Back to Journals » Journal of Inflammation Research » Volume 10
Protein phosphatase 2Cδ/Wip1 regulates phospho-p90RSK2 activity in lesional psoriatic skin
Authors Rasmussen MK ![]() , Nielsen J, Kjellerup RB, Andersen SM, Rittig AH, Johansen C
, Nielsen J, Kjellerup RB, Andersen SM, Rittig AH, Johansen C ![]() , Iversen L
, Iversen L ![]() , Gesser B
, Gesser B ![]()
Received 29 September 2017
Accepted for publication 23 November 2017
Published 15 December 2017 Volume 2017:10 Pages 169—180
DOI https://doi.org/10.2147/JIR.S152869
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Mads K Rasmussen, Jakob Nielsen, Rasmus B Kjellerup, Stine M Andersen, Anne H Rittig, Claus Johansen, Lars Iversen, Borbala Gesser
Department of Dermatology, Aarhus University Hospital, Aarhus, Denmark
Objectives: P90 ribosomal S6 kinase (RSK) 1 and 2 are serine/threonine protein kinases believed to mediate proliferation and apoptosis via the extracellular signal-regulated kinases (ERK1/2) signaling pathway. Macrophage migration inhibitory factor (MIF) and epidermal growth factor (EGF) are activators of this pathway and are elevated in the serum of patients with psoriasis compared with healthy controls. Studies on COS-7 cell cultures have shown that protein phosphatase 2Cδ (PP2Cδ) decreases the activity of RSK2 following EGF stimulation. We therefore hypothesize that PP2Cδ regulates RSK2 activity in psoriasis.
Methods: In paired biopsies from nonlesional (NL) and lesional (L) skins, we analyzed the level of RSK1, 2 phosphorylation and the expression of PP2Cδ isoforms, integrin-linked kinase-associated serine/threonine phosphatase (ILKAP) and wild-type p53-induced phosphatase 1 (Wip1) by Western blotting, immunofluorescence and coimmunoprecipitation with monoclonal antibody for RSK2. The induction of Wip1 by MIF or EGF was studied in cultured normal human keratinocytes.
Results: The protein level of RSK1, 2 phosphorylated at T573/T577 was significantly increased in L compared with NL psoriatic skin, while phosphorylation at S380/S386 was reduced in L compared with NL psoriatic skin when assayed by Western blotting and immunofluorescence microscopy. ILKAP expression was significantly higher in L than in NL skin, whereas Wip1 was expressed in similar amounts but showed increased coimmunoprecipitation with RSK2 in L compared with NL psoriatic skin. In cultured normal human keratinocytes stimulated with MIF, Wip1 phosphorylation and Wip1 expression were increased after 24 hours, but not when costimulated with dimethyl fumarate (DMF). The increased coimmunoprecipitation of Wip1 with RSK2 was significantly induced by EGF or MIF activation at 24 hours and could be significantly inhibited by DMF or the ERK1/2 inhibitor PD98059.
Conclusion: The complex formation of Wip1 with RSK2 indicates a direct interaction reducing P-RSK2 (S386) activation in L skin and indicates that Wip1 has a role in the pathogenesis of psoriasis.
Keywords: P90 RSK1, 2, EGF, MIF, PP2Cδ/ILKAP, PP2Cδ/Wip1
Introduction
It has been suggested that extracellular signal-regulated kinase (ERK1/2) and mitogen- and stress-activated kinase (MSK1/2) are involved in the pathogenesis of psoriasis.1,2 These kinases are activated by macrophage migration inhibitory factor (MIF) and epidermal growth factor (EGF); both are overexpressed in serum from psoriasis patients.3–5
The p90 ribosomal S6 kinases, RSK1–3, are a family of serine/threonine kinases activated by phosphorylation through ERK1/2 signaling. The phosphorylation sites in RSK1–3 are highly conserved amino acid sequences.6–8 Phospho-RSK1 was induced by MIF and EGF in cultured human keratinocytes,9 but the expression of phospho-RSK1, 2 in psoriatic skin has not been tested.
RSK1, 2, and 3 isoforms are present in most tissues; they activate genes involved in proliferation and inactivate proapoptotic proteins like the BCL2-associated agonist of cell death.10,11 In resting cells, ERK1/2 is bound to the C-terminal site of RSK kinases, and upon stimulation, RSK1, 2 become phosphorylated at T573/T577. This is followed by autophosphorylation of homologous sites at S380/S386 in the middle region of RSK1, 2. The middle region cooperates with phosphoinositide-dependent protein kinase to dock and subsequently phosphorylate the N-terminal kinase domain of RSK1, 2 at S221/S227, respectively, which is needed for full RSK activation.12–15
Any inhibition of these steps will prohibit the effect of RSK on substrates and block the feedback reaction from the N-terminal kinase domain, controlling binding to P-ERK1/2.
Dimethyl fumarate (DMF) is a well-known oral drug for the treatment of psoriasis.16 DMF inhibits the induced phosphorylation of MSK1/2 and nuclear factor-κB (NF-κB)/p65 in cultured human keratinocytes, which is needed for cytokine transcription and is believed to be important in the pathogenesis of psoriasis.5,17,18 The structurally related MSK1/2 and RSK1, 2 are both activated by the ERK1/2 signaling pathway. In keratinocytes, preincubation with DMF inhibited the MIF- or EGF-induced activation of P-MSK1 and P-RSK1 at multiple sites, similar to the ERK1/2 inhibitor PD98059.9 These specific effects led to the inhibition of keratinocyte proliferation mediated by the induction of p-p53 (S15).
Members of the protein phosphatase family (PP2C) play a role in reversing protein kinase activation. PP2C is a highly conserved family of serine/threonine phosphatases.19–22 PP2Cα, PP2Cβ, and PP2Cδ/wild-type p53-induced phosphatase 1 (Wip1) isoforms inhibit the activation of Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) kinases and apoptosis. In contrast, the human PP2Cδ/integrin-linked kinase-associated serine/threonine phosphatase (ILKAP; 46 kDa) isoform inhibits proliferation and oncogenic transformation by suppressing integrin-linked kinase 1 (ILK1) activity, causing the activation of p38 MAPK and JNK/c-Jun kinases and apoptosis.21–23
The mammalian PP2Cδ/Wip1 isoform is induced by ionizing radiation in a p53-dependent manner.24,25 Wip1 (67 kDa) is considered an oncogene as it inactivates the tumor suppressor protein p53.26
In COS-7 cells transfected with empty vector or human influenza hemagglutinin-tagged RSK2 or myc-PP2Cδ/ILKAP plasmids, RSK2 activity was regulated by complex formation with PP2Cδ. The overexpression of PP2Cδ/ILKAP decreased RSK2 kinase activity by 50% by dephosphorylating p-RSK2 at S386. This complex formation could, in turn, be induced by phorbol 12-myristate 13-acetate through ERK1/2-mediated phosphorylation of PP2Cδ/ILKAP.20
Thus, the interaction of RSK2 and PP2Cδ/ILKAP appears to play an important role in the modulation of the ERK1/2 signaling cascade, at least in vitro, and could potentially play a role in psoriasis. To investigate this further, we therefore determined the protein expression of total and phosphorylated RSK1 and RSK2 and the expression of the PP2Cδ isoforms ILKAP and Wip1 in lesional (L) and nonlesional (NL) psoriatic skin. In addition, we used coimmunoprecipitation to investigate the complex formation between RSK2 and Wip1 and ILKAP.
The Regional Ethical Committee of Region Midtjylland, Denmark, approved the experiments with psoriatic patients (M20090102). A signed consent was obtained from each patient who donated skin samples, according to the Declaration of Helsinki principles.
Materials and methods
Cell cultures
Normal adult human keratinocytes were obtained by trypsinization of skin samples, as described before.27 Second-passage keratinocytes were trypsinated and seeded in 10 cm petri dishes, 4.5×106 cells/plate in keratinocyte basal medium (Gibco 17005; Thermo Fisher Scientific, Waltham, MA, USA), with only bovine pituitary growth hormone added from the supplement9 Gibco 37000-015, 50 µg/mL), gentamicin (Gibco 15710, 5 µg/mL), and 2% fetal calf serum (FCS; Gibco 160000 44). After 24 hours, the cells were either left alone or preincubated with DMF (140 µM) or 50 µM of PD98059 for 1 hour and stimulated with recombinant MIF (100 ng/mL; R&D Systems, Oxon, UK) or h-EGF (2 ng/mL; PeproTech, London, UK) for 24 hours. Alternatively, the cells were stimulated with MIF for 0 and 30 minutes and 2, 12, 24, 48, and 96 hours alone or after preincubation with 140 µM DMF (47967-25G-F; Sigma-Aldrich, Brøndby, Denmark).
DMF was dissolved as described before.5,9 The ERK1/2 inhibitor PD98059 and MSK1/RSK1 inhibitor R0-318220 from Calbiochem (La Jolla, CA, USA) were dissolved in dimethyl sulfoxide (DMSO; 1% vol/vol). No-drug controls contained an equal volume of DMSO.
Human breast cancer cell line MCF-7 from the European Collection of Cell Cultures (Wiltshire, UK) was cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco 61965-026) with 10% FCS, gentamicin (Gibco 15710), penicillin, streptomycin (Gibco 15140-148), and 2.5% Hepes until 60% confluence. Cells were trypsinated and seeded in 10 cm petri dishes, 4.0×106 cells/plate in DMEM, supplemented with pituitary growth hormone (50 µg/mL), 2% FCS, and 2.5% Hepes.
Tissue material from donors
Keratome and punch skin biopsies were obtained from volunteers with moderate-to-severe plaque psoriasis. The biopsies were collected during two different time periods. In the first group, 5 patients’ paired biopsies were analyzed by Western blotting (Figures 1A and B and 2A and B) and immunohistochemistry (results similar to those seen in Figure 3A and B). Biopsies from the second group of 6 patients were analyzed by immunohistochemistry (Figure 3A and B), Western blotting (Figure 4A and C) and immunoprecipitation (Figure 5A–C).
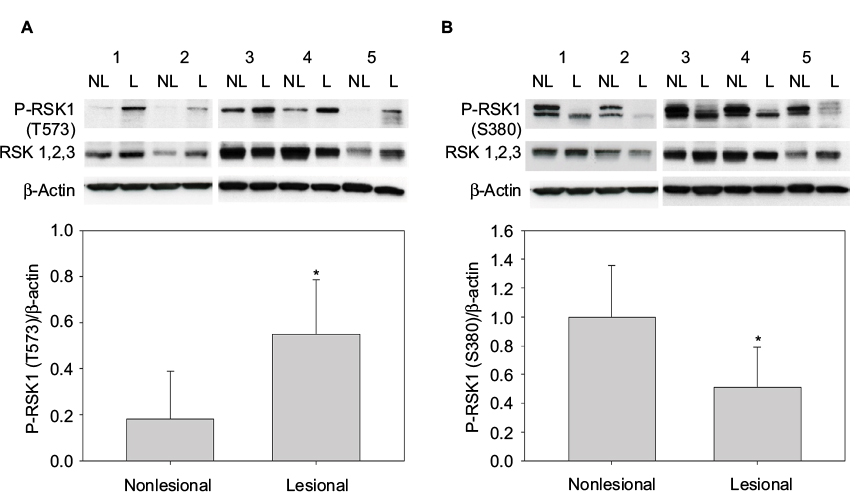 | Figure 1 P-RSK1 (T573) and P-RSK1 (S380) are differentially expressed in L psoriatic skin compared with NL psoriatic skin. Western blot analyses of whole cell extracts from paired biopsies, Patients 1–5 from NL and L psoriatic skin with anti-P-RSK1 (T573) p*=0.006 (A) and anti-P-RSK1 (S380) p*=0.049 (B) antibodies, reprobed with anti-RSK1, 2, and 3 and anti-β-actin. P-RSK activity was normalized to β-actin. Abbreviations: L, lesional; NL, nonlesional; RSK, p90 ribosomal S6 kinase. |
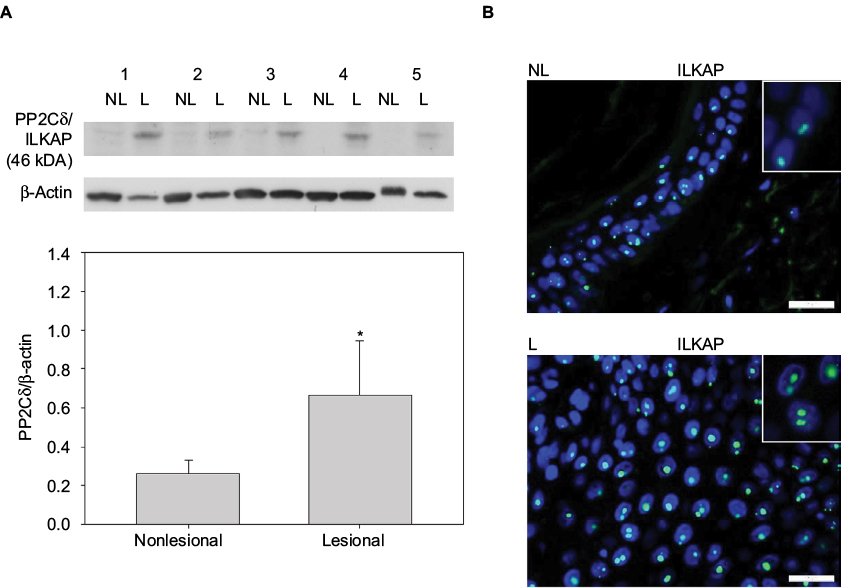 | Figure 2 (A) The expression of ILKAP is higher (p*=0.008) in L than in NL psoriatic skin. Western blot analyses of whole cell extracts from paired biopsies, Patients 1–5 from NL and L skin with anti-PP2Cδ/ILKAP antibodies. The expression of ILKAP was normalized to β-actin. (B) Nuclear staining of keratinocytes in the epidermis from NL and L skin tested with anti-PP2Cδ/ILKAP and developed with Alexa Flour 488 (green) and nuclear staining DAPI (blue) merged (bars 25 µm). Abbreviations: ILKAP, integrin-linked kinase-associated serine/threonine phosphatase; L, lesional; NL, nonlesional; PP2Cδ, protein phosphatase 2Cδ. |
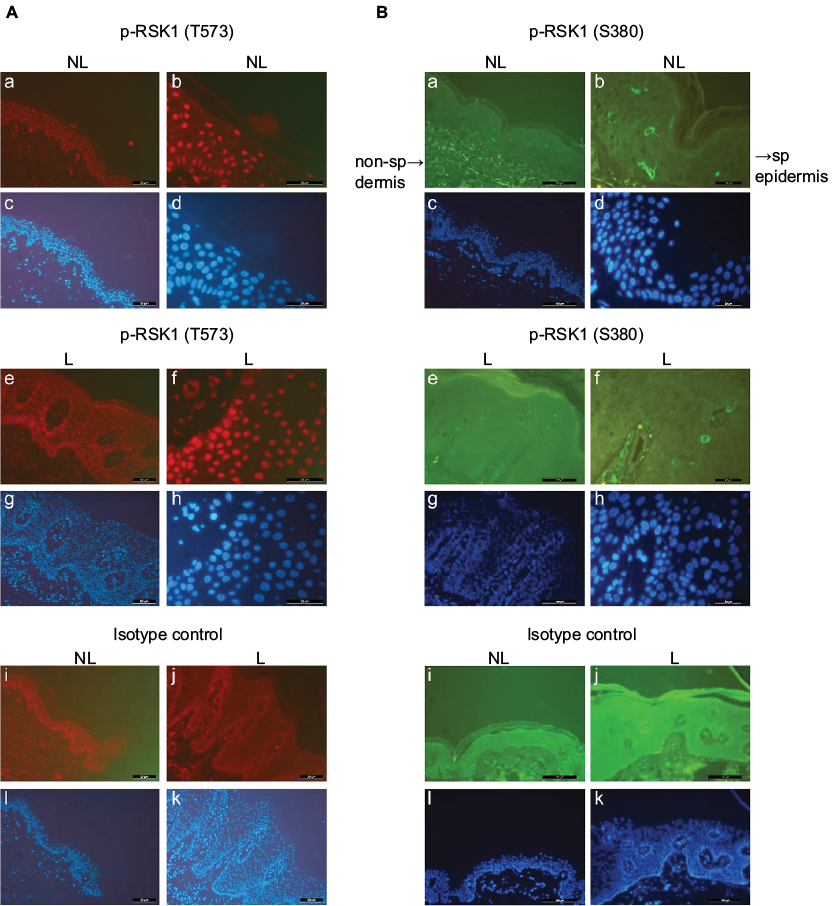 | Figure 3 (A) Nuclear staining of keratinocytes in biopsies from NL and L skin by P-RSK1 (T573) antibodies, NL (a, b) and plaque-type L (e, f) psoriatic skin, isotype control Ab (i, j) and Alexa Flour 594 (red) (bars 50 µm; resp. 20 µm). (B) Cytosolic staining of keratinocytes in NL (a, b) and L (e, f) skin by P-RSK1 (S380) antibodies, and Alexa Flour 488 (green), nuclear staining DAPI (blue) (c, d, g, h, l, k) (bars 100 µm a, c, e, g, i, l; resp. 20 µm b, d, f, h, j, k). Abbreviations: DAPI, 4’,6-diamidine-2’-phenylindole; L, lesional; NL, nonlesional; non-sp, nonspecific; RSK, p90 ribosomal S6 kinase; sp, specific. |
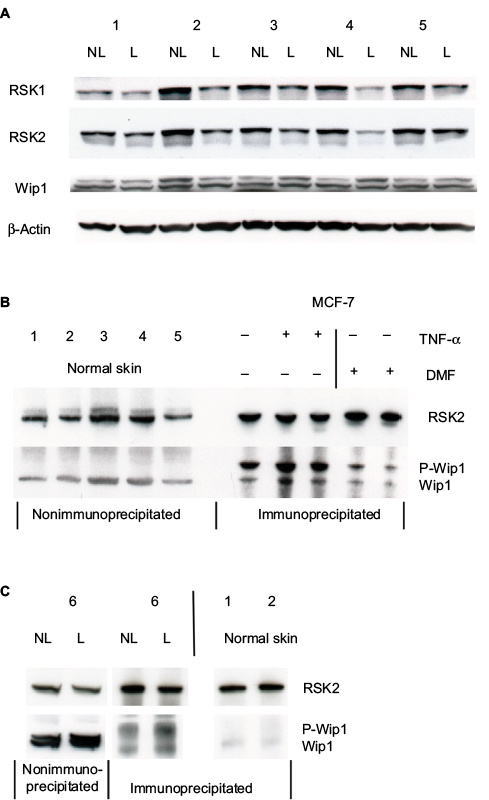 | Figure 4 (A) Whole cell extracts (50 µg proteins) from paired biopsies NL and L skin were analyzed for the expression of Wip1 by Western blotting with antibodies for Wip1 and reprobed with anti-RSK2 or anti-RSK1 and anti-β-actin. (Patients 1–5). (B) Whole cell extracts from healthy normal skin (50 µg proteins), n=5, were tested for the level of Wip1 by Western blotting. MCF-7 cells were either left alone or preincubated with DMF (140 µM) for 1 hour and stimulated with TNF-α (10 ng/mL) for 24 hours. Protein extracts were immunoprecipitated with anti-RSK2 antibody and tested by Western blotting with anti-Wip1 and anti-RSK2 Ab. Normal skin samples expressed Wip1, while MCF-7 cells expressed P-Wip1/Wip1, which was inhibited by DMF. (C) Cell extracts were analyzed as before. The level of immunoprecipitated P-Wip1/Wip1 in psoriatic skin compared with normal skin was tested. Abbreviations: DMF, dimethyl fumarate; L, lesional; NL, nonlesional; RSK, p90 ribosomal S6 kinase; TNF-α, tumor necrosis factor-α; Wip1, wild-type p53-induced phosphatase 1. |
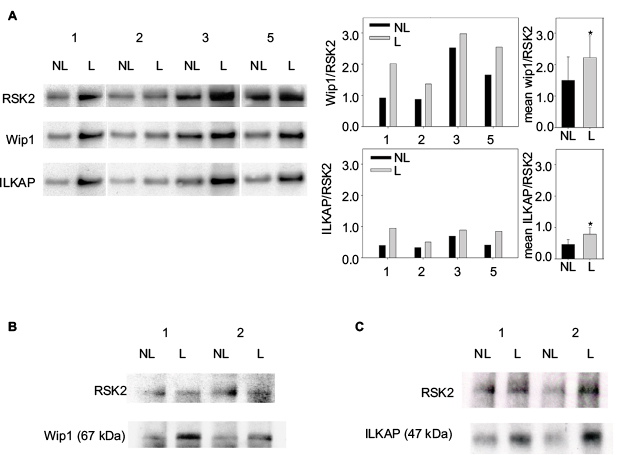 | Figure 5 (A) RSK2 forms complexes with both Wip1 and ILKAP in psoriatic skin. Proteins from NL and L skin from Patients 1–3 and 5 were immunoprecipitated with anti-RSK2 antibodies and analyzed by Western blotting with anti-Wip1 and anti-ILKAP. The ratios of the immunoreactive Wip1 to RSK2 in NL (black bar) and in L skin (grey bar) are shown in graphs. The mean ratios of Wip1 to RSK2 (p*=0.0168) and mean ILKAP to RSK2 (p*=0.033) were significant (single measurements from 4 patients) L compared with NL skin. (B) Proteins from 2 new patients were immunoprecipitated with anti-Wip1 (H-300: sc-20712) antibody and tested with anti-Wip1 epitope center AA 182-212 (AP12875c) and anti-RSK2. (C) Similarly, proteins from 2 new patients were immunoprecipitated with anti-ILKAP antibodies and tested with anti-ILKAP and anti-RSK2 antibodies. Abbreviations: ILKAP, integrin-linked kinase-associated serine/threonine phosphatase; L, lesional; NL, nonlesional; RSK, p90 ribosomal S6 kinase; Wip1, wild-type p53-induced phosphatase 1. |
The third group of 7 normal healthy donors who had donated keratome biopsies were analyzed by Western blotting and immunoprecipitation (Figure 4B and C). Each participant with psoriasis was untreated for at least 1 month prior to the time of biopsy.
Immunohistochemistry
Punch biopsies from NL and L plaque-type psoriatic skin were obtained as described before.1,2 Then, 4-µm sections were heated in 10 mM sodium citrate buffer for 10 minutes in a microwave oven (750 W). The sections were further permeabilized in Tris-buffered saline (TBS) with 1% Triton X-100 for 10 minutes and washed with TBS with 0.3% Triton X-100. Nonspecific binding sites were blocked with Image-iT FX signal enhancer (136933; Thermo Fisher Scientific, Waltham, MA, USA). Tissue sections were incubated with the primary antibody dissolved in TBS med 0.3% Triton X-100 (T-9284; Sigma-Aldrich) with 5% of goat nonimmune serum (DAKO X0907) at 4°C overnight. Antibodies were rabbit anti-P-RSK1 (T573) or P-RSK1 (S380; Cell Signalling Technology, Beverly, MA, USA), isotype control (sc-2027) normal rabbit IgG, and goat anti-rabbit Alexa Flour 594 (A-11037) or Alexa Flour 488 (A 31628; Invitrogen, Molecular Probes®). The samples were mounted in ProLong anti-fade reagent with 4’,6-diamidine-2’-phenylindole (DAPI; P36935; Invitrogen, Roskilde, Denmark). Samples were viewed by using an epifluorescence microscope (Leica Microsystems, Wetzlar, Germany).
In other experiments, tissue sections were incubated with rabbit anti-ILKAP (ab70019; Abcam, Cambridge, UK) at 4°C overnight, developed with goat anti-rabbit Alexa Flour 488 (A-31628; Invitrogen) for 1 hour, and mounted with DAPI.
Immunoprecipitation
Immunoprecipitation was performed using protein isolated from keratome biopsies or cultured human keratinocytes, extracted in 1× lysis buffer (#9803; Cell Signalling Technology) with added 22 µL/mL protease inhibitor cocktail (ethylenediaminetetraacetic acid-free complete; Hoffman-La Roche Ltd., Basel, Switzerland), 10 µL/mL of 100 nM phenylmethylsulfonyl fluoride (PMSF)/mL, and nuclear lysis benzonase (1 µL/100 µL).5 An amount of protein (500 µg) in 500 µL of lysis buffer was added to 2 µg of mouse monoclonal RSK2 antibody (Santa Cruz, CA, USA) and incubated overnight at 4°C on a rotating table. Then, 40 µL of the suspended volume of Protein A/G Plus-Agarose (Santa Cruz, CA, USA) was added at 4°C for 4 hours. The pellets were collected by centrifugation at 2500 rpm for 5 minutes, washed 4 times with sterile PBS, and resuspended in 40 µL of sample buffer containing 10 mM dithiothreitol and 2.5% sodium dodecyl sulfate (SDS).9 Samples were boiled for 2–3 minutes and centrifuged to pellet the agarose beads. Then, they (20 µL) were separated on gels.
Western blotting
Cell extracts from keratome biopsies or cell cultures were prepared in 1× lysis buffer supplemented with 22 µL/mL of protease inhibitor and 10 µL/mL of 100 mM PMSF/mL buffer. Equal loads of protein (50 µg) were added to 3× SDS lysis buffer and separated on SDS-polyacrylamide gel electrophoresis (PAGE) 8%–16% Tris–Glycine Gels (Invitrogen) or on NuPAGE 4%–12% Bis-Tris Gel, (Novex; Life Technology; Thermo Fisher Scientific). Proteins were blotted and incubated as described before.9 Equal load was controlled by anti-β-actin (A-1978; Sigma-Aldrich). Densitometric analysis of the band intensity was performed on a scanner (Epson PERFECTION V75 PRO) and quantitated by Jelly Quant (Mads Rasmussen 2004, Aarhus, Denmark).
The following antibodies were used: anti-P-RSK1 (T573) and (S380), anti-RSK1, 2, 3, and HRP anti-rabbit Ab #7074 (Cell Signalling Technology); rabbit anti-RSK1 and monoclonal anti-RSK2 (E-1), HRP anti-mouse (sc-2060), and HRP anti-rabbit (sc-2054); immunoprecipitation: monoclonal anti-RSK2 (E-1): sc-9986 and rabbit anti-Wip1 (H-300; sc-20712) epitope C-terminus AA 306-605 (Santa Cruz Biotech, Santa Cruz, CA, USA); rabbit anti-PP2Cδ/ILKAP (ab700019; Abcam); HRP-conjugated goat anti-rabbit #7074, HRP-conjugated horse anti-mouse #7076 (Cell Signalling Technology); and rabbit anti-PP2Cδ/Wip1:PPM1D Center AA 182-212 (Cat. No. AP13875c) that does not recognize P-Wip1 (Abgent Europe, London, UK).
Statistical methods and calculation of the experimental data
Antibody binding analyzed by Western blotting and densitometry analysis of immune reactive band intensity were carried out as described previously.5 Graphs were made with Sigma Plot Version 11. STATA was used to test for normal distribution and for performing Student’s t-tests. A probability of p<0.05 was regarded as statistically significant.
Results
P-RSK1 (T573) and P-RSK1 (S380) are differentially expressed in L compared with NL psoriatic skin
Whole cell extracts from paired biopsies from 5 patients (Patient No. 1–5) of NL and L psoriatic skin were analyzed by Western blotting using anti-P-RSK1 (T573) or anti-P-RSK1 (S380) antibodies and reincubated with an anti-RSK1-3 antibody. P-RSK1 (T573) immunoactive bands were normalized to β-actin. Data were represented as mean ± standard deviation (SD) from 5 patients. As seen in Figure 1A, anti-P-RSK1 (573) and anti-P-RSK1 (S380) antibodies recognize a conserved area expressed in all of RSK1, 2, and 3; therefore, an anti-RSK1, 2, or 3 antibody was used.8
The level of P-RSK1 (T573) was significantly higher (p*=0.006) in paired samples of L skin than in NL psoriatic skin (Figure 1A). In contrast, the P-RSK1 (S380) immunoactive bands normalized to β-actin showed that P-RSK1 (S380; n=5) was significantly lower (p*=0.049) in L skin than in NL skin (Figure 1B). In order to localize P-RSK1 (T573) by immunostaining, sections of paraffin-embedded skin from NL and L skin were incubated with anti-P-RSK1 (T573) antibodies and anti-rabbit Alexa Flour 594 (red color) and DAPI (blue color). This showed nuclear staining of keratinocytes in the epidermis (Figure 3A). The number of stained nuclei (manual counting) in (20×20) µm area/patients was higher in L skin (L, 10.2±1.2) than in NL skin (NL, 7.8±1.2). The number of cytosolic-stained cells in the epidermis with anti-P-RSK1 (S380) and anti-rabbit Alexa Flour 488 (green color) in (100×150) µm area/patients was lower in L skin (L, 2±0) than in NL skin (NL, 5±1; Figure 3B). One of four patients is shown. Isotype control showed that immunostaining with anti-P-RSK1 (S380) Ab was nonspecific in the dermis and specific in the epidermis.
Expression of ILKAP is higher in L than in NL psoriatic skin
To investigate the presence of ILKAP, paired biopsies from NL and L skin were analyzed by Western blotting using anti-ILKAP antibodies and anti-β-actin for normalization. Data from 5 patients were represented as mean values ± SD. These values show that the level of ILKAP (46 kDa) expression was significantly higher (p*=0.008) in L skin than in NL skin (Figure 2A).
In order to localize ILKAP by immunostaining, paired biopsies from NL and L skin were incubated with anti-ILKAP, anti-rabbit Alexa Flour 488 (green), and DAPI. ILKAP staining in the nuclei in L skin was 53 dots/equal area compared with NL skin with 32 dots/equal area (Figure 2B).
Wip1 was expressed at a similar level in NL and L psoriatic skin
Whole cell protein extracts (50 µg/lane) from paired biopsies were prepared as before and analyzed by Western blotting using anti-Wip1 antibodies. The blots were reincubated with a monoclonal anti-RSK2 or with polyclonal anti-RSK1 and anti-β-actin. This showed that the level of Wip1 (67 kDa) was not significantly different in NL compared with L skin when quantitated to β-actin (Figure 4A). Wip1 was expressed as a double band in psoriatic skin. Because RSK1, 2 but not RSK3 are involved in the regulation of proliferation, we had to confirm the amount of RSK1 and RSK2.10 The levels of RSK1 and RSK2 were not equal in NL and L skin, as seen before.
The level of Wip1 in healthy normal skin biopsies was tested. Because MCF-7 breast cancer cells have previously been shown to express constitutive high levels of Wip1,24,26 we used these cells as a positive control and tested for association with RSK2 (Figure 4B).
MCF-7 cells were preincubated with vehicle or DMF (140 µM) for 1 hour and stimulated with tumor necrosis factor-α (TNF-α; 10 ng/mL) for 24 hours. Whole cell protein extracts were immunoprecipitated with anti-RSK2 Ab and analyzed by Western blotting using anti-Wip1 and anti-RSK2 Ab. Wip1 was expressed as a single band in normal, healthy skin, at the molecule weight of Wip1 when compared with MCF-7 cells expressing P-Wip1/Wip1 (antibody recognizing both forms) in complex with RSK2. Preincubation with DMF strongly reduced the complex formation of P-Wip1/Wip1 with RSK2 (Figure 4B). We tested the Wip1 and RSK2 complex formation in psoriatic and normal skin biopsies. As before, proteins were immunoprecipitated with anti-RSK2 antibody and analyzed by Western blotting (Figure 4C). The analysis showed that there was a complex formation of Wip1 with RSK2 in psoriatic skin, but not in normal skin samples.
RSK2 forms more complexes with both Wip1 and ILKAP in L psoriatic skin than in NL skin
Complex formation between PP2Cδ/ILKAP and RSK2 was previously identified in immunoprecipitates from COS-7 cells.20 We therefore investigated the complex formation between RSK2 and Wip1 or ILKAP in psoriatic skin. Whole cell protein extracts (see “Materials and methods” section) from NL and L skin (n=4) were immunoprecipitated with monoclonal antibodies for RSK2 and analyzed with antibodies for Wip1, ILKAP, and RSK2 by Western blotting. The expression of Wip1 and ILKAP was normalized against the expression of RSK2. The ratio of Wip1 to RSK2 indicated a significant association of Wip1 in complex with RSK2 in L skin (grey graphs) compared with NL skin (black graphs; p*=0.0168). The ratio of ILKAP to RSK2 indicated that there was also significant complex formation between ILKAP and RSK2 in L skin (p*=0.033) compared with NL skin (Figure 5A).
In order to confirm the association between Wip1 or ILKAP and RSK2, proteins from NL and L skin from two new patients were immunoprecipitated with anti-Wip1 Ab (H-300: sc-20712)28 and tested with anti-Wip1 Ab (AP12876c; which does not recognize P-Wip1) and anti-RSK2 Ab. This confirmed that the amount of Wip1 associated with RSK2 was higher in L skin than in NL skin (Figure 5B). Immunoprecipitation with anti-ILKAP Ab and testing with anti-ILKAP and anti-RSK2 Ab confirmed that the amount of ILKAP associated with RSK2 was higher in L than in NL skin (Figure 5C).
Activation and new synthesis of Wip1 are regulated by MIF and DMF in normal human keratinocytes
The induction of P-Wip1/Wip1 was studied in a time-dependent manner. Keratinocytes were preincubated with either vehicle or DMF (140 µM) for 1 hour and then stimulated with MIF (100 ng/mL) for 30 minutes or 2 or 24 hours (Figure 6A). Alternatively, keratinocytes were stimulated with MIF (100 ng/mL) for 12, 24, 48, and 96 hours (Figure 6B). Whole cell extracts were analyzed with anti-Wip1 Ab (H-300: sc20712)28 and Western blotting. MIF stimulation induced P-Wip1 and Wip1 at 24 hours, which was inhibited by DMF (Figure 6A). MIF stimulation induced a second new synthesis of Wip1 at 48 hours, which was inhibited by DMF (Figure 6B). Because MIF stimulation has been shown to inhibit the expression of p-P53 (S15) from 24 to 96 hours, we tested the effect of DMF on the new synthesis of Wip1. Preincubation with DMF reduced the new synthesis of Wip1 (Figure 6B) and simultaneously activated the expression of p-P53 (S15), as seen before.9
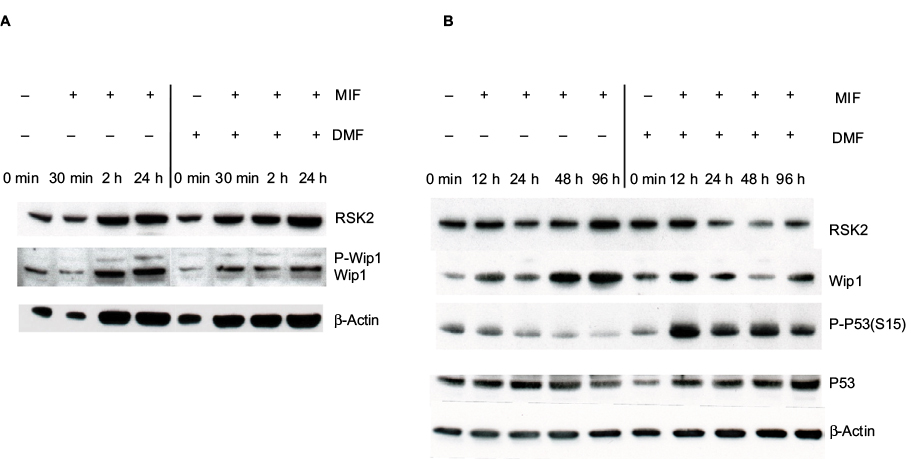 | Figure 6 (A) MIF stimulation activated P-Wip1 at 24 hours and the new synthesis of Wip1 at 48 hours, which were inhibited by costimulation with DMF. Keratinocytes were preincubated with vehicle or DMF (140 µg/mL) and stimulated with MIF (100 ng/mL) at 30 minutes and 2 and 24 hours. (B) Keratinocytes were preincubated with vehicle or DMF (140 µM) and stimulated with MIF (100 ng/mL) for 12, 24, 48, and 96 hours. Whole cell extracts were analyzed by Western blotting with anti-Wip1 and reproved with anti-p-P53 (S15), anti-p53, and anti-RSK2 antibodies. Abbreviations: DMF, dimethyl fumarate; MIF, macrophage migration inhibitory factor; RSK, p90 ribosomal S6 kinase; Wip1, wild-type p53-induced phosphatase 1. |
Complex formation of Wip1 and RSK2 is induced by EGF and MIF in normal human keratinocytes
In order to investigate the role of the ERK signaling pathway in the association of Wip1 with RSK2, keratinocytes were preincubated with DMF (140 µM) or PD98059 (50 µM) for 1 hour and then stimulated with EGF (2 ng/mL) or MIF (100 ng/mL) for 24 hours. Proteins (400 µg/ sample) were immunoprecipitated with anti-RSK2 Ab and analyzed by Western blotting by anti-Wip1 and anti-RSK2 antibodies. Stimulation with EGF or MIF induced the immunoprecipitated complexes of P-Wip1/Wip1 with RSK2 at 24 hours. Figure 7A shows a representative blot. Densitometry of 6 different experiments showed that the ratio of P-Wip1/Wip1 (both) complexes with RSK2 was significantly increased by EGF as well as by MIF compared with controls (co; anti-Wip1 (H-300) sc-20712, epitope C-terminus). Preincubation with the ERK1/2 inhibitor PD98059 significantly inhibited the ratio of EGF- or MIF-induced association of total Wip1 with RSK2. Preincubation with DMF significantly reduced the EGF-induced association of total Wip1 with RSK2, while the inhibitory effect of DMF on the MIF-induced association of total Wip1 with RSK2 was only borderline significant (Figure 7B). The slow-migrating band P-Wip1 (Figure 7A) was not inhibited by DMF, similar to Ro318220, which did not inhibit P-PP2Cδ/ILKAP.20
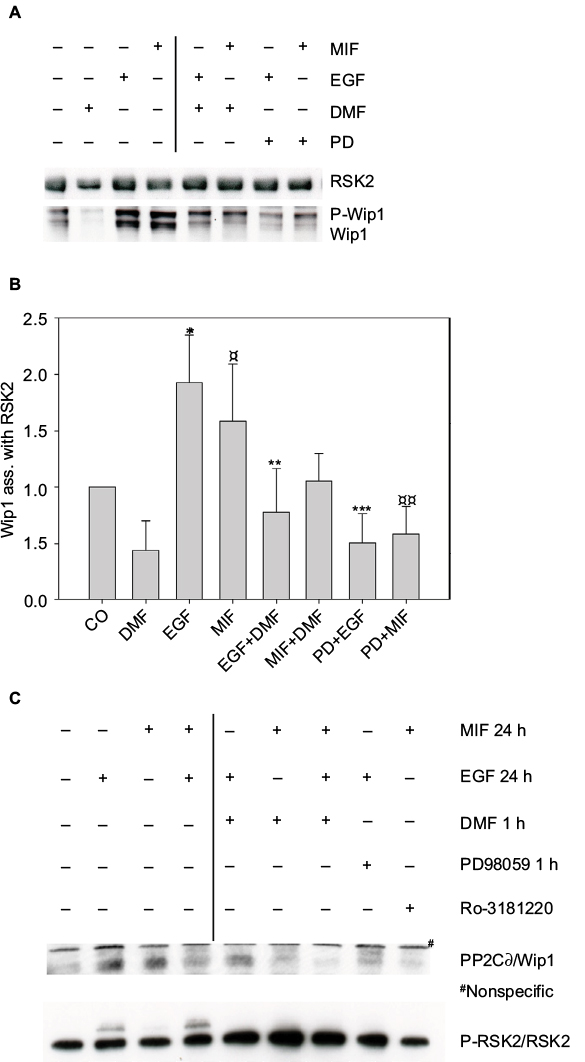 | Figure 7 (A) Complex formation of Wip1 with RSK2 in human keratinocytes was induced by EGF and MIF. Keratinocytes were either left alone or preincubated with DMF (140 µM) or PD98059 (50 µM) for 1 hour and stimulated with EGF (2 ng/mL) or MIF (100 ng/mL) for 24 hours. Whole cell extracts were immunoprecipitated with anti-RSK2 antibody and analyzed by WB with anti-Wip1 (H-300) (sc-20712 epitope C-terminus) and anti-RSK2; 1 representative gel is shown. (B) Densitometry of 6 different experiments from pooled data shows the ratio of P-Wip1/Wip1 (both) associated with RSK2. EGF-stimulated versus (co) untreated cells *p=0.003; MIF stimulated versus (co) untreated cells ¤p=0.037; EGF-stimulated versus (DMF and EGF) **p=0.008; MIF-stimulated versus (DMF and MIF) p=0.059; EGF-stimulated versus (PD98059 and EGF) ***p<0.001; MIF-stimulated versus (PD98059 and MIF) ¤¤p=0.0045. (C) Phosphorylation status of RSK2 at 24 hours. Keratinocytes were preincubated with vehicle, DMF, PD98059, or Ro-318220 and stimulated as before and immunoprecipitated with anti-RSK2 Ab. The protein complexes were analyzed with anti-PP2Cδ/Wip1 (epitope Center AA 182-212; antibody does not recognize P-Wip1). The activation of P-RSK2 was tested by anti-RSK2 antibody. Abbreviations: DMF, dimethyl fumarate; EGF, epidermal growth factor; MIF, macrophage migration inhibitory factor; PP2Cδ, protein phosphatase 2Cδ; RSK, p90 ribosomal S6 kinase; Wip1, wild-type p53-induced phosphatase 1. |
To identify the phosphorylation status of RSK2 at 24 hours, keratinocytes were stimulated as before, immunoprecipitated with anti-RSK2 antibodies, and analyzed with anti-PP2Cδ/Wip1 (epitope PPMID Center AA 182-212 [Cat. No. AP13875c]) and anti-RSK2 Ab. This showed that EGF or MIF with EGF activated P-RSK2 expression at 24 hours in complex with PP2Cδ/Wip1, and this was inhibited by DMF, PD98059, and Ro-318220 (Figure 7C). Anti-PP2Cδ/Wip1 (epitope Center AA 182-212) Ab did not recognize the slow-migrating band of P-Wip1.
Discussion
In this study, we made 3 novel observations that provide information on how RSK2 activity is regulated by PP2Cδ/Wip1 or ILKAP in L skin of patients with psoriasis. First, the level of P-RSK1, 2 (T573)/(T577) was significantly higher in L than in NL psoriatic skin, while the opposite was true for P-RSK1, 2 S380/S386 (Figure 1A and B). Second, the level of ILKAP was significantly higher in L than in NL skin (Figure 2A and B). Third, although the level of Wip1 was similar in L and NL psoriatic skin (Figure 4A), we observed a pronounced complex formation between Wip1 and RSK2 in L skin compared with NL skin (Figure 5A and B). There was no complex formation in normal skin (Figure 4C). ILKAP also formed significant complexes with RSK2 in L skin (Figure 5A and C). Based on these findings, we propose that Wip1 and ILKAP regulate the activity of RSK2 and the phosphorylation of RSK2 (S386) in L psoriatic skin. Because phosphorylation sites of RSK1-3 are highly conserved amino acid sequences,8 antibodies for P-RSK1 (T573) or P-RSK1 (S380) (double band, cleavage product) detect also P-RSK2 (T577) or P-RSK2 (S386) (Figure 1A and B). Because phosphorylation is considered to stabilize protein expression, we normalized P-RSK1, 2 to β-actin and not to RSK1, 2, and 3.30
Previous research shows that nonactive RSK1-3 is localized in cytoplasm, while active RSKs are translocated to the nucleus.10 Ultraviolet type B stimulation induced the nuclear expression of P-RSK2 (T577) in normal skin and skin cancer cells.31 In psoriatic skin, the number of keratinocytes expressing nuclear P-RSK1, 2 T573/T577 was higher in L than in NL epidermis (Figure 3A). The number of cells expressing cytoplasmic P-RSK1, 2 S380/S386 in the epidermis was reduced (Figure 3B), pointing to dephosphorylation by Wip1 in psoriatic epidermis.
Two different gene loci are described for the PP2Cδ/Wip1 and ILKAP isoforms.21 ILKAP (46 kDa) has been shown to regulate ILK1 activity physiologically.23 Nuclear localization of ILK in keratinocytes was associated with increased DNA synthesis.32 In accordance with this, we found more anti-ILKAP staining in the nuclei of keratinocytes in L than in NL epidermis (Figure 2B). Two anti-ILKAP positive dots/ nucleus indicate cell division.
Phosphorylation at Ser and Thr residues in peptides was important for binding ILKAP or Wip1 to RSK2. Affinity was higher for pSer386 peptides than for pThr577, which indicates that ILKAP in partnership with Wip1 controls P-RSK2 activity.33,34
Unlike ILKAP, Wip1 is an oncogene overexpressed in breast and ovarian carcinoma, and Wip1 enhances growth by dephosphorylating tumor suppressor proteins (p53, AIM, INK4A, and ARF).24,25 Wip1 mediates negative feedback of p38 MAPK/p53 signaling by dephosphorylating p-p53 (S15) in response to ultraviolet radiation.28 The p53 gene and protein have been previously reported to be reduced in L psoriatic skin compared with NL normal skin.35,36 This is in accordance with our findings that Wip1 is active in L psoriatic skin, forms complexes with p-RSK2, deregulates P-RSK2 (S386) activity, and dephosphorylates p-p53 (S15).
In cultured keratinocytes, MIF stimulation increased the new synthesis of Wip1 and inhibited P-53 (S15), but costimulation with DMF inhibited Wip1 and induced p-P53 (S15) in accordance with the function of Wip1 and our earlier results9 (Figure 6B).
Deleting Wip1 phosphatase activity inhibited tumor growth.37 In MCF-7 cells, immunoprecipitation with RSK2 confirmed the high level of association of RSK2 with P-Wip1/Wip1. Earlier, TNF-α induced the transcription of Wip1 by activating NF-κB in MCF-7 cells.29 In the promoter region of Wip1, potential binding sites were found for transcription factors NF-κB, c-Jun, and CREB/ATF.38
MSK1 activation was shown to induce NF-κB gene transcription, which is in accordance with the new synthesis of Wip1.5 Along that line, preincubating cells with DMF resulted in the inhibition of P-MSK1/2, P-RSK1-2, and NF-κB activation,5,9 reducing the MIF-induced transcription of Wip1 and the association of Wip1 with RSK2 in keratinocytes and in MCF-7 cells, according to our hypothesis (Figure 8).
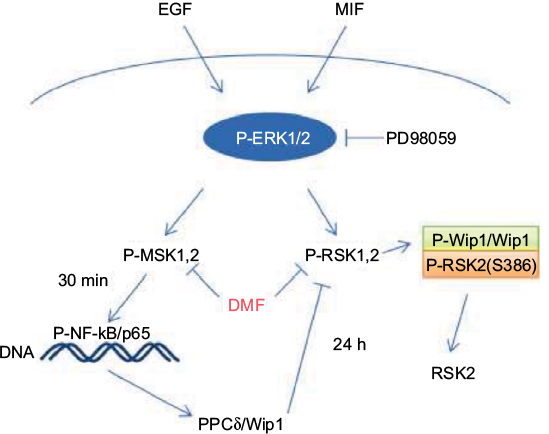 | Figure 8 Schematic representations of EGF- and MIF-induced activation of P-ERK1, 2 and the phosphorylation of MSK1/2 and RSK1/2 kinases. P-MSK1/2 induces P-NF-κB/p65 (S276) and the transcription of PP2Cδ/Wip1, which is inhibited by DMF; 24 hours stimulation by EGF and MIF induces P-Wip1/Wip1, resulting in dephosphorylation of P-RSK2 (S386) in complex formation with P-RSK2. This is inhibited by costimulation with the ERK1/2 inhibitor PD98059 or DMF. Abbreviations: DMF, dimethyl fumarate; EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; MIF, macrophage migration inhibitory factor; MSK, mitogen- and stress-activated kinase; NF-κB, nuclear factor-κB; PP2Cδ, protein phosphatase 2Cδ; RSK, p90 ribosomal S6 kinase; Wip1, wild-type p53-induced phosphatase 1. |
IL-20 is produced by suprapapillary keratinocytes in L psoriatic skin, and IL-20 polymorphism has been suggested to determine susceptibility to plaque-type psoriasis.40,41 The gene expression of IL-20 is induced through the activation of MSK1 and NF-κB in keratinocytes, and DMF inhibited IL-20 mRNA expression in vitro.2,5 Therefore, treatment with DMF will also reduce the level of IL-20 in the psoriatic skin and inhibit psoriasis-like morphological changes and inflammation.
Conclusion
Our results indicate that the constitutive, high levels of EGF and MIF in psoriatic patients3,4,39 result in the activation of the ERK signaling pathway, phosphorylation of MSK1/2 and RSK1/2 kinases, and activation and new synthesis of Wip1. The complex formation with Wip1 results in disruption of the full-length activation of RSK2 at S386, significantly interfering with the regulation of proliferation and apoptosis in keratinocytes in L psoriatic skin.
Acknowledgments
The work was supported by research grants from the Danish Psoriasis Foundation, Aage Bang Foundation DK, and the Novo Nordisk Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Johansen C, Kragballe K, Westergaard M, Henningsen J, Kristiansen K, Iversen L. The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin. Br J Dermatol. 2005;152:37–42. | ||
Funding AT, Johansen C, Kragballe, et al. Mitogen- and stress-activated protein kinase 1 is activated in lesional psoriatic epidermis and regulates the expression of pro-inflammatory cytokines. J Invest Dermatol. 2006;126:1784–1791. | ||
Shimizu T, Nishihira J, Mizue Y, et al. High macrophage migration inhibitory factor (MIF) serum levels associated with extended psoriasis. J Invest Dermatol. 2004;116:989–990. | ||
Anderson KS, Petersson S, Wong J, et al. Elevation of serum epidermal growth factor and interleukin 1 receptor antagonist in active psoriasis vulgaris. Br J Dermatol. 2010;163:1085–1089. | ||
Gesser B, Johansen C, Rasmussen MK, et al. Dimethylfumarate specifically inhibits the mitogen and stress-activated kinases 1 and 2 (MSK1/2): possible role for its anti-psoriatic effect. J Invest Dermatol. 2007;127:1326–1336. | ||
Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol Cell Biol. 2003;23:4796–4804. | ||
Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mo Cell Biol. 2008;9:747–758. | ||
Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. | ||
Gesser B, Rasmussen MK, Raaby L, et al. Dimethylfumarate inhibits MIF-induced proliferation of keratinocytes by inhibiting MSK1 and RSK1 activation and by inducing nuclear p-c-Jun (S63) and p.p53 (S15) expression. Inflamm Res. 2011;60:643–653. | ||
Carriere A, Ray J, Blenis J, Roux PP. The RSK factors activating the Ras/MAPK signalling cascade. Front Biosci. 2008;13:4258–4275. | ||
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signalling pathway by transcription dependent and independent mechanism. Science. 1999;286:1358–1362. | ||
Anjum R, Roux PP, Ballif BA, Gygi SP, Blenis J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signalling. Curr Biol. 2005;15(19):1762–1767. | ||
Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem. 1999;274:2893–2898. | ||
Richards SA, Dreisbach VC, Murphy LO, Blenis J. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol Cell Biol. 2001;21:7470–7480. | ||
Frödin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–2934. | ||
Mrowietz U, Asadulla K. Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 2005;11:43–48. | ||
Stoof TJ, Flier J, Sampat S, Nieboer C, Tensen CP, Boorsma DM. The antipsoriatic drug dimethylfumarate strongly suppresses chemokine production in human keratinocytes and peripheral blood mononuclear cells. Br J Dermatol. 2001;144:114–120. | ||
Meissner M, Valesky EM, Kippenberger S, Kaufmann R. [Dimethyl fumarate – only an anti-psoriatic medication?] J Dtsch Dermatol Ges. 2012;10:793–801. German. | ||
Tong Y, Quirion R, Shen SH. Cloning and characterization of a novel mammalian PP2C isozyme. J Biol Chem. 1998;273:35282–35290. | ||
Doehn U, Gammeltoft S, Shen SH, Jensen CJ. p90 ribosomal S6 kinase 2 is associated with and dephosphorylated by protein phosphatase 2Cδ. Biochem J. 2004;382:425–431. | ||
Lammers T, Lavi S. Role of type 2C protein phosphatases in growth regulation and in cellular stress signaling. Crit Rev Biochem Mol Biol. 2007;42:437–461. | ||
Tamura S, Toriumi S, Saito J, Awano K, Kudo TA, Kobayashi T. PP2C family members play key roles in cell survival and apoptosis. Cancer Sci. 2006;97:563–567. | ||
Leung-Hagesteijn, C, Mahendra A, Naruszewicz I, Hannigan GE. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. EMBO J. 2001;20:2160–2170. | ||
Fischele M, Zhang HL, Fan S, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–6053. | ||
Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–135. | ||
Yu E, Ahn YE, Jang SJ, et al. Overexpression of the Wip1 gene abrogates the p38 MAPK/p53/Wip1 pathway and silences p16 expression in human breast cancer. Breast Cancer Res Treat. 2007;101:269–278. | ||
Kjellerup RB, Kragballe K, Iversen L, Johansen C. Pro-inflammatory cytokines release in keratinocytes is mediated through the MAPK signal integrated kinases. Exp Dermatol. 2008;17:498–504. | ||
Takekawa M, Adachi M, Nakahata A, et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19(23): 6517–6526. | ||
Lowe JM, Cha H, Yang Q, Fornace AJ, Jr. Nuclear factor-κB (NF-κB) is a novel transcriptional regulator of the oncogenic WiP1 phosphatase. J Biol Chem. 2010;285(8):5249–5257. | ||
Mouravlev A, Young D, During MJ. Phosphorylation-dependent degradation of transgenic CREB protein initiated by heterodimerization. Brain Res. 2007;1130(1):31–37. | ||
Cho YY, Lee MH, Lee CJ, et al. RSK2 as a key regulator in human skin cancer. Carcinogenesis. 2012;33(12):2529–2537. | ||
Nakrieko KA, Vespa V, Mason D, Irvine TS, D’Souza SJ, Dagnino L. Modulation of integrin-linked kinase nucleo-cytoplasmic shuttling by ILKAP and CRM1. Cell Cycle. 2008;7(14):2157–2166. | ||
Højlys-Larsen KB, Sørensen KK, Jensen KJ, Gammeltoft S. Probing protein phosphatase substrates binding: affinity pull-down of ILKAP phosphatase 2C with phosphopeptides. Mol Biosyst. 2012;8:1452–1460. | ||
Yamaguchi H, Minopoli G, Demidov ON, et al. Substrate specificity of the human protein phosphatase 2C∂, Wip1. Biochemistry. 2005;44:5285–5294. | ||
Michel G, Auer H, Kemeny L, Böcking A, Ruzicka T. Antioncogene P53 and mitogenic cytokine interleukin-8 aberrantly expressed in psoriatic skin are inversely regulated by the antipsoriatic drug tacrolimus (FK506). Biochem Pharmacol. 1996;51:1315–1320. | ||
Johansen C, Flindt E, Kragballe K, et al. Inverse regulation of the nuclear factor-κB binding to the p53 and interleukin-8 κB response elements in lesional psoriatic skin. J Invest Dermatol. 2005;124:1284–1292. | ||
Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the WiP1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16 Ink4a-p19Arf pathway. Nat Genet. 2004;36:343–350. | ||
Choi J, Appella E, LA Donehower. The structure and expression of the murine wild-type p53-induced phosphatase 1 (Wip1) gene. Genomics. 2000;64:298–306. | ||
Steinhoff M, Meinhardt A, Steinhoff A, Gemsa D, Bucala R, Bacher M. Evidence for a role of macrophage migration inhibitory factor in psoriatic skin diseases. Br J Dermatol. 1999;141:1061–1066. | ||
Bech R, Otkjær K, Birkelund S, et al. Interleukin 20 protein locates to distinct mononuclear cells in psoriatic skin. Exp Dermatol. 2014;23:345–368. | ||
Kingo K, Koks S, Nikopensius T, Silm H, Vasar E. Polymorphisms in the interleukin-20 gene: relation to plaque-type psoriasis. Genes Immun. 2004;5:117–121. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.