Back to Journals » Journal of Inflammation Research » Volume 14
Protective Effects of Dexmedetomidine on Sepsis-Induced Vascular Leakage by Alleviating Ferroptosis via Regulating Metabolic Reprogramming
Authors She H ![]() , Hu Y, Zhou Y, Tan L, Zhu Y, Ma C, Wu Y, Chen W, Wang L, Zhang Z, Wang L, Liu L, Li T
, Hu Y, Zhou Y, Tan L, Zhu Y, Ma C, Wu Y, Chen W, Wang L, Zhang Z, Wang L, Liu L, Li T
Received 27 September 2021
Accepted for publication 3 December 2021
Published 10 December 2021 Volume 2021:14 Pages 6765—6782
DOI https://doi.org/10.2147/JIR.S340420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Monika Sharma
Han She,1,2,* Yi Hu,1,* Yuanqun Zhou,2 Lei Tan,1 Yu Zhu,2 Chunhua Ma,2 Yue Wu,2 Wei Chen,1 Li Wang,2 Zisen Zhang,2 Li Wang,1 Liangming Liu,2 Tao Li2
1Department of Anesthesiology, Daping Hospital, Army Medical University, Chongqing, 400042, People’s Republic of China; 2State Key Laboratory of Trauma, Burns and Combined Injury, Second Department of Research Institute of Surgery, Daping Hospital, Army Medical University, Chongqing, 400042, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liangming Liu; Tao Li Email [email protected]; [email protected]
Introduction: Vascular leakage plays a vital role in sepsis-induced multi-organ dysfunction. Currently, no specific measures are available for vascular leakage. Ferroptosis, as a recently recognized form of cell death, plays a crucial role in cell dysfunction. It is still unknown whether ferroptosis participates in the occurrence of organ dysfunction following sepsis. Our previous study showed that dexmedetomidine (Dex) could alleviate sepsis-induced organ dysfunction. However, whether the mechanism is related to ferroptosis is not clear.
Methods: The publicly available datasets of septic patients were reanalyzed, and septic models in vivo and vitro by cecal ligation and puncture and lipopolysaccharide-stimulated vascular endothelial cells (VECs) were applied. The occurrence of ferroptosis in septic patients and rats was observed, and the protective effects of Dex on ferroptosis, and related mechanisms on regulating metabolic reprogramming and mitochondrial fission were further studied.
Results: The transcriptomics data of patients from the GEO database showed that ferroptosis was closely related to sepsis. Sepsis induced significant ferroptosis in VECs by metabolomics analysis. The level of lipid peroxidation was increased in VECs, and the mitochondrial cristae was decreased after sepsis. Metabolomics analysis showed that Dex activated the pentose phosphate pathway and increased glutathione in VECs via up-regulation of G6PD expression. Dex could antagonize sepsis-induced the decrease in the level of Nrf2. The Nrf2 inhibitor reversed the protective effect of Dex on ferroptosis. Further study showed that Dex significantly alleviated sepsis-induced mitochondrial over-division, improved mitochondrial function, and decreased ROS, further inhibiting the ferroptosis of VECs. Dex alleviated the permeability of vessels by reducing ferroptosis and enhanced the intercellular junction of VECs.
Conclusion: Dex protects vascular leakage following sepsis by inhibiting ferroptosis. The mechanism is mainly related to metabolic reprogramming via Nrf2 up-regulation and inhibition of mitochondrial fission.
Keywords: dexmedetomidine, sepsis, ferroptosis, metabolic reprogramming, mitochondrial fission
Introduction
Sepsis is a dysregulated systemic response to infection that causes organ dysfunction.1 As a common critical illness in clinical practice, sepsis has a very high fatality rate. The global epidemiological survey showed that sepsis affected around 31.5 million people per year globally, of which approximately 5.3 million died. Sepsis is still a huge task all around the world.2 Sepsis was also among the leading causes of death associated with COVID-19 infection.3 In the progression of sepsis, vascular leakage with characterization of endothelial barrier dysfunction is a critical pathophysiological step, which results in tissue edema and organ dysfunction, and there is no effective therapy.4 It is of crucial clinical significance to find more effective approaches to treating sepsis.
Ferroptosis was recently discovered as a nonapoptotic cell death and was first reported by Dixon.5 It is distinct from other cell death pathways, including apoptosis, necrosis, and pyroptosis. An excessive amount of intracellular free iron interact with hydrogen peroxide by Fenton reaction, resulting in lipid peroxidation of polyunsaturated fatty acids of cell membranes, which is the initiating mechanism of ferroptosis. Additionally, GPX4 (glutathione peroxidase 4) and system Xc- are two key regulators of ferroptosis. As a selenoprotein, GPX4 could reduce lipid peroxides using GSH (glutathione) and prevent ferroptosis.6 Up to date, ferroptosis was involved in multiple pathological processes, including neurotoxicity, acute kidney failure, and heart disease.7,8 Besides, several studies indicated that ferroptosis was closely related to the development of sepsis.9,10 Li et al found that ferroptosis played a vital role in septic myocardial dysfunction. However, it is still unknown whether ferroptosis participate in the vascular endothelial barrier function after sepsis.
Dexmedetomidine (Dex) is a novel, highly selective α2-adrenoceptor agonist with analgesic, sedative, anxiolytic and anti-sympatholytic properties.11 Several reports demonstrated that Dex could protect the heart, brain, and kidney after sepsis. Previous studies showed that the mechanism of Dex protecting organ function was related to the inhibition of oxidative stress and inflammatory response.12,13 And a recent study showed that Dex could exert cardioprotective effects via ferroptosis inhibition,14 which reduced CLP-induced heart injury by increasing SOD and GSH levels. However, it is unknown whether Dex alleviates vascular leakage by reducing ferroptosis of VECs.
Mitochondria are energy factories and a major source of oxidative stress in cells. Large studies have demonstrated that mitochondrial morphology plays a key role in the determinant of mitochondrial function.15,16 Our previous study17 found that the mitochondria of VECs were excessively fissioned in sepsis, and that mitochondrial morphology presented fragmentation, leading to mitochondrial dysfunction. ∆Ψm and ATP levels significantly reduced, and the generation of ROS increased. It is unclear whether Dex could inhibit ferroptosis of VECs in sepsis by improving the morphology of mitochondria.
In the present study, the clinical data were re-analyzed on the relationship with ferroptosis in patients with sepsis using the GEO database. Cecal ligation and puncture (CLP) induced sepsis model and lipopolysaccharide (LPS) stimulated vascular endothelial cells (VECs) were used to explore the protective effect of Dex on ferroptosis of sepsis, and the underlying mechanism was further observed.
Materials and Methods
Ethical Statement
All research protocols were conducted by the National Institutes of Health (Publication No. 85–23, revised 1996) and approved by the Laboratory animal welfare and ethics committee of Army Medical University (No. AMUWEC20171288). Adult male and female Sprague Dawley (SD) rats, weighing 200–220 g, were purchased from the animal center of the Research Institute of surgery. They were fasted for 12 h before the experiment and could drink water freely.
Reagents
Dexmedetomidine was purchased from Hengrui (Jiangsu, China). Albumin-fluorescein isothiocyanate conjugate (FITC-BSA) and Lipopolysaccharide (LPS) were purchased from Sigma (St. Louis, MO, US). Antibodies for G6PD, GPX4, ZO-1, β-actin, and ROS detection kit were purchased from Abcam (Cambridge, MA, America). Antibody for Nrf2 was purchased from GeneTEX (San Antonio, TX, America). Lipid Peroxidation kit, MITO-SOX detection kit, Mito-tracker and Thiol-tracker were purchased from Thermo Scientific (Waltham, MA, America). Glutathione, MDA, and ATP detection kit were purchased from Beyotime Biotechnology (Shanghai, China). Brusatol and Erastin were purchased from MedChemExpress (Monmouth, NJ, America). All other chemicals were purchased from Sigma unless specifically mentioned otherwise.
Sepsis Model Establishment
According to a previous study,18 cecal ligation and puncture (CLP) was used to replicate the sepsis model in rats. Rats were anesthetized with pentobarbital sodium (45mg/kg) by intraperitoneal, and then the cecum was exposed and ligated. The hole was punctured 0.7 cm from the distal end with a triangular needle (the needle was approximately 1.5 mm diameter), and feces were allowed to flow into the abdominal cavity freely. Cut off both sides of the omentum and sutured the wound, the rats were returned to the cages. 12 hours after CLP, the vascular tissues were taken for the following experiments. Rats in the Dex group were intraperitoneally injected with Dex (10 µg/kg) 30 min prior to the operation. Another dose of Dex (10µg/kg) was administered 6 h after the operation. In the Bru group, 0.4 mg/kg of brusatol was intraperitoneal injection for rats 10 min before injecting dexmedetomidine.
Vascular Endothelial Cell Preparation and Treatment
Vascular endothelial cells were obtained from pulmonary veins of SD rats.4 After anesthesia and sterilized with iodine, the thoracic cavity of rat was opened and then separated the pulmonary vein. Vascular tissues were cut up to 1 mm × 1 mm pieces and attached to the bottom of the culture flask and placed in the incubator at 37°C. After the tissues were completely adhered to the wall, the ECM (Sciell, America; 5% fetal bovine serum) medium was added. After 72 hours, the pieces were removed from the flask, and the cells crawling on at the bottom of the culture flask were VECs. The passage 3–5 endothelial cells were used for the follow-up study.17 LPS (1μg/mL) or Erastin (5µM) were used to incubate VECs for 12 hours to induce an in vitro vascular endothelial injury model.8 In order to explore the effects of Dex on ferroptosis, VECs were treated with 0.1µM Dex after LPS or Erastin stimulation. Brusatol (40 nM) was used to incubate the cells for 10 min before Dex treatment. In the present study, samples in an experiment represented one independent replicate, and each experiment was repeated at least 3 times.
Detection of Vascular Permeability in Septic Rats
FITC-BSA (9mg/kg) was injected via the jugular vein in rats. 1 h after the administration, the abdominal cavity was opened, and the abdominal aorta was cut open, slowly infusing the normal saline solution through the jugular vein until the lung tissue became white. The right-lung tissue was embedded with OCT glue, and a frozen section (section thickness 10–15μm) was performed. Tissues were fixed with 4% paraformaldehyde for 15 min, permeated with 0.1% Triton-X for 5 min, and incubated with DAPI (1:50) at 37°C for 30 min. The FITC-BSA exudation in lung tissue was observed under a laser confocal microscope. A 2 cm incision was made along the middle of the abdomen for other rats to select mesenteric fixation with abundant micro-vessels. FITC-BSA was injected into the femoral vein. After 6 minutes, the leakage of FITC-BSA into mesenteric micro-vessels was observed by an inverted microscope (Hamamatsu, Japan).19
Transmembrane Electrical Resistance (TER) and BSA Leakage Detection
Endothelial cells were seeded on transwell plates (pore size 0.4 µm, BD Biosciences, Franklin Lakes, USA). The TER values of cells were measured every 30 minutes by voltmeter (World Precision Inc., America) for 24 hours. The TER of the non-cell chamber was regarded as control. The resistivity of cells = (actual TER – blank control)/actual TER. After the detection of TER, 20 μL (10 μg/mL) FITC-BSA was added into the upper insert of the transwell, and 200 µL of the medium of the lower chamber at 10, 20, 30, 40, 50, and 60 min was collected for the measurement of fluorescence intensity. An equal volume of culture medium was added into the lower chamber after each collection. The permeability of endothelial cells was (A10min + A20min + A30min + A40min + A50min + 15 × A60min)/total fluorescence intensity.19
Transmission Electronic Microscopy Observation
Pulmonary veins of rats were quickly separated and fixed under the microscope and then soaked in acetone and resin (1:1) for 3 h. The tissues were polymerization for 48 h at high temperature after being embedded. Then, the specimens were taken out and stained. Finally, a transmission electron microscope (H-7700, Hitachi, Japan) was used to observe and image.
Mitochondrial Oxygen Consumption Rate Detection
The oxygen consumption rate (OCR) was measured using a 24-well XFe plate (Seahorse, Agilent Cell Analysis technology, USA).20 Endothelial cells were seeded at a density of 1 × 104 per well, and put the cell plate in the super-clean bench for 1 h to make the cells settle naturally. Then, the cell plate was put back into the cell incubator overnight to make the cells adhere to the wall. When the cell confluence reaches 70–80%, LPS and Dex were added to treat cells for 12 h. Before detection, the basic assay medium contains 2.5μM glucose and 2mM glutamine was used to culture cells for 50 min. Then, 2μM oligomycin, 1μM FCCP, and 0.5μM rotenone/antimycin A were performed sequentially. The OCR was measured by an extracellular flux analyzer under the mitochondrial stress test condition.
Metabolomics Profiling
Cells were rapidly frozen in liquid nitrogen to halt metabolism and stored at −80°C. For extraction, the extract solution (acetonitrile: methanol: water = 2: 2: 1, with the isotopically labeled internal standard mixture) was added to cells. Then, the samples were incubated for 1 h at −40°C and centrifuged at 12,000 rpm for 15 min at 4°C. Then, a UHPLC system (Vanquish, Thermo Fisher Scientific) was used to analyze the samples. The raw data were converted to the mzXML format and processed with an in-house program for peak detection, extraction, and integration. Then, an in-house MS2 database (BiotreeDB) was applied in metabolite annotation. The cutoff for annotation was set at 0.3.21
Bioinformatics Analysis
Principal component analysis (PCA) was performed to examine intrinsic clusters of metabolomics data. A 95% confidence interval (CI) was used as the threshold to identify potential outliers in all samples. In addition, heat maps were displayed by the R software package Pheatmap to visualize the metabolite difference within the data set. Volcano and violin plots were made using GraphPad Prism V.8.0.0 Software, which directly showed the up-regulated and down-regulated metabolites. MetaboAnalyst 2 was applied for pathway enrichment analysis.
Immunofluorescence Staining
VECs were seeded into the confocal chamber and incubated with a Mito-tracker (1:10, 000) at 37°C for 30 min. The morphology of mitochondria was observed by a laser confocal microscope (Leica SP5, Germany). The mitochondrial length was analyzed using a mitochondrial network analysis (MiNA) toolset included in ImageJ software (https://fiji.sc/). Cells were incubated with DCFH-DA (10μM) and Mito-SOX (5μM) at 37°C for 30 min. Then, the average fluorescence intensity of intracellular ROS and mitochondrial ROS was calculated by ImageJ.17
Lipid Peroxidation Detection
VECs were incubated with 10 μM lipid peroxidation kit (C10445; Invitrogen)22 for 30 min at 37°C. FITC fluorescence was detected at excitation/emission of 488/510 nm, and TRITC fluorescence was detected at excitation/emission of 581/591 nm by confocal microscopy (Leica TCS SP5, Wetzlar, Germany). The ratio of the emission fluorescence intensities at 510 nm to 590 nm detected by Image J software (https://fiji.sc/) gives a read-out for lipid peroxidation in cells.
Statistical Analysis
Data from animal studies were repeated in at least 6 independent experiments, data from cell studies were repeated in at least 3 independent experiments (3 times for Western blot experiments), and data from one representative experiment were shown. SPSS 20.0 (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. The data were represented as means± SD. An independent sample t-test was used to analyze the difference between the two groups. One-way analysis of variance (ANOVA) was used for experiments with more than two groups, followed by Tukey’s post hoc analysis and (SNK/LSD) comparison. P < 0.05 was considered statistically significant.
Results
The Occurrence of Ferroptosis and the Metabolic Reprogramming in Septic Patients
The microarray data for septic patients were collected from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database to re-analyze ferroptosis-related and metabolism-related genes. GSE5706523 dataset contains 107 blood samples (Healthy (healthy volunteers, n = 25), sepsis (patients with sepsis, n = 82). Additionally, 6 healthy control blood monocytes samples and 8 blood monocytes samples from patients with sepsis were involved in the GSE4695524 dataset. Metabolism-related genes were obtained from the Molecular Signatures Database v7.1 (MSigDB) (http://software.broadinstitute.org/gsea/msigdb) by searching for the term “metabolism.” Then, a gene expression matrix consisting of 863 gene expression values was obtained from GSE57065 by R software. LIMMA package of R software was used to analyze the differential expression of mRNAs. Principal component analysis (PCA) indicated substantial diversity between the two groups, and almost all samples were within the 95% confidence interval (Figure 1A). The volcano plot showed there were 558 different metabolism-related genes between the healthy and sepsis group in GSE57065 (Figure 1B) (metabolism-related genes were selected by P-value <0.05). Biological pathways of the differently expressed metabolism-related genes were explored by Funrich software.25 The results showed that glutathione synthesis and recycling, pentose phosphate pathway, and NADPH regeneration were involved (Figure 1C).
 |
Figure 1 Transcriptomics analysis of metabolism-related genes and ferroptosis-related genes in septic patients. (A) Principal component analysis between healthy and sepsis group of GSE57065. (B) Volcano plot of differentially expressed metabolism-related genes of GSE57065. (C) Biological pathways of differentially expressed metabolism-related genes were analyzed by Funrich software. (D) Kegg pathways of differentially expressed genes between healthy and sepsis group of GSE46955. (E) Heatmap of ferroptosis-related genes between healthy and septic patients. (F) ROC curve of ACSL4. (G) ROC curve of GPX4. |
Furthermore, we analyzed the differentially expressed genes (DEGs) of monocytes between the healthy and sepsis group in GSE46955 (A total of 938 DEGs were identified by P-value <0.05 and |Log(FC)|>1). KEGG pathway analysis of DEGs showed that enriched pathways included glutathione metabolism and the pentose phosphate pathway, similar to the results obtained from GSE57065 (Figure 1D). Consensus pathway analysis (CPA) (https://bioinformatics.cse.unr.edu/.software/cpa/)26 was used to analyze the ferroptosis-related genes between healthy and septic patients and found that there were 40 kinds of genes that changed after sepsis (Figure 1E). Acyl-CoA synthetase long-chain family member 4 (ACSL4), a key indicator and regulator of ferroptosis, which could activate long-chain polyunsaturated fatty acids to participate in the synthesis of membrane phospholipids, was increased significantly in the septic patients with an AUC of 0.854 (95% CI = 0.638–1.000) (Figure 1F). The expression of Glutathione peroxidase (GPX4), which could reduce lipid peroxidation by transforming GSH into GSSG,27 was decreased after sepsis with an AUC of 1.000 (95% CI = 1.000–1.000) (Figure 1G). These results indicated that sepsis induced metabolic reprogramming and ferroptosis.
The Ferroptosis of Vascular Endothelial Cells Were Involved by Metabolomics Analysis in Rats Following Sepsis
Ferroptosis, a new form of cell death, can cause severe organ dysfunction.14 The expression of GPX4 in VECs was observed between Normal and LPS groups. The incubation with LPS resulted in the decrease in expression of GPX4 in VECs (Figure 2A and B). Furthermore, the metabolomics analysis in hierarchical clustering heatmap and chord plot showed that the biosynthesis of unsaturated fatty acids such as arachidonic acid was significantly increased after sepsis, which was an important factor leading to ferroptosis28 (Figure 2C and D, Supplementary Figure 1). The lipid peroxidation levels were enhanced in the LPS-stimulated VECs (Figure 2E and F), and the MDA levels of superior mesenteric vein tissues in septic rats also increased by 82% (Figure 2G). The structures of mitochondria in pulmonary veins were observed with a TEM and found that the mitochondrial morphology in septic rats presented the hallmarks of ferroptosis. The mitochondria presented a fragmented morphology with loss of cristae (Figure 2H). The following study focused on the restraint effects of Dex on ferroptosis. Western blot showed that Dex abolished LPS stimulation-induced the reduction of GPX4 in VECs, and the lipid peroxidation was attenuated significantly as compared with the LPS group (Figure 2E and F). The level of MDA in rats with Dex treatment was decreased, and mitochondrial morphology was improved significantly (Figure 2G and H). Erastin is a known inducer of ferroptosis, resulting in cell death, ROS accumulation and GSH reduction. Dex also could attenuate erastin-induced ferroptosis which was shown with the decrease in lipid peroxidation in VECs (Figure 2E and F). The above results suggest that ferroptosis is present in VECs after sepsis, and Dex can inhibit the ferroptosis.
 |
Figure 2 The measurement of ferroptosis in vascular endothelial cells by metabolomics analysis. (A and B) Western blot analysis of GPX4 expression in the VECs after sepsis, n=3. (C) Heatmap of differentially expressed metabolites between LPS and Normal group, n=6. (D) Chord plot of differentially expressed metabolites for group LPS vs Normal. (E) Confocal images to observe lipid peroxidation of VECs (Bar, 10μm), n=3. (F) Statistic graph of lipid peroxidation. (G) Effects of Dex on the level of MDA, n=8. (H) Representative mitochondria TEM images and ratio of mitochondria with vacuolar structure to total mitochondria in per unit area by ImageJ (Bar, 0.5μm), n=6. a: P<0.05 compared with the sham or normal group, b: P<0.05 compared with CLP or LPS group, c: P<0.05 compared with Erastin group. Abbreviation: Mito, mitochondria. |
Dex Alleviated Ferroptosis for Endothelial Cells Following LPS Incubation via Modulating Metabolic Reprogramming
The metabolomics profiling of VECs following LPS and Dex treatments was examined to explore whether Dex suppresses ferroptosis in VECs after sepsis via metabolic reprogramming. Principal component analysis (PCA) plots showed that the samples were almost entirely different (all samples were within the confidence interval) (Figure 3A). The results of OPLS-DA confirmed the stability and reliability of the model (Figure 3B). The volcano plot showed that there were 274 different metabolites between the LPS and Dex group (Figure 3C) (Metabolites were selected by VIP value >1 and P-value <0.05). Hierarchical clustering heatmap and radar chart results showed that Dex administration promoted the expressions of D-Ribose5-phosphate, Gamma-Glutamylcysteine, L-Glutamic acid, and L-Malic acid significantly (Figure 3D–G). The pentose phosphate pathway (PPP) is the main source for producing D-Ribose5-phosphate.29 The up-regulation of D-Ribose5-phosphate indicated that the PPP following Dex treatment was activated and which led to metabolic reprogramming from glycolysis to PPP. In addition, the results of the KEGG pathway analysis in the bubble chart revealed that many different metabolites were remarkably concentrated in the pentose phosphate pathway, glutathione metabolism, and TCA cycle (Figure 3H, Supplementary Figure 2).
 |
Figure 3 Effects of Dex on the metabolomics of LPS treated vascular endothelial cells. (A) Cell metabolites were significantly different between the LPS and Dex group by principal component analysis, n=6. (B) OPLS-DA permutation test to assess the favorable stability of the sample data. (C) Volcano plot of metabolomics of VECs. (D) Heatmap of differentially expressed metabolites affected by Dex and LPS. (E) The intensity of our concerned metabolites was detected by metabolomics mass spectrometry. (F) Radar chart analysis of differentially expressed metabolites for group Dex vs LPS. (G) Correlation analysis of metabolites for group Dex vs LPS. (H) Pathway analysis of differentially enriched metabolites affected by Dex and LPS. Each bubble represents a metabolic pathway. a: P<0.05 compared with the LPS group. |
Dex Reprogrammed Pentose Phosphate Pathway to Synthesize Glutathione Through G6PD
The pentose phosphate pathway is an essential source of NADPH (Figure 4C), an electron donor that participates in many metabolic pathways, such as maintaining the redox status of glutathione. We found that the several differently expressed metabolites were related to glutathione metabolism by building metabolic networks through Cytoscape software (http://www.cytoscape.org/download.php)30 and KEGG database (Figure 4A and B, Supplementary Figure 3). Glutathione is a tripeptide consisting of three amino acids (glutamic acid, cysteine, and glycine), scavenging superoxide anion radicals, and other peroxyl-derived radicals. It is one of the most important substances to fight against ferroptosis by protecting cell membrane integrity and against lipid peroxidation.31 Previous research demonstrated that NADPH from PPP participated in the anti-ferroptosis via maintaining GSH level. GSH is the necessary cofactor for GPX4. The inhibition of GSH synthesis leads to the downregulation of activity of GPX4, eventually resulting in ferroptosis. Therefore, we speculate that Dex may inhibit ferroptosis by regulating glutathione synthesis through metabolic reprogramming of VECs. The results showed that compared with the sham-operated rats, levels of GSH in pulmonary veins after sepsis decreased, accompanied by a decrease in GSH/GSSG. Dex treatment improved both GSH and GSH/GSSG levels (Figure 4D and E). A similar trend of the expression of GSH in VECs was observed by immunofluorescence (Figure 4F). Glucose 6-phosphate dehydrogenase (G6PD) is a rate-limiting enzyme that catalyzes the first reaction in the pentose phosphate pathway.32 We further investigated whether Dex participated in metabolic reprogramming by regulating G6PD. The results showed that G6PD significantly down-regulated after sepsis and enhanced after Dex treatment (Figure 4G and H). The results indicate that Dex inhibits ferroptosis by improving the GSH-GPX4 axis via metabolic reprogramming.
 |
Figure 4 Dex up-regulated glutathione through G6PD in sepsis. (A and B) The metabolic network of gamma-L-Glutamyl-L-cysteine, L-Glutamate, and Taurine was built by Cytoscape software. (C) Schematic diagram of pentose phosphate pathway. (D and E) The levels of glutathione and GSH/GSSG on pulmonary vein tissues in sepsis, n=6. (F) Confocal images to measure glutathione expression of VECs in sepsis (Bar, 25μm), n=3. (G and H) Western blot analysis of G6PD in the VECs treated with LPS, n=3. a: P<0.05 compared with the sham or normal group, b: P<0.05 compared with CLP or LPS group. |
Dex Alleviated Ferroptosis Through Reprogramming Pentose Phosphate Pathway via the Activation of the Nrf2/G6PD Signaling Pathway
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a critical transcription factor in maintaining redox homeostasis. Previous studies have reported that Nrf2 can be directly regulated by Dex.33 In order to determine the role of Nrf2 in Dex modulating PPP, the binding mode of Nrf2 and G6PD was predicted by molecular docking analysis. There was no structure of G6PD reported in rats. Protein sequences of G6PD were compared between rats and humans. And the sequence alignments showed an identity of 88.7% and a similarity of 92.6%. We selected the most complete and highest-resolution structure as the receptor protein (PDB ID: 6E08). Similar consideration was also valid for the human protein structure of Nrf2 (PDB ID: 2FLU) (82.4% identity, 89.6% similarity). Protein structures were downloaded from RCSB Protein Data Bank (https://www.rcsb.org/) and submitted to the ZDOCK server. Images were generated using Pymol. Nrf2 could interact with G6PD and formed six hydrogen bonds by amino acid residues. These interactions provided theoretical support for the possibility of Nrf2 regulating G6PD (Figure 5A). And the data analyzed from GEO database (GSE18344)34 showed that the expression of G6PD was decreased in Nrf2 knockout mice as compared with wild type (Figure 5B). In the following study, the results showed a significantly lower expression of Nrf2 in sepsis, and Dex improved the expression of Nrf2 (Figure 5C). The Nrf2 inhibitor, brusatol (Bru, 40nM),35 was used to incubate the cells, and the results showed that the inhibition of Nrf2 significantly antagonized Dex treatment induced the up-regulation of G6PD in VECs (Figure 5D). Next, the effects of Nrf2 inhibition on Dex protecting sepsis-related ferroptosis were further observed. Immunofluorescence results showed that the extent of lipid peroxidation was significantly abolished after being treated with Bru as compared with the Dex group (Figure 5F and G). At the same time, the levels of MDA in the superior mesenteric vein were higher than that in the Dex group by 57% (Figure 5E), and the levels of ROS following Dex treatment in VECs reversed by Nrf2 inhibition (Figure 5H). The results indicate that Dex inhibits ferroptosis by modulating the metabolic reprogramming via Nrf2/G6PD pathway.
 |
Figure 5 Dexmedetomidine inhibited ferroptosis through Nrf2 in sepsis. (A) Molecular docking of Nrf2 to G6PD. (B) Genomic analysis of G6PD between WT and Nrf2 KO mice based on GEO database (GSE18344). (C) The expression and distribution of Nrf2 in superior mesenteric veins (Bar, 50μm), n=6. (D) Western blot analysis of Nrf2 and G6PD expression in the VECs, n=3. Bru = brusatol, the Nrf2 inhibitor. (E) Effects of Nrf2 on the level of MDA, n=8. (F and G) Confocal images to observe lipid peroxidation of VECs after being treated with Bru (Bar, 10μm), n=3. (H) Effects of Dex and Bru on ROS (Bar, 20μm) of VECs, n=3. a: P<0.05 compared with the sham or normal group, b: P<0.05 compared with CLP or LPS group, c: P<0.05 compared with the CLP+Dex or LPS+Dex group. |
Dex Inhibited the Ferroptosis of VECs by Suppressing Oxidative Damage via Inhibiting Mitochondrial Fission
Previous studies have demonstrated that ferroptosis, mainly due to the destruction of redox balance and mitochondrial dysfunction, is a major source of ROS, which can cause oxidative damage. Moreover, mitochondria fusion and fission balance play an essential role in mitochondrial function. In order to explore whether Dex protects ferroptosis by inhibiting mitochondrial fission, the mitochondria structure was observed. Each group randomly selected cells and the mitochondrial morphology was blindly scored and classified into two categories: Long (>3 µm), Short (≤3 µm).15 The results showed that LPS stimulation led to the mitochondria fragment and presented punctate distribution. The proportion of short mitochondria distinctly increased compared with the normal group. The length of mitochondria shortened from 3.84 ± 5.95 µm in the normal group to 0.53 ± 0.63 µm in sepsis (Figure 6A–C). Dex treatment could improve the proportion of long mitochondria, and the length of mitochondria elongated to 2.95 µm ± 4.65 µm (Figure 6A–C).
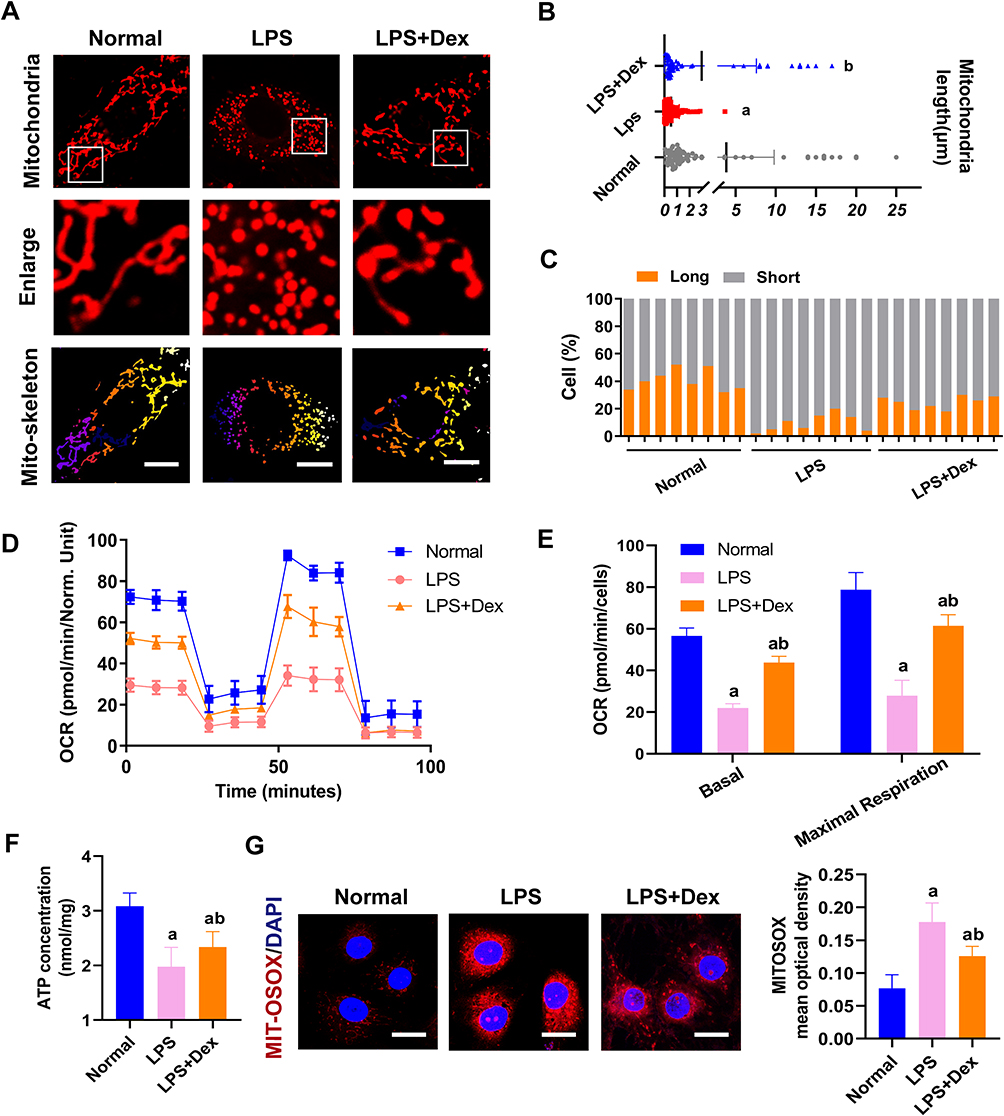 |
Figure 6 Dexmedetomidine ameliorated mitochondrial fission and function in sepsis. (A) Confocal images to observe mitochondrial morphology of VECs (Bar, 25 µm). (B) The length of mitochondria of VECs in each group. (C) The statistical analysis of mitochondrial morphology of VECs in different groups, n=8. (D and E) Effects of Dex on the mitochondrial respiration in VECs, n=3. (F) Effects of Dex on the level of ATP of VECs after sepsis by the luciferase method, n=3. (G) Representative confocal images of MITO-SOX (Bar, 15µm) in sepsis in VECs, n=3. a: P<0.05 compared with the normal group, b: P<0.05 compared with the LPS group. |
The mitochondrial functions after Dex treatment were detected. Mitochondrial oxygen consumption rate (OCR) was measured by the Agilent Seahorse XF technology. LPS stimulation reduced the OCR of VECs, and Dex could improve the mitochondrial respiration of LPS treated VECs (Figure 6D and E). Mito-SOX staining was used to examine the mitochondrial ROS content. After LPS stimulation, a significant increase of Mito-SOX fluorescence was observed. Dex treatment significantly attenuated ROS generation in mitochondria of VECs (Figure 6G). Meanwhile, the ATP levels were significantly reduced after LPS stimulation, and Dex increased the production of ATP (Figure 6F). These results indicate that Dex alleviates the ferroptosis of VECs by reducing ROS via inhibiting mitochondrial fission.
Dex Reduces Vascular Leakage in Sepsis by Inhibiting Ferroptosis
Vascular endothelial cells play an important role in the endothelial barrier, and vascular endothelial injury can directly lead to vascular leakage. Similar to the results reported from our previous study,17 the present study showed that the pulmonary vascular permeability of septic rats (after 12 h of CLP) significantly increased by 72% as compared with the sham group (Figure 7A and B), and the vascular leakage of FITC-BSA from mesenteric micro-vessels showed the same tendency as well (Figure 7C). In VECs, LPS stimulation decreased trans-endothelial electrical resistance (TER) of VECs by 88% and increased the BSA leakage of VECs (Figure 7D and E). Dex could notably alleviate the vascular leakage of sepsis. The administration of Dex alleviated the leakage of FITC-BSA by 20% compared with the CLP group (Figure 7A and B) and decreased the exudation of mesenteric micro-vessels significantly (Figure 7C). Dex also reversed LPS-induced reduction of TER in VECs and alleviated BSA permeability of VECs (Figure 7D and E). The results were further demonstrated on cell connections. The transmission electron microscope (TEM) results showed that the tight junctions between VECs were closed and dense in the sham group. The cells were swelling, and the tight junctions were destroyed after sepsis. Following Dex treatment, tight junctions were restored to a normal degree (Figure 7F). The results of ZO-1 by immunofluorescence and Western blot revealed that LPS stimulation decreased the expression of ZO-1 by 63%, and the structure was loosely distributed with gaps. Following Dex administration, the distribution of ZO-1 improved, and the expression of ZO-1 increased by 52% compared with the LPS group (Figure 7G and H). These results suggest that Dex protects sepsis-induced vascular leakage of rats by alleviating ferroptosis.
 |
Figure 7 Effects of Dex on vascular leakage in sepsis. (A and B) Vascular permeability of the lung, measured by the optical density of injected FITC-BSA in vivo (Bar, 50μm), n=8. (C) The FITC-BSA leakage of mesentery micro-vessels in rats, measured by inverted intravital microscopy in vivo (Bar, 100μm), n=6. (D) The infiltration rate of FITC-BSA in VECs in sepsis, n=3. (E) Effects of Dex on the TER of VEC monolayers in sepsis, n=3. (F) Representative transmission electron microscope microphotographs of tight junctions of pulmonary venules (Bar, 200nm), n=6. (G) The expression of ZO-1 (green) in VECs was measured by immunofluorescence (Bar, 10μm), n=3. (H) Effects of Dex on the level of ZO-1 detected by Western Blotting, n=3. a: P<0.05 compared with the sham or normal group, b: P<0.05 compared with CLP or LPS group. Abbreviation: TJ, tight junction. |
Discussion
The present study showed that Dex was beneficial to vascular leakage by inhibiting ferroptosis via modulating metabolic reprogramming and inhibiting mitochondria fission. The mechanisms were related to the fact that Dex up-regulated the expression of G6PD by Nrf2 and enhanced the metabolic pathway of PPP, led to an increase of GSH, enhanced the ability to scavenge free radicals, and reduced lipid peroxidation. Furthermore, Dex antagonized sepsis-induced excessive mitochondrial fission to improve mitochondrial function, reduced ROS production, and protected VECs from inhibiting ferroptosis (Figure 8). The present study clarifies the protective effects of Dex on vascular leakage by alleviating ferroptosis and its mechanisms, and the study provides a new direction for prevention and treatment of sepsis.
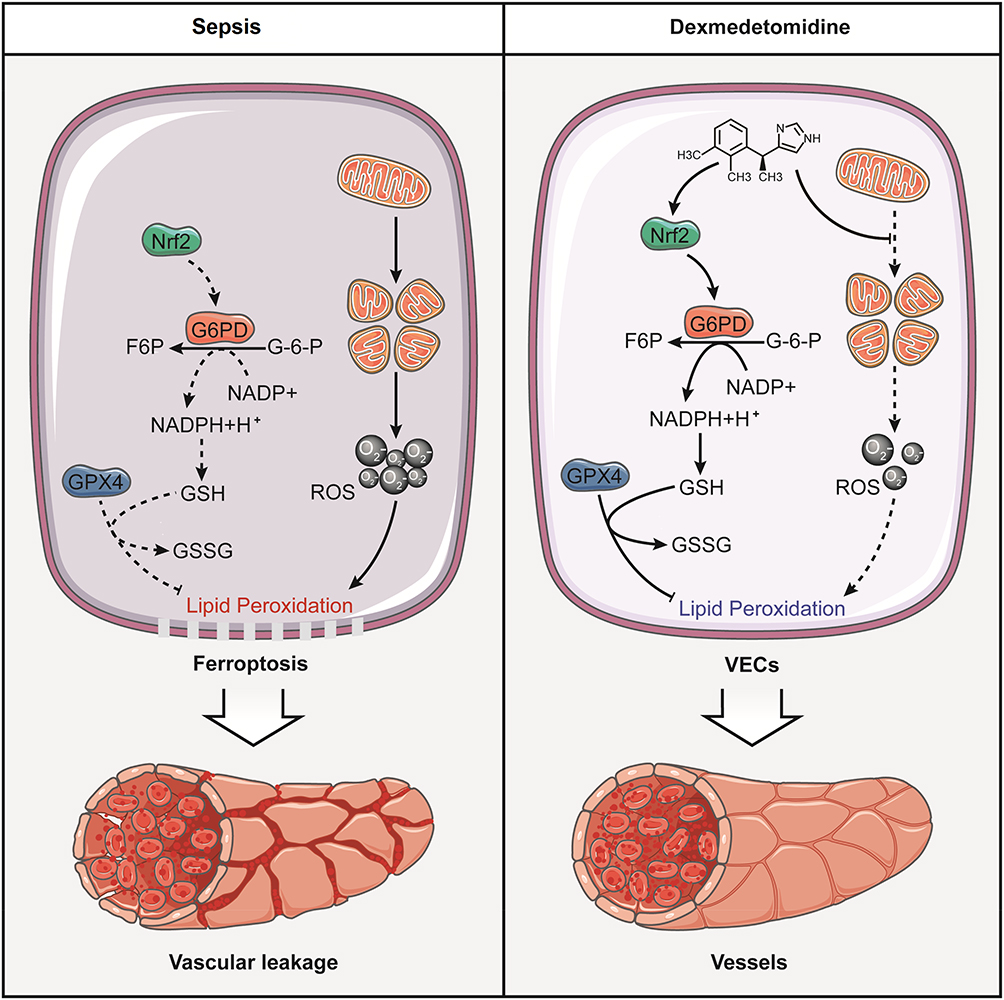 |
Figure 8 Schematic diagram for the mechanism of dexmedetomidine regulating vascular leakage by inhibiting ferroptosis via modulating metabolic reprogramming and inhibiting mitochondria fission. Dexmedetomidine enhanced the expression of G6PD by Nrf2 and activated the metabolic pathway of PPP, led to an increase of GSH, and reduced lipid peroxidation. Meanwhile, Dex antagonized sepsis-induced excessive mitochondrial fission to reduce ROS production. |
Vascular endothelial cells are located on the surface of the vascular endothelium, forming a barrier between circulating blood and tissues and avoiding tissues damage from various noxious agents and inflammatory injury.36 Sepsis-induced endothelial damage resulted in vascular leakage, which led to multiple organ dysfunction syndromes. Previous studies reported that apoptosis and pyroptosis participated in sepsis-induced organ dysfunction. Yang et al reported that the inhibition of pro-apoptotic protein Bax could alleviate the injury of H9C2 cells induced by LPS.37 Sun et al also found that the release of inflammatory cytokines by activated astrocytes can exacerbate sepsis-associated encephalopathy.13 Recent research has implicated that ferroptosis is distinct from apoptosis and pyroptosis, which is involved in developing sepsis.9,10 Ferroptosis is a lipid peroxidation-related cell death and was recently shown also to be ROS‑dependent.25 Liu et al38 found that LPS could cause ferroptosis in human bronchial epithelial cells and acute lung injury in septic mice. Another study also found that ferroptosis occurred in myocardial cells after sepsis. Inhibition of ferroptosis could effectively reduce myocardial dysfunction.9 It is not yet known whether endothelial damage following sepsis is associated with ferroptosis. The present study demonstrated that lipid peroxidation, one of the critical features of ferroptosis, increased significantly after sepsis, and the mitochondria cristae of cells after sepsis were swollen, sparse, and even vacuolated. Meanwhile, the vascular permeability also increased significantly in septic rats, which indicated that ferroptosis of VECs participated in the sepsis-induced vascular leakage. Dex administration reduced lipid peroxidation and MDA production, improved mitochondrial morphology, reduced vascular permeability, and alleviated vascular leakage. This study provides a novel direction for the prevention and treatment of sepsis-induced vascular leakage and organ dysfunction.
The production and the clearance of ROS in vivo are in a dynamic equilibrium. When the ROS homeostasis is destroyed, macromolecules such as DNA, proteins, and lipids are damaged,39 and the level of lipid peroxidation can be considered as a key indicator of ferroptosis. Studies indicated that the protective effects of Dex on organ functions were related to the inhibition of lipid peroxidation. For example, Sha et al40 found that Dex could increase superoxide dismutase (SOD) activity and the level of GSH in septic liver cells, but the specific mechanism is not known. The present study found that Dex activated the pentose phosphate pathway in VECs of sepsis by metabolomics analysis. Previous studies demonstrated that the pentose phosphate pathway played a central role in cellular redox balance, which prevented oxidative stress by maintaining the function of antioxidants. Furthermore, our metabolomics analysis results also found that the expression of metabolites related to glutathione syntheses such as gamma-Glutamylcysteine and L-glutamic acid increased after Dex treatment. The pentose phosphate pathway was the primary source of NADPH, which quickly responded to the need for NADPH to maintain cellular redox. NADPH supplied H+ to glutathione reductase, which reduced glutathione disulfide (GSSG) to glutathione (GSH).29 Further findings in the present study suggested that Dex could increase GSH by activating the pentose phosphate pathway via up-regulating G6PD. G6PD is a key enzyme involved in the PPP, catalyzing the first step (also the rate-limiting step) of generating NADPH.32 Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox-sensitive transcription factor. When the level of pro-oxidant increases, Nrf2 can no longer be sequestered by Keap-1 and is subsequently translocated to the nucleus, which then binds to the promoter and activates the transcription of target genes. Our data also found that Dex regulated metabolic reprogramming by activating G6PD through augmenting the expression of Nrf2, and that the regulation of G6PD and lipid peroxidation by Dex was abolished by Nrf2 inhibitor. Currently, the mechanism of Nrf2 activation by Dex is unknown, but a study has reported that Nrf2 is able to bind with a2-AR.41 Therefore, the agonism of a2-AR induced by Dex may be responsible for the upregulation of Nrf2. However, the detailed mechanism requires additional investigation.
GPX4 is an essential regulator of ferroptosis that can maintain the balance of lipid oxidation and effectively repair the damage to lipid peroxidation.42 GPX4 is the critical enzyme capable of converting toxic lipid hydroperoxides into nontoxic lipid alcohols that require GSH as the substrate during ferroptosis. The lack of GSH in sepsis directly decreases GPX4 activity. In the present study, through transcriptomics analysis, we found that the expression of GPX4 decreased in septic patients, and Western blot also found the same trend in VECs of septic rats. Dex up-regulated the expression of GPX4, which indicated that Dex could improve the activity of GPX4 by regulating the generation of GSH.
Mitochondria are the regulatory centers of cellular metabolism and energy metabolism with continuously remodeled by fission and fusion. And mitochondria can be involved in cellular functions, such as ATP generation, intracellular calcium regulation, and ROS production.16 Our previous research found that the mitochondria of VECs were over division after sepsis, and the morphology changes from filamentous to punctate.17 The present study found that mitochondrial dysfunction after sepsis led to more production of ROS. Dex alleviated mitochondrial fission and improved mitochondrial function. Our previous study found that Dex inhibited mitochondrial division by regulating the translocation of dynamin-related protein1 (Drp1) and reducing ER-MITO contacts of VECs via reducing actin polymerization.17 However, detailed mechanisms need further study. Many reports showed that mitochondrial morphology and mitochondrial metabolism interacted. For example, Song et al43 inhibited mitochondrial fission by silencing Drp1 and found an increase in the fatty acid content of lipid droplets and a decrease in mitochondrial fatty acid oxidation, which clarified the role of mitochondrial dynamics in the control of intracellular fuel utilization and partitioning. The change of mitochondrial metabolism could model mitochondrial morphology. A study44 reported that lipid infusion increased Drp1 phosphorylation at serine 616 in skeletal muscle while reducing mitochondrial membrane potential. The present study found that Dex could protect VECs from sepsis by regulating the metabolism and inhibiting mitochondrial fission. However, further research is necessary regarding the mutual correlation of the two parts.
There remain some limitations in this study. First of all, the present study showed that Dex could inhibit ferroptosis and vascular leakage in septic rats in vivo and in vitro. However, it is still unknown whether Dex has a consistent protective effect in large animals and humans. Second, there are many types of regulated cell death in sepsis, such as apoptosis, pyroptosis and ferroptosis. These forms of cell death play an important role in the process of organ dysfunction induced by sepsis. Previous studies have shown that the treatment of apoptosis and pyroptosis can effectively improve the organ dysfunction of sepsis.13 In the present study, we found that sepsis can cause ferroptosis of endothelial cells, Besides, the inhibition of ferroptosis of Dex can effectively inhibit vascular leakage. In the future research, the ferroptosis inhibitor may be used to observe the protective effect of vascular leakage in sepsis, and the combination therapy for multiple types of cell death may be an important way to treat sepsis.
Conclusion
In summary, the present study revealed that Dex could effectively inhibit the ferroptosis and vascular leakage of sepsis. The mechanism is related to metabolic reprogramming via Nrf2/G6PD and remodeling of mitochondrial morphology. These findings may provide a new direction for the treatment of sepsis.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval for the version to be published; and agreed to be accountable for all aspects of the work.
Funding
This study was supported by the Key Program of the National Natural Science Foundation of China (No.81730059).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
2. Fleischmann C, Scherag A, Adhikari NKJ, et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–272. doi:10.1164/rccm.201504-0781OC
3. Roberton T, Carter ED, Chou VB, et al. Early estimates of the indirect effects of the COVID-19 pandemic on maternal and child mortality in low-income and middle-income countries: a modelling study. Lancet Glob Health. 2020;8(7):e901–e908. doi:10.1016/S2214-109X(20)30229-1
4. Zheng DY, Zhang J, Zhang ZS, et al. Endothelial micro-vesicles induce pulmonary vascular leakage and lung injury during sepsis. Front Cell Dev Biol. 2020;8:643. doi:10.3389/fcell.2020.00643
5. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
6. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. 2018;29(1):61–74. doi:10.1089/ars.2017.7115
7. Guo LX, Zhang TP, Wang F, et al. Targeted inhibition of Rev-erb-α/β limits ferroptosis to ameliorate folic acid-induced acute kidney injury. Br J Pharmacol. 2021;178(2):328–345. doi:10.1111/bph.15283
8. Fang XX, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
9. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
10. Wei SS, Bi JB, Yang LF, et al. Serum irisin levels are decreased in patients with sepsis, and exogenous irisin suppresses ferroptosis in the liver of septic mice. Clin Transl Med. 2020;10(5):e173. doi:10.1002/ctm2.173
11. Nelson LE, Lu J, Guo TZ, et al. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98(2):428–436. doi:10.1097/00000542-200302000-00024
12. Yu TY, Liu D, Gao M, et al. Dexmedetomidine prevents septic myocardial dysfunction in rats via activation of α7nAChR and PI3K/Akt-mediated autophagy. Biomed Pharmacother. 2019;120:109231. doi:10.1016/j.biopha.2019.109231
13. Sun YB, Zhao HL, Mu DL, et al. Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 2019;10(3):167. doi:10.1038/s41419-019-1416-5
14. Wang CY, Yuan WL, Hu A, et al. Dexmedetomidine alleviated sepsis‑induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22(1):175–184. doi:10.3892/mmr.2020.11114
15. Brooks C, Wei QQ, Cho SG, et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119(5):1275–1285. doi:10.1172/JCI37829
16. Roy S, Kim D, Sankaramoorthy A. Mitochondrial structural changes in the pathogenesis of diabetic retinopathy. J Clin Med. 2019;8(9):1363. doi:10.3390/jcm8091363
17. She H, Zhu Y, Deng HY, et al. Protective effects of dexmedetomidine on the vascular endothelial barrier function by inhibiting mitochondrial fission via ER/Mitochondria contact. Front Cell Dev Biol. 2021;9:636327. doi:10.3389/fcell.2021.636327
18. Zhu Y, Wu HL, Wu Y, et al. Beneficial effect of intermedin 1-53 in septic shock rats: contributions of Rho kinase and Bkca pathway-mediated improvement in cardiac function. Shock. 2016;46(5):557–565. doi:10.1097/SHK.0000000000000639
19. Zhao HL, Zhu Y, Zhang J, et al. The beneficial effect of HES on vascular permeability and its relationship with endothelial glycocalyx and intercellular junction after hemorrhagic shock. Front Pharmacol. 2020;11:597. doi:10.3389/fphar.2020.00597
20. Zhao L, Wu Q, Wang XL, et al. Reversal of abnormal CD4+ T cell metabolism alleviates thyroiditis by deactivating the mTOR/HIF1a/Glycolysis pathway. Front Endocrinol. 2021;12:659738. doi:10.3389/fendo.2021.659738
21. Zhang S, Lu X, Hu CX, et al. Serum metabolomics for biomarker screening of esophageal squamous cell carcinoma and esophageal squamous dysplasia using gas chromatography-mass spectrometry. ACS Omega. 2020;5(41):26402–26412. doi:10.1021/acsomega.0c02600
22. Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1):3145. doi:10.1038/s41467-019-10991-7
23. Cazalis M-A, Lepape A, Venet F, et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp. 2014;2(1):20. doi:10.1186/s40635-014-0020-3
24. Shalova IN, Lim JY, Chittezhath M, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity. 2015;42(3):484–498. doi:10.1016/j.immuni.2015.02.001
25. Fonseka P, Pathan M, Chitti SV, et al. FunRich enables enrichment analysis of OMICs datasets. J Mol Biol. 2021;433(11):166747. doi:10.1016/j.jmb.2020.166747
26. Nguyen H, Tran D, Galazka JM, et al. CPA: a web-based platform for consensus pathway analysis and interactive visualization. Nucleic Acids Res. 2021;49(W1):W114–W124. doi:10.1093/nar/gkab421
27. Zhu H, Santo A, Jia ZQ, et al. GPx4 in bacterial infection and polymicrobial sepsis: involvement of ferroptosis and pyroptosis. React Oxyg Species. 2019;7(21):154–160. doi:10.20455/ros.2019.835
28. Gregorio AC, Rivera AKA, Lozano AJO, et al. Lipid metabolism and oxidative stress in HPV-related cancers. Free Radic Biol Med. 2021;172:226–236. doi:10.1016/j.freeradbiomed.2021.06.009
29. Tchouagué M, Grondin M, Glory A, et al. Heat shock induces the cellular antioxidant defenses peroxiredoxin, glutathione and glucose 6-phosphate dehydrogenase through Nrf2. Chem Biol Interact. 2019;310:108717. doi:10.1016/j.cbi.2019.06.030
30. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
31. Duan CY, Kuang L, Xiang XM, et al. Activated Drp1-mediated mitochondrial ROS influence the gut microbiome and intestinal barrier after hemorrhagic shock. Aging. 2020;12(2):1397–1416. doi:10.18632/aging.102690
32. Tang YC, Hsiao JR, Jiang SS, et al. c-MYC-directed NRF2 drives malignant progression of head and neck cancer via glucose-6-phosphate dehydrogenase and transketolase activation. Theranostics. 2021;11(11):5232–5247. doi:10.7150/thno.53417
33. Zhao Y, Kong GY, Pei WM, et al. Dexmedetomidine alleviates hepatic injury via the inhibition of oxidative stress and activation of the Nrf2/HO-1 signaling pathway. Eur Cytokine Netw. 2019;30(3):88–97. doi:10.1684/ecn.2019.0431
34. Gebel S, Diehl S, Pype J, et al. The transcriptome of Nrf2-/- mice provides evidence for impaired cell cycle progression in the development of cigarette smoke-induced emphysematous changes. Toxicol Sci. 2010;115(1):238–252. doi:10.1093/toxsci/kfq039
35. Kim EH, Ridlo MR, Lee BC, et al. Melatonin-Nrf2 signaling activates peroxisomal activities in porcine cumulus cell-oocyte complexes. Antioxidants. 2020;9(11):1080. doi:10.3390/antiox9111080
36. Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence. 2013;4(6):507–516. doi:10.4161/viru.24530
37. Yang D, Jiang YZ, Qian HX, et al. Silencing cardiac troponin i-interacting kinase reduces lipopolysaccharide-induced sepsis-induced myocardial dysfunction in rat by regulating apoptosis-related proteins. Biomed Res Int. 2021:5520051. doi:10.1155/2021/5520051
38. Liu PF, Feng YT, Li HW, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. 2020;25:10. doi:10.1186/s11658-020-00205-0
39. Duan CY, Wang L, Zhang J, et al. Mdivi-1 attenuates oxidative stress and exerts vascular protection in ischemic/hypoxic injury by a mechanism independent of Drp1 GTPase activity. Redox Biol. 2020;37:101706. doi:10.1016/j.redox.2020.101706
40. Sha JC, Zhang HY, Zhao Y, et al. Dexmedetomidine attenuates lipopolysaccharide-induced liver oxidative stress and cell apoptosis in rats by increasing GSK-3β/MKP-1/Nrf2 pathway activity via the α2 adrenergic receptor. Toxicol Appl Pharmacol. 2019;364:144–152. doi:10.1016/j.taap.2018.12.017
41. Xu DM, Zhou CB, Lin JY, et al. Dexmedetomidine provides protection to neurons against OGD/R-induced oxidative stress and neuronal apoptosis. Toxicol Mech Methods. 2021;31(5):374–382. doi:10.1080/15376516.2021.1888363
42. Ma HD, Wang XD, Zhang WL, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in Type 2 diabetic osteoporosis. Oxid Med Cell Longev. 2020:9067610. doi:10.1155/2020/9067610
43. Song JE, Alves TC, Stutz B, et al. Mitochondrial fission governed by Drp1 regulates exogenous fatty acid usage and storage in hela cells. Metabolites. 2021;11(5):322. doi:10.3390/metabo11050322
44. Axelrod CL, Fealy CE, Erickson ML, et al. Lipids activate skeletal muscle mitochondrial fission and quality control networks to induce insulin resistance in humans. Metabolism. 2021;121:154803. doi:10.1016/j.metabol.2021.154803
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.