")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 7
Profile of rociletinib and its potential in the treatment of non-small-cell lung cancer
Authors Tran P, Klempner S
Received 25 March 2016
Accepted for publication 2 May 2016
Published 18 July 2016 Volume 2016:7 Pages 91—97
DOI https://doi.org/10.2147/LCTT.S94337
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
Phu N Tran,1 Samuel J Klempner2,3
1Division of Hematology/Oncology, University of California Irvine, Irvine, CA, 2Angeles Clinic and Research Institute, 3Cedars-Sinai Medical Center, Los Angeles, CA, USA
Abstract: Patients with non-small-cell lung cancer (NSCLC) harboring activating mutations in EGFR benefit from treatment with EGFR small-molecule tyrosine-kinase inhibitors. However, the development of acquired resistance to EGFR inhibitors is universal and limits treatment efficacy. Over half of patients receiving first-generation EGFR inhibitors (erlotinib and gefitinib) develop resistance via the gatekeeper EGFR T790M (EGFRT790M) mutation, and therapies able to overcome T790M-mediated resistance have been an unmet need in NSCLC. Rociletinib (CO-1686) is a third-generation small-molecule EGFR inhibitor with potent activity against EGFRT790M currently in advanced clinical development in NSCLC. Early clinical data suggested significant activity in EGFR-mutant NSCLC harboring T790M alterations. However, important questions regarding side-effect profile, comparability to competitor compounds, acquired resistance, EGFR-therapy sequencing, and combination therapies remain. Here, we review the available preclinical and clinical data for rociletinib, highlight the comparison to other third-generation EGFR inhibitors, and discuss resistance implications and future directions in NSCLC.
Keywords: lung cancer, rociletinib, EGFR, T790M, CO-1686, resistance, tyrosine-kinase inhibitor
Introduction
Non-small-cell lung cancer (NSCLC) remains the leading cause of global cancer-related mortality, with a 5-year survival rate of less than 20% in US patients.1 The discovery of activating mutations in EGFR and subsequent development of small-molecule EGFR inhibitors revolutionized the management of EGFR-mutant NSCLC. Activating mutations in EGFR are detected in approximately 30%–40% of NSCLC in Asian patients and 10% of NSCLC in Caucasian patients.2 An in-frame deletion in exon 19 (exon19del) and the L858R point mutation in exon 21 constitute nearly 90% of known EGFR alterations, though more sensitive and comprehensive methods continue to identify new therapeutically relevant EGFR alterations.3,4 The IPASS and EURTAC trials established and confirmed the benefit of the first-generation EGFR tyrosine-kinase inhibitors (TKIs) gefitinib and erlotinib over chemotherapy in patients harboring sensitizing EGFR alterations.5,6 Despite response rates of 50%–70% for first-generation EGFR inhibitors in EGFR exon19del and L858R patients, the median progression-free survival (PFS) is 9–13 months, highlighting the need for additional therapies.6–8
Progression-biopsy samples from patients no longer responding to erlotinib and gefitinib elucidated the landscape of resistance to first-generation EGFR TKIs. Resistance mechanisms can be classified broadly into target modification, bypass signaling, and phenotypic transformation.9 Target modification via development of an EGFRT790M mutation is responsible for 50%–60% of acquired resistance to first-generation inhibitors.10 EGFR-activating mutations, such as L858R, G719S, and exon 19 deletions decrease the affinity of EGFR for adenosine triphosphate (ATP), rendering them more susceptible to competitive inhibition by TKIs relative to wild-type (WT) EGFR (EGFRWT). The threonine (T) at position 790 (T790) lies near the passage to the kinase hydrophobic back pocket (gatekeeper), and substitution with the bulkier methionine (M) (T790M) mediates resistance partly by inducing steric hindrance in the ATP-binding pocket and preventing inhibitor binding.11 Additionally, the cis (same allele) addition of T790M increases the affinity for ATP of mutant EGFR, restoring it to the level of the WT kinase, leading to reduced TKI efficacy.12 Beyond a T790M-mediated mechanism, preclinical work and progression biopsies from patients on first-generation EGFR TKIs have revealed MET amplification, ERBB2 amplification, SCLC transformation, epithelial–mesenchymal changes, and acquired RET rearrangements as resistance mechanisms.13–18 Tumor heterogeneity, clonal evolution, and the role of small populations with de novo EGFRT790M alterations are also emerging as an important biologic underpinning to clinical resistance in EGFR-mutant NSCLC.19–21
Prior to the advent of third-generation EGFR inhibitors, patients found to harbor an EGFRT790M mutation after progression on first-generation EGFR inhibitors had limited options, including standard second-line chemotherapy, platinum doublets, and EGFR TKIs combined with chemotherapy.22–24 The second-generation EGFR TKIs, including afatinib, a highly selective and irreversible pan-HER inhibitor, demonstrated preclinical activity in EGFRT790M models; however, in the Lux-Lung 4 trial, 62 patients with advanced NSCLC who progressed on either erlotinib and/or gefitinib received afatinib with limited benefit.25–27 A single patient with EGFRT790M achieved stable disease for 9 months. The addition of cetuximab to afatinib improved the overall response rate (ORR) to 29% (32% RR in T790M+ and 25% in T790M– tumors) with a median PFS of 4.7 months.28 However, this regimen was associated with significant cutaneous, mucosal, and gastrointestinal toxicities, limiting its clinical impact. More recently, the addition of bevacizumab to erlotinib has demonstrated activity in EGFRT790M, but ongoing trials are needed for confirmation.29 Given the underwhelming activity of earlier approaches, novel compounds capable of overcoming EGFRT790M remain a significant unmet need in NSCLC.
Rociletinib characterization and preclinical development
Rociletinib (CO1686) is a potent 5-CF3 2,4-diaminopyrimidine-based molecule that consists of a reactive acrylamide group, an aminopyrimidine group, and a piperazine ring.30 The acrylamide group forms a covalent bond with cysteine (C) 797 in the ATP-binding pocket of the EGFR kinase and is responsible for the irreversible inhibition. The aminopyrimidine binds to the hinge residue methionine (M) 793 through hydrogen bonding, and the 5-substituent of the aminopyrimidine points to the gatekeeper residue (methionine in T790M) and forms strong hydrophobic interaction.31 In the EGFRWT kinase, the smaller threonine (T) residue at position 790 results in weaker hydrophobic forces, and contributes to the selectivity for the T790M mutant over EGFRWT.
In preclinical kinase assays, compound EGFRL858R/T790M mutant kinases were 22-fold more sensitive to rociletinib versus EGFRWT, with inactivation rate constant to binding constant ratios of 2.41×105 M–1S–1 and 1.12×104 M–1S–1, respectively.30 In the in vitro kinase assay, the half-maximal inhibitory concentration (IC50) of rociletinib for EGFRL858R/T790M and EGFRWT were <0.51 nM and 6 nM, respectively. Cellular growth assays reported IC50 in the 100–140 nM range (Table 1). Kinase profiling demonstrated activity against EGFR exon 19 deletion, T790M, L858R/T790M, and L858R, with weak inhibition of other kinases, including FAK, CHK2, ERBB4, and JAK3. In EGFRT790M, rociletinib demonstrated dose-dependent tumor response and improved activity compared to erlotinib and afatinib, a finding confirmed in transgenic mouse studies.32 Mice carrying EGFRL858R mutant tumors had responses to both erlotinib and rociletinib, while mice carrying EGFRL858R/T790M tumors displayed responses to rociletinib only. Overall, preclinical studies indicate strong efficacy signals with favorable early toxicity profile, warranting further clinical investigation.
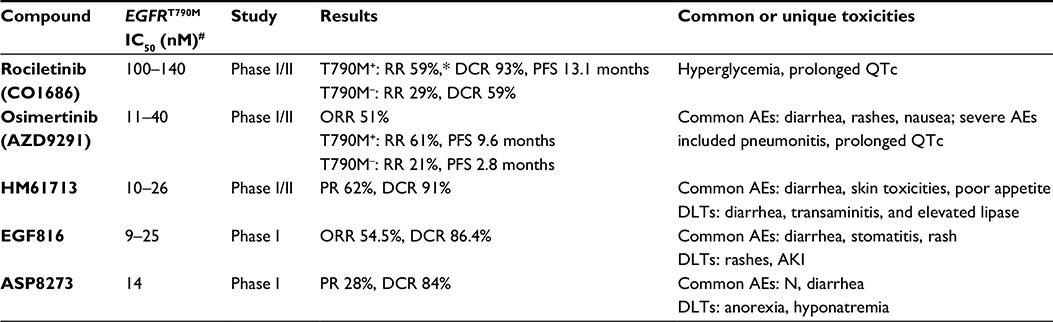 | Table 1 Comparison of the preclinical and clinical features of small-molecule EGFR inhibitors with activity against the EGFRT790M resistance mutation Notes: *As reported in the original publication,29 and subsequently revised down; #range reported to reflect testing in differing cell lines and compound mutants L858R/T790M and exon19del/T790M. Abbreviations: AKI, acute kidney injury; IC50, half-maximal inhibitory concentration (to achieve 50% inhibition in cellular growth assays); N, nausea; RR, response rate; AEs, adverse events; DCR, disease-control rate; PFS, progression-free survival; QTc, corrected QT; ORR, overall response rate; DLTs, dose-limiting toxicities; PR, partial response. |
Rociletinib in NSCLC: clinical activity
A Phase I/II rociletinib clinical trial investigated the safety, maximum tolerated dose, recommended Phase II dose, and early antitumor activity in patients with advanced EGFR-mutated NSCLC who had been previously treated and progressed on either first- or second-generation EGFR TKIs.33 This study enrolled 130 patients at ten institutions, and patients received oral rociletinib on daily, twice-daily, or thrice-daily dosing schedules. The maximum tolerated dose was not reached, and the most consistently effective dose was 900 mg twice daily in the initial free-base formulation. The study subsequently switched to a newer rociletinib hydrobromic acid form, which had improved bioavailability: 92 of 130 patients received therapeutic doses and were included in the efficacy analysis. Among 46 patients harboring EGFRT790M mutations, the investigator-assessed RR was 59% and disease-control rate (DCR) 93% (Table 1).33 RRs were similar between patients with original exon19del and L858R EGFR mutations. The median PFS in the T790M+ subset was 13.1 months at the time of analysis. In EGFR-mutant but T790M– (n=17) patients, the RR was 29% and DCR 59%. The initially reported data showed comparable activity to the simultaneously published competitor compound osimertinib (AZD9291).34 The most common reported toxicities from the Phase I/II study included nausea (35%), fatigue (24%), diarrhea (22%), and poor appetite (20%). Although rociletinib was generally well tolerated in early studies, several interesting side effects were noted, including hyperglycemia, corrected QT (QTc) prolongation (12%), and development of cataracts. Hyperglycemia occurred in 43% of patients, caused by a rociletinib metabolite (M502) that inhibited insulin-like growth factor (IGF)-receptor and insulin-receptor kinases and induced glucose intolerance in rodents in preclinical studies.33,35 Rociletinib quickly undergoes amide hydrolysis into the metabolite M502 (69%), which can be converted to the M460 (3%) metabolite by the same reaction.36 Time-dependent exposure to M502 and M460 is responsible for inducing hyperglycemia and QTc prolongation, respectively. While the half-life of rociletinib is 3.7 hours, the half-lives of M502 and M460 are up to 20 hours and 51 hours, respectively. Both M502 and M460 are acetylated to subsequent metabolites by N-acetyltransferase (NAT2).36 Interestingly, NAT2 exhibits significant polymorphisms across different racial/ethnic groups, leading to different rates of acetylation (eg, slow versus rapid acetylators).37 Asians are generally faster acetylators, while Caucasians, Hispanics, and African-Americans are slower acetylators. Therefore, Asian populations may theoretically experience fewer metabolite-mediated toxicities from rociletinib. The current published data do not report stratification of RRs or toxicities by race/ethnicity. Overall, there was no study discontinuation due to toxicities, though dose reduction was needed in 44 of 92 (48%) patients.
The clinical activity of rociletinib was refined in an updated release from Clovis Oncology, in which the confirmed RR was revised down to 28% for the 79 patients in the 500 mg-dose arm and 34% for the 170 patients in the 625 mg-dose group. As reported, a significant portion of the initially described responses were not confirmed on subsequent imaging, largely due to disease progression (commonly brain metastases).38 Stratification by T790M status was not reported in conjunction with this release. In the updated release, hyperglycemia and prolonged QTc remained important treatment-related toxicities, with rates of grade 3–4 adverse events at 34% and 33%, respectively.36 Additionally, cases of potentially life-threatening arrhythmias, including torsades de pointes (n=1), ventricular arrhythmia (n=4), atrial fibrillation (n=5), and supraventricular tachycardia (n=4), were observed. The toxicity profile was similar across the dose range (500–1,000 mg twice daily), although the proportion of patients requiring three or four dose reductions was higher for patients given 625 mg twice daily or higher.
In April 2016, based on available data, an independent panel of experts voted 12-1 against giving rociletinib an accelerated approval and recommended waiting for the results and safety analysis from the ongoing Tiger 3 trial, comparing rociletinib to chemotherapy in both T790M+ and T790M– patients with acquired TKI resistance after failure of one or more previous EGFR TKIs and platinum-doublet chemotherapy.
Additional third-generation EGFR inhibitors
Several other EGFR TKIs able to overcome the EGFRT790M resistance mutation are in advanced stages of development, and on November 13, 2015 the US Food and Drug Administration (FDA) approved osimertinib (AZD9291) for patients with EGFRT790M+ NSCLC who have progressed on prior EGFR TKI therapy. AZD9291 is a structurally distinct mutant EGFR-selective monoanilinopyrimidine that covalently binds the EGFRL858R/T790M cysteine at position 797.39 In a large Phase I trial, the ORR was 51% in patients who had failed prior EGFR TKIs.40 In patients with centrally confirmed EGFRT790M, RR and median PFS were 61% and 9.6 months (95% confidence interval [CI]: 8.3–not reached), respectively. In contrast, the RR and median PFS were 21% (95% CI: 12–34) and 2.8 months (95% CI: 2.1–4.3) for patients with EGFRT790M– tumors (Table 1). AZD9291 was generally well tolerated, with common toxicities including diarrhea (47%), rashes (40%), nausea (22%), anorexia (21%), xerosis (20%), pruritus (19%), and fatigue (17%). There were six patients with hyperglycemia, and eleven patients had non-clinically significant QTc prolongation. Six patients developed pneumonitis-like toxicity requiring drug discontinuation.40
HM61713 (BI1482694) is an irreversible EGFRT790M inhibitor with potent and selective inhibition in EGFR-mutant models (Table 1).41 Results from an ongoing Phase I/II HM-EMSI-101 study evaluating the safety and efficacy of HM61713 on 173 patients with centrally confirmed T790M+ NSCLC who had failed prior TKIs were presented at the 2015 American Society of Clinical Oncology Annual Meeting.42 Most common adverse events included diarrhea, rash, skin exfoliation, nausea, pruritus, decreased appetite, and dry skin. Dose-limiting toxicities included abdominal pain, diarrhea, idiosyncratic drug reaction, transaminitis, and elevated lipase. Over half (58.5%) of patients achieved a partial response (PR), and the DCR was 97.1% in a subset of 37 evaluable patients. Updated interim data presented at the European Society for Medical Oncology meeting in December 2015 showed PR and DCR rates of 62% and 91%, respectively, among 69 evaluable patients.43,44 Consequently, the FDA granted HM61713 breakthrough-therapy designation, and the global second-line ELUXA 1 (HM-EMSI-202) study is ongoing (NCT02485652).
EGF816 is an irreversible EGFR TKI that has nanomolar inhibitory potency against L858R/T790M+ and ex19del/T790M+, with up to 60-fold selectivity over EGFRWT in vitro (Table 1). EGF816 suppresses phosphorylated EGFR levels and inhibits proliferation of cells harboring EGFRT790M mutations at IC50 of 3 nM and 25 nM, respectively.45 In an ongoing Phase I multicenter dose-escalation study in advanced NSCLC patients with confirmed EGFRT790M status, the most common reported toxicities were diarrhea (25%), stomatitis (22.5%), rash (17.5%), and pruritus (15%) in 40 patients who were evaluated for safety. Among 22 evaluable patients, the ORR and DCR were 54.5% and 86.4%, respectively.46 Other trials are investigating the efficacy and safety of combining EGF816 and INC280 (MET inhibitor) (NCT02335944) and EGF816 with the anti-PD1 antibody nivolumab (NCT02323126).
The third-generation EGFR TKI ASP8273 has activity in T790M+ tumors with IC50 values of 8–33 nM (Table 1).47 ASP8273 selectively inhibits mutant EGFR by covalently binding to C797 (like rociletinib), and has demonstrated complete tumor regression in mouse xenograft models. Importantly, ASP8273 has demonstrated activity in a cell line resistant to AZD9291 and CO1686.48 In a Phase I study, 35 previously treated NSCLC patients with activating EGFR mutations were enrolled, and side effects were generally mild, including nausea (25.7%) and diarrhea (17.1%).49 Dose-limiting toxicities at 400 mg were grade 3 hyponatremia and grade 3 anorexia. Hyperglycemia and QTc prolongation were not observed. Among 25 subjects evaluable for response, seven (28%) patients had PR and 15 (56%) achieved stable disease. Astellas is investigating the role of ASP8273 as first-line therapy compared to erlotinib or gefitinib in an ongoing Phase III trial (NCT02588261). Other compounds in earlier stages of development have shown comparable early efficacy, and therapeutic choices are likely to be impacted by side-effect profiles, PFS, and ultimately overall survival data.
Rociletinib resistance
Despite the reported clinical efficacy of the third-generation EGFR inhibitors, acquired resistance is expected to be universal, and has recently been described. The acquisition of a substitution in EGFR at serine (S) 797 for cysteine (C) (C797S) causes resistance to C01686, AZD9291, and HM61713 in patients.50–52 The C797S substitution interferes with the covalent bond formation between the cysteine residue of the acrylamide group of rociletinib, reducing inhibitor binding and efficacy, and would be predicted as a vulnerability. Despite limited data, the C797S mutation appears more common in exon19del/T790M+ tumors, and the C797S mutation was observed in seven of ten exon19del/T790M+ patients but none of the six patients with L858R/T790M+ mutations who progressed on AZD9291.51,53 Interestingly, a small series suggests that osimertinib may be able to induce response after progression on rociletinib, highlighting differences between these two compounds.54
Recently, Niederst et al indicated that the genomic configuration (cis versus trans) of the T790M and C797S mutations could impact response to TKI therapy. Specifically, when the mutations were in trans (on different alleles), the combination of first- and third-generation EGFR TKIs abrogated the resistance. If the mutations were in cis (on the same allele), then the cells were resistant to a combination of EGFR TKIs.55 While this level of genomic detail is not yet used to guide therapies, one can appreciate how cis and trans conformation may impact therapeutic sequence.
EGFR mutations L718Q and K844V, which appear to create steric hindrance with rociletinib (but not AZD9291) have been observed in vitro, and may ultimately impact clinical treatment choices.50 Quinazoline-based EGFR inhibitors, such as gefitinib and afatinib, are able to overcome the C797S, L781Q, and K844V alterations, and appear active in T790M– tumors harboring these rociletinib-resistant mutations.50 In models studying compound resistance mutations involving EGFRT790M with concurrent C797S, L718Q, or K844V alterations, the compound mutants were resistant to all known EGFR inhibitors in vitro. Cetuximab appeared to retain some activity in a subset of cells with tertiary EGFRL858R/T790M/C797S mutations, but further in vivo studies are required to confirm this observation.50
Treatment of T790M+ patients with rociletinib may also result in “loss” of the T790M+ population or selection/emergence of T790 WT clones, as has been observed in progression samples from six of 12 patients in one series.56 This phenomenon was also observed in some T790M+ patients who had progressive disease on AZD9291.51 Other mechanisms of rociletinib resistance include histologic transformation and bypass resistance similar to other EGFR TKIs.30,57,58 Walter et al reported rociletinib-resistant cells exhibited a spindle-like morphology, overexpressed genes involved in epithelial–mesenchymal transition, had higher level of basal phosphorylated AKT, and were sensitive to a combination of the AKT inhibitors MK2206 and GDC0068.30 SCLC transformation was observed in two of 12 patients who had progression on rociletinib.
Resistance observations have important implications for sequential EGFR TKI therapy. For example, treatment of EGFR-mutant NSCLC tumor cells with reversible quinazoline-based TKIs leads mainly to secondary T790M mutation, whereas up-front treatment with T790M-potent EGFR TKIs prevents emergence of the EGFRT790M mutation and shifts toward resistance mechanisms involving IGF1-receptor and mitogen-activated protein kinases (MAPK)-pathway activation.57,58 These preclinical models suggest that combining T790M-potent EGFR TKIs with either MEK or IGF1T inhibitors may prevent/delay the emergence of drug-resistant clones. However, it may be advantageous to begin with first-generation inhibitors, given the emerging efficacy of third-generation compounds. The wide array of potential resistance mechanisms to T790M-active EGFR TKIs underscores the importance of sequential biopsies and extended molecular testing to identify the underlying mechanism of resistance and guide subsequent therapy.
Conclusion and future directions
Here, we have provided an overview of the preclinical and clinical data for rociletinib (CO1686) in NSCLC (Tables 1 and 2). In the rapidly evolving landscape of EGFR-mutant, T790M+ NSCLC, rociletinib showed significant early promise, but the revised response and toxicity data highlight the importance of rigorous prospective trials to validate early data in drug development. Although direct head-to-head comparison among third-generation inhibitors is not available, the absolute RR and side-effect profile of rociletinib has raised concerns about the optimal pathway forward. Data addressing NAT2-polymorphism impact on rociletinib response and toxicity may suggest populations that derive benefit with limited toxicity. Incorporation of early response biomarkers, including circulating tumor DNA for EGFRT790M burden are likely to further refine optimal patient populations. Early toxicity-recognition and management strategies may reduce the hyperglycemia rates in the ongoing rociletinib studies. Ultimately, larger-outcome data sets from Phase II and III studies will define the role of rociletinib in EGFRT790M+ NSCLC.
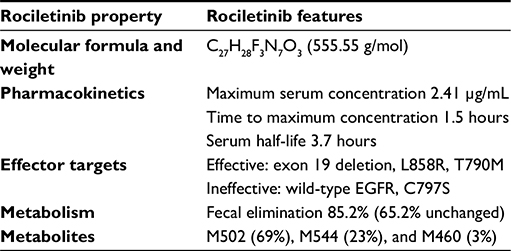 | Table 2 Structural and pharmacologic properties of the third-generation EGFR inhibitor rociletinib |
Acknowledgment
The authors would like to recognize the important contributions from researchers whose work could not be cited due to space constraints.
Disclosure
The authors report no conflicts of interest in this work. Subsequent to the acceptance of this manuscript Clovis Oncology stopped further clinical development of rociletinib.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–967. | ||
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. | ||
Schrock AB, Frampton GM, Herndon D, et al. Comprehensive genomic profiling identifies frequent drug sensitive EGFR exon 19 deletions in NSCLC not identified by prior molecular testing. Clin Cancer Res. Epub 2016 Mar 1. | ||
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. | ||
Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. | ||
Zhou C, Wu YL, Chen G, et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann Oncol. 2015;26:1877. | ||
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. | ||
Lovly CM, Shaw AT. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res. 2014;20(9):2249–2256. | ||
Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | ||
Yu HA, Riely GJ, Lovly CM. Therapeutic strategies utilized in the setting of acquired resistance to EGFR tyrosine kinase inhibitors. Clin Cancer Res. 2014;20(23):5898–5907. | ||
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | ||
Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. | ||
Chung JH, Rho JK, Xu X, et al. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer. 2011;73(2):176–182. | ||
Suda K, Tomizawa K, Fujii M, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6(7):1152–1161. | ||
Klempner SJ, Bazhenova LA, Braiteh FS, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer. 2015;89(3):357–359. | ||
Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. | ||
Piotrowska Z, Niederst MJ, Karlovich CA, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5(7):713–722. | ||
Soucheray M, Capelletti M, Pulido I, et al. Intratumoral heterogeneity in EGFR-mutant NSCLC results in divergent resistance mechanisms in response to EGFR tyrosine kinase inhibition. Cancer Res. 2015;75(20):4372–4383. | ||
Gridelli C, Ciardiello F, Gallo C, et al. First-line erlotinib followed by second-line cisplatin-gemcitabine chemotherapy in advanced non-small-cell lung cancer: the TORCH randomized trial. J Clin Oncol. 2012;30(24):3002–3011. | ||
Halmos B, Pennell NA, Fu P, et al. Randomized phase II trial of erlotinib beyond progression in advanced erlotinib-responsive non-small cell lung cancer. Oncologist. 2015;20(11):1298–1303. | ||
Soria JC, Wu YL, Nakagawa K, et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 2015;16(8):990–998. | ||
Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102(21):7665–7670. | ||
Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. | ||
Katakami N, Atagi S, Goto K, et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol. 2013;31(27):3335–3341. | ||
Janjigian YY1, Smit EF2, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4(9):1036–1045. | ||
Stahel RA, Dafni U, Gautschi O, et al. A phase II trial of erlotinib (E) and bevacizumab (B) in patients with advanced non-small-cell lung cancer (NSCLC) with activating epidermal growth factor receptor (EGFR) mutations with and without T790M mutation: the Spanish Lung Cancer Group (SLCG) and the European Thoracic Oncology Platform (ETOP) BELIEF trial. Poster presented at: 2015 European Cancer Congress; September 25–29, 2015; Vienna, Austria. | ||
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. | ||
Sjin RT, Lee K, Walter AO, et al. In vitro and in vivo characterization of irreversible mutant-selective EGFR inhibitors that are wild-type sparing. Mol Cancer Ther. 2014;13(6):1468–1479. | ||
Simmons AD, Haringsma HJ, Walter AO, Tham Sjin RT, Weaver Z, Allen A, Harding TC. Abstract B120: Preclinical assessment of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Mol Cancer Ther. 2013;12(Suppl 11):B120–B120. | ||
Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–1709. | ||
Janne, PA, Ramalingam SS, Yang JCH, et al. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients (pts) with EGFR inhibitor-resistant non-small cell lung cancer (NSCLC). ASCO Annual Meeting Proceedings. 2014:32(15)suppl. Available from: http://meetinglibrary.asco.org/content/129721-144. Accessed July 5, 2016. | ||
Villadolid J, Ersek JL, Fong MK, Sirianno L, Story ES. Management of hyperglycemia from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) targeting T790M-mediated resistance. Transl Lung Cancer Res. 2015;4(5):576–583. | ||
US Food and Drug Administration. FDA briefing document: NDA 208542 – rociletinib. 2016. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM494782.pdf. Accessed May 27, 2016. | ||
Walker K, Ginsberg G, Hattis D, Johns DO, Guyton KZ, Sonawane B. Genetic polymorphism in N-acetyltransferase (NAT): population distribution of NAT1 and NAT2 activity. J Toxicol Environ Health B Crit Rev. 2009;12(5–6):440–472. | ||
BusinessWire. Clovis Oncology announces regulatory update for rociletinib NDA filing. 2015. Available from: http://www.businesswire.com/news/home/20151116005513/en. Accessed May 27, 2016. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. | ||
Ou SH, Soo RA. Dacomitinib in lung cancer: a “lost generation” EGFR tyrosine-kinase inhibitor from a bygone era? Drug Des Devel Ther. 2015;9:5641–5653. | ||
Park KC. Updated safety and efficacy results from phase I/II study of HM61713 in patients (pts) with EGFR mutation positive non-small cell lung cancer (NSCLC) who failed previous EGFR-tyrosine kinase inhibitor (TKI). 2015. Available from: http://meetinglibrary.asco.org/content/113498?media=vm. Accessed May 27, 2016. | ||
Lee JS, Park KC, Han JY, et al. Clinical activity and safety of the EGFR mutant-specific inhibitor, BI1482694, in patients (pts) with T790M-positive NSCLC. Ann Oncol. 2015;26 Suppl 9:ix128–ix129. | ||
Jänne PA, Son J, Voccia I, Uttenreuther-Fische M, Park K. Phase II study of BI1482694 in patients (pts) with T790M-positive non-small cell lung cancer (NSCLC) after treatment with an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI). Ann Oncol. 2015;26 Suppl 9:ix145. | ||
Jia Y, Juarez J, Li J, et al. EGF816 exerts anticancer effects in non-small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res. 2016;76(6):1591–1602. | ||
Tan DS, Seto T, Leighl NB, et al. First-in-human phase I study of EGF816, a third generation, mutant-selective EGFR tyrosine kinase inhibitor, in advanced non-small cell lung cancer (NSCLC) harboring T790M. J Clin Oncol. 2015;33 Suppl:8013. | ||
Sakagami H, Konagai S, Yamamoto H, et al. ASP8273, a novel mutant-selective irreversible EGFR inhibitor, inhibits growth of non-small cell lung cancer (NSCLC) cells with EGFR activating and T790M resistance mutations. Cancer Res. 2014;74(19 Suppl):1728. | ||
Konagai S, Sakagami H, Yamamoto H, et al. ASP8273 selectively inhibits mutant EGFR signal pathway and induces tumor shrinkage in EGFR mutated tumor models. Cancer Res. 2015;75(15 Suppl):2586. | ||
Yu HA. Phase I dose escalation study of ASP8273, a mutant-selective irreversible EGFR inhibitor, in subjects with EGFR mutation positive NSCLC. J Clin Oncol. 33, 2015(suppl; abstr 8083). | ||
Ercan D, Choi HG, Yun CH, et al. EGFR mutations and resistance to irreversible pyrimidine based EGFR inhibitors. Clin Cancer Res. 2015;21(17):3913–3923. | ||
Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–562. | ||
Song HN, Jung KS, Yoo KH, et al. Acquired C797S mutation upon treatment with a T790M-specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancer. J Thorac Oncol. 2016;11(4):e45–e47. | ||
Yu HA, Tian SK, Drilon AE, et al. Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. 2015;1(7):982–984. | ||
Sequist LV, Piotrowska Z, Niederst MJ, et al. Osimertinib responses after disease progression in patients who had been receiving rociletinib. JAMA Oncol. 2016;2(4):541–543. | ||
Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924–3933. | ||
Piotrowska Z, Niederst MJ, Karlovich CA, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5(7):713–722. | ||
Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2(10):934–947. | ||
Cortot AB, Repellin CE, Shimamura T, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73(2):834–843. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.