")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 19
Prevalence of Cardiovascular Disease and Rate of Major Adverse Cardiovascular Events in Severe Alpha-1 Antitrypsin Deficiency COPD
Authors Ellis P, Bailey E, Choate R , Holm KE , Sandhaus RA, Turner AM , Newnham M
Received 27 August 2023
Accepted for publication 5 December 2023
Published 17 January 2024 Volume 2024:19 Pages 149—159
DOI https://doi.org/10.2147/COPD.S419846
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Paul Ellis,1,2 Emily Bailey,2 Radmila Choate,3 Kristen E Holm,4,5 Robert A Sandhaus,5,6 Alice M Turner,1,2 Michael Newnham1,2
1Institute of Applied Health Research, University of Birmingham, Birmingham, UK; 2University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK; 3University of Kentucky College of Public Health, Lexington, KY, USA; 4Division of Neurology and Behavioural Health, National Jewish Health, Denver, CO, USA; 5AlphaNet, Kissimmee, FL, USA; 6Division of Pulmonary, Critical Care and Sleep Medicine, National Jewish Health, Denver, CO, USA
Correspondence: Paul Ellis, Email [email protected]
Aim: Alpha-1 antitrypsin deficiency is an autosomal co-dominant condition that predisposes individuals to early-onset emphysema. As with COPD, AATD-COPD is associated with pulmonary exacerbations, which impacts on overall mortality and quality of life. Though there is evidence that COPD is associated with a higher prevalence of cardiovascular disease and major adverse cardiovascular events (MACE), it is unclear if this is true for patients with AATD-COPD.
Methods: Prevalence of cardiovascular disease was determined in two separate severe AATD cohorts: AlphaNet, USA and the Birmingham AATD registry, UK. All patients had preexisting lung disease. Cardiovascular disease was defined as presence of any of the following: heart failure, ischaemic heart disease, atrial fibrillation, stroke, and myocardial infarction. A Cox proportional hazards model was used to assess the impact of prior cardiovascular disease and frequent exacerbator phenotype on risk of future MACE.
Results: Out of 3493 patients with severe AATD, 14.7% had prior cardiovascular disease, including stroke (2.3%), myocardial infarction (2.2%), and heart failure (2.5%). Frequent exacerbators were more likely to have preexisting cardiovascular disease compared with those with one or no exacerbations in the preceding year (63% vs 44.8%, p = 0.001). There was increased risk of future MACE in frequent exacerbators (HR 1.85, 95% CI 1.24 to 2.75), former and current smokers (HR 1.80, 95% CI 1.07 to 3.02, p = 0.026, and HR 4.04, 95% CI 1.44 to 11.32, p = 0.008, respectively), and those with prior cardiovascular disease (HR 3.81, 95% CI 2.60 to 5.58, p < 0.001).
Conclusion: In severe AATD-COPD, MACE are associated with an increased exacerbation frequency, previous cardiovascular disease, and a history of smoking.
Keywords: chronic obstructive pulmonary disease, COPD, alpha-1 antitrypsin deficiency, AATD, cardiovascular disease
Introduction
Alpha-1 antitrypsin deficiency (AATD) is an autosomal co-dominant disorder characterised by reduced serum levels of alpha-1 antitrypsin (AAT).1 AAT is an important serine protease inhibitor which has a role in limiting the destructive effects of proteases. Patients with severe forms of AATD, most commonly PiZZ, have significantly reduced levels of circulating AAT.2 Uninhibited activity of proteolytic enzymes leads to accelerated elastin degradation within alveolar tissue, resulting in early-onset pulmonary emphysema.3 The presenting features of AATD-COPD are like that of usual COPD and as such are treated in a similar way. AATD patients with progressive disease may be offered AAT augmentation therapy to slow the rate of emphysema decline.4 As with usual COPD, patients with AATD-COPD suffer with pulmonary exacerbations, which are longer in duration and feature higher levels of inflammation compared to usual COPD.5
Cardiovascular disease (CVD) is the leading cause of mortality worldwide, with over 85 million people affected by CVD in Europe alone.6 CVD is associated with an increased prevalence in patients with COPD, independent of common cardiovascular risk factors such as smoking and sedentary lifestyle.7 The prevalence of ischaemic heart disease in patients with COPD was 2.28 times higher compared with non-COPD individuals in one meta-analysis.8
COPD patients with co-existing CVD often experience higher rates of morbidity, including worsening quality of life, dyspnoea, and exercise tolerance9 in addition to a higher rate of hospital admissions.10 The association between CVD and COPD has been summarised previously7 and may be the result of shared risk factors (genetic, environmental exposures) or shared pathophysiological mechanisms, including increased inflammation during stable COPD and pulmonary exacerbations.
The role of increased inflammation in COPD is widely cited as a potential biological mechanism to explain this association.11–13 Translocation of inflammatory cytokines, such as metalloproteinases and IFN-γ, from inflamed lungs into the systemic circulation may activate macrophages and other inflammatory cells within the vascular intima and accelerate atherosclerosis development,14 and reduce plaque stability.15 Thrombosis at the site of plaque rupture within coronary arteries leads to acute coronary syndrome.15 In addition, enhanced systemic elastin degradation is an important contributing factor that may link emphysema and CVD in both non-AATD and AATD-COPD.16
Major adverse cardiovascular events (MACE), such as cardiovascular death and myocardial infarction, are associated with pulmonary exacerbations17,18 which feature in both AATD-COPD and usual COPD.19
Despite the growing evidence of CVD in COPD, there are a limited number of studies investigating the natural history of CVD in patients with AATD-COPD.
We aim to investigate the prevalence of CVD and the relationship between exacerbation frequency and future rates of MACE for patients with severe AATD-related COPD.
Methods
We analysed cardiovascular disease prevalence and associations of major adverse cardiovascular events (MACE), with particular focus on the frequent exacerbator phenotype (≥2 exacerbations per year), in adult patients (age ≥18 years) with severe AATD (PiZZ or other rarer phenotypes with severe AATD) with COPD from two prospective AATD cohorts: AlphaNet, based in the USA, and the Birmingham AATD registry, UK. Methods for data collection in both cohorts have been summarised previously.20
Study Cohorts
AlphaNet provides disease management for patients prescribed AAT augmentation therapy in the USA. All individuals self-reported at baseline that they have lung disease. History of self-reported cardiovascular disease and incidence of MACE were collected via quarterly structured telephone interviews in addition to information provided at baseline. Baseline telephone interviews were conducted between 2008 and the end of 2016.
The Birmingham AATD registry includes augmentation-naïve patients with AATD. Patients were referred from primary or secondary care for annual review. Follow-up occurred between 1996 and 2016 and included medical history, physical exam, post bronchodilator lung function tests, and quality of life scores. Evidence of COPD was defined with FEV1/FVC ratio <0.7. Further details can be found in the Supplementary Methods. Separate consent was gained from both cohorts, and the study protocol was approved by the Western Institutional Review Board (WIRB; IRB tracking number 20181997) following independent ethical review in both the United States and United Kingdom (REC 3359a, South Birmingham Research Ethics Committee). All data accessed complied with HIPAA and NIHR Good Clinical Practice guidelines.
Definition of Cardiovascular Disease and MACE
For this analysis, baseline is defined as the time of enrolment in AlphaNet services or the first review at a specialist referral appointment. Follow-up includes the duration of which data was available for analysis.
The frequent exacerbator phenotype was defined as two or more pulmonary exacerbations in the year prior to baseline assessment, which aligns with the standard definition in COPD literature.21,22
Prevalence of cardiovascular disease was taken as the percentage of patients with a history of any of the following conditions at baseline assessment: ischaemic heart disease, previous myocardial infarction, heart failure, atrial fibrillation, stroke, and transient ischaemic attack (TIA). AlphaNet do not collect specific data for ischaemic heart disease, and as such this was not included in the definition for cardiovascular disease in the US cohort.
MACE was defined as the first occurrence of either myocardial infarction, non-fatal stroke, or decompensated heart failure during the follow-up period. The definition of MACE is variable in the literature but usually includes cardiovascular mortality.23,24 Cause of death was not captured and as such is not included in our definition of MACE.
Statistical Analysis
All statistical analysis was performed using R version 4.2.3.25 Baseline characteristics were described using mean and standard deviation for normally distributed continuous values or median (with 1st and 3rd quartiles) for non-normally distributed continuous variables. Frequency and percentage values were used for categorical variables.
Baseline characteristics were compared between patients with and without MACE during the follow-up period, and patients with and without prior cardiovascular disease. For continuous variables, t-tests were used for normally distributed data and the Kruskal–Wallis test for non-normally distributed data. Chi-square tests were used to compare categorical variables.
To explore the impact of key variables on prevalence of cardiovascular disease at baseline, logistic regression was performed. Cardiovascular disease was set as the outcome variable, and sex, smoking history, and age as predictors.
Crude incidence rates were calculated for the entire cohort and stratified by exacerbation frequency phenotype. Kaplan–Meier survival curves26 were calculated, with MACE defined as the event of interest and stratified by patients with ≥2 annual exacerbations in the year prior to baseline assessment vs those with 1 or no exacerbations in the same time period. For the AlphaNet cohort, an end date of 31/12/2016 was set as the censor date for MACE to comply with a prior data sharing agreement. For the Birmingham AATD registry, a censor date of 31st December 2020 was used.
The effect of exacerbation phenotype on rates of MACE was assessed with a Cox proportional hazards model and adjusted for the following variables: sex, age, smoking status (never, ever, or current smoker), prior diagnosis of CVD (heart failure, ischaemic heart disease, atrial fibrillation, stroke, and myocardial infarction), and presence of diabetes and hypertension at baseline. The risk of MACE was deemed proportional throughout the follow-up period by graphical means.
To assess any potential confounding from missing data or influence of prior cardiovascular disease, we performed two sensitivity analyses which included only patients without prior cardiovascular disease or those with an available FEV1 measurement at baseline assessment. In addition, a competing risk model was performed to explore the impact of death during follow-up on the hazard of MACE during follow-up.
Results
Study Population and Prevalence of Cardiovascular Disease
Table 1 summarises baseline characteristics for the whole cohort, and for those with and without prior CVD. In total 3493 patients were included in the analysis: AlphaNet, 2561, Birmingham AATD Registry, 932. The mean age was 50.7 years old with 52.8% males. Most patients were either previous (69.8%) or never smokers (26.5%), with very few current smokers (3.7%). The mean FEV1% predicted was 50.4% as calculated from Global Lung Function Initiative reference values.27 Overall, 14.7% of patients had a history of CVD at baseline, with 84 patients (2.4%) having a history of 2 or more cardiovascular diseases. The median rate of exacerbations was higher in those with previous cardiovascular disease. Almost a third of frequent exacerbators (30.4%) had a history of heart failure compared to 7.2% of infrequent exacerbators (p < 0.001). The prevalence ratio (PR) of CVD, as assessed by logistic regression, was higher in frequent exacerbators (PR 1.49, 95% CI 1.20 to 1.85) and older patients (PR 1.02, 95% CI 1.01 to 1.03), after adjustment for age, sex, smoking history, and exacerbation frequency (Table S1) of the study cohort including cardiovascular disease.
 |
Table 1 Baseline Characteristics |
Risk of Major Cardiovascular Events (MACE)
A flow diagram of patients included in the study is shown in Figure 1. From the original dataset, 2844 patients had valid follow-up data that included MACE for a median of 4.1 years (1.9 to 8.0). During follow-up, 122 (4.3%) patients reported a major adverse cardiovascular event which included congestive heart failure, 55 (1.9%); myocardial infarction, 40 (1.4%); and stroke, 27 (0.95%). Death occurred in 946 patients. For those who died, there was a higher proportion of deaths in the frequent exacerbator group (349 (26.5%) vs 304 (22.7%) and those with prior cardiovascular disease (172 (37.4%) vs 530 (22.2%)). Table 2 summarises baseline characteristics of patients with and without MACE during the follow-up period. Patients who developed MACE had a higher rate of prior cardiovascular disease (15.0% vs 43.4%, p < 0.001). Exacerbation frequency was higher in the MACE group with a median of 1.0 more exacerbation per year (44.8% vs 63.1%, p = 0.001); 63% of patients in the MACE group were frequent exacerbators compared to 44.8% in those not suffering MACE (p < 0.001). There was also a higher prevalence of hypertension and diabetes in the MACE group (hypertension 34.0% vs 49.2%, p = 0.001, diabetes 5.1% vs 9.8%, p = 0.036). There was a higher prevalence of ex-smokers in the MACE group with similar rates of current smokers.
 |
Table 2 Baseline Characteristics of Grouped by Patients with and without MACE During Follow-Up Period |
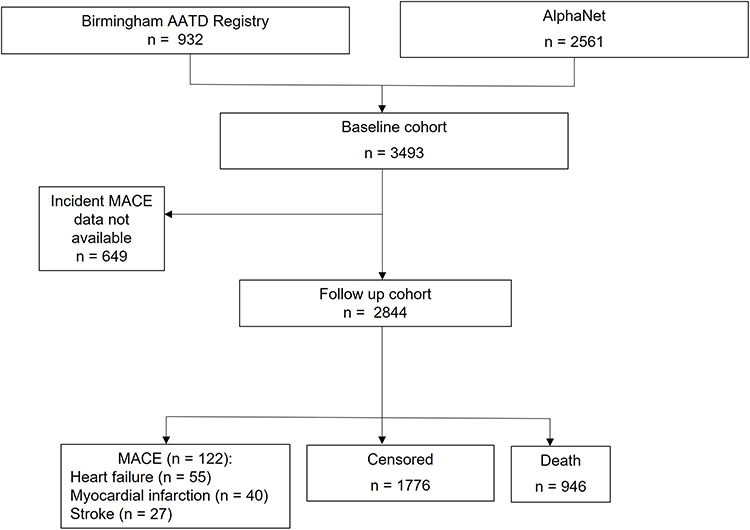 |
Figure 1 Flow diagram for study participants. |
The crude incidence rate (IR) for MACE was 9.31 per 1000 person-years (95% CI 5.7 to 13.0). Frequent exacerbators had a higher IR (12.3 per 1000 person-years, 95% CI 6.3 to 18.2) compared to those with one or no exacerbations (6.1 per 1000 person-years, 95% CI 2.1 to 10.6).
Patients with a frequent exacerbator phenotype were more likely to develop MACE during the study follow-up period (Figure S1 and Supplementary Material). After 5 years, 93.9% (95% CI 92.4 to 95.5) of patients had not suffered a MACE in the frequent exacerbator group compared with 97.3% (95% CI 96.3 to 98.4) in the infrequent exacerbator group (log rank p < 0.001). In patients without a history of cardiovascular disease there were 64 MACE with a crude incidence rate of 6.07 per 1000 person-years (95% CI 2.84 to 9.31).
Using a Cox proportional hazard model (Figure 2), after adjustment for age, sex, smoking status at baseline, previous CVD, hypertension, and diabetes, the rate of MACE was almost double in those with ≥2 annual exacerbations at baseline (HR 1.85, 95% CI 1.24 to 2.75, p < 0.002). There was a 4-fold increase in rate of MACE in current smokers compared to never smokers (HR 4.04, 95% CI 1.44 to 11.3, p = 0.008). Due to missing key variables, 211 patients were excluded from the model with a total of 115 MACE. A competing risks model was performed to assess the impact of death by any cause which showed a similar impact of exacerbator phenotype on rates of MACE (HR 1.64, 95% CI 1.00 to 2.69, p = 0.05)
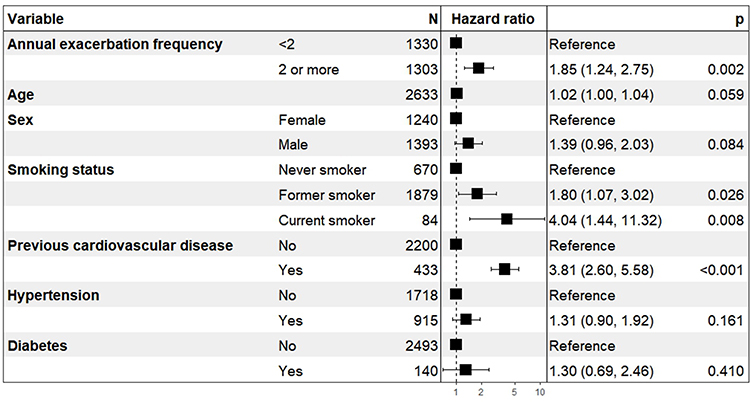 |
Figure 2 Cox proportional hazard model for MACE incidence adjusted for annual exacerbation frequency (<2 or 2 or more exacerbations/year), age, sex, smoking status, previous cardiovascular disease, hypertension, and diabetes. |
Two sensitivity analyses were performed to explore the impact of prior cardiovascular disease and FEV1% predicted on rates of MACE in frequent exacerbators. For patients with no prior cardiovascular disease there was an increased rate of MACE in those with frequent exacerbations (HR 2.12, 1.38 to 3.23 p < 0.001). With adjustment for baseline FEV1% predicted, HR for exacerbation status was 1.51 (95% CI 0.89 to 2.58, p = 0.127) though this only included 1421 patients with 61 MACE.
Discussion
Prevalence of Cardiovascular Disease in AATD
This is the largest study to date describing prevalence of cardiovascular disease in patients with severe AATD-COPD. It has shown an overall unadjusted prevalence of 14.7% for cardiovascular disease, with significantly lower numbers for previous major cardiovascular events such as stroke (2.3%), myocardial infarction (2.2%), and heart failure (2.5%). Those with CVD were on average 2.9 years older than those without CVD, with similar gender distribution and smoking history. Frequent exacerbators were up to 50% more likely to have previous cardiovascular disease.
These results align with findings from other studies of cardiovascular disease prevalence in AATD.28–30 Fahndrich et al reported on 139 patients with AATD-COPD, the majority of whom were receiving AAT augmentation therapy. Prevalence rates were 1.8% for myocardial infarction and 3.0% for heart failure with no recorded strokes. They also found lower rates of coronary artery disease compared to non-AATD COPD. Another study by Tanash et al assessed the cardiovascular risk of 1545 PiZZ patients from the Swedish National AATD registry. AATD was associated with a lower risk of ischaemic heart disease compared to controls. A sub analysis of 711 PiZZ patients with COPD found a more than 3-fold increase in rate of ischaemic heart disease compared to non-COPD AATD. A similar risk increase was seen in COPD vs controls without AATD, suggesting the presence of COPD ameliorates any potential cardiovascular protective effects of AATD. Notably there were far more never smokers in the AATD group. Head-to-head comparison of rates of ischaemic heart disease in AATD-COPD vs COPD was not performed in this study.
The observed low prevalence of cardiovascular disease in AATD-COPD somewhat contradicts the hypothesis that increased inflammation associated with COPD drives development of cardiovascular disease. Several studies have indicated that increased serine protease activity, the main feature of AATD,31 may accelerate atherosclerosis and vascular injury.14,32 Indeed, AATD is linked with higher prevalence of panniculitis,33 polyangiitis with granulomatosis,34 and other inflammatory conditions.35 Vascular stiffness, a surrogate marker of cardiovascular risk, is also elevated in patients with AATD compared to controls.36
There are possible explanations for the contradiction between the perceived low prevalence of cardiovascular disease in AATD and proposed biological mechanisms.
Patients with AATD develop COPD at a much younger age with typically more modest smoking histories compared to patients with usual COPD.37 In this study only 3.7% continued to smoke at baseline assessment, though this may be underestimated in our cohort since a prerequisite for initiation and maintenance of augmentation therapy is smoking abstinence. This rate of current smoking is significantly lower than reports in COPD; for comparison, 36% of the ECLIPSE cohort were current smokers.21
Atherosclerosis may develop more slowly than emphysema in AATD and require higher rates of exposure to tobacco to cause clinically relevant disease,38 though there is no evidence for this in AATD to date. In addition, we speculate that AATD patients have a more potent motivating factor for lifestyle modification than those with usual COPD since they are told they have a rare and severe form of COPD. Lifestyle modification may include increased exercise, earlier smoking cessation, and improved diet, all of which reduce risk of cardiovascular disease.39 Though three-quarters of our cohort were prescribed augmentation therapy, this is unlikely to have impacted on the prevalence of cardiovascular disease as most patients started augmentation therapy within 1 year of baseline assessment.
There may also be underdiagnosis of cardiovascular disease in patients with known AATD. In AATD, early symptoms of cardiovascular disease (such as progressive breathlessness and peripheral oedema) may be attributed to progression of their known rare and severe lung disease or to complications of lung disease such as secondary pulmonary hypertension. There may also be patients who unknowingly have AATD and were included in studies of usual COPD,29 though this is unlikely to impact the overall results of this study.
Biomarker studies support an association between an increased inflammatory signature and cardiovascular disease. Serum desmosine, a cleavage product of elastin from lung and vascular tissue in response to elastolysis,40 is raised in patients with cardiovascular disease41 and during acute COPD exacerbations.42
Serum desmosine is higher in patients with AATD-COPD compared with usual COPD.43 Equally, administration of pooled human AAT has been shown to reduce serum levels of desmosine44 and has been suggested as a possible treatment for atherosclerosis in the context of myocardial injury.45
It is plausible that predominantly basal emphysema distribution in AATD-COPD may have a cardioprotective role. Since ventilation perfusion ratios are greatest at the lung apices,46 diversion of pulmonary blood flow away from areas of basal emphysema to well-ventilated upper zone lung may lead to less impact on overall gas exchange and reduced hypoxia relative to the magnitude of emphysema.47 In usual COPD, where emphysema is typically apical,48 the reverse would be true. Over time, regional differences in emphysema and gas exchange may impact on overall cardiovascular health. There is little in the literature exploring this hypothesis. Disparities in gas trapping and dynamic hyperinflation may also have an impact on pulmonary vascular dynamics.49
Risk of MACE in AATD
This study also provides evidence for a link between frequent exacerbations and increased risk of future MACE in patients with AATD-COPD, independent of underlying smoking status and other cardiovascular risk factors. The strongest risk factor for future MACE, as expected, was previous cardiovascular disease. However, the association between frequent exacerbations and MACE was sustained when excluding prior cardiovascular disease. There was a 4.5-fold increase in MACE episodes in current smokers, giving further weight to the hypothesis that reduced cardiovascular risk in AATD relates to reduced rate of smoking. These results are in keeping with the biological theory of increased inflammation and increased risk of future MACE as discussed above. The authors acknowledge that unknown variables that increase both risk of exacerbations and MACE cannot be accounted for in the models performed in this study.
In usual COPD, the impact of exacerbations on future MACE is well reported in the literature.50 In addition to the increased inflammatory burden from frequent exacerbations, it is plausible that early cardiovascular disease mimics or magnifies exacerbation events. Occult cardiovascular disease in those with frequent exacerbations may later manifest as MACE. As such, clinical scrutiny of exacerbation episodes for features of cardiovascular pathology may identify cardiovascular disease earlier and allow treatment and risk factor modification. There were higher rates of heart failure in frequent exacerbators in this study, which supports this recommendation. The heterogeneous nature of pulmonary exacerbations in AATD-COPD will continue to present a diagnostic challenge.51
The impact of COPD severity was explored in our study. A sensitivity analysis, which included FEV1% predicted, suggested an increased risk of MACE in frequent exacerbators, though rates of MACE were low which reduced statistical power.
It is unclear if rates of MACE were impacted by augmentation therapy prescription. There is inconsistency of results pertaining to AECOPD rate and augmentation therapy, however, with observational work52 being suggestive of a reduction in rate, and meta-analysis of trials53 being suggestive of an increase. The EARCO group54 have further work underway in this area.
Strengths and Limitations
The main strength of this study is the large number of severe AATD patients with well characterised baseline and follow-up data, the largest studied to date. We acknowledge several limitations of this study. The most significant limitation is lack of a comparator non-AATD group which was not available for this analysis. In addition, the presence of COPD in AlphaNet was largely self-reported.55 A prerequisite for AAT prescription in the United States is presence of AATD-related lung disease, though we acknowledge that a small number of patients may not have met physiological criteria for COPD. The methods with which data were collected in both cohorts were slightly different, though this is unlikely to change the reported prevalence or incidence of cardiovascular disease and MACE, respectively. The definition of MACE did not include cardiovascular-related death, and therefore some MACE were not captured. We have demonstrated that there were more deaths in patients with prior cardiovascular disease and in frequent exacerbators, which suggests that if data on cardiovascular death were available it may strengthen our findings. We also performed a competing risk analysis which demonstrated a higher risk of MACE in frequent exacerbators, and it is likely that a proportion of deaths were cardiovascular deaths and therefore MACE underestimated. In addition, some MACE were self-reported. There were high levels of missing data for key variables including FEV1% predicted and prevalence of ischaemic heart disease and hypercholesterolaemia in the US cohort. Sensitivity analyses were used to mitigate against these issues, though these were limited by reduced number of MACE. Notably, data were not captured for family history of cardiovascular disease or smoking pack year history.
Conclusion
In severe AATD-COPD, MACE are associated with increased exacerbation frequency, previous cardiovascular disease, and a history of smoking. Further studies examining the relationship between pulmonary exacerbations and cardiovascular disease are needed to further clarify the risk of cardiovascular disease in severe AATD-COPD, in addition to a comparison to matched non-AATD COPD patients.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was supported by a grant from CHEST foundation.
Disclosure
PE has received speaker fees from Chiesi, GSK, and AstraZeneca.
AT has had grants and/or honoraria from AstraZeneca, GSK, Chiesi, CSLBehring, Vertex, and Grifols. KEH has received consulting income from AlphaNet. RS is employed by AlphaNet, reports personal fees from Grifols and CSL Behring, non-financial support from Inhibrx and Arrowhead, and grants from Matrx, outside the submitted work. EB, RC, and MN have no conflicts of interest.
References
1. Laurell CB, Eriksson S. The serum alpha-L-antitrypsin in families with hypo-alpha-L-antitrypsinaemia. Clin Chim Acta. 1965;11(5):395–398. doi:10.1016/0009-8981(65)90184-1
2. Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110(11):1585–1590. doi:10.1172/JCI0216782
3. Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax. 2004;59(5):441–445. doi:10.1136/thx.2003.006510
4. Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):3608. doi:10.1016/S0140-6736(15)60860-1
5. Hill AT, Campbell EJ, Bayley DL, Hill SL, Stockley RA. Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha(1)-antitrypsin deficiency (PiZ). Am J Respir Crit Care Med. 1999;160(6):1968–1975. doi:10.1164/ajrccm.160.6.9904097
6. European Heart Network. European cardiovascular disease statistics 2017 edition; 2017.
7. Balbirsingh V, Mohammed AS, Turner AM, Newnham M. Cardiovascular disease in chronic obstructive pulmonary disease: a narrative review. Thorax. 2022;77(9):939–945. doi:10.1136/thoraxjnl-2021-218333
8. Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Respir Med. 2015;3(8):631–639. doi:10.1016/S2213-2600(15)00241-6
9. Morgan AD, Zakeri R, Quint JK. Defining the relationship between COPD and CVD: what are the implications for clinical practice? Therap Adv Respir Dis. 2018;12:1753465817750524. doi:10.1177/1753465817750524
10. Curkendall SM, DeLuise C, Jones JK, et al. Cardiovascular disease in patients with chronic obstructive pulmonary disease, Saskatchewan Canada cardiovascular disease in COPD patients. Ann Epidemiol. 2006;16(1):63–70. doi:10.1016/j.annepidem.2005.04.008
11. Sin DD, Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107(11):1514–1519. doi:10.1161/01.CIR.0000056767.69054.B3
12. Maclay JD, MacNee W. Cardiovascular disease in COPD: mechanisms. Chest. 2013;143(3):798–807. doi:10.1378/chest.12-0938
13. Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1(1):73–83. doi:10.1016/S2213-2600(12)70060-7
14. Van Eeden S, Leipsic J, Paul Man SF, Sin DD. The relationship between lung inflammation and cardiovascular disease. Am J Respir Crit Care Med. 2012;186(1):11–16. doi:10.1164/rccm.201203-0455PP
15. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–1695. doi:10.1056/NEJMra043430
16. Maclay JD, McAllister DA, Rabinovich R, et al. Systemic elastin degradation in chronic obstructive pulmonary disease. Thorax. 2012;67(7):606–612. doi:10.1136/thoraxjnl-2011-200949
17. Donaldson GC, Hurst JR, Smith CJ, Hubbard RB, Wedzicha JA. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest. 2010;137(5):1091–1097. doi:10.1378/chest.09-2029
18. Reilev M, Pottegård A, Lykkegaard J, Søndergaard J, Ingebrigtsen TS, Hallas J. Increased risk of major adverse cardiac events following the onset of acute exacerbations of COPD. Respirology. 2019;24(12):1183–1190. doi:10.1111/resp.13620
19. Smith DJ, Ellis PR, Turner AM. Exacerbations of Lung Disease in Alpha-1 Antitrypsin Deficiency. Chronic Obstr Pulm Dis. 2020;8(1):162.
20. Ellis PR, Holm KE, Choate R, et al. Quality of life and mortality outcomes for augmentation naïve and augmented patients with severe Alpha-1 antitrypsin deficiency. Chronic Obstr Pulm Dis. 2023;10(2):139–147. doi:10.15326/jcopdf.2022.0339
21. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. doi:10.1056/NEJMoa0909883
22. McGarvey L, Lee AJ, Roberts J, Gruffydd-Jones K, McKnight E, Haughney J. Characterisation of the frequent exacerbator phenotype in COPD patients in a large UK primary care population. Respir Med. 2015;109(2):228–237. doi:10.1016/j.rmed.2014.12.006
23. Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380(4):347–357. doi:10.1056/NEJMoa1812389
24. Kelshiker MA, Seligman H, Howard JP, et al. Coronary flow reserve and cardiovascular outcomes: a systematic review and meta-analysis. Eur Heart J. 2022;43(16):1582–1593. doi:10.1093/eurheartj/ehab775
25. R Studio Team. Integrated Development for R. Boston, MA: RStudio, Inc.; 2015. Available from: http://www.rstudio.com/.
26. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457–481. doi:10.1080/01621459.1958.10501452
27. Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40(6):1324–1343. doi:10.1183/09031936.00080312
28. Fähndrich S, Biertz F, Karch A, et al. Cardiovascular risk in patients with alpha-1-antitrypsin deficiency. Respir Res. 2017;18(1):171. doi:10.1186/s12931-017-0655-1
29. Nakanishi T, Forgetta V, Handa T, et al. The undiagnosed disease burden associated with alpha-1 antitrypsin deficiency genotypes. Eur Respir J. 2020;56(6):2001441. doi:10.1183/13993003.01441-2020
30. Greulich T, Nell C, Hohmann D, et al. The prevalence of diagnosed α1-antitrypsin deficiency and its comorbidities: results from a large population-based database. Eur Respir J. 2017;49(1):1600154. doi:10.1183/13993003.00154-2016
31. Lomas DA. 1-antitrypsin deficiency* 4: molecular pathophysiology. Thorax. 2004;59(6):529–535. doi:10.1136/thx.2003.006528
32. Yao J, Guihard PJ, Blazquez-Medela AM, et al. Serine protease activation essential for endothelial-mesenchymal transition in vascular calcification. Circ Res. 2015;117(9):758–769. doi:10.1161/CIRCRESAHA.115.306751
33. Johnson EF, Tolkachjov SN, Gibson LE. Alpha-1 antitrypsin deficiency panniculitis: clinical and pathologic characteristics of 10 cases. Int J Dermatol. 2018;57(8):952–958. doi:10.1111/ijd.14012
34. Stone H, Pye A, Stockley RA. Disease associations in alpha-1-antitrypsin deficiency. Respir Med. 2014;108(2):338–343. doi:10.1016/j.rmed.2013.10.006
35. Ellis PR, Campbell EJ, Turner AM, Stockley RA. Alpha-1 antitrypsin deficiency: a predisposing factor for the development of pulmonary langerhans cell histiocytosis. Chronic Obstr Pulm Dis. 2019;6(3):206–209.
36. Duckers JM, Shale DJ, Stockley RA, et al. Cardiovascular and musculskeletal co-morbidities in patients with alpha 1 antitrypsin deficiency. Respir Res. 2010;11(1):173. doi:10.1186/1465-9921-11-173
37. Bernhard N, Lepper PM, Vogelmeier C, et al. Intensive smoking diminishes the differences in quality of life and exacerbation frequency between the alpha-1-antitrypsin deficiency genotypes PiZZ and PiSZ. Respir Med. 2017;130:1–8. doi:10.1016/j.rmed.2017.07.004
38. Auerbach O, Hammond EC, Garfinkel L. Smoking in relation to atherosclerosis of the coronary arteries. N Engl J Med. 1965;273(15):775–779. doi:10.1056/NEJM196510072731501
39. Joseph JJ, Deedwania P, Acharya T, et al. Comprehensive management of cardiovascular risk factors for adults with type 2 diabetes: a scientific statement from the American Heart Association. Circulation. 2022;145(9):e722–e59. doi:10.1161/CIR.0000000000001040
40. Luisetti M, Stolk J, Iadarola P. Desmosine, a biomarker for COPD: old and in the way. Eur Respir J. 2012;39(4):797–798. doi:10.1183/09031936.00172911
41. Rabinovich RA, Miller BE, Wrobel K, et al. Circulating desmosine levels do not predict emphysema progression but are associated with cardiovascular risk and mortality in COPD. Eur Respir J. 2016;47(5):1365–1373. doi:10.1183/13993003.01824-2015
42. Huang JT, Chaudhuri R, Albarbarawi O, et al. Clinical validity of plasma and urinary desmosine as biomarkers for chronic obstructive pulmonary disease. Thorax. 2012;67(6):502–508. doi:10.1136/thoraxjnl-2011-200279
43. Ma S, Lin YY, Turino GM. Measurements of desmosine and isodesmosine by mass spectrometry in COPD. Chest. 2007;131(5):1363–1371. doi:10.1378/chest.06-2251
44. Ma S, Lin YY, Cantor JO, et al. The effect of Alpha-1 proteinase inhibitor on biomarkers of elastin degradation in Alpha-1 antitrypsin deficiency: an analysis of the RAPID/RAPID extension trials. Chronic Obstr Pulm Dis. 2016;4(1):34–44. doi:10.15326/jcopdf.4.1.2016.0156
45. Feng Y, Hu L, Xu Q, et al. Cytoprotective role of Alpha-1 antitrypsin in vascular endothelial cell under hypoxia/reoxygenation condition. J Cardiovasc Pharmacol. 2015;66(1):96–107. doi:10.1097/FJC.0000000000000250
46. West JB. Blood-flow, ventilation and gas exchange in the lung. Lancet. 1963;2(7316):1055–1058. doi:10.1016/S0140-6736(63)90024-2
47. Mostafa AB, Tulley NJ, Harding LK, Stockley RA. Regional distribution of ventilation and perfusion in patients with obstructive pulmonary disease and alpha 1-antitrypsin deficiency. Eur J Nucl Med. 1983;8(8):338–341. doi:10.1007/BF00253541
48. Angelini ED, Yang J, Balte PP, et al. Pulmonary emphysema subtypes defined by unsupervised machine learning on CT scans. Thorax. 2023;78(11):1067–1079. doi:10.1136/thorax-2022-219158
49. Barr RG, Bluemke DA, Ahmed FS, et al. Percent emphysema, airflow obstruction, and impaired left ventricular filling. N Engl J Med. 2010;362(3):217–227. doi:10.1056/NEJMoa0808836
50. Kunisaki KM, Dransfield MT, Anderson JA, et al. Exacerbations of chronic obstructive pulmonary disease and cardiac events. A post hoc cohort analysis from the SUMMIT Randomized Clinical Trial. Am J Respir Crit Care Med. 2018;198(1):51–57. doi:10.1164/rccm.201711-2239OC
51. Ejiofor SI, Stolk J, Fernandez P, Stockley RA. Patterns and characterization of COPD exacerbations using real-time data collection. Int J Chron Obstruct Pulmon Dis. 2017;12:427–434. doi:10.2147/COPD.S126158
52. Barros-Tizon JC, Torres ML, Blanco I, Martinez MT. Reduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-antitrypsin augmentation therapy. Therap Adv Respir Dis. 2012;6(2):67–78. doi:10.1177/1753465812438387
53. Edgar RG, Patel M, Bayliss S, Crossley D, Sapey E, Turner AM. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chron Obstruct Pulmon Dis. 2017;12:1295–1308. doi:10.2147/COPD.S130440
54. Miravitlles M, Chorostowska-Wynimko J, Ferrarotti I, et al. The European Alpha-1 Research Collaboration (EARCO): a new ERS Clinical Research Collaboration to promote research in alpha-1 antitrypsin deficiency. Eur Respir J. 2019;53(2):1900138. doi:10.1183/13993003.00138-2019
55. Choate R, Mannino DM, Holm KE, Sandhaus RA. Comparing patients with ZZ versus SZ Alpha-1 antitrypsin deficiency: findings from AlphaNet’s Disease Management Program. Chronic Obstr Pulm Dis. 2018;6(1):29–39.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.