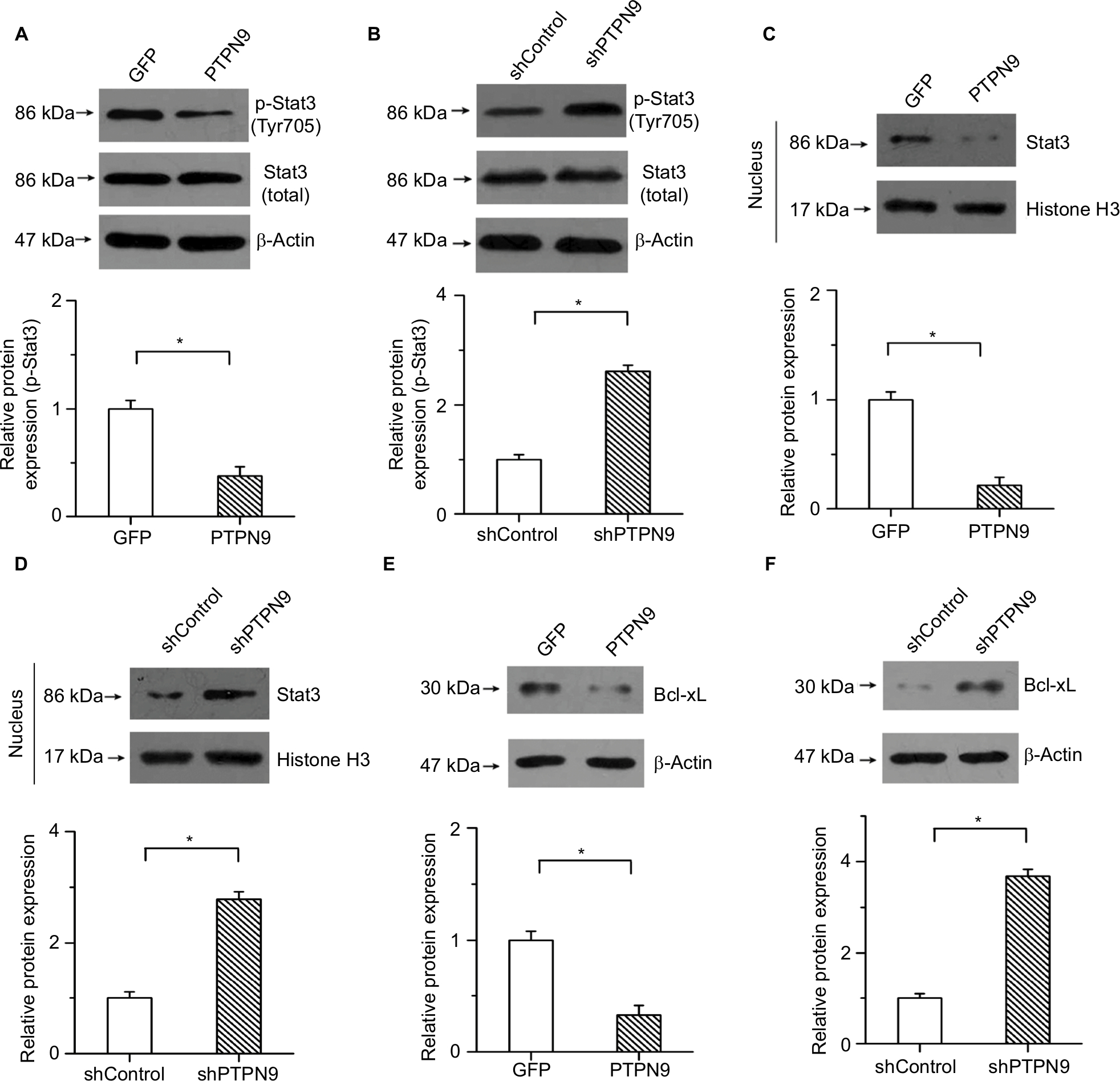 | Figure 4 PTPN9 negatively regulated the activation of Stat3 in colorectal cancer. Notes: Overexpression of PTPN9 significantly inhibited the phosphorylation of Stat3 (A), while the phosphorylation of Stat3 was increased by knockdown of PTPN9 (B). (C) PTPN9 overexpression depressed the nuclear translocation of Stat3. (D) Expression of Stat3 in the cellular nucleus was significantly enhanced by knockdown of PTPN9. (E) PTPN9 mitigated the protein level of Bcl-xL. (F) Knockdown of PTPN9 facilitated Bcl-xL expression. An unpaired Student’s t-test (for comparisons of two groups) was used to evaluate statistical significance (*P<0.05). Abbreviations: GFP, green fluorescent protein; PTPN9, protein tyrosine phosphatase nonreceptor type 9; sh, short hairpin; Stat3, signal transducer and activator of transcription 3. |
In addition, Bcl-xL, a familiar transcription target of Stat3, plays an important role in regulating the opening of the outer mitochondrial membrane channel and cell apoptosis. To demonstrate that PTPN9 facilitated cell apoptosis by affecting the transcriptional activation of Stat3, the expression of Bcl-xL was examined. Our results showed that PTPN9 mitigated the protein level of Bcl-xL and that Bcl-xL expression was enhanced by PTPN9 knockdown (Figure 4E, F). These results imply that PTPN9 negatively regulates the nuclear translocation and activation of Stat3 in colorectal cancer.
Effects of PTPN9 on cell apoptosis are carried out by regulating the activation of Stat3
To explore the roles of Stat3 in PTPN9 knockdown-inhibited cell apoptosis, we carried out the study under conditions of H2O2-induced cell apoptosis and examined whether knockdown of PTPN9-inhibited cell apoptosis was attenuated by the inhibition of Stat3. An inhibitor of Stat3 (Stattic) was utilized to block this pathway. As shown in Figure 5A, the phosphorylation of Stat3 induced by PTPN9 knockdown was obviously suppressed by 10 μM Stattic. The CCK-8 results showed that PTPN9 knockdown-increased cell viability was suppressed by Stattic in colorectal cancer cells (Figure 5B). Moreover, the inhibitory effects of PTPN9 knockdown on H2O2-induced activation of caspase-9 and caspase-3 were attenuated by the inhibition of Stat3; there was no significant difference between shControl + Stattic and shPTPN9 + Stattic (Figure 5C, D). Additionally, knocking down PTPN9 expression mitigated the release of cytochrome c into the cytoplasm, which was reversed by the inhibitor of Stat3 (Figure 5E). These results indicate that PTPN9 knockdown inhibits cell apoptosis and promotes cell survival, at least in part, by activating the Stat3 pathway.
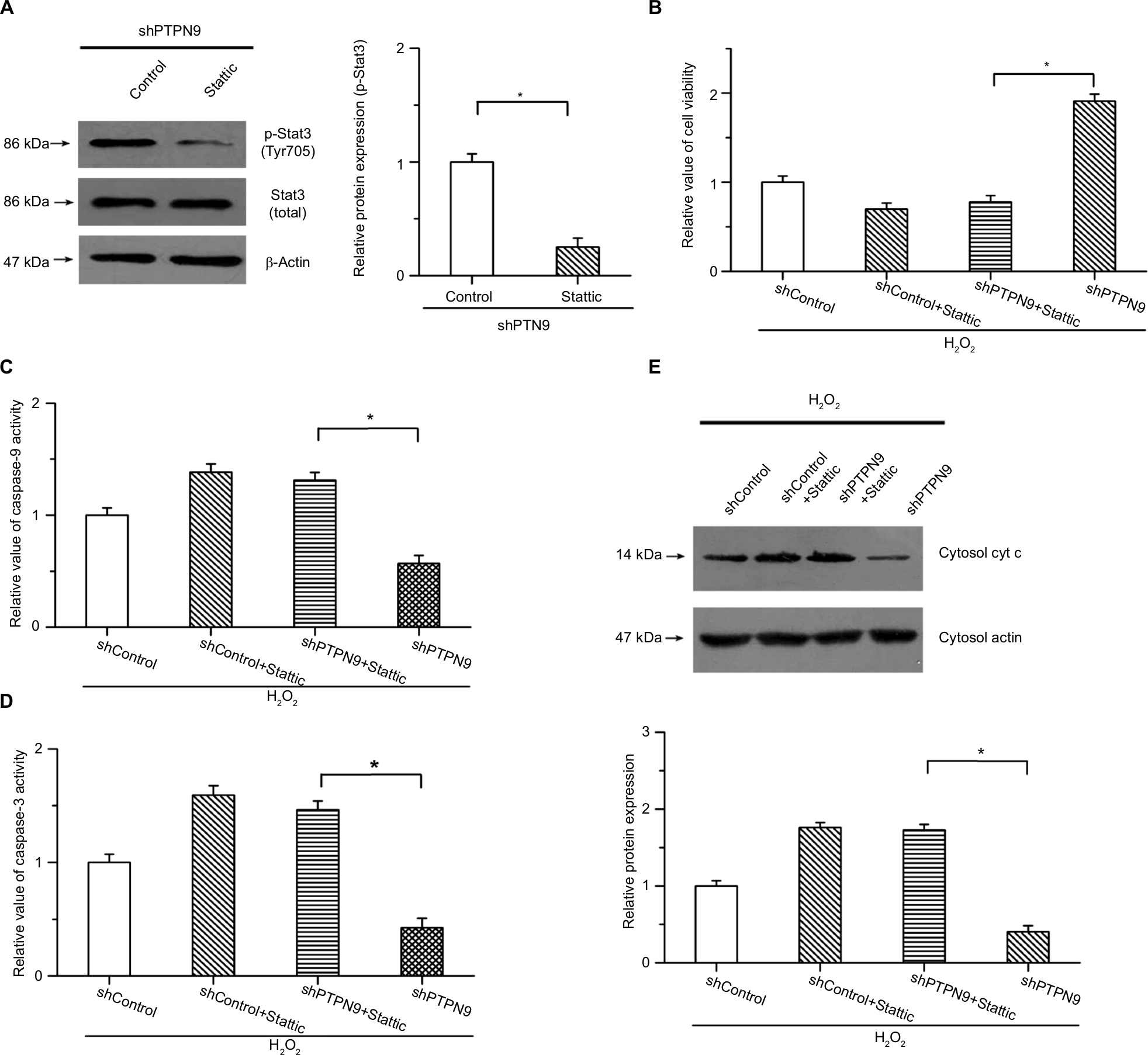 | Figure 5 The roles of PTPN9 in cell apoptosis were carried out by regulating the activation of Stat3. Notes: (A) The phosphorylation of Stat3 was obviously suppressed by Stattic in SW480 cells. (B) Increased cell viability induced by knockdown of PTPN9 was suppressed by Stattic. (C and D) Inhibitory effects of PTPN9 knockdown on H2O2-induced activation of caspase-9 and caspase-3 were mitigated by inhibition of Stat3 in SW480 cells. (E) PTPN9 knockdown repressed the release of cytochrome c into the cytoplasm, which was reversed by the inhibitor of Stat3. An unpaired Student’s t-test (for comparisons of two groups) or one-way ANOVA followed by Tukey’s multiple comparison test was used to evaluate statistical significance (*P<0.05). Abbreviations: cyt c, cytochrome c; PTPN9, protein tyrosine phosphatase nonreceptor type 9; sh, short hairpin; Stat3, signal transducer and activator of transcription 3. |
Discussion
Colorectal cancer is one of the most common types of cancer, especially in the Western world. Although surgical and medical therapy has been greatly improved, the 5-year survival rate of patients with colorectal cancer is still poor.1,2 An important reason for the largely unchanged overall mortality is the lack of effective therapeutic targets in colorectal cancer. Thus, exploring critical factors involved in regulating the progression of colorectal cancer and elucidating the regulatory mechanisms involved are necessary for reducing the development of this disease and improving the survival rate. In the present study, our results show that PTPN9 overexpression induces cell apoptosis and inhibits the growth of cancer cells by repressing the activation of Stat3, implying that PTPN9 functions as a tumor suppressor in the progression of colorectal cancer. To the best of our knowledge, this is the first study to validate the role of PTPN9 in colorectal carcinogenesis.
Accumulating evidence has implied that protein-tyrosine phosphatase enzymes, which catalyze the dephosphorylation of phosphotyrosyl residues of proteins, have direct effects on the delicate regulation of signaling pathway transduction and play important roles in modulating the progression of some cancers.25,26 Previous studies have shown that ErbB2-induced mammary tumorigenesis and lung metastasis are prevented by the inhibition of PTPN1B, and PTPN12 protects against cell growth and transformation by interacting with multiple oncogenic tyrosine kinases in triple-negative breast cancer.6,7 Upregulation of PTPN11 is frequently observed in hepatocellular carcinoma; higher levels of PTPN11 predict the poor prognosis of patients with hepatocellular carcinoma, and overexpression of PTPN11 facilitates hepatocellular carcinoma cell growth and metastasis.8 Furthermore, PTPN9, which is localized to the cytoplasm, has been reportedly dysregulated in some kinds of human cancers. However, its roles in different cancers are not always consistent. It has been reported that PTPN9 inhibits the development of breast cancer by repressing the phosphorylation of ErbB2 and EGFR or the tyrosine phosphorylation of Stat3, and low expression of PTPN9 in patients with breast cancer is associated with increased metastasis of cancer cells.14,17,18 In hepatocellular carcinoma, downregulation of PTPN9 promotes cell growth and tumor development.27 Decreased expression of PTPN9 caused by miR-613 upregulation facilitates cell growth and metastasis in cervical cancer.19 In gastric cancer cells, PTPN9 negatively regulated by miR-181a-5p inhibited cell proliferation and migration and decelerated tumor growth.28 In contrast, in esophageal squamous cell carcinoma, the expression of PTPN9 was positively correlated with tumor node metastasis stage and tumor classification. The survival time is shorter in patients with positive PTPN9 expression. PTPN9 knockdown suppresses cell proliferation and invasion.21 However, the roles of PTPN9 in colorectal cancer are still inconclusive and need to be elucidated. In our study, we found that PTPN9 expression was downregulated in patients with colorectal cancer. Overexpression of PTPN9 led to growth inhibition and proapoptotic changes, while PTPN9 knockdown had the opposite effects. These results indicate that PTPN9 inhibits cell survival and tumor progression in colorectal cancer.
Apoptosis is an important negative regulator of cell growth. It is believed that the fast and unlimited growth of cancer cells results from the dysfunction of cell apoptosis, and facilitating the apoptosis of cancer cells is considered an effective treatment for blocking the development of cancers. Stat3, as a crucial and well-known transcription factor, regulates various cellular physiological processes, such as cell proliferation, apoptosis, and differentiation.29,30 The oncogenic functions of Stat3 have been proven in various types of cancers, including colorectal cancer. Stat3 in the cytoplasm is phosphorylated and is then translocated into the cellular nucleus to activate the transcription of some genes, including Bcl-xL, cyclin D1, and c-Myc.31 In this study, we found that PTPN9 overexpression inhibited the phosphorylation of Stat3 and repressed its nuclear translocation. In contrast, the phosphorylation of Stat3 was significantly enhanced by PTPN9 knockdown, which was accompanied by the increased accumulation of Stat3 in the cellular nucleus. These results were consistent with previous studies showing that PTPN9 is a potential regulator that can inhibit the activation of Stat3.32 Furthermore, Bcl-xL, a transcription target of Stat3, plays an important role in regulating mitochondrial function and apoptosis. Downregulation of Bcl-xL expression leads to a decrease in mitochondrial membrane potential and reduced opening of the outer mitochondrial membrane channel. Then, cytochrome c is released into the cytoplasm and results in the activation of caspase-9 and caspase-3. Our results showed that PTPN9 overexpression inhibited the expression of Bcl-xL and that Bcl-xL expression was facilitated by knockdown of PTPN9, which was consistent with the results shown in Figures 2 and 3. These results imply that PTPN9 induces cell apoptosis and inhibits cell survival by regulating the activation of Stat3.
Conclusion
Our results have shown that PTPN9, which is frequently downregulated in the tissues of colorectal cancer, inhibits cell growth and induces cell apoptosis as a tumor suppressor. Moreover, the inhibitory effects of PTPN9 on cell survival are carried out by repressing the activation of Stat3. This finding demonstrates an important mechanism of regulating the growth of cancer cells and provides a new potential therapeutic target for reducing the progression of colorectal cancer.
Acknowledgments
This work was supported by grants from the China Postdoctoral Science Foundation, People’s Republic of China (grant no. .2012M520769 to DW), the Youth Fund of National Natural Science Fund, People’s Republic of China (grant no. 81600492 to DW), the Heilongjiang Province Department of Education Science and Technology Research Fund Project (grant no. 12541814 to ZC), and the Postdoctoral Scientific Research Start Fund Project in Heilongjiang Province (grant no. LBH-Q16219 to ZC).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014; 383(9927):1490–1502. | ||
Zhao S, Sedwick D, Wang Z. Genetic alterations of protein tyrosine phosphatases in human cancers. Oncogene. 2015;34(30):3885–3894. | ||
Labbé DP, Hardy S, Tremblay ML. Protein tyrosine phosphatases in cancer: friends and foes! Prog Mol Biol Transl Sci. 2012;106:253–306. | ||
Zhu Z, Liu Y, Li K, et al. Protein tyrosine phosphatase receptor U (PTPRU) is required for glioma growth and motility. Carcinogenesis. 2014;35(8):1901–1910. | ||
Julien SG, Dubé N, Read M, et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39(3):338–346. | ||
Sun T, Aceto N, Meerbrey KL, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144(5):703–718. | ||
Han T, Xiang DM, Sun W, et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J Hepatol. 2015;63(3):651–660. | ||
Gu M, Warshawsky I, Majerus PW. Cloning and expression of a cytosolic megakaryocyte protein-tyrosine-phosphatase with sequence homology to retinaldehyde-binding protein and yeast Sec14p. Proc Natl Acad Sci USA. 1992;89(7):2980–2984. | ||
Saito K, Williams S, Bulankina A, Höning S, Mustelin T. Association of protein-tyrosine phosphatase MEG2 via its Sec14p homology domain with vesicle-trafficking proteins. J Biol Chem. 2007;282(20):15170–15178. | ||
Zhang D, Marlin MC, Liang Z, et al. The protein tyrosine phosphatase MEG2 regulates the transport and signal transduction of tropomyosin receptor kinase A. J Biol Chem. 2016;291(46):23895–23905. | ||
Cho CY, Koo SH, Wang Y, et al. Identification of the tyrosine phosphatase PTP-MEG2 as an antagonist of hepatic insulin signaling. Cell Metab. 2006;3(5):367–378. | ||
Hao Q, Samten B, Ji HL, Zhao ZJ, Tang H. Tyrosine phosphatase PTP-MEG2 negatively regulates vascular endothelial growth factor receptor signaling and function in endothelial cells. Am J Physiol Cell Physiol. 2012;303(5):C548–C553. | ||
Su F, Ren F, Rong Y, et al. Protein tyrosine phosphatase MEG2 dephosphorylates signal transducer and activator of transcription 3 and suppresses tumor growth in breast cancer. Breast Cancer Res. 2012;14(2):R38. | ||
Xu MJ, Sui X, Zhao R, Dai C, Krantz SB, Zhao ZJ. PTP-MEG2 is activated in polycythemia vera erythroid progenitor cells and is required for growth and expansion of erythroid cells. Blood. 2003;102(13):4354–4360. | ||
Wang Y, Vachon E, Zhang J, et al. Tyrosine phosphatase MEG2 modulates murine development and platelet and lymphocyte activation through secretory vesicle function. J Exp Med. 2005;202(11):1587–1597. | ||
Yuan T, Wang Y, Zhao ZJ, Gu H. Protein-tyrosine phosphatase PTPN9 negatively regulates ErbB2 and epidermal growth factor receptor signaling in breast cancer cells. J Biol Chem. 2010;285(20):14861–14870. | ||
Du WW, Fang L, Li M, et al. MicroRNA miR-24 enhances tumor invasion and metastasis by targeting PTPN9 and PTPRF to promote EGF signaling. J Cell Sci. 2013;126(Pt 6):1440–1453. | ||
Li WT, Wang BL, Yang CS, Lang BC, Lin YZ. MiR-613 promotes cell proliferation and invasion in cervical cancer via targeting PTPN9. Eur Rev Med Pharmacol Sci. 2018;22(13):4107–4114. | ||
Ma X, Shi W, Peng L, Qin X, Hui Y. MiR-96 enhances cellular proliferation and tumorigenicity of human cervical carcinoma cells through PTPN9. Saudi J Biol Sci. 2018;25(5):863–867. | ||
Zhu J, Li H, Ma J, Huang H, Qin J, Li Y. PTPN9 promotes cell proliferation and invasion in Eca109 cells and is negatively regulated by microRNA-126. Oncol Lett. 2017;14(2):1419–1426. | ||
Ahmad KA, Iskandar KB, Hirpara JL, Clement MV, Pervaiz S. Hydrogen peroxide-mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells. Cancer Res. 2004;64(21):7867–7878. | ||
Zhao LC, Li J, Liao K, et al. Evodiamine induces apoptosis and inhibits migration of HCT-116 human colorectal cancer cells. Int J Mol Sci. 2015;16(11):27411–27421. | ||
Bu Y, Su F, Wang X, et al. Protein tyrosine phosphatase PTPN9 regulates erythroid cell development through STAT3 dephosphorylation in zebrafish. J Cell Sci. 2014;127(Pt 12):2761–2770. | ||
Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7(11):833–846. | ||
den Hertog J, Ostman A, Böhmer FD. Protein tyrosine phosphatases: regulatory mechanisms. Febs J. 2008;275(5):831–847. | ||
Hu B, Yan X, Liu F, et al. Downregulated expression of PTPN9 contributes to human hepatocellular carcinoma growth and progression. Pathol Oncol Res. 2016;22(3):555–565. | ||
Liu Z, Sun F, Hong Y, et al. MEG2 is regulated by miR-181a-5p and functions as a tumour suppressor gene to suppress the proliferation and migration of gastric cancer cells. Mol Cancer. 2017;16(1):133. | ||
Ray S, Zhao Y, Jamaluddin M, Edeh CB, Lee C, Brasier AR. Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell Signal. 2014;26(7):1445–1455. | ||
Wang P, Xue Y, Han Y, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344(6181):310–313. | ||
Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle. 2005;4(9):1131–1133. | ||
Kim M, Morales L, Jang IS, Cho YY, Kim DJ. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int J Mol Sci. 2018;19(9):E2708. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.