Back to Journals » International Journal of Nanomedicine » Volume 20
Pleiotropic Multi-Drug Co-Assembled Nanocomposites Offer Protection Against Doxorubicin-Induced Cardiotoxicity
Authors Yin D, Li C, Li J, Zeng J, Jia M, Yuan W, Liu L, Tang Y, Wang Y, Tian J
Received 27 March 2025
Accepted for publication 18 July 2025
Published 23 July 2025 Volume 2025:20 Pages 9311—9326
DOI https://doi.org/10.2147/IJN.S528349
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Dan Yin,1,2 Can Li,3 Jiajin Li,1 Jieying Zeng,4 Mengjie Jia,4 Wenjing Yuan,1 Lingjuan Liu,1 Yi Tang,2 Yi Wang,4 Jie Tian1
1National Clinical Key Cardiovascular Specialty, Key Laboratory of Children’s Important Organ Development and Diseases of Chongqing Municipal Health Commission, Chongqing, People’s Republic of China; 2Department of Ultrasound, Children’s Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 3Department of Cardiology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 4School of Basic Medical Science, Chongqing Medical University, Chongqing, 400016, People’s Republic of China
Correspondence: Jie Tian; Yi Wang, Email [email protected]; [email protected]
Background: Doxorubicin (DOX) is a first-line chemotherapeutic agent, yet its clinical utility is limited by doxorubicin-induced cardiotoxicity (DIC), a dose-dependent side effect. Current drug delivery strategies fail to prevent off-target accumulation of DOX in cardiac tissue, necessitating innovative therapeutic approaches that protect the heart without compromising antitumor efficacy through targeted cardioprotection.
Methods: We developed a multifunctional self-assembled nanoplatform (PGPP/NPs) via co-assembly of ginsenoside Rb1, probucol (PB), and a phosphoinositide 3-kinase gamma (PI3Kγ) inhibitor (PI), with surface modification using PCM peptide for cardiomyocyte-targeted delivery. In vitro and in vivo models of DIC were used to evaluate targeting specificity and therapeutic efficacy. Mechanistic investigations included ROS detection (DCFH-DA assay), inflammatory phenotype analysis (immunohistochemistry for CD68 and CD206), and autophagy flux assessment (immunofluorescence for LC3-II).
Results: PGPP/NPs demonstrated selective accumulation in cardiomyocytes compared to non-targeted GPP/NPs (p< 0.05). This nanocomposite significantly alleviated DIC through antioxidative, anti-inflammatory, and autophagy-promoting mechanisms. Notably, it reduced DIC severity while preserving the antitumor efficacy of DOX.
Conclusion: This novel therapeutic strategy shows great promise for mitigating chemotherapy-induced cardiotoxicity and may be extended to other chemotherapeutic agents with cardiac side effects.
Keywords: doxorubicin, cardiotoxicity, detoxification, self-assembled nanoparticles, antioxidative
Corrigendum for this paper has been published.
Introduction
Doxorubicin (DOX), an anthracycline chemotherapy agent, remains the cornerstone in the treatment of hematologic malignancies and solid tumors, significantly contributing to improvements in the average 5-year cancer survival rate1. However, due to the nonspecific distribution, most DOX accumulates in normal cells and organs, especially in the heart, which can induce dose-dependent cardiotoxicity and subsequent heart failure.2 The pathogenesis of DOX-induced cardiotoxicity (DIC) is complex, involving multiple cellular and signaling pathways.3–7 Extensive studies have revealed that oxidative stress and inflammation play crucial roles in DIC progression.8–10 Notably, recent studies have highlighted that phosphoinositide 3-kinase gamma (PI3Kγ) acts as a vital regulator of tumorigenesis and cardiac function.11 PI3Kγ inhibitors have emerged as promising therapeutic targets for DIC, as they promote autophagy to mitigate DOX-induced cardiotoxicity while enhancing DOX’s antitumor efficacy. Leveraging the critical roles of oxidative stress and inflammation in DIC pathogenesis, combined with PI3Kγ inhibition as a novel target, we aimed to design a novel cardiac detoxification strategy.
With the rapid development of nanobiotechnology and materials science, researchers have explored numerous methods against DIC.12–15 However, the major drawbacks of this strategy are the single-function platform design and the requirement on synthetic nanocarriers for drug delivery. Moreover, some nanoscale detoxification platforms are designed to competitively bind DOX to alleviate DIC.16 Whereas, these platforms are unable to distinguish between tumor and normal tissues, potentially compromising the antitumor effect. Therefore, it is imperative to develop a pleiotropic and convenient therapeutic strategy that alleviates DIC without compromising the antitumor efficacy of DOX.
In recent decades, accumulating evidence has revealed that certain active compounds of traditional Chinese medicine (TCM) can self-assemble or co-assemble into nanoparticles (NPs) through non-covalent intermolecular forces, such as electrostatic forces, hydrogen bonding, hydrophobic interactions.17–20 These self-assembled NPs offer promising drug carriers for drug delivery, as they are composed exclusively of drugs, simplifying preparation methods, enhancing drug loading capacity, improving biocompatibility, and increasing safety compared to synthetic NPs.21–24 Although this self-assembly strategy has been widely applied in cancer targeted therapy, its application in alleviating DIC has not been reported to our knowledge. Ginsenoside Rb1, a major bioactive compound extracted from ginseng, exhibits multiple pharmacological properties, including antioxidative stress, anti-inflammatory, and anticancer effects.25–29 Additionally, Ginsenoside Rb1 can co-assemble with other drugs to form multifunctional nanoplatforms. Consistently, probucol (PB), an FDA-approved drugs, is acknowledged for its anti-oxidative and anti-inflammatory effects.30–33 It is capable of synergizing with the Ginsenoside Rb1 to alleviate oxidative stress and inflammatory response of DIC.
Herein, we propose a multifunctional and convenient strategy to relieve DIC without compromising antitumor efficacy. This strategy involves co-assembling Ginsenoside Rb1 with PB and PI3Kγ inhibitor (PI) to generate self-assembled nanoparticles (GPP/NPs). These GPP/NPs are further modified with a cardiomyocyte-targeting peptide (PCM) to enhance cardiac-specific targeting (termed PGPP/NPs). The developed PGPP/NPs selectively target myocardial tissue, and alleviate DIC through antioxidative, anti-inflammatory, and autophagy-promoting mechanisms.
Method and Materials
Materials
Ginsenoside Rb1, probucol (PB), PI3Kγ inhibitor, and 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-Poly (ethylene glycol) (DSPE-PEG) were purchased from China National Pharmaceutical Group Corporation (Beijing, China). PCM (WLSEAGPVVTVRALRGTGSW) was synthesized by Shanghai Bioengineering Co. Dulbecco’s modified Eagle’s medium (DMEM), Roswell Park Memorial Institute 1640 medium (RPMI 1640), collagenase II, fetal bovine serum (FBS), penicillin– streptomycin, and trypsin were obtained from Gibco (Carlsbad, CA, USA). 1,19-Dioctadecyl-3,3,39,39-tetramethylindodicarbocyanine perchlorate (DiD), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), 4′,6-diamidino-2-phenylindole (DAPI), Reactive Oxygen Species (ROS) assay kit, and a cell counting kit 8 (CCK-8) were purchased from Beyotime Biotechnology (Shanghai, China). LIVE/DEAD kit was purchased from Thermo Fisher Scientific (Mainland China). 1,1′-Dioctadecyl- 3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was purchased from Sigma-Aldrich (Saint Louis, MO, USA). N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and lipopolysaccharide (LPS) was purchased from Solarbio (Beijing, China).
LC3A/B Antibody (#4108) and rhodamine phalloidin were obtained from Cell Signaling Technology (USA). Mouse cardiac troponin I (cTnI) and mouse Brain Natriuretic Peptide (BNP) ELISA kits were purchased from Wuhan Colorful Gene Biological Technology Co. Ltd (Wuhan, China). All chemicals used in this study were of analytical grade and used as received.
Preparation and Characterization of Self-Assembled GPP/NPs and PGPP/NPs
Nanoprecipitation was employed to prepare self-assembling NPs. Specifically, the mass ratio of GI/PB/PI is 10:20:1. The mass of DSPE-PEG was 15% of the total mass of all materials. The above materials were dissolved with dimethyl sulfoxide (DMSO), then thoroughly mixed together under sonication. The mixture was quickly and dropwise added to 4 mL ultrapure water to obtain GPP/NPs solutions. Subsequently, the GPP/NPs solutions were transferred to a dialysis bag (MWCO = 3500 Da) for 12 h to remove DMSO. Finally, the GPP/NPs solutions were collected and stored at 4 °C for later use. Consistently, PGPP/NPs solutions were prepared following the identical method and the mass ratio of GI/PB/PI/PCM is 10:20:1:3. DiI or DiD-labeled GPP/NPs and PGPP/NPs were prepared by adding free DiI (50 μM) or DiD (50 μM) and stirring for 30 min.
The morphology of GPP/NPs and PGPP/NPs were observed by transmission electron microscopy (TEM) at 100 kV (JEM-1200EX, Japan). The particle size and Zeta potentials of GPP/NPs and PGPP/NPs were measured by dynamic light scattering (DLS; Malvern Instruments Ltd., UK). The encapsulation efficiency (EE) of PI3Kγ inhibitor in the PGPP/NPs was obtained at 341 nm by the ultraviolet spectrophotometer(UVS) (UV1700, Milford, USA). PBS solution (PH 7.4) containing 30% absolute ethanol and 2% Tween-80 was selected as the release medium. The in vitro drug release profile of PGPP/NPs was assessed using a dialysis method (MWCO = 3500 Da). Briefly, PGPP/NPs solutions (2 mg/mL) were loaded into dialysis bags and immersed in 30 mL of release medium (pH 7.4 PBS), then incubated at 37°C with continuous shaking at 100 rpm. At predetermined time intervals (0, 2, 4, 8, 10 h), 1 mL of the release medium was withdrawn and replaced with an equal volume of fresh pre-warmed medium to maintain sink conditions. The concentration of the released PI3Kγ inhibitor was quantified by UV-Vis spectroscopy (UV1700, Milford, USA) at 320 nm.
Cell Culture
The isolation and culture of primary cardiomyocytes from mouse followed previously reported protocols.34 Briefly, primary mouse cardiomyocytes were obtained from neonatal mice (days 0–2), digested with 0.2% collagenase II, neutralized with twice the volume of serum, and filtered with a 100 μM screen. Then, these cells were routinely cultured in Dulbecco′s modified Eagle′s medium (DMEM) supplemented with 20% foetal bovine serum(FBS), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C with 5% CO2 in a humidified cell incubator. Furthermore, H9C2 cells (rat cardiomyocytes) and 4T1 cells (mouse breast cancer cells) were purchased from the American Type Culture Collection (ATCC). They were respectively cultured in DMEM and Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS and 2% penicillin−streptomycin. The cells were incubated at 37 °C with 5% CO2 and 95% air.
Hemolytic Test of GPP/NPs and PGPP/NPs
The blood was collected from the abdominal aorta of Sprague-Dawley rats and placed in tubes containing EDTA anticoagulant. The collected blood was centrifuged and then the red blood cells (RBCs) were washed with saline for five times. RBCs were then diluted to 2% (v/v) hematocrit with saline to prepare RBCs suspensions for subsequent usage. Saline was selected as negative control and distilled water as positive control, in addition, GPP/NPs (1 mg/mL) and PGPP/NPs (1 mg/mL) solutions as experimental groups. In the assay, 1 mL of GPP/NPs, PGPP/NPs, saline, and distilled water respectively added to 20 μL of RBCs suspensions, the mixtures were incubated at 37 °C for 60 min. After the mixtures were centrifuged, the absorbance value of the supernatants was determined by ultraviolet spectrophotometer.
Hemolysis (%) = (As-Ab)/(Ac-Ab) *100%21
(Ab is the absorbance value of the negative control, Ac the absorbance value of positive control, and as the absorbance value of experimental groups).
Biocompatible Evaluation of PGPP/NPs in H9C2 Cells in vitro
The viability of H9C2 cells was measured using a Cell Counting Kit-8 (CCK-8) assay. H9C2 cells were seeded in a 96-well plate (5 × 103 cells/well) and incubated until approximately 80% confluence. Followed the incubation, PGPP/NPs solutions with various concentrations were add to each well for 24 h. After removal of the original medium, the CCK-8 agents were added to each well and cultured for 2 h. Finally, the absorbance was measured at a wavelength of 490 nm. All concentrations were performed in sextuplicate. Besides, to determine IC50 value, H9C2 cells were seeded and grown in 96-well plates. After culture medium removal, the fresh mediums in a fixed 100 μL volume containing various concentrations of DOX were added and incubated for 24 h. Afterwards, H9C2 cells were subjected to CCK-8 assay. IC50 values were calculated by a nonlinear regression equation.
Cellular Uptake of GPP/NPs and PGPP/NPs in vitro
H9C2 cells (1×106 cells/well) were seeded in 6-well plates and cultured for 24 h. DiO-labelled GPP/NPs and PGPP/NPs solutions (25 μg/mL, nanoparticle weight) were added to the 6-well plates to investigate the intracellular fluorescence intensity at the 2 h and 4 h time point. The intracellular fluorescence intensity was positively correlated with the targeting ability. Simultaneously, the cytoskeleton of H9C2 cells was labelled by rhodamine phalloidin and the cell nucleus labelled by DAPI. The results were observed by confocal laser microscopy (TCS SP8, Leica, Germany) and analyzed by flow cytometry (Cytoflex, Beckman, CA, USA).
Vitro Detoxification Tests of PGPP/NPs
To evaluate the cardioprotective capacity of PGPP/NPs against DIC, a CCK-8 test was used to determine cell viability. In a 96-well plate, H9C2 cells were seeded at a density of 5×103 cells per well. After the cell confluence reached 80–90%, the different concentrations of PGPP/NPs were added to the cell plate for 4 h. All concentrations were performed in sextuplicate. Then, the medium was discarded and 200 μL of fresh medium containing IC50 dosages of DOX were added into each well, followed by 24 h of incubation. Subsequently, H9C2 cells were subjected to CCK-8 assay.
Furthermore, LIVE/DEAD kit and Reactive Oxygen Species (ROS) Assay Kit were used to investigate the detoxification effect against DIC. H9C2 cells (1×106 cells well) were seeded into 6-well plates and incubated overnight. Then, GPP/NPs (25 μg/mL), PGPP/NPs (25 μg/mL), and PBS solutions were added for 4 h of incubation. Thereafter, DOX at IC50 dosages were added into each well and incubated for another 24 h. The cells were stained for 30 min with the LIVE/DEAD kit and ROS kit. Finally, the excess dye was washed 3 times with PBS, and the samples were imaged by fluorescence microscope. In addition, flow cytometry was used to quantify the fluorescence intensity of intracellular ROS. Finally, primary cardiomyocytes were incubated and intervened in the same manner as above. LC3-II (LC3A/B Antibody) was selected as the primary antibody for cellular immunofluorescence staining to investigate cardiac autophagy. Observation and quantitative analysis were performed by confocal laser microscopy.
Animal Model Construction
The animal protocols were authorized by the Committee on the Ethics of Animal Experiments of Chongqing Medical University (Permit Number: IACUC-CQMU-2023-10,037). BALB/c mice (6−8 weeks old, 16–18 g, female) were obtained from Chongqing Medical University, China. To determine the maximal tolerance dosage (MTD) of DOX, eighteen BALB/c mice were equally divided into six groups and injected with different DOX doses including 40, 30, 20, 15, 10, 0 mg/kg. Then, the survival rates and body weights of all mice were monitored daily for 15 consecutive days. The MTD of DOX was defined as the dose of DOX that resulted in significantly weight loss while still maintaining 100% survival.
4T1 cells were incubated to approximately 80% confluence and collected. Female BALB/c mice were inoculated with 1×106 4T1 cells by subcutaneous injection. At the tumor volume reached roughly 70–80 mm3, the mice were randomly divided into five groups (n=5). The groups concluded the control group, the DOX group, the GPP/NPs + DOX group, the PGPP/NPs + DOX group, and the unassembled drug + DOX group. The unassembled drug represented a simple mixture of GI, PB, and PI3Kγ inhibitor and did not form nanoparticles by self-assembled. Each group received intraperitoneal injection of DOX at a dose of 2 mg/kg each time. The above treatments were repeated every 3 days, and tumor volume were measured with a caliper.
In vivo Blood Circulation and Biodistribution of GPP/NPs and PGPP/NPs
DiR-labeled GPP/NPs and PGPP/NPs were injected intravenously into mice bearing 4T1 tumors at a dose of 25 mg/kg. At 0 min, 1 min, 6 h, 12 h, 24 h, 36 h, 48 h, and 72 h after administration, 30 μL of whole blood was collected and added into 96-well plates containing 70 μL of water per well. The DiR fluorescence intensities were measured by multifunctional microplate reader (Tecan Infinite 200Pro, Austria, Switzerland) at an excitation/emission of 745/778 nm. Each time point contained three mice. Besides, to investigate the biodistribution, 25 mg/kg of DiR-labeled GPP/NPs or PGPP/NPs were intravenously injected into the 4T1 tumors bearing mice. The control group was injected with the same volume of saline. Major organs (heart, liver, and tumor) of tumor-bearing mice were collected at 24 h after injection. Ex vivo fluorescence imaging was performed using a fluorescence system (CRi Inc., Woburn, MA, USA), and the fluorescence intensities were calculated. Each group contained five mice.
In vivo Therapeutic Efficacy of PGPP/NPs
In order to verify the cardiac detoxification effect of DOX at its MTD, BALB/c mice were respectively intravenously preinjected with 25 mg/kg of GPP/NPs, PGPP/NPs, and unassembled drug, followed 4 h later by 15 mg/kg of DOX. Mice were treated with single DOX (15 mg/kg) or PBS respectively as a positive or negative control. On the day after the twenty-one treatment, in vivo cardiac function was assessed by transthoracic echocardiography in sedated mice using a Vevo 550 System (Fujifilm Visualsonics Inc). M-mode imaging was performed on the short-axis view of the left ventricle when heart rate fluctuated between 400 bpm and 500 bpm. Ejection fraction (EF), fractional shortening (FS), stroke volume (SV), cardiac output (CO), and heart rate (HR) were determined separately. Afterwards, blood samples and vital organ samples (heart and liver) were collected from all mice for next detection. Serum was collected after centrifugation of fresh blood and detected by mouse cardiac troponin I (cTnI) and mouse Brain Natriuretic Peptide BNP) ELISA kits. Cardiac tissues were removed for H&E, TUNEL, Sirius red, and immunohistochemistry (IHC) staining. Histochemistry score (H-Score) was used to represent semiquantitative results of tissue staining for each slice. H-Score = ∑(pi × i)= (percentage of weak intensity × 1) + (percentage of moderate intensity× 2) + (percentage of strong intensity × 3).35 To study the cardiac autophagic flux in vivo, autophagic marker LC3-II was selected as the primary antibody and fluorescein (FITC)-labeled antibody as the secondary antibody. After washing with PBS, the sections were stained with DAPI and then imaged by fluorescence microscopy.
Statistical Analysis
All data were analyzed using Prism software (GraphPad software Inc) and presented as mean ± SD. Statistical significance was determined by unpaired two-tailed t-test between two groups and one way analysis of variance (ANOVA) between multiple groups. P<0.05 was considered statistically significant.
Results
Characterization of GPP/NPs and PGPP/NPs
A graphical abstract shows the fabrication route of GPP/NPs and PGPP/NPs (Figure 1). GPP/NPs and PGPP/NPs were fabricated via a nanoprecipitation method, leveraging π-π stacking and hydrophobic interactions to self-assemble hydrophobic drugs (ginsenoside Rb1, PB, and PI3Kγ inhibitor) into a carrier-free core, with DSPE-PEG conjugated to the surface via PCM peptide for cardiomyocyte targeting. TEM imaging confirmed homogeneous spherical morphology (Figure 2A). DLS analysis revealed uniform particle sizes (111.43 ± 1.48 nm for GPP/NPs; 109.87 ± 0.95 nm for PGPP/NPs) with narrow polydispersity indices (0.11 ± 0.01 and 0.14 ± 0.01, respectively) and stable zeta potentials (−25.5 ± 1.81 mV for GPP/NPs; −30.23 ± 0.32 mV for PGPP/NPs) (Figure 2B). Dimensional stability assays demonstrated that PGPP/NPs maintained uniform size and stability in DMEM (10% FBS), PBS, or pure water for up to 7 days (Figure 2C). UV spectroscopy confirmed the characteristic absorption peak of the PI3Kγ inhibitor at 341 nm, and a standard curve was generated (Supplementary Figure S1). The EE of the PI3Kγ inhibitor in PGPP/NPs reached 72.55%. Drug release assays showed controlled release in physiological environments, ensuring efficient drug delivery to target regions (Figure 2D). CCK-8 assays revealed cytotoxicity only at concentrations >100 μg/mL (Figure 2E), and in vitro hemolysis assays demonstrated significantly lower hemolysis rates for GPP/NPs and PGPP/NPs compared to the positive control (Figure 2F). Fluorescent labeling consistency between GPP/NPs and PGPP/NPs using equivalent fluorescent agents was confirmed via supplementary experiments (Supplementary Figure S2). Biocompatibility was validated by normal hematological parameters and H&E staining of key organs (Supplementary Figure S3). Collectively, these results indicate successful preparation of GPP/NPs and PGPP/NPs with favorable size, stability, cytocompatibility, and targeting potential.
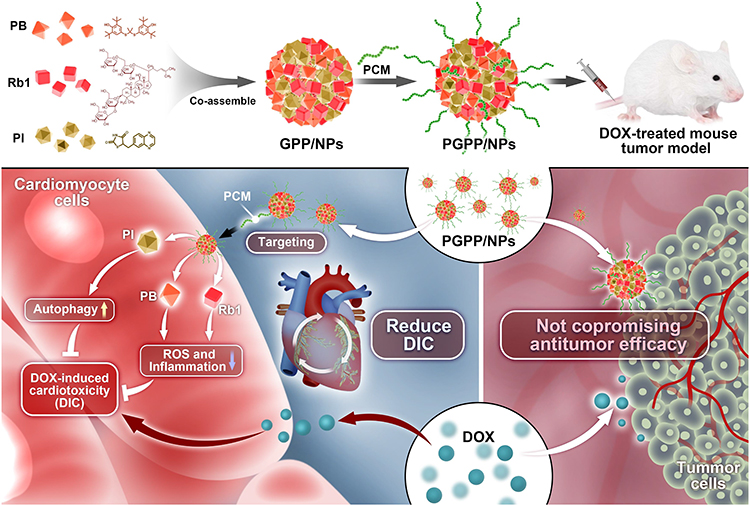 |
Figure 1 Schematic illustration of the preparation and therapeutic mechanism of PGPP/NPs. |
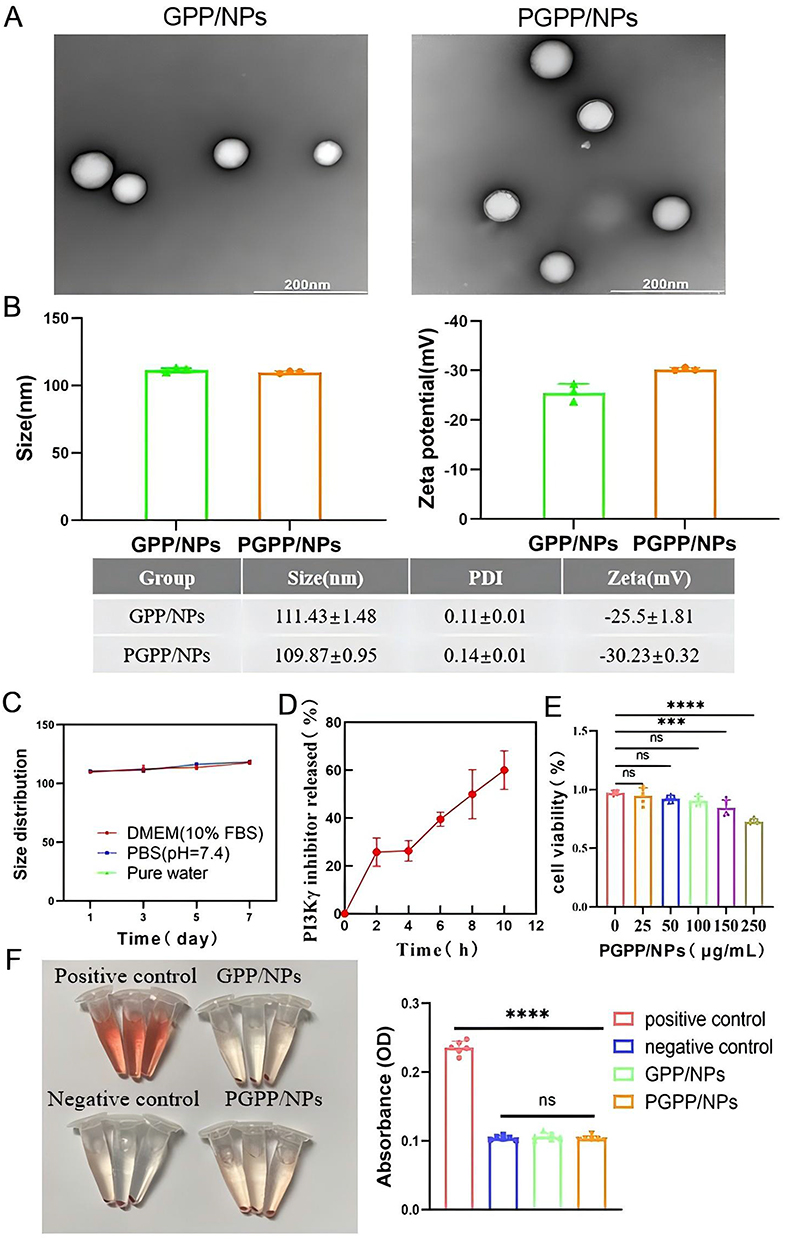 |
Figure 2 Synthesis and characterization. (A) TEM images of GPP/NPs and PGPP/NPs. Scale bars, 200 nm. (B) The particle size and Zeta potential analysis of GPP/NPs and PGPP/NPs (n=3). (C) The particle size of PGPP/NPs in DMEM supplemented with 10% fetal bovine serum (FBS), PBS, or pure water within 7 days (n = 3). (D) Drug release curve of PI3Kγ inhibitor in PGPP/NPs (n=3). (E) Cell viability evaluation of PGPP/NPs in H9C2 cells (n = 6). (F) Hemolysis assays of GPP/NPs and PGPP/NPs (n=6). Data are presented as the mean ± SD. ***p < 0.001; ****p < 0.0001. |
In vitro Cellular Uptake in H9C2 Cells
The PCM peptide (WLSEAGPVVTVRALRGTGSW), a cardiomyocyte-targeting peptide, was identified by in vivo phage display technology.36 PCM-modified NPs can enhance the uptake of cardiomyocytes and improve the targeting of the heart. The cellular uptake of PCM-modified particles by H9C2 cells was evaluated in vitro. Flow cytometry (FCM) and confocal laser scanning microscopy (CLSM) both confirmed that the PGPP/NPs group exhibited stronger intracellular fluorescence than the GPP/NPs group, implying that the PCM modification enhanced the targeting effect of cardiomyocytes. Moreover, the intracellular uptake was time-dependent (Figure 3A–C). In addition, the cellular uptake of particles increased with the rising PGPP/NPs concentrations, indicating a concentration-dependent characteristic (Figure 3D–F).
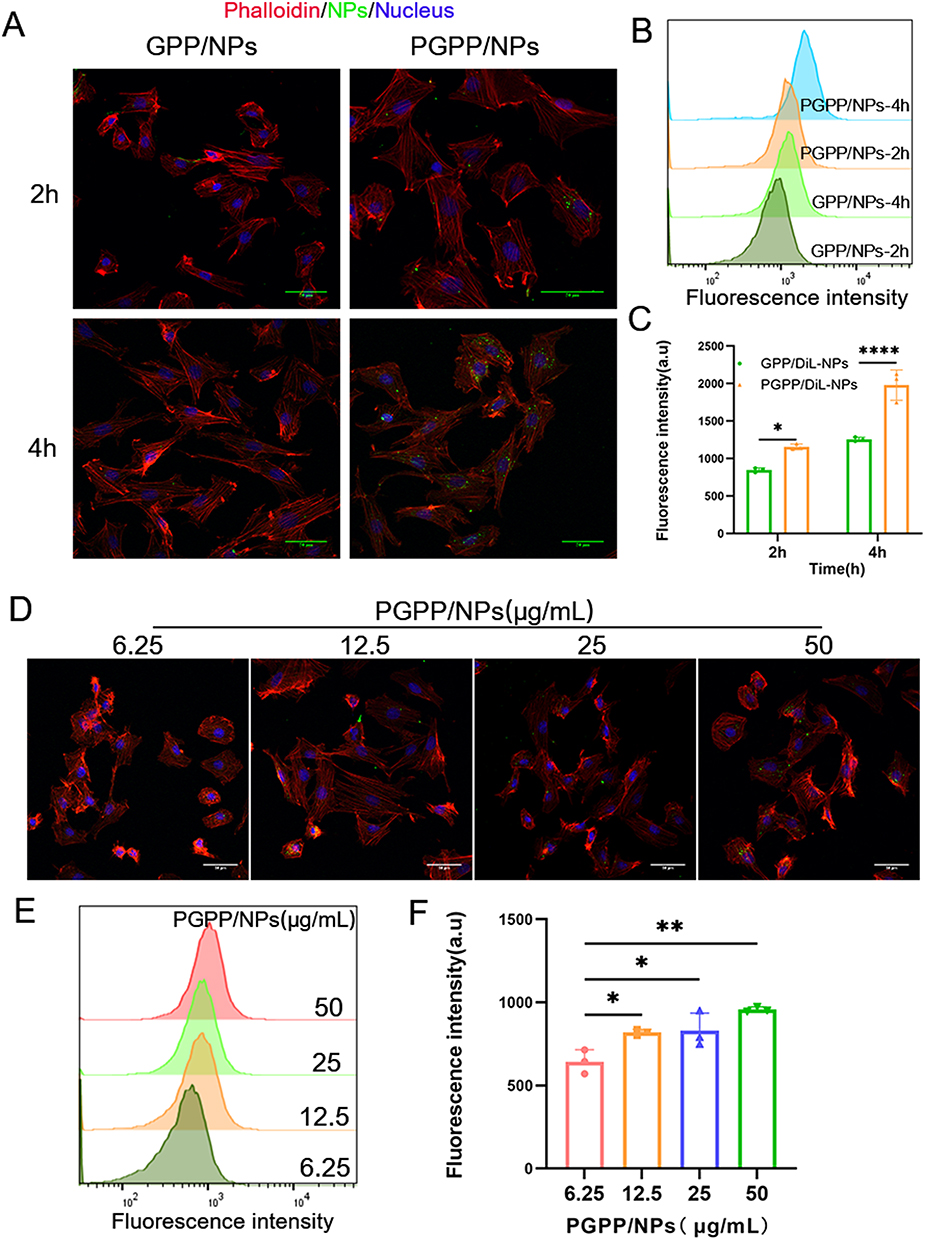 |
Figure 3 Cellular uptake assays by H9C2 cells in vitro. (A) CLSM images and (B and C) Flow cytometry analysis for internalization ability among of GPP/NPs and PGPP/NPs at 2 h and 4 h after incubation with H9C2 cells (n=3). (D) CLSM images and (E and F) Flow cytometry analysis for internalization ability of PGPP/NPs at various concentrations after incubation with H9C2 cells (n=3). Data are presented as the mean ± SD. *p < 0.05; **p < 0.01; ****p < 0.0001. |
In vitro Therapeutic Efficacy of PGPP/NPs
Then, we assessed the capacity of PGPP/NPs to attenuate Dox-induced cytotoxicity. Initially, the 50% inhibitory concentration (IC50) of DOX in H9C2 cells was confirmed, which was 1.58 μM (Supplementary Figure S4). Inflammatory marker (IL-6, TNF-α) results indicated successful construction of DOX-induced H9C2 inflammatory cells (Supplementary Figure S5). As shown in the CLSM image of Figure 4A, DOX (IC50) induced H9C2 cells to produce a large amount of ROS, which was manifested as strong green fluorescence. The green fluorescence intensity was significantly reduced after pretreatment with PGPP/NPs, which confirmed the anti-oxidative effects of PGPP/NPs. Consistently, FCM results also verified above conclusion (Figure 4B). LIVE/DEAD cell-staining exhibited H9C2 cells apoptosis caused by DOX (IC50), with red fluorescence represented dead cells and green fluorescence represented live cells. Pretreatment with PGPP/NPs significantly inhibited DOX-induced apoptosis in cells (Figure 4C). Furthermore, as shown in Figure 4D, H9C2 cells experienced approximately a 50% loss in viability under the IC50 concentration of DOX alone. Pretreatment with PGPP/NPs at concentrations below the safe limit (100 μg/mL) gradually restored cell viability, with near complete recovery observed at a concentration of 50 μg/mL.
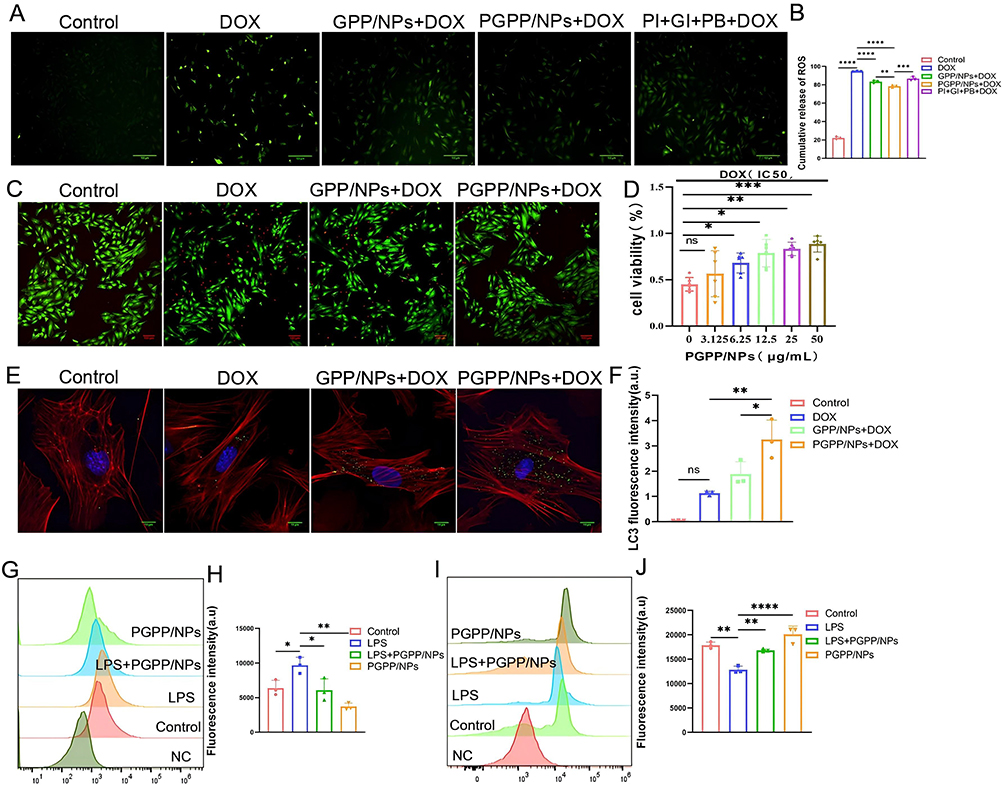 |
Figure 4 PGPP/NPs conferred protection to normal cells against DOX-induced cytotoxicity in vitro. (A) CLSM images and (B) Flow cytometry analysis of ROS expression in H9C2 cells after treating with PBS, DOX, GPP + DOX, PGPP + DOX, and PI + GI + PB + DOX (n = 3). Scale bars, 100 μm. (C) LIVE–DEAD cell-staining of H9C2 cells after treating with PBS, DOX, DOX + GPP/NPs, and DOX + PGPP/NPs (Green: Live,Red: Dead). Scale bars, 100 μm. (D) Cell viabilities of H9C2 incubated with Dox at IC50 value after preincubation with increasing concentrations of PGPP/NPs (n = 6). (E) CLSM images and (F) fluorescence quantification of LC3 expression in primary cardiomyocytes after treating with PBS, DOX, DOX + GPP/NPs, and DOX + PGPP/NPs (n = 3). Scale bars, 10 μm. Flow cytometry results of the expression of (G and H) CD86 and (I and J) CD206 in RAW 264.7 cells after treating with PBS, LPS, PGPP/NPs, and LPS + PGPP/NPs (n = 3). Data are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
In this study, it was observed that PI3Kγ inhibitor-loaded PGPP/NPs could enhance autophagy in primary cardiomyocytes (Figure 4E and F). In addition, we evaluated the anti-inflammatory effect of PGPP/NPs in vitro by FCS. The results showed a higher expression of CD206 (M2 marker) and a lower expression of CD86 (M1 marker) in the PGPP/NPs + LPS group compared to the LPS group (Figure 4G–J). These results implied that PGPP/NPs effectively reduced intracellular ROS levels, promoted autophagy of cardiomyocytes, and ameliorated inflammation by inhibiting M1(CD86) while favoring M2(CD206) polarization of macrophages.37
The Biodistribution and Blood Circulation of GPP/NPs and PGPP/NPs in vivo
To investigate the biodistribution and blood circulation of NPs in vivo, 4T1 tumors bearing mice were injected with GPP/NPs or PGPP/NPs via the caudal vein, and then the major organs (heart, liver, and tumor) were removed for ex vivo fluorescence imaging. GPP/NPs or PGPP/NPs were gathered distinctly in the liver or heart regions 24 h post-administration (Figure 5A–D). Most notably, the cardiac fluorescence intensity was higher in the PGPP/NPs group compared to the GPP/NPs group (Figure 5E). These results demonstrated that after PCM modification, PGPP/NPs significantly improved the targeting of cardiomyocytes in vivo. As shown in Figure 5F, the trend of long blood circulation was generally consistent between GPP/NPs and PGPP/NPs groups. The blood fluorescence signal increased from the time of administration and began to decline after 6 h. These data indicated that PGPP/NPs could efficiently accumulate in the heart and be internalized by target cardiomyocytes.
 |
Figure 5 The blood circulation and biodistribution of GPP/NPs and PGPP/NPs in vivo. (A and B) Ex vivo fluorescence imaging and quantitative analysis of fluorescent intensity of heart after injection of GPP/NPs or PGPP/NPs for 24 h (n = 5). (C and D) Ex vivo fluorescence imaging and quantitative analysis of fluorescent intensity of liver after injection of GPP/NPs or PGPP/NPs for 24 h (n = 5). (E) CLSM images showing the distribution of DiO-labeled GPP/NPs and PGPP/NPs. (F) Relative serum signal of GPP/NPs and PGPP/NPs intravenously administered (n = 3). Data are presented as the mean ± SD. *p < 0.05; ****p < 0.0001. |
In vivo Therapeutic Efficacy of PGPP/NPs
Firstly, we determined the maximal tolerated dosage (MTD) of DOX for BALB/c mice. During treatment with various DOX doses, mice in high-dose groups (20, 30, 40 mg/kg) died sequentially, while the mice in low-dose groups (15, 10 mg/kg) did not die. In addition, complete survival and significant body weight loss were observed simultaneously only at a concentration of 15 mg/kg (Supplementary Figure S6). Hence, 15 mg/kg was determined to be the MTD of DOX for BALB/c mice. Subsequently, we evaluated these four biomarkers (AST, ALT, LDH, and CK-MB) in mice, and the results confirmed the successful construction of the mouse cardiac injury model (Supplementary Figure S7).
Transthoracic echocardiography was performed after 21 days of treatment. Echocardiographic assessment revealed that DOX induced significant left ventricular systolic dysfunction (Figure 6A). Quantitative analysis of left ventricular ejection fraction (LV-EF) (Figure 6B) and left ventricular fraction shortening (LV-FS) (Figure 6C) further revealed heart dysfunction in the 4T1 tumors bearing mice receiving DOX intervention. Significantly, PGPP/NPs treatment restored the LV-EF and LV-FS indicators to normal. Mice administered with the free drug or GPP/NPs showed no significant differences compared to control. Subsequently, serum markers of myocardial injury were measured. Blood samples were collected from different groups to detect the content of Cardiac troponin I (cTnI) (Figure 6D) and brain natriuretic peptide (BNP) (Figure 6E) in serum. The results showed significantly elevated levels of cTnI and BNP in the DOX group compared to control. Treatment with PGPP/NPs significantly prevented the rise in cTnI and BNP levels in serum.
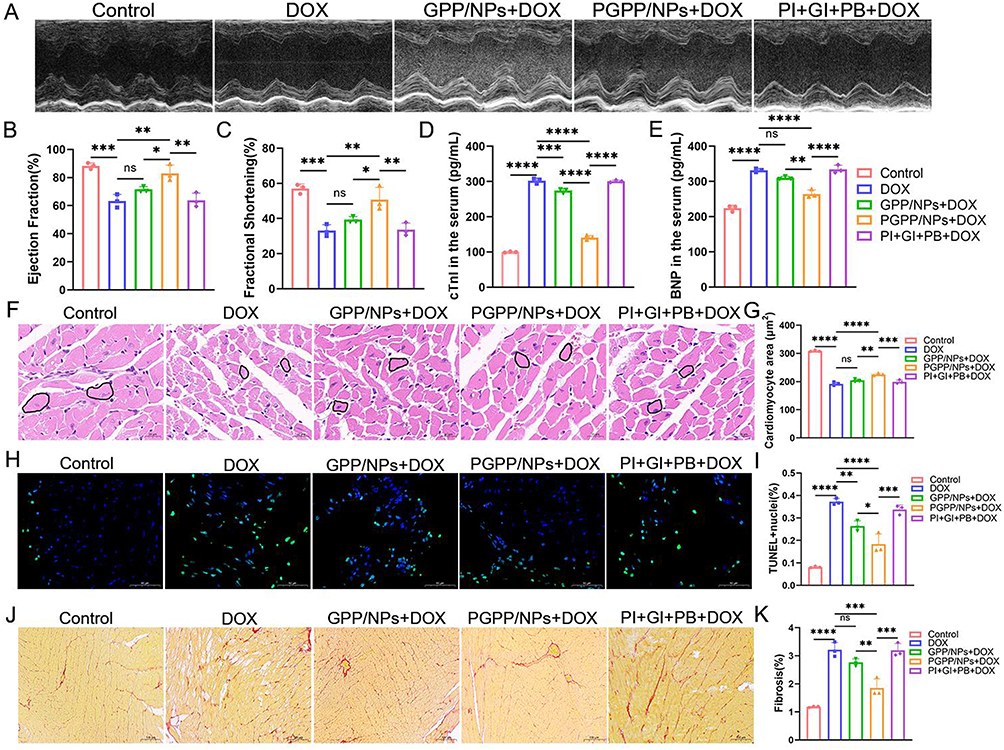 |
Figure 6 PGPP/NPs detoxification of Dox in vivo. (A) Representative M-mode echocardiographic images 21 days after treating with PBS, DOX, GPP + DOX, PGPP + DOX, and PI + GI + PB + DOX. (B) Left ventricular ejection fraction (LVEF) and (C) fractional shortening (FS) 21 days after treating as in (A) (n = 3). The content of (D) cTnI and (E) BNP in serum 21 days after treating as in (A) (n = 3). (F and G) Representative images of H&E staining and quantification of cardiomyocyte area treated as in (A) (n = 3). Scale bars, 20 μm. (H and I) Representative images of TUNEL staining and quantification of TUNEL-positive nuclei per field in whole hearts treated as in (A) (n = 3). Scale bars, 50 μm. (J and K) Representative images of PicroSirius Red staining and relative quantification of collagen deposition in heart sections treated as in (A) (n = 3). Scale bars, 100 μm. Data are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
It was worth mentioning that, cardiomyocyte size, apoptosis, and cardiac fibrosis were three key hallmarks of DOX-induced cardiotoxicity.38 H&E staining revealed that PGPP/NPs maintained normal cardiomyocyte size after DOX injection (Figure 6F and G). Besides, TUNEL staining results indicated that PGPP/NPs could extensively alleviate myocardial apoptosis, and thus attenuating DIC (Figure 6H–I). Furthermore, Sirius red staining showed a remarkable increase of cardiac fibrosis in the DOX group, which was prevented by PGPP/NPs (Figure 6J and K).
To further validate our findings, we performed cardiac ROS detection in mice. After harvesting the hearts, they were stained with DHE, and ROS fluorescence intensity and distribution were visualized (Figure 7A). Additionally, flow cytometry was used to quantify intracellular ROS levels (Figure 7B). The results revealed that PGPP/NPs treatment significantly reduced cardiac ROS production compared to controls. These results were complemented by assessments of inflammatory and autophagic markers. Immunohistochemistry revealed higher CD206 and lower CD86 expression in PGPP/NP-treated hearts, indicating reduced inflammation via M1-to-M2 macrophage polarization (Figure 7C–E). Autophagic activity, assessed by LC3-II immunofluorescence, was enhanced in PGPP/NP-treated hearts (Figure 7F and G), consistent with PI3Kγ inhibitor-mediated autophagy promotion.
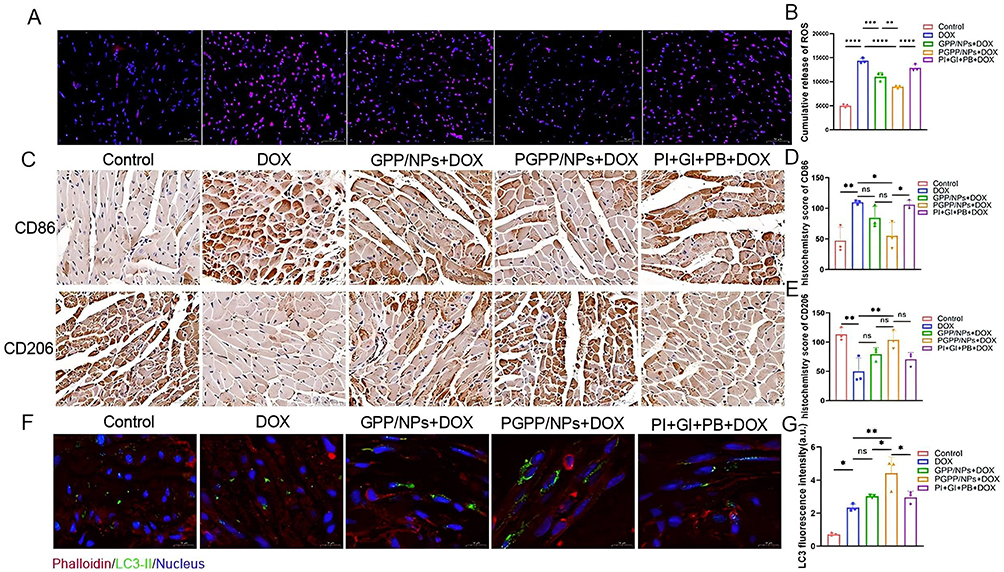 |
Figure 7 PGPPNPs alleviate oxidative stress, promote M2 macrophage polarization, and enhance cardiac autophagy. (A) Immunofluorescence staining images and (B) Flow cytometry analysis of ROS expression in cardiac tissue after treating with PBS, DOX, GPP + DOX, PGPP + DOX, and PI + GI + PB + DOX (n = 3). Scale bars, 50 μm. (C) Immunohistochemical staining images and (D and E) Histochemistry score of CD86 (M1) and CD206 (M2) after treating as in (A). (n = 3). Scale bars, 50 μm. (F) Immunofluorescence staining images and (G) relative quantification of LC3 puncta in heart sections from animals treated as in (A) (n = 3). Scale bars, 10 μm. Data are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
Effect of PGPP/NPs on the Anticancer Activity of DOX
We further investigated tumor growth during the treatment period. In the in vivo experiments, tumor volume was significantly reduced following DOX administration. Importantly, the therapeutic efficacy of DOX was not diminished by the addition of PGPP/NPs (Supplementary Figure S8). Although there was no significant difference in tumor volume among the DOX, GPP/NPs + DOX, and PGPP/NPs + DOX groups, mice in the PGPP/NPs + DOX group had the smallest tumor volume. Overall, PGPP/NPs effectively reduce DIC while maintaining, and even enhancing, the tumor-killing efficacy of DOX, thereby demonstrating a synergistic effect against tumor progression.
Discussion
The development of effective strategies to mitigate doxorubicin-induced cardiotoxicity (DIC) while preserving the antitumor efficacy of doxorubicin (DOX) remains a critical challenge in oncology, as DIC often limits treatment duration and dosage, ultimately compromising patient outcomes. Our study introduces PGPP/NPs, a multifunctional nanoparticle system engineered with cardiomyocyte-targeting capability and loaded with a PI3Kγ inhibitor, to address this unmet need. Below, we contextualize our findings within the broader literature, explore their mechanistic underpinnings, and discuss their implications for clinical translation, while also addressing limitations and future directions.
The carrier-free core design of GPP/NPs and PGPP/NPs, driven by π-π stacking and hydrophobic interactions, represents a departure from traditional nanoparticle systems that rely on polymer or lipid carriers. This approach minimizes potential off-target effects by avoiding carrier-related toxicity and simplifies formulation complexity, as it eliminates the need for synthetic polymers or lipids that may induce immune responses or interfere with drug bioactivity. The use of DSPE-PEG as a minimal excipient for surface conjugation of the PCM peptide further enhances biocompatibility; PEGylation is well-documented to reduce macrophage clearance via the “stealth” effect, prolonging nanoparticle circulation time and improving target tissue accumulation. This design choice aligns with emerging trends in nanomedicine, where simplicity and biocompatibility are prioritized to enhance therapeutic index.
Central to the success of PGPP/NPs is their precise cardiomyocyte targeting, mediated by the PCM peptide identified via in vivo phage display. Flow cytometry and confocal microscopy confirmed that PCM modification significantly increased intracellular fluorescence in H9C2 cells compared to non-targeted GPP/NPs, with uptake kinetics showing time- and concentration-dependence. This specificity is pivotal for DIC treatment, as precise delivery of therapeutic agents to cardiac tissue minimizes systemic exposure and reduces off-target toxicity. In vivo biodistribution studies using 4T1 tumor-bearing mice corroborated this targeting effect: PGPP/NPs accumulated preferentially in the heart (versus liver for GPP/NPs), aligning with clinical needs for cardiac-specific drug delivery. Notably, the circulation profile of PGPP/NPs—maintaining fluorescence intensity similar to GPP/NPs—indicates that PCM modification does not compromise nanoparticle stability or circulation time, a common concern with ligand-conjugated nanoparticles. This balance between targeting efficiency and circulation stability is critical, as premature clearance would reduce cardiac accumulation, while excessive circulation could increase off-target effects.
The in vitro experiments revealed that PGPP/NPs mitigate DOX-induced cytotoxicity through three interrelated mechanisms: antioxidative stress, anti-inflammatory modulation, and autophagy promotion. At the cellular level, ROS overproduction is a central driver of DOX-induced cardiomyocyte damage, disrupting mitochondrial function and inducing oxidative stress. PGPP/NPs significantly reduced ROS levels in H9C2 cells, as evidenced by decreased green fluorescence in CLSM and flow cytometry assays. This aligns with prior studies demonstrating that nanoparticle-based antioxidant delivery can scavenge ROS and protect cardiac cells. By encapsulating ginsenoside Rb1—a natural antioxidant—as part of the carrier, PGPP/NPs likely synergize with the PI3Kγ inhibitor to enhance redox balance, though the exact contribution of Rb1 requires further investigation.
Chronic inflammation exacerbates DIC by promoting cardiomyocyte apoptosis and fibrosis. PGPP/NPs modulated macrophage polarization toward an M2 phenotype (CD206⁺/CD86−), a key anti-inflammatory state. This shift is critical, as M1 macrophages (CD86⁺) secrete pro-inflammatory cytokines (eg, TNF-α, IL-6) that amplify tissue damage, while M2 macrophages secrete reparative factors (eg, IL-10) that support healing. Our findings extend previous reports linking nanoparticle-mediated M2 polarization to cardiac repair, suggesting PGPP/NPs create a pro-healing microenvironment in the heart. This anti-inflammatory effect likely complements the antioxidative mechanism, as reducing inflammation can further lower oxidative stress by dampening pro-inflammatory signaling cascades.
Dysregulated autophagy is another hallmark of DIC, with excessive or insufficient autophagic flux contributing to cardiomyocyte death. The PI3Kγ inhibitor loaded in PGPP/NPs enhanced autophagic flux, as indicated by increased LC3-II expression (implied by immunofluorescence). LC3-II is a marker of autophagosome formation, and its upregulation suggests enhanced degradation of damaged organelles (eg, mitochondria) and misfolded proteins. By promoting autophagic clearance, PGPP/NPs likely prevent the accumulation of toxic cellular components, thereby reducing apoptosis. This multi-faceted approach—targeting ROS, inflammation, and autophagy—reflects a systems-level strategy to protect cardiac tissue, which is often more effective than single-target therapies.
The in vivo results further validate PGPP/NPs as a promising DIC therapeutic. Transthoracic echocardiography revealed that PGPP/NPs restored left ventricular ejection fraction (LV-EF) and fractional shortening (LV-FS) to near-normal levels in DOX-treated mice, indicating improved cardiac contractility. These functional improvements correlate with reduced serum levels of cardiac troponin I (cTnI) and brain natriuretic peptide (BNP)—biomarkers of myocardial injury and heart failure, respectively. Histological analyses (H&E, TUNEL, Sirius red staining) provided structural evidence of protection: PGPP/NPs preserved cardiomyocyte size, reduced apoptotic nuclei, and limited collagen deposition, all of which are critical for maintaining cardiac architecture and function.
Notably, PGPP/NPs did not compromise DOX’s antitumor efficacy. Tumor volume in the PGPP/NPs + DOX group was comparable to (or slightly smaller than) the DOX-only group, suggesting that the nanoparticle carrier does not interfere with DOX’s ability to kill cancer cells. This is a critical observation, as many cardioprotective agents (eg, dexrazoxane) are limited by dose-dependent cardiotoxicity or reduced chemotherapeutic efficacy. The lack of antagonism may arise from the spatial separation of free DOX (systemically distributed) and PGPP/NPs (cardiac-targeted), or from differential pharmacokinetics that prevent competitive binding at tumor or cardiac targets. This finding addresses a major barrier in developing dual-purpose therapeutics for cancer and cardiac protection.
Despite these promising results, several limitations warrant discussion. First, while we confirmed the EE of the PI3Kγ inhibitor, the loading efficiency of ginsenoside Rb1 (a carrier component) was not measured. Although Rb1’s role as a structural stabilizer was validated indirectly through nanoparticle characterization, its potential contribution to cardioprotection (eg, via anti-inflammatory or cytoprotective effects) remains to be elucidated. Future studies should quantify Rb1 EE and explore its biological activity in vitro and in vivo, as this could optimize formulation design by adjusting Rb1 loading to enhance efficacy. Second, our in vivo studies focused on a 4T1 breast cancer model, which may not fully recapitulate DIC pathophysiology in other cancer types (eg, lung or liver cancer) or in patients with comorbidities. Validation in additional models (eg, human xenografts or genetically engineered mouse models) would strengthen the translational relevance of our findings. For example, testing PGPP/NPs in a lung cancer model could reveal whether the targeting mechanism generalizes to other tissues, while studying aged or diabetic mice might uncover interactions between DIC and comorbid conditions. Third, while LC3-II upregulation indicates enhanced autophagic flux, we did not directly visualize autophagosomes or assess autophagic degradation (eg, via p62 Western blotting or electron microscopy). Additional studies using these methods would confirm the role of autophagy in PGPP/NP-mediated cardioprotection. For instance, electron microscopy could reveal the number and morphology of autophagosomes, while p62 degradation assays would confirm the completion of the autophagic process (ie, lysosomal degradation of autophagosomes). Finally, clinical translation considerations include nanoparticle scalability, long-term toxicity, and immunogenicity. While DSPE-PEG is generally well-tolerated, prolonged circulation may increase the risk of anti-PEG immune responses, which can reduce nanoparticle efficacy over time. Further preclinical studies evaluating these parameters—such as repeated dosing studies to assess cumulative toxicity and immune reactions—are necessary to advance PGPP/NPs toward clinical application. Additionally, optimizing the manufacturing process to ensure reproducibility and scalability will be critical for translating this research from the bench to the bedside.
In summary, our study demonstrates that PGPP/NPs, a cardiomyocyte-targeted nanoparticle system loaded with a PI3Kγ inhibitor, effectively mitigate DIC through antioxidative, anti-inflammatory, and autophagy-promoting mechanisms while preserving DOX’s antitumor activity. These findings provide a compelling rationale for further preclinical development of PGPP/NPs as a dual-purpose nanotherapeutic for cancer therapy and cardiac protection. By addressing key challenges in DIC management—including off-target toxicity and chemotherapeutic efficacy preservation—this work bridges the gap between nanotechnology and precision oncology, offering a promising platform for future therapeutic innovation.
Conclusion
Despite the indispensable role of doxorubicin (DOX) in cancer therapy, its clinical application is severely limited by dose-dependent cardiotoxicity, particularly diastolic dysfunction (DIC). Here, we report a multifunctional self-assembling nanoparticle platform (PGPP/NPs) engineered through a co-assembly strategy to simultaneously mitigate DIC and preserve antitumor efficacy. By incorporating ginsenoside Rb1, probucol, and a PI3Kγ inhibitor, PGPP/NPs synergistically scavenged reactive oxygen species, activated autophagy, and suppressed apoptosis in DOX-exposed cardiomyocytes, while PCM-functionalization enhanced myocardial-specific delivery. In 4T1 tumor-bearing mice, PGPP/NPs demonstrated cardiac accumulation and dual therapeutic benefits: serum troponin I levels decreased and left ventricular ejection fraction was preserved, confirming cardioprotection. Notably, tumor growth inhibition rates promote, demonstrating maintained antitumor efficacy. This pharmacological synergy, coupled with tumor-targeted delivery via macrophage polarization, positions PGPP/NPs as a first-in-class platform to address DOX-induced cardiotoxicity. While further validation in large-animal models is required, our findings provide a translational framework for safer DOX-based chemotherapy.
Ethics Statements: Studies Involving Animal Subjects
All procedures involving experimental animals were carried out according to the guidelines approved by the Institutional Animal Care and Use Committee of Chongqing Medical University (IACUC-CQMU-2023-10037).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32201150), the Natural Science Foundation of Chongqing (CSTB2022NSCQ-MSX0096).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Ozcan M, Guo Z, Valenzuela Ripoll C, et al. Sustained alternate-day fasting potentiates doxorubicin cardiotoxicity. Cell Metab. 2023;35(6):928–942.e4. doi:10.1016/j.cmet.2023.02.006
2. Fan M, Li H, Shen D, et al. Decoy Exosomes Offer Protection Against Chemotherapy-Induced Toxicity. Adv Sci. 2022;9(32):e2203505. doi:10.1002/advs.202203505
3. Magdy T, Jouni M, Kuo HH, et al. Identification of Drug Transporter Genomic Variants and Inhibitors That Protect Against Doxorubicin-Induced Cardiotoxicity. Circulation. 2022;145(4):279–294. doi:10.1161/CIRCULATIONAHA.121.055801
4. Han D, Wang Y, Wang Y, et al. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ Res. 2020;127(4):e108–e125. doi:10.1161/CIRCRESAHA.119.316061
5. Lo Sardo V, Kamp TJ. Preventing Anthracycline-Induced Cardiotoxicity Using Functional Genomics and Human-Induced Pluripotent Stem Cell-Derived. Circulation. 2022;145(4):295–298. doi:10.1161/CIRCULATIONAHA.121.058128
6. Seara FAC, Kasai-Brunswick TH, Nascimento JHM, Campos-de-Carvalho AC. Anthracycline-induced cardiotoxicity and cell senescence: new therapeutic option? Cell Mol Life Sci. 2022;79(11):568. doi:10.1007/s00018-022-04605-7
7. Bernstein D. Anthracycline Cardiotoxicity: worrisome Enough to Have You Quaking? Circ Res. 2018;122(2):188–190. doi:10.1161/CIRCRESAHA.117.312395
8. Jahng JWS, Liu L, Wu JC. Tumor Repressor Circular RNA as a New Target for Preventative Gene Therapy Against Doxorubicin-Induced Cardiotoxicity. Circ Res. 2020;127(4):483–485. doi:10.1161/CIRCRESAHA.120.317568
9. Magdy T, Jiang Z, Jouni M, et al. RARG variant predictive of doxorubicin-induced cardiotoxicity identifies a cardioprotective therapy. Cell Stem Cell. 2021;28(12):2076–2089.e7. doi:10.1016/j.stem.2021.08.006
10. Gupta SK, Garg A, Bär C, et al. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity Through Regulation of Cardiac Circular RNA Expression. Circ Res. 2018;122(2):246–254. doi:10.1161/CIRCRESAHA.117.311335
11. Li M, Sala V, De Santis MC, et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation. 2018;138(7):696–711. doi:10.1161/CIRCULATIONAHA.117.030352
12. Jiang Q, Chen M, Yang X, et al. Doxorubicin Detoxification in Healthy Organs Improves Tolerability to High Drug Doses for Enhanced Antitumor Therapy. ACS Nano. 2023;17(8):7705–7720. doi:10.1021/acsnano.3c00195
13. Moutabian H, Ghahramani-Asl R, Mortezazadeh T, et al. The cardioprotective effects of nano-curcumin against doxorubicin-induced cardiotoxicity: a systematic review. Biofactors. 2022;48(3):597–610. doi:10.1002/biof.1823
14. Santin Y, Formoso K, Haidar F, et al. Inhalation of acidic nanoparticles prevents doxorubicin cardiotoxicity through improvement of lysosomal function. Theranostics. 2023;13(15):5435–5451. doi:10.7150/thno.86310
15. Geng S, Feng Q, Wang C, et al. A Versatile PDA(DOX) Nanoplatform for Chemo-Photothermal Synergistic Therapy against Breast Cancer and Attenuated Doxorubicin-Induced Cardiotoxicity. J Nanobiotechnology. 2023;21(1):338. doi:10.1186/s12951-023-02072-1
16. Zhao R, Liu X, Yang X, et al. Nanomaterial-Based Organelles Protect Normal Cells against Chemotherapy-Induced Cytotoxicity. Adv Mater. 2018;30(27):e1801304. doi:10.1002/adma.201801304
17. Yang Y, Li D, Liu B. The preparation of a boronate affinity-based controlled oriented imprinting coating on a silica nanoparticle surface for the separation and purification of shikimic acid in herbal medicine. Anal Methods. 2024;16(18):2878–2887. doi:10.1039/d4ay00219a
18. Singh D. Activated carbon nanoparticles generated from coffee husk: a pioneering biomaterial for improving drug targeting in cancer. Nano-Struct Nano-Objects. 2024;38101134:2352–507X. doi:10.1016/j.nanoso.2024.101134
19. Zhong D, Cheng H, Liu H, et al. Bibliometric analysis of Traditional Chinese Medicine nanoparticles research from 2005 to 2023. Electrophoresis. 2024;45(3–4):288–299. doi:10.1002/elps.202300207
20. Cui Y, Li Q, Yang D, Yang Y. Colorimetric-SERS dual-mode sensing of Pb(II) ions in traditional Chinese medicine samples based on carbon dots-capped gold nanoparticles as nanozyme. Spectrochim Acta A. 2024;313:124100. doi:10.1016/j.saa.2024.124100
21. Cui J, Wang X, Li J, et al. Immune Exosomes Loading Self-Assembled Nanomicelles Traverse the Blood-Brain Barrier for Chemo-immunotherapy against Glioblastoma. ACS Nano. 2023;17:1464–1484. doi:10.1021/acsnano.2c10219
22. Zhu J, Zhang K, Zhou Y, et al. Correction to “A Carrier-Free Nanomedicine Enables Apoptosis-Ferroptosis Synergistic Breast Cancer Therapy by Targeting Subcellular Organelles”. ACS Appl Mater Interfaces. 2023;15(24):29653. doi:10.1021/acsami.3c06803
23. Xu Z, Zhang X, Dong W, et al. Self-assembling and pH-responsive protein nanoparticle as potential platform for targeted tumor therapy. Front Mol Biosci. 2023;10:1172100. doi:10.3389/fmolb.2023.1172100
24. Voltà-Durán E, Alba-Castellón L, Serna N, et al. High-precision targeting and destruction of cancer-associated PDGFR-β+ stromal fibroblasts through self-assembling, protein-only nanoparticles. Acta Biomater. 2023;170:543–555. doi:10.1016/j.actbio.2023.09.001
25. Balusamy SR, Perumalsamy H, Huq MA, et al. A comprehensive and systemic review of ginseng-based nanomaterials: synthesis, targeted delivery, and biomedical applications. Med Res Rev. 2023;43(5):1374–1410. doi:10.1002/med.21953
26. Xu Y, Zhu BW, Li X, Yf L, Ye XM, Hu JN. Glycogen-based pH and redox sensitive nanoparticles with ginsenoside Rh2 for effective treatment of ulcerative colitis. Biomaterials. 2022;280:121077. doi:10.1016/j.biomaterials.2021.121077
27. Ren Z, Chen X, Hong L, et al. Nanoparticle Conjugation of Ginsenoside Rg3 Inhibits Hepatocellular Carcinoma Development and Metastasis. Small. 2020;16(2):e1905233. doi:10.1002/smll.201905233
28. Park HH, Kim H, Lee HS, et al. PEGylated nanoparticle albumin-bound steroidal ginsenoside derivatives ameliorate SARS-CoV-2-mediated hyper-inflammatory responses. Biomaterials. 2021;273:120827. doi:10.1016/j.biomaterials.2021.120827
29. Dai L, Liu K, Si C, et al. Ginsenoside nanoparticle: a new green drug delivery system. J Mater Chem B. 2016;4(3):529–538. doi:10.1039/c5tb02305j
30. Liang X, Li H, Li X, et al. Highly sensitive H2O2-scavenging nano-bionic system for precise treatment of atherosclerosis. Acta Pharm Sin B. 2023;13(1):372–389. doi:10.1016/j.apsb.2022.04.002
31. Wagle SR, Ionescu CM, Kovacevic B, et al. Pharmaceutical characterization of probucol bile acid-lithocholic acid nanoparticles to prevent chronic hearing related and similar cellular oxidative stress pathologies. Nanomedicine. 2023;18(12):923–940. doi:10.2217/nnm-2023-0092
32. Wang X, Zhang W, Zeng S, Wang L, Wang B. Collagenase Type I and Probucol-Loaded Nanoparticles Penetrate the Extracellular Matrix to Target Hepatic Stellate Cells for Hepatic Fibrosis Therapy. Acta Biomater. 2024;175:262–278. doi:10.1016/j.actbio.2023.12.027
33. Wagle SR, Kovacevic B, Ionescu CM, et al. Polymer-Based Nanoparticles with Probucol and Lithocholic Acid: a Novel Therapeutic Approach for Oxidative Stress-Induced Retinopathies. Mol Pharm. 2024;21(7):3566–3576. doi:10.1021/acs.molpharmaceut.4c00269
34. Song Z, Song H, Liu D, et al. Overexpression of MFN2 alleviates sorafenib-induced cardiomyocyte necroptosis via the MAM-CaMKIIδ pathway in vitro and in vivo. Theranostics. 2022;12(3):1267–1285. doi:10.7150/thno.65716
35. Wang P, Xu S, Xu J, et al. Elevated MCU Expression by CaMKIIδB Limits Pathological Cardiac Remodeling. Circulation. 2022;145(14):1067–1083. doi:10.1161/CIRCULATIONAHA.121.055841
36. Wang Z, Yang N, Hou Y, et al. L-Arginine-Loaded Gold Nanocages Ameliorate Myocardial Ischemia/Reperfusion Injury by Promoting Nitric Oxide Production and Maintaining Mitochondrial Function. Adv Sci. 2023;10(26):e2302123. doi:10.1002/advs.202302123
37. Duan BW, Liu YJ, Li XN, et al. An Autologous Macrophage-Based Phenotypic Transformation-Collagen Degradation System Treating Advanced Liver Fibrosis. Adv Sci. 2024;11(7):e2306899. doi:10.1002/advs.202306899
38. Russo M, Guida F, Paparo L, et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur J Heart Fail. 2019;21(4):519–528. doi:10.1002/ejhf.143
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of Ameliorative Effect of Quercetin and Candesartan in Doxorubicin-Induced Cardiotoxicity
Majhi S, Singh L, Yasir M
Vascular Health and Risk Management 2022, 18:857-866
Published Date: 13 December 2022
Analysis and Validation of Critical Signatures and Immune Cell Infiltration Characteristics in Doxorubicin-Induced Cardiotoxicity by Integrating Bioinformatics and Machine Learning
Huang C, Pei J, Li D, Liu T, Li Z, Zhang G, Chen R, Xu X, Li B, Lian Z, Chu XM
Journal of Inflammation Research 2024, 17:669-685
Published Date: 2 February 2024
Irisin Mitigates Doxorubicin-Induced Cardiotoxicity by Reducing Oxidative Stress and Inflammation via Modulation of the PERK-eIF2α-ATF4 Pathway
Zhang Z, Yu X, Li J, Shen X, Fu W, Liu Y, Dong X, Wang Z
Drug Design, Development and Therapy 2025, 19:1067-1081
Published Date: 15 February 2025
Mechanistic Insights into Flavonoid Subclasses as Cardioprotective Agents Against Doxorubicin-Induced Cardiotoxicity: A Comprehensive Review
Shang W, Li XH, Zeng LH, Li Z, Hu Y, Wen HM, Cao FJ, Wan GX
Drug Design, Development and Therapy 2025, 19:5553-5596
Published Date: 1 July 2025
Emerging Targets and Treatments for Doxorubicin-Induced Cardiac Inflammation
Khuanjing T, Chattipakorn SC, Chattipakorn N
Journal of Inflammation Research 2026, 19:546243
Published Date: 18 February 2026