Back to Journals » Journal of Inflammation Research » Volume 18
Predicting Risk of Post-Treatment Relapse in Patients with Inflammatory Bowel Disease Based on Intestinal Microbiota
Authors Zhang T ![]() , He B, Lan C, Liu J, Zeng Q, Pu W, Zhou L, Zhou Q, Hu D, Chen Y, Peng Y, Li G, Wang Q, Chen L, Du Z, Li S, Tang X, Chen J, Xiao C
, He B, Lan C, Liu J, Zeng Q, Pu W, Zhou L, Zhou Q, Hu D, Chen Y, Peng Y, Li G, Wang Q, Chen L, Du Z, Li S, Tang X, Chen J, Xiao C
Received 13 August 2025
Accepted for publication 7 November 2025
Published 19 November 2025 Volume 2025:18 Pages 16079—16092
DOI https://doi.org/10.2147/JIR.S558136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Fatih Türker
Tao Zhang,1,* Binbo He,1,* Chao Lan,1,* Jie Liu,1 Qingyu Zeng,1 Wenfeng Pu,1 Lifeng Zhou,1 Qian Zhou,1 Dan Hu,1 Yanan Chen,1 Yiming Peng,1 Guobing Li,1 Qing Wang,1 Long Chen,1 Zonghan Du,1 Shiqing Li,1 Xiaobo Tang,1 Jian Chen,1 Chuanxing Xiao2
1Department of Gastroenterology, Nanchong Central Hospital, The Second Clinical Medical College, North Sichuan Medical College, Nanchong, Sichuan, 637000, People’s Republic of China; 2Xiamen Institutes of Respiratory Health, Xiamen, Fujian, 361000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tao Zhang, Department of Gastroenterology, Nanchong Central Hospital, The Second Clinical Medical College, North Sichuan Medical College, Nanchong, Sichuan, 637000, People’s Republic of China, Email [email protected] Chuanxing Xiao, Xiamen Institutes of Respiratory Health, Xiamen, Fujian, 361000, People’s Republic of China, Email [email protected]
Background: Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract. Post-treatment relapse is a major clinical challenge, and gut microbiota dysbiosis is hypothesized to be involved.
Methods: We enrolled 88 patients with IBD (46 UC, 42 CD) to investigate gut microbiota features associated with post-treatment relapse. Fecal samples collected before and after therapy were analyzed by 16S rRNA sequencing. A random forest (RF) model was developed to evaluate the predictive value of microbiota signatures for recurrence.
Results: At baseline (pre-treatment), no differences were observed in gut microbiota diversity between patients with UC and CD. However, significant compositional differences were observed, with Fusobacterium and Parabacteroides enriched in CD patients, and Anaerostipes and Enterococcus enriched in UC patients. Post-treatment, there was no significant difference in the α-diversity across IBD patients; however, β-diversity exhibited significant alterations, marked by enrichment of Akkermansia and Lachnoclostridium. Patients maintaining remission exhibited significant post-treatment beta-diversity shifts and enrichment of Erysipelatoclostridium, Delftia, Tyzzerella, Sphingomonas, Subdoligranulum, Proteus, and Enterococcus. Conversely, patients experiencing recurrence showed a significant reduction in Shannon alpha-diversity post-treatment and enrichment of UCG-002, Odoribacter, Delftia, Flavonifractor, and Erysipelotrichaceae_UCG-003. Post-treatment microbiota composition differed significantly between recurrent and non-recurrent patients, with higher alpha-diversity in the non-recurrent group. Non-recurrent patients exhibited enrichment of Eubacterium_hallii_group, Clostridioides, UCG-002, Paraprevotella, Bilophila, Desulfovibrio, Butyricimonas, Clostridium_sensu_stricto_1, Megamonas, Romboutsia, Parabacteroides, and Enterococcus, while Delftia was predominantly enriched in recurrent patients. The RF model, built using differentially abundant genera to distinguish recurrence status, achieved an area under the curve (AUC) of 0.721 in the validation set and 0.861 in the test cohort, indicating good predictive performance.
Conclusion: Our findings suggest that gut microbiota composition may hold clues for predicting IBD relapse. The RF model is a proof-of-concept that warrants external validation in prospective, multi-center studies before clinical application.
Keywords: inflammatory bowel disease, relapse, gut microbiota, random forest model, 16S rRNA sequencing
Introduction
Inflammatory Bowel Disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract characterized by a complex pathophysiology involving genetic predisposition, immune dysregulation, intestinal microbiota dysbiosis, and environmental factors.1 The clinical manifestations of IBD are heterogeneous, commonly including bloody diarrhea, anemia, and abdominal pain.2 IBD is primarily categorized into two subtypes: Crohn’s disease (CD) and ulcerative colitis (UC). UC is predominantly characterized by inflammation of the colonic mucosa,3 whereas CD is distinguished by transmural inflammation.3 Globally, the incidence of IBD is increasing, with reported annual incidence ranging from 7.6 to 246 per 100,000 individuals for UC and from 3.6 to 214 per 100,000 individuals for CD.4,5 Notably, relapse following discontinuation of anti-tumor necrosis factor (TNF) therapy remains a significant concern, with studies indicating that approximately 55% of patients experience recurrence; the median time to relapse is reported as 32 months for CD patients and 18 months for UC patients.6 Effectively managing IBD relapse poses a substantial clinical challenge. Both perturbations in the intestinal microbiota composition (dysbiosis) and systemic infections are recognized triggers that can contribute to recurrence by activating the gastrointestinal mucosal immune system.7
Current therapeutic strategies primarily target suppression of the hyperactive immune system and inflammatory response modulation through pharmacological interventions; however, these approaches largely overlook the critical role of the intestinal microbiota.8 Furthermore, a substantial proportion of patients exhibit primary non-response to existing therapies or develop secondary resistance over time.9 These agents may also induce adverse effects and potentially contribute to disease recurrence. Compared with healthy individuals, patients with IBD demonstrate reduced gut microbiota α-diversity, elevated abundance of pathobionts, and significant alterations in microbial metabolite profiles.10–13 Specifically, enrichment of Rothia and Ruminococcus has been associated with intestinal stricture formation, while increased Collinsella abundance correlates with penetrating disease development.10 Mucosal inflammation in IBD correlates with depletion of anaerobic bacteria, including Bacteroides, Eubacterium, and Lactobacillus.11 Notably, reduced abundance of F. prausnitzii is linked to impaired intestinal mucosal barrier function12 and shorter time to recurrence following infliximab therapy discontinuation.13
The association between gut microbiota dysbiosis and IBD pathogenesis is well-established. While several studies have explored microbial signatures for discriminating IBD diagnoses or predicting treatment response,14–16 the development of models specifically for forecasting post-therapeutic relapse remains comparatively limited and represents a critical unmet need. While established clinical biomarkers like C-reactive protein (CRP) and fecal calprotectin (FC) are invaluable for monitoring current inflammatory activity, their ability to reliably predict future relapse is suboptimal. This limitation underscores the potential value of complementary approaches. Recent findings suggested that certain microbial taxa, such as the reduction of Faecalibacterium prausnitzii and enrichment of Escherichia/Shigella, have been associated with disease activity and relapse risk,17,18 establishing a foundation for predictive efforts. Building upon this existing knowledge, our study aims to validate and extend these observations by identifying a comprehensive, relapse-associated microbial signature within a well-defined cohort of UC and CD patients. We focus on the post-treatment microbiota state and develop a random forest model to assess its predictive value for recurrence. Our work therefore seeks not only to confirm previously reported associations but also to uncover novel candidate taxa and construct an integrated model that may eventually aid in clinical risk stratification.
Methods
Study Cohorts
A total of 88 patients (46 with UC and 42 with CD) were recruited from the Gastroenterology Department of Nanchong Central Hospital between November 2022 and October 2023. The study was approved by the Ethical Committee of Nanchong Central Hospital (Approval No. 2022–075), and written informed consent was obtained from all participants or their guardians in accordance with the Declaration of Helsinki. Patients were treated according to standard clinical guidelines, which primarily included 5-aminosalicylates, corticosteroids, immunomodulators, such as azathioprine or 6-mercaptopurine, and/or biologic agents (anti-TNF therapy). Clinical remission was defined as the resolution of IBD-related symptoms (absence of bloody diarrhea and abdominal pain) and objective evidence of mucosal healing from endoscopic evaluation.
Patients’ Clinical Characteristics
Demographic and clinical data were systematically collected from all participants, including: (1) Baseline characteristics: name, age, age at disease onset, gender, and body mass index (BMI); (2) Lifestyle factors: smoking/alcohol status and dietary patterns; (3) Clinical parameters: IBD-related complications, comorbidities, and current medication use. Gastrointestinal symptom severity was assessed using standardized grading scales. Routine hematological parameters were analyzed from venous blood samples. Dietary intake was quantified through a validated food frequency questionnaire (FFQ).
Inclusion and Exclusion Criteria
Inclusion criteria: (1) Confirmed IBD diagnosis according to the 2018 Consensus on Diagnosis and Treatment of Inflammatory Bowel Disease, based on comprehensive assessment of clinical manifestations, laboratory investigations, imaging findings, endoscopic features, and histopathological evidence;19 (2) Mentally competent and able to communicate normally; (3) Fully informed about the study content and have provided written consent. Exclusion Criteria: (1) Patients without provided consent; (2) History of cancer; (3) Use of antibiotics or probiotics, or prebiotics within three months before data collection.
Group Classification
Patients were dichotomized based on post-therapeutic disease recurrence status: relapse group and non-relapse group. Patients were followed up every six months after achieving remission to monitor for disease recurrence. Relapse group: Re-emergence of IBD-related clinical symptoms during the follow-up period after achieving clinical remission through pharmacotherapy. Non-relapse group: Sustained clinical remission without symptom recurrence during follow-up after pharmacotherapy-induced remission.
Fecal Sample and DNA Extraction
Fecal samples were collected from participants before and after treatment. Uncontaminated portions of these samples were then selected and preserved in a stabilization solution at ambient temperature prior to being dispatched to Xiamen Treatgut Biotechnology Co., Ltd (China) for 16S rRNA gene sequencing. Genomic DNA was subsequently extracted from stabilized fecal samples using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Cat. No. 51604, Germany) according to the manufacturer’s protocol.
16S rRNA Gene Sequencing
The V3-V4 region of bacterial 16S ribosomal RNA genes was amplified using primers 341F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACNVGGGTWTCTAAT-3′). PCR amplification protocol consisted of an initial denaturation at 95°C for 2 min, followed by 25 cycles of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 30 sec, and a final extension at 72°C for 10 min. PCR products were quantified using the Quant-iT™ PicoGreen™ dsDNA Assay Kit (ThermoFisher Scientific, Cat. No. P7589) on QuantiFluor fluorometer (Promega), and the libraries were subsequently pooled. Negative controls (no-template controls) were included during the PCR amplification and library preparation steps to monitor for potential reagent contamination. Paired-end sequencing was performed on the Illumina sequencing platform 250PE (Illumina) following manufacturer’s standardized protocols.
Bioinformatic Processing of Sequencing Data
Bioinformatics analysis was performed using QIIME2 (version 2023.5.1). The workflow initiated with demultiplexing and quality control via the q2-demux plugin, followed by sequence denoising employing the DADA2 algorithm. Potential contaminants were bioinformatically monitored by inspecting the prevalence of taxa in the negative controls. Any taxa predominantly found in controls were removed from subsequent analyses. For taxonomic classification, amplicon sequence variants (ASVs) were classified using the q2-feature-classifier classify-sklearn tool, which leveraged a naive Bayes classifier with the SILVA database release 138 as a reference. The analysis was refined by including only bacterial genera that met a threshold of at least 0.01% of the mean relative abundance across a minimum of 10% of the samples for subsequent analyses. Alpha diversity, encompassing richness and the Shannon index, was determined using the R package microbiome version 1.22.0. Beta diversity was evaluated via Principal Coordinate Analysis (PCoA), which was based on Bray-Curtis dissimilarity metrics or unweighted Unifrac distance and calculated with the R package vegan version 2.6.4. These diversity indices were computed following rarefaction to the lowest sequencing depth. This study focused on taxonomic profiling to identify microbial signatures associated with relapse. Functional prediction analysis was not performed, as reliable inference of gene content from 16S rRNA data is limited. Additionally, in this study, the designation “UCG” in taxonomic assignments, such as Erysipelotrichaceae_UCG-003, refers to “Uncultured Group”, indicating phylogenetic clusters that are known from sequence data but have not yet been formally isolated and cultivated.
Biomarker Identification Using Linear Discriminant Analysis Effect Size (LEfSe)
Linear Discriminant Analysis Effect Size (LEfSe) was employed to identify taxa exhibiting differential abundance across distinct disease states. This analysis was performed using the R package microeco version 1.5.0, which employed a non-parametric factorial Kruskal–Wallis test coupled with linear discriminant analysis to detect biologically relevant features. The LEfSe algorithm generated effect size estimates quantified by Linear Discriminant Analysis (LDA) scores, where higher absolute values indicate greater discriminatory power between groups. Taxa that achieved statistical significance (Kruskal–Wallis p < 0.05) with LDA scores>2.0 were considered potential biomarkers, satisfying both statistical significance and biological effect magnitude thresholds.
Random Forest Classifier Model
To evaluate the discriminatory capacity of microbial biomarkers for inter-group differentiation, we implemented a random forest classifier using the randomForest R package (v4.6.14). To ensure the reproducibility of the random partitioning and model training, a random seed was set (set.seed(123)) prior to data splitting and model building. Moreover, to ensure model rigor and prevent overfitting, the following structured pipeline was adhered to: First, the entire dataset was randomly partitioned into a training set (70%) and a hold-out test set (30%). A strict separation between these sets was maintained throughout the entire model-building process to prevent data leakage. Specifically, the differential genera (biomarkers) used as input features for the Random Forest model were identified exclusively from the training set using LEfSe analysis with stringent thresholds (Kruskal–Wallis p < 0.05, LDA score > 2) or, for clinical models, least absolute shrinkage and selection operator (LASSO) regression via the glmnet package (v4.1.8). The test set was never involved in this feature discovery phase. Subsequently, the Random Forest model was trained and optimized using only the data from the training set, with hyperparameter tuning assisted by five-fold cross-validation implemented via the R package caret (v6.0.88). Finally, the fully-trained model, with its features and parameters fixed, was evaluated only once on the independent hold-out test set. Classifier efficacy was assessed through receiver operating characteristic (ROC) analysis (pROC v1.17.0.1), with the area under the curve (AUC) quantifying predictive accuracy. Feature importance was determined by Gini index scores.
Statistical Analysis
Statistical analyses were conducted using R (version 4.3.1). For graphical representation, boxplots and barplots were generated with the aid of the R packages ggpubr (v0.6.0) and ggplot2 (v3.5.0), respectively. Heatmaps were crafted using the R package pheatmap (v1.0.12). Spearman correlation analysis was executed with the R package corrplot (v0.92). The selection of statistical tests was guided by the study’s design, which encompassed both longitudinal (within-subject) and cross-sectional (between-group) comparisons. For longitudinal comparisons of the same group at different time points (eg, pre- vs post-treatment microbiota within all IBD patients, or within the relapse/non-relapse subgroups), the paired Wilcoxon signed-rank test was applied. For cross-sectional comparisons between different groups at the same time point (eg, baseline microbiota between patients who subsequently relapsed and those who maintained remission, post-treatment microbiota between relapse vs non-relapse patients, or all UC vs CD comparisons at baseline), the unpaired Wilcoxon rank-sum test (Mann–Whitney U-test) was employed. The Kruskal–Wallis test was utilized for comparisons across multiple groups. Multiple testing correction was applied using the Benjamini-Hochberg false discovery rate (FDR) procedure. Statistical significance was defined as: *P < 0.05, **P < 0.01, ***P < 0.001, and NS for not significant (p ≥ 0.05).
Results
Clinical Characteristics of Study Cohorts
This study comprised 40 IBD recurrence patients (24 male and 16 female). The median age was 37 years, ranging from 14 to 75 years, while the median BMI was 20.8, varying between 16.6 and 28.6, the median disease duration was 3 years ranging from 1 to 17 years. And comprised 48 non-relapse IBD patients, including 29 male and 19 female. The median age of the non-relapse was 40.5 years, with an age range of 16 to 80 years, and a median BMI of 21.1, ranging from 15.6 to 28.0, the median disease duration was 3 years ranging from 1 to 17 years. No significant differences in gender, age, disease duration, or BMI were observed between groups (Table 1).
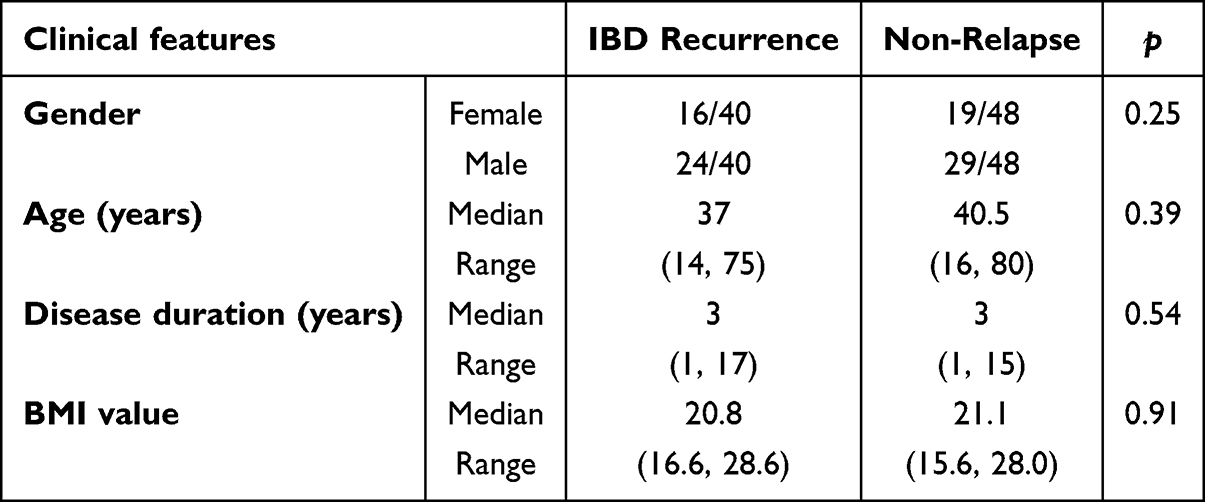 |
Table 1 Baseline Clinical Characteristics of the Study Cohorts |
No Significant Differences in α- and β-Diversity of Gut Microbiota Were Observed Between UC and CD Patients at Baseline
The study cohort included 46 UC and 42 CD patients, with 40 experiencing recurrence and 48 maintaining remission. Histopathological evaluation (hematoxylin and eosin staining) and endoscopic imaging were performed at three timepoints: baseline, post-treatment improvement, and post-recurrence (Figure S1A and B). Comparative analysis of pretreatment gut microbiota revealed no significant difference in species richness or Shannon diversity index (Figure S2A). PCoA based on Bray-Curtis dissimilarity showed no significant separation between UC and CD groups (Figure S2B). Notwithstanding distinct clinical presentations, UC and CD patients exhibited comparable gut microbiota profiles at baseline, with no significant differences in α-diversity indices or β-diversity structure.
Dynamic Shifts in Gut Microbiota Following Therapeutic Intervention
Comparative analysis of gut microbiota from IBD patients before and after treatment revealed no significant differences in α-diversity metrics, including species richness and the Shannon index (Figure 1A). However, PCoA plots based on Bray-Curtis dissimilarity revealed significant shifts in microbial community structure (P = 0.01; Figure 1B). To identify treatment-responsive taxa, we performed LEfSe analysis (LDA score>2.0), which revealed significant pre-treatment enrichment of Gemella and post-treatment enrichment of Akkermansia and Lachnoclostridium (Figure 1C).
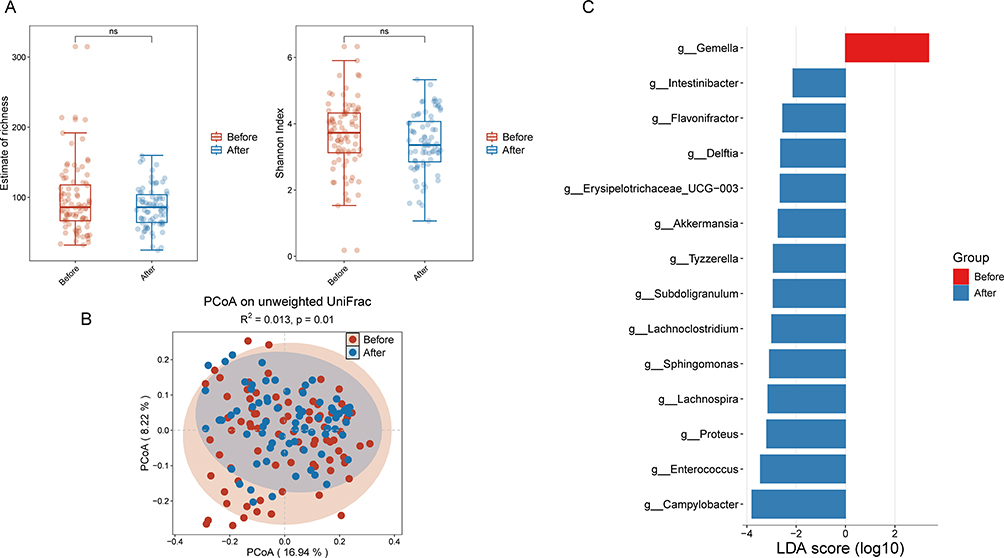 |
Figure 1 (A) Alpha diversity indices of gut microbiota before and after treatment in IBD patients. (B) Beta diversity indices of gut microbiota before and after treatment in IBD patients. (C) Enrichment analysis of differential bacterial genera in the gut microbiota before and after treatment in IBD patients. Abbreviation: NS, not significant. |
Post-Therapeutic Microbial Restructuring in Non-Relapsed IBD Patients and Relapsed Patients
Analysis of gut microbiota dynamics in non-relapsed patients revealed no significant differences in α-diversity metrics (species richness and Shannon index) between pre- and post-treatment states (Figure 2A). However, PCoA demonstrated significant shifts in microbial community structure (P = 0.049; Figure 2B). Further LEfSe analysis (LDA score>2.0) identified significant post-treatment enrichment of the genera Erysipelatoclostridium, Delftia, Tyzzerella, Sphingomonas, Subdoligranulum, Proteus, and Enterococcus in these non-relapsed patients (Figure 2C).
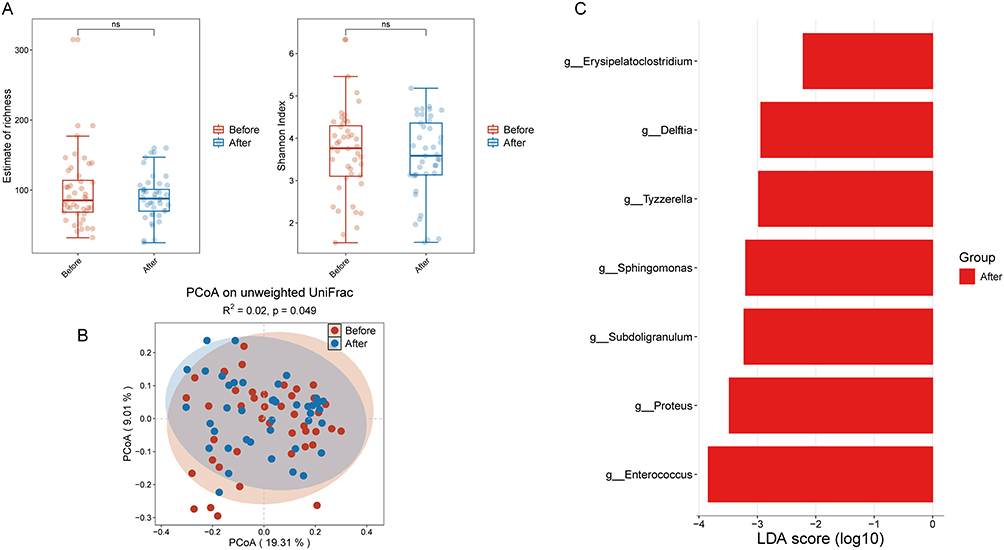 |
Figure 2 (A) Alpha diversity indices of gut microbiota pre- and post-treatment in non-relapsed IBD patients. (B) Beta diversity indices of gut microbiota before and after treatment in non-relapsed IBD patients. (C) Enrichment analysis of differential bacterial genera in the gut microbiota before and after treatment in non-relapsed IBD patients. Abbreviation: NS, not significant. |
In relapsed patients, analysis of gut microbiota diversity before and after treatment showed no significant differences in species richness or β-diversity. Nevertherless, a notable reduction in the Shannon index, indicative of α-diversity, was observed post-treatment (Figure 3A and B). To identify differentially bacterial genera, LEfSe analysis (LDA score>2.0) revealed significant enrichment of the genera Escherichia-Shigella, Clostridium_sensu_stricto_1, Romboutsia, and Campylobacter prior to treatment, whereas the genera UCG-002, Odoribacter, Delftia, Flavonifractor, and Erysipelotrichaceae_UCG_003 were significantly enriched following treatment in these patients (Figure 3C).
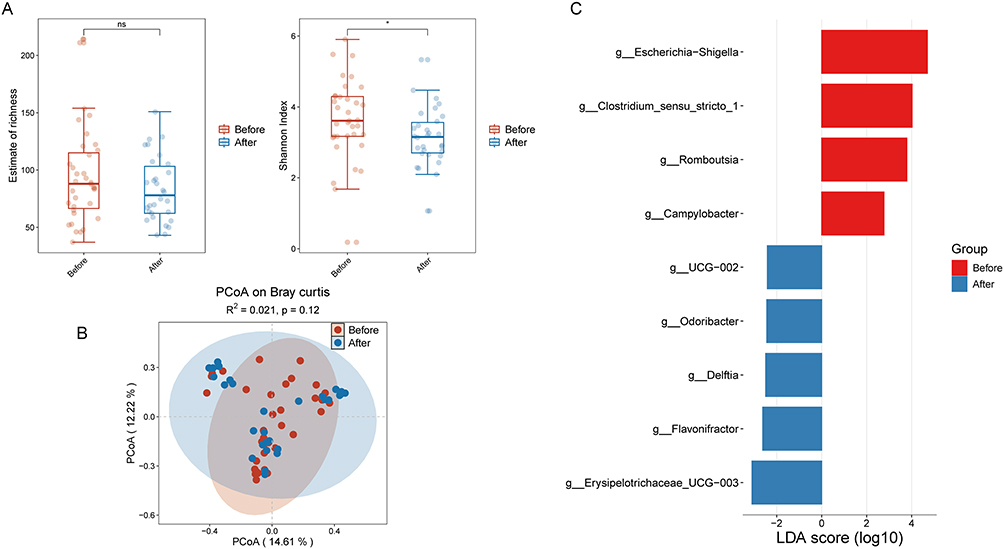 |
Figure 3 (A) Alpha diversity indices of gut microbiota before and after treatment in relapsed IBD patients. (B) Beta diversity indices of gut microbiota in relapsed IBD patients before and after treatment. (C) Enrichment analysis of differential bacterial genera in the gut microbiota in relapsed IBD patients pre- and post-treatment. *P < 0.05. Abbreviation: NS, not significant. |
Significant Differences Existed in the Baseline Gut Microbiota Between Patients Who Later Experienced Recurrence and Those Who Remained Non-Recurrent
Analysis of baseline alpha diversity indicated no significant difference between the two groups (Figure 4A). However, PCoA demonstrated significant differences in beta diversity (P = 0.012; Figure 4B). The nine most abundant phyla identified in the baseline gut microbiota of all IBD patients were Actinobacteriota, Bacteroidota, Campilobacterota, Desulfobacterota, Firmicutes, Fusobacteriota, Proteobacteria, Synergistota, and Vermicomicrobiota (Figure 4C). To identify genus-level differences at baseline, we performed LEfSe analysis (LDA score>2.0). It was found that the bacterial genera Haemophilus and Anaerostipes were significantly enriched in patients who experienced recurrence, whereas the genera Monoglobus, Actinobacillus, Aggregatibacter, Delftia, and Phascolarctobacterium were significantly enriched in patients who remained non-recurrent (Figure 4D). These results indicated that the structure (but not the diversity) of the baseline gut microbiota differentiates between relapsed and non-relapsed IBD patients. Specifically, enrichment of genera such as Haemophilus was associated with relapse risk, whereas genera such as Phascolarctobacterium may confer protective effects, offering potential biomarkers for relapse prediction.
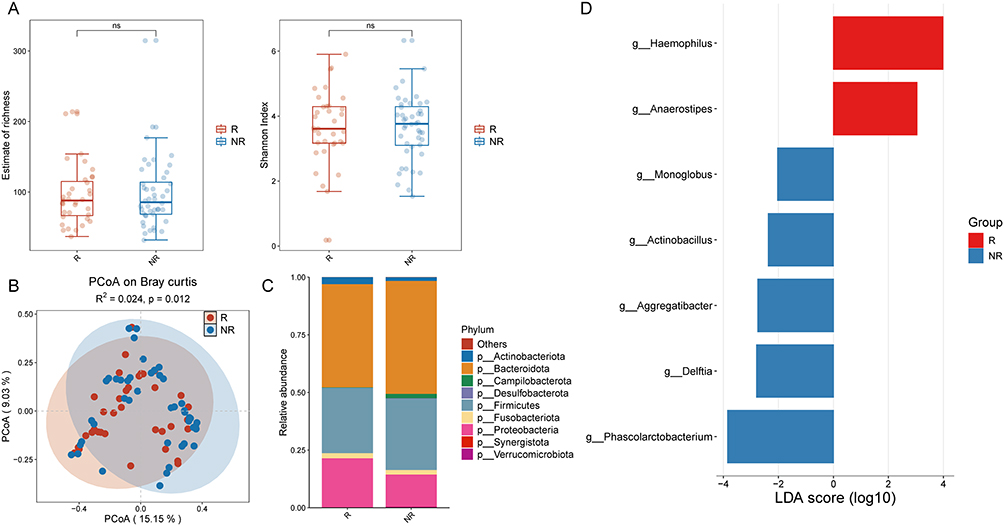 |
Figure 4 (A) Alpha diversity indices of gut microbiota at baseline in relapsed and non-relapsed IBD patients. (B) Beta diversity indices of gut microbiota at baseline in relapsed and non-relapsed IBD patients. (C) Phylum-level distribution of gut microbiota at baseline in relapsed and non-relapsed IBD patients. (D) Enrichment analysis of differential bacterial genera in the gut microbiota at baseline between relapsed and non-relapsed IBD patients. Abbreviation: NS, not significant. |
Post-Treatment Gut Microbiota Composition Differed Significantly Between Recurrent and Non-Recurrent IBD Patients
Analysis of post-treatment α-diversity revealed significantly higher diversity in non-recurrent patients compared to recurrent patients (Figure 5A). PCoA demonstrated significant differences in β-diversity (P = 0.019; Figure 5B). The nine most abundant phyla in the post-treatment gut microbiota of all IBD patients were Actinobacteriota, Bacteroidota, Campilobacterota, Desulfobacterota, Firmicutes, Fusobacteriota, Proteobacteria, Synergistota, and Vermucomicrobiota (Figure 5C). To identify differentially abundant bacterial genera post-treatment, we performed LEfSe analysis (LDA score>2). Non-recurrent patients exhibited significant enrichment of the genera Eubacterium_hallii_group, Clostridioides, UCG-002, Paraprevotella, Bilophila, Desulfovibrio, Butyricimonas, Clostridium_sensu_stricto_1, Megamonas, Romboutsia, Parabacteroides, and Enterococcus. Conversely, Delftia was significantly enriched in recurrent patients (Figure 5D). Collectively, following treatment, non-relapsed IBD patients exhibited significantly higher gut microbiota diversity and enrichment of multiple potentially beneficial genera capable of producing short-chain fatty acids (SCFAs). Conversely, relapsed patients displayed reduced microbial diversity alongside enrichment of pro-inflammatory genera such as Delftia. these differences provide a critical foundation for predicting relapse and developing personalized therapeutic strategies.
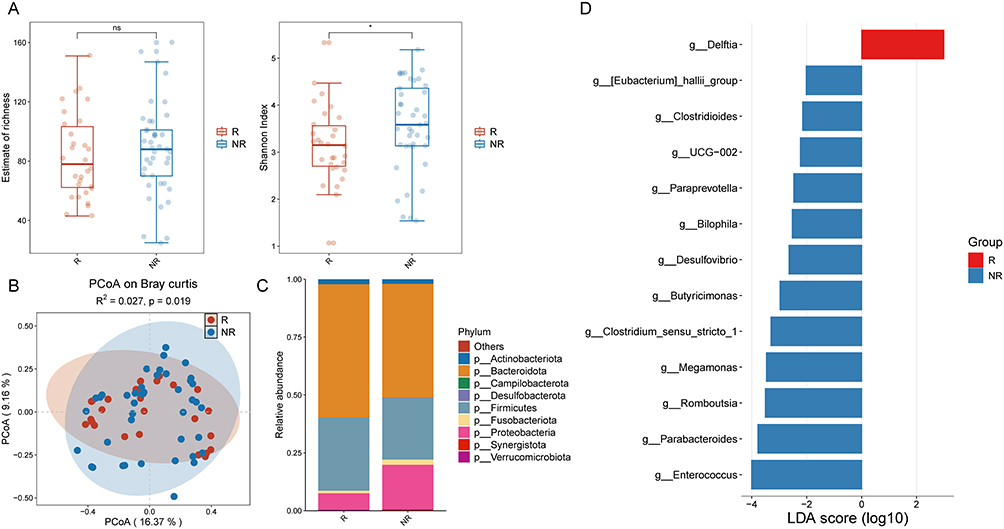 |
Figure 5 (A) Alpha diversity indices of gut microbiota in relapsed and non-relapsed IBD patients after treatment. (B) Beta diversity indices of gut microbiota in relapsed and non-relapsed IBD patients after treatment. (C) Phylum-level distribution of gut microbiota in relapsed and non-relapsed IBD patients after treatment. (D) Enrichment analysis of differential bacterial genera in relapsed and non-relapsed IBD patients after treatment. *P < 0.05. Abbreviation: NS, not significant. |
Correlations Between Post-Treatment Differential Genera and Clinical Indicators in Recurrent versus Non-Recurrent Patients
Correlation analysis showed significant associations between treatment-responsive bacterial genera and key clinical indicators—including albumin (ALB), derived neutrophil-to-lymphocyte ratio (dNLR), and eosinophil counts—in both patient groups. At baseline, ALB was positively correlated with Anaerostipes and Monoglobus, but negatively correlated with Delftia. Following treatment, ALB demonstrated a positive association with Enterococcus and Clostridioides. No bacterial genera exhibited correlations with dNLR; however, post-treatment, dNLR was negatively associated with Paraprevotella. In terms of eosinophil count, no significant correlations with bacterial genera were identified at baseline, but post-treatment, a significant positive association was observed with the genus Paraprevotella (Figures 6A and B, S3A and B).
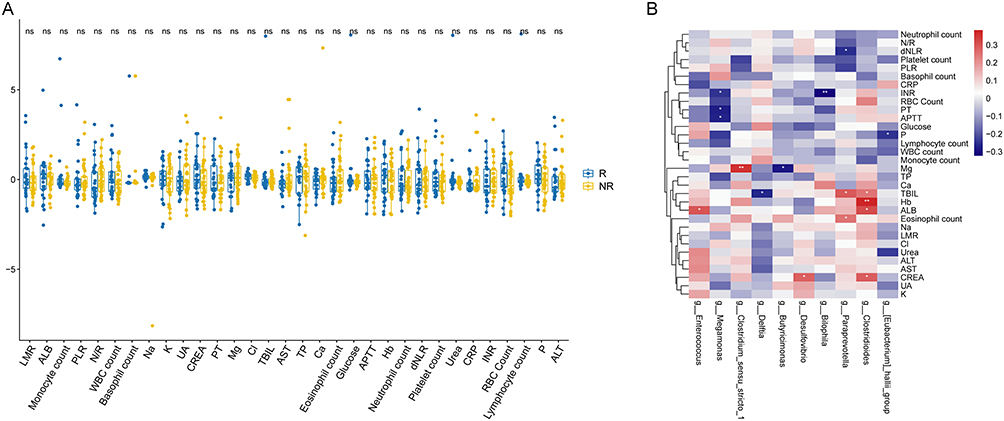 |
Figure 6 (A) Correlation analysis of clinical indicators after treatment in IBD patients with and without relapse. (B) Correlation analysis between clinical indicators after treatment and differential bacterial genera in patients with and without relapse. *P < 0.05; **P < 0.01. |
Predicting IBD Relapse Risk Using Baseline Differential Genera and Clinical Indicators in Recurrent versus Non-Recurrent Patients
To evaluate the prognostic value of baseline gut microbiota for treatment response, we constructed a random forest (RF) classifier to predict post-treatment relapse risk in recurrent versus non-recurrent IBD patients. This model integrated LEfSe-identified differential genera distinguishing patient outcomes: Haemophilus, Anaerostipes, Phascolarctobacterium, Delftia, Monoglobus, Aggregatibacter, and Actinobacillus (Figure S4A). The microbiota-based classifier achieved AUCs of 0.724 (validation cohort) and 0.677 (test cohort) (Figure S4B). Additionally, an RF model incorporating LASSO-selected clinical indicators—platelet count, chloride (Cl), urea, hemoglobin (Hb), and magnesium (Mg) (Figure S5A) —yielded AUCs of 0.661 (validation) and 0.662 (test) (Figure S5B). Furthermore, we constructed an integrated RF classifier combining LEfSe-derived genera and LASSO-selected clinical features (Figure 7A). This combined model demonstrated significantly improved performance with AUCs of 0.676 (validation) and 0.769 (test) (Figure 7B).
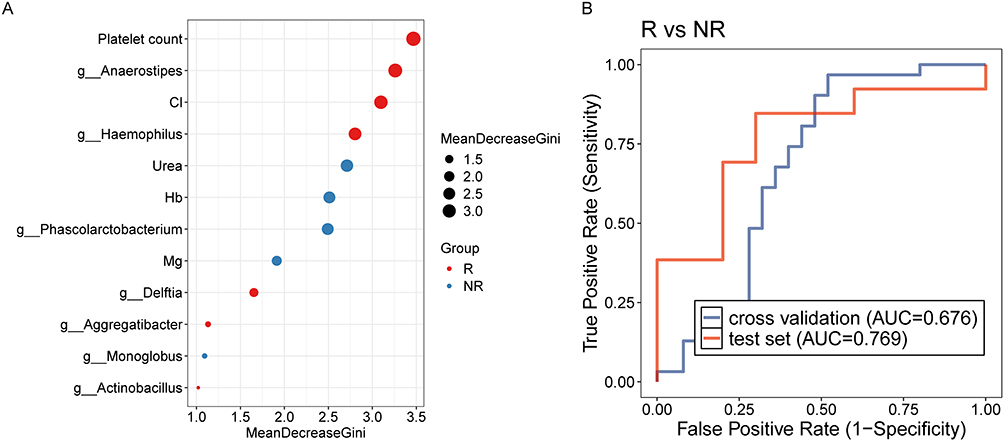 |
Figure 7 (A) Lefse analysis for identifying bacterial genera based on gut microbiota and clinical indicators at baseline between patients with and without relapse. (B) Random forest model to predict IBD relapse post-treatment, utilizing differential bacterial genera and clinical indicators at baseline. |
Predicting Post-Treatment Relapse Risk Using Differential Genera and Clinical Indicators in Recurrent versus Non-Recurrent IBD Patients
To assess the prognostic value of the post-treatment gut microbiota for relapse prediction, we developed a RF model leveraging LEfSe-identified differential genera between recurrent and non-recurrent patients. The model incorporated 13 genera: Clostridium_sensu_stricto_1, Parabacteroides, Eubacterium_hallii_group, Romboutsia, Delftia, Enterococcus, Bilophila, Clostridioides, Butyricimonas, Desulfovibrio, UCG-002, Megamonas, and Paraprevotella (Figure 8A). This microbiota-based predictor achieved AUCs of 0.721 (validation cohort) and 0.861 (test cohort) (Figure 8B), demonstrating robust discriminatory capacity. We subsequently established a clinically-driven RF model using LASSO-selected features. LASSO regression identified 19 clinical indicators for inclusion (Figure S6A and B), yet the model delivered minimal predictive utility with AUCs of 0.485 (validation) and 0.489 (test) (Figure S6C). Finally, an integrated RF classifier combining LEfSe-derived genera and LASSO-selected clinical variables (Figure S7A) attained AUCs of 0.747 (validation) and 0.688 (test) (Figure S7B). Overall, post-treatment microbial signatures, particularly those encompassing 13 key genera, exhibit considerable discriminatory power in predicting relapse risk, significantly surpassing the predictive performance of models based solely on conventional clinical indicators. These findings underscore the potential of gut microbiota as independent, robust biomarkers for relapse stratification in IBD patients.
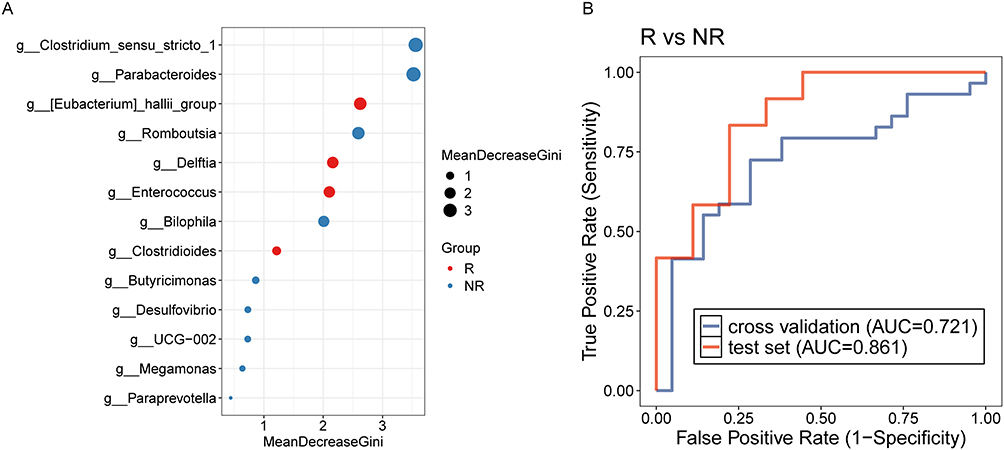 |
Figure 8 (A) Lefse analysis for discriminating bacterial taxa in the gut microbiota after treatment between patients with and without relapse. (B) Random forest model to predict IBD relapse post-treatment, based on differential bacterial taxa identified after treatment. |
Discussion
In this study, we employed 16S rRNA gene sequencing to compare gut microbiota dynamics in relapsed versus non-relapsed IBD patients before and after treatment. Significant intergroup differences emerged in microbial diversity, community structure, and dominant taxa post-treatment. Furthermore, correlation analyses identified significant associations between relapse-associated differential genera and key clinical indicators. We subsequently developed an RF classifier for post-treatment relapse risk prediction, utilizing LEfSe-derived genera and/or LASSO-selected clinical features. The microbiota-based model encompassing 13 key genera (Clostridium_sensu_stricto_1, Parabacteroides, Eubacterium_hallii_group, Romboutsia, Delftia, Enterococcus, Bilophila, Clostridioides, Butyricimonas, Desulfovibrio, UCG-002, Megamonas, and Paraprevotella) achieved an AUC of 0.861, demonstrating substantial clinical utility for relapse monitoring and early intervention in IBD management.
Intestinal mucosal barrier, composed of epithelial cells secreted by goblet cells and a mucus layer, harboring commensal bacteria, serves as the first line of defense against pathogenic intestinal flora.20 Impairment of the intestinal barrier resulting from gut microbiota dysbiosis may be a significant contributing factor to the recurrence of IBD.21 In comparison to healthy individuals, patients with IBD in remission display a persistently reduced α-diversity and significant alterations in gut microbiota composition, potentially increasing their susceptibility to external and internal perturbations. During such disturbances, certain pathogenic bacteria associated with the onset and progression of IBD may proliferate rapidly, leading to disease recurrence.22 Additionally, in patients with a stabilized gut microecology following treatment, beneficial bacteria in the intestine can regulate bile acid metabolism and exert a positive influence on intestinal immunity by activating bile acid receptors.23 Our study demonstrated that there was no significant change in gut microbiota diversity among non-relapsed patients before and after treatment. In contrast, a notable reduction in gut microbiota diversity was observed in relapsed patients. These finding suggested that post-treatment gut microbiota dysbiosis occurred in relapsed patients, with a potential increase in the abundance of bacteria linked to IBD recurrence during this period, thereby influencing the relapse of IBD.
In patients with IBD, gut microbiota dysbiosis contributed to the disruption of immune tolerance, potentially triggering disease relapses.24 There was a direct association between the chronic inflammatory response characteristic of IBD and an imbalance in the immune system. Innate immune cells, including neutrophils and eosinophils, exacerbated inflammation in the intestinal mucosa through the release of cytotoxic proteins.25 Research has demonstrated that the high recurrence rate of CD following surgical intervention was linked to elevated tissue eosinophil activity and the overexpression of interleukin-5 (IL-5).26 Our study identified a significant correlation between eosinophil counts in peripheral blood post-treatment and the presence of the genus Paraprevotella. Notably, Paraprevotella was the predominant genus in patients who did not experience disease recurrence following treatment. Dysbiosis of the Prevotellaceae family was linked to the progression of IBD in experimental mouse models.27 Prevotellaceae are known to activate the Wnt signaling pathway, leading to the upregulation of pro-inflammatory cytokines and the downregulation of anti-inflammatory cytokines. This activation promoted lymphocytic inflammatory infiltration and played a significant role in the pathogenesis of colorectal cancer (CRC).28 Our study indicated a potential association between Prevotellaceae and eosinophil count in the mechanism of IBD recurrence. However, these findings must be interpreted with caution. The cross-sectional nature of our analysis cannot determine whether microbial shifts drive clinical changes or vice versa. The clinical relevance of these correlations remains speculative and requires validation in longitudinal studies.
IBD is characterized by a high risk of relapse after treatment, and gut microbiota differences as well as specific clinical indicators can be used for predicting IBD relapses.29,30 We constructed Random Forest models based on the differential gut microbiota and selected clinical indicators, either individually or in combination, from patients who experienced relapse versus those who did not, both before and after treatment. Results indicated that the Random Forest model built upon the differential gut microbiota of post-treatment relapse versus non-relapse patients yielded an AUC value of 0.721 in the validation cohort and 0.861 in the test cohort, demonstrating the highest predictive accuracy among all models examined in this study. This predictive model shows potential in aiding the cessation of anti-TNF therapy for patients at reduced risk of relapse, equipping physicians with a valuable instrument for making informed decisions concerning the management of anti-TNF therapy in patients with IBD. Nonetheless, before its integration into clinical practice, it is crucial for the model to be validated through extensive prospective studies. Beyond its standalone predictive value, a critical next step involves evaluating how our microbiota-based model complements existing clinical biomarkers, particularly fecal calprotectin.31 FC is a robust indicator of current intestinal inflammation but has limited reliability in predicting future relapse.32 We hypothesize that our model, which reflects the underlying microbial ecology, provides a complementary and prospective dimension to risk stratification. Integrating the microbial risk profile with FC levels has the potential to facilitate more nuanced and personalized management decisions, ultimately improving long-term outcomes for patients with IBD.
Despite these findings, this study has several limitations that warrant consideration. First, the relatively modest sample size (n=88) may limit the statistical power and generalizability of our conclusions. A larger cohort would enhance the robustness of the identified microbial signatures and allow for more sophisticated subgroup analyses, such as stratifying by IBD subtype (UC vs CD) or specific treatment regimens, to uncover more nuanced associations. Second, and critically, our predictive model was developed and validated within a single-center cohort, lacking an independent external validation set. Although our model demonstrated encouraging performance in the test cohort (AUC = 0.861), its more moderate performance in the validation set (AUC = 0.721) and the lack of external validation indicate that its generalizability requires further confirmation. Future studies involving multi-center, prospective cohorts are essential to externally validate and refine these microbial biomarkers, confirming their clinical utility for predicting IBD relapse risk across diverse demographics and healthcare settings. Moreover, unaccounted confounders, including dietary patterns, lifestyle behaviors, and environmental exposures, could introduce bias in gut microbiota analyses. In addition, it is important to note that genera such as Delftia are also common environmental contaminants. While we observed a statistically significant association with relapse status, the biological relevance of this finding warrants further validation in studies that include negative control samples to definitively rule out contamination. Furthermore, building upon the promising but preliminary findings of this 16S rRNA-based study, Shotgun metagenomic sequencing would provide superior taxonomic resolution to the species or strain level and directly elucidate the functional potential of the microbial community, allowing for a more precise validation of genera like UCG-002 and Delftia. Furthermore, metabolomic profiling of fecal and serum samples could identify microbial metabolites that serve as functional effectors in IBD relapse, bridging the gap between microbial presence and host physiology. The integration of these multi-omics datasets using advanced machine learning frameworks is a critical next step. Despite these limitations, our investigation offers meaningful insights into the gut microbiota-relapse relationship in IBD and lays a foundation for future mechanistic research.
Conclusions
In summary, our study provides preliminary evidence of distinct gut microbiota structures between IBD patients who relapse and those who maintain remission after treatment. We observed statistical associations between specific microbial taxa and clinical indicators, although these relationships remain speculative and their clinical utility as biomarkers is not established. The predictive model built on post-treatment microbiota showed promising but preliminary discriminatory power, requiring external validation. Its primary contributions lie in identifying microbial candidates for future investigation and in constructing a foundational model that must be rigorously tested in larger, multi-center cohorts. Ultimately, our findings do not confirm but rather propose a potential link between the post-treatment microbiota and IBD relapse, setting the stage for future research to elucidate any causal relationships.
Abbreviations
IBD, Inflammatory bowel disease; UC, Ulcerative colitis; CD, Crohn’s disease; AUC, Area under the curve; ALB, Albumin; dNLR, derived neutrophil-to-lymphocyte ratio.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are available in the Sequence Read Archive (SRA) repository BioProject: PRJNA1256227 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1256227/).
Ethics Approval and Consent to Participate
The study was approved by the Ethical Committee of Nanchong Central Hospital (Approval No. 2022-075), and written informed consent was obtained from all participants or their guardians in accordance with the Declaration of Helsinki.
Acknowledgments
We would like to thank the patients who enrolled in the study.
Funding
This study was supported by Health Commission of Sichuan Province (No. 24LCYJPT04).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Qiu P, Ishimoto T, Fu L, Zhang J, Zhang Z, Liu Y. The gut microbiota in inflammatory bowel disease. Front Cell Infect Microbiol. 2022;12. doi:10.3389/fcimb.2022.733992
2. Höie O, Wolters F, Riis L, et al. Ulcerative colitis: patient characteristics may predict 10-yr disease recurrence in a European-wide population-based cohort. Am J Gastroenterol. 2007;102(8):1692–1701. doi:10.1111/j.1572-0241.2007.01265.x
3. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12(4):205–217. doi:10.1038/nrgastro.2015.34
4. Ghouri YA, Tahan V, Shen B. Secondary causes of inflammatory bowel diseases. World J Gastroenterol. 2020;26(28):3998–4017. doi:10.3748/wjg.v26.i28.3998
5. Sýkora J, Pomahačová R, Kreslová M, Cvalínová D, Štych P, Schwarz J. Current global trends in the incidence of pediatric-onset inflammatory bowel disease. World J Gastroenterol. 2018;24(25):2741–2763. doi:10.3748/wjg.v24.i25.2741
6. Bots SJ, Kuin S, Ponsioen CY, et al. Relapse rates and predictors for relapse in a real-life cohort of IBD patients after discontinuation of anti-TNF therapy. Scand J Gastroenterol. 2019;54(3):281–288. doi:10.1080/00365521.2019.1582693
7. Miner PB Jr. Factors influencing the relapse of patients with inflammatory bowel disease. Am J Gastroenterol. 1997;92(12 Suppl):1s–4s.
8. Vindigni SM, Zisman TL, Suskind DL, Damman CJ. The intestinal microbiome, barrier function, and immune system in inflammatory bowel disease: a tripartite pathophysiological circuit with implications for new therapeutic directions. Therap Adv Gastroenterol. 2016;9(4):606–625. doi:10.1177/1756283X16644242
9. Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev. 2014;13(1):24–30. doi:10.1016/j.autrev.2013.06.002
10. Kugathasan S, Denson LA, Walters TD, et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet. 2017;389(10080):1710–1718. doi:10.1016/S0140-6736(17)30317-3
11. Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53(5):685–693. doi:10.1136/gut.2003.025403
12. Sokol H, Seksik P, Furet JP, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. doi:10.1002/ibd.20903
13. Rajca S, Grondin V, Louis E, et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm Bowel Dis. 2014;20(6):978–986. doi:10.1097/MIB.0000000000000036
14. Yu S, Li J, Ye Z, et al. Identification of a 10-species microbial signature of inflammatory bowel disease by machine learning and external validation. Cell Regener. 2025;14(1):32. doi:10.1186/s13619-025-00246-w
15. Feng J, Wu S, Yang H, et al. Microbe-bridged disease-metabolite associations identification by heterogeneous graph fusion. Briefings Bioinf. 2022;23(6). doi:10.1093/bib/bbac423
16. Ning L, Zhou YL, Sun H, et al. Microbiome and metabolome features in inflammatory bowel disease via multi-omics integration analyses across cohorts. Nat Commun. 2023;14(1):7135. doi:10.1038/s41467-023-42788-0
17. Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105(43):16731–16736. doi:10.1073/pnas.0804812105
18. Russo E, Cinci L, Di Gloria L, et al. Crohn’s disease recurrence updates: first surgery vs. surgical relapse patients display different profiles of ileal microbiota and systemic microbial-associated inflammatory factors. Front Immunol. 2022;13:886468.
19. Inflammatory Bowel Disease Group CSoG, Chinese Medical Association. Chinese consensus on diagnosis and treatment in inflammatory bowel disease (2018, Beijing). Chin J Inflamm Bowel Dis. 2018;2(3):173–190.
20. Cai R, Cheng C, Chen J, Xu X, Ding C, Gu B. Interactions of commensal and pathogenic microorganisms with the mucus layer in the colon. Gut Microbes. 2020;11(4):680–690. doi:10.1080/19490976.2020.1735606
21. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):1–9. doi:10.1038/s12276-018-0126-x
22. Pisani A, Rausch P, Bang C, et al. Dysbiosis in the gut microbiota in patients with inflammatory bowel disease during remission. Microbiology Spectrum. 2022;10(3):e00616–00622. doi:10.1128/spectrum.00616-22
23. Li L, Liu T, Gu Y, et al. Regulation of gut microbiota-bile acids axis by probiotics in inflammatory bowel disease. Front Immunol. 2022;13:974305.
24. Haneishi Y, Furuya Y, Hasegawa M, Picarelli A, Rossi M, Miyamoto J. Inflammatory Bowel Diseases and Gut Microbiota. Int J Mol Sci. 2023;24(4):3817. doi:10.3390/ijms24043817
25. Ling LM, Peterson C, Hedin CRH, et al. Faecal biomarkers for diagnosis and prediction of disease course in treatment-naïve patients with IBD. Aliment Pharmacol Ther. 2024;60(6):765–777. doi:10.1111/apt.18154
26. Alhmoud T, Gremida A, Steele DC, et al. Outcomes of inflammatory bowel disease in patients with eosinophil-predominant colonic inflammation. BMJ Open Gastroenterol. 2020;7(1):S. doi:10.1136/bmjgast-2020-000373
27. Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156(5):1045–1059. doi:10.1016/j.cell.2014.01.026
28. Lu Y, Cui A, Zhang X. Commensal microbiota-derived metabolite agmatine triggers inflammation to promote colorectal tumorigenesis. Gut Microbes. 2024;16(1):2348441. doi:10.1080/19490976.2024.2348441
29. Zhou Y, Xu ZZ, He Y, et al. Gut microbiota offers universal biomarkers across ethnicity in inflammatory bowel disease diagnosis and infliximab response prediction. mSystems. 2018;3(1). doi:10.1128/msystems.00188-17
30. Serrano-Gómez G, Mayorga L, Oyarzun I, et al. Dysbiosis and relapse-related microbiome in inflammatory bowel disease: a shotgun metagenomic approach. Comput Struct Biotechnol J. 2021;19:6481–6489. doi:10.1016/j.csbj.2021.11.037
31. Burri E, Beglinger C. IBD: faecal calprotectin testing--the need for better standardization. Nat Rev Gastroenterol Hepatol. 2014;11(10):583–584. doi:10.1038/nrgastro.2014.154
32. Jukic A, Bakiri L, Wagner EF, Tilg H, Adolph TE. Calprotectin: from biomarker to biological function. Gut. 2021;70(10):1978–1988. doi:10.1136/gutjnl-2021-324855
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Mesenchymal Stem Cells Ameliorate DSS-Induced Experimental Colitis by Modulating the Gut Microbiota and MUC-1 Pathway
Wang H, Sun Y, Xiao FJ, Zhao X, Zhang WY, Xia YJ, Wang LS
Journal of Inflammation Research 2023, 16:2023-2039
Published Date: 11 May 2023
Serological Biomarker-Based Machine Learning Models for Predicting the Relapse of Ulcerative Colitis

Pang W, Zhang B, Jin L, Yao Y, Han Q, Zheng X
Journal of Inflammation Research 2023, 16:3531-3545
Published Date: 21 August 2023
Microbial Disruptions in Inflammatory Bowel Disease: A Comparative Analysis
Ma J, Wang K, Wang J, Zeng Q, Liu K, Zheng S, Chen Y, Yao J
International Journal of General Medicine 2024, 17:1355-1367
Published Date: 6 April 2024
Unveiling the Therapeutic Potential of Berberine in Rheumatoid Arthritis: A Comprehensive Study of Network Pharmacology, Metabolomics, and Intestinal Flora

Li B, Liu J, He C, Deng Z, Zhou X, Peng R
Journal of Inflammation Research 2024, 17:10849-10869
Published Date: 10 December 2024
The Role of Gut Microbiota on Intestinal Fibrosis in Inflammatory Bowel Disease and Traditional Chinese Medicine Intervention
Fang L, Peng H, Tan Z, Deng N, Peng X
Journal of Inflammation Research 2025, 18:5951-5967
Published Date: 7 May 2025