Back to Journals » Journal of Inflammation Research » Volume 15
Platelets, Macrophages, and Thromboinflammation in Chagas Disease
Authors Choudhuri S, Garg NJ ![]()
Received 2 July 2022
Accepted for publication 24 August 2022
Published 4 October 2022 Volume 2022:15 Pages 5689—5706
DOI https://doi.org/10.2147/JIR.S380896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Subhadip Choudhuri, Nisha J Garg
Department of Microbiology and Immunology, Institute for Human Infections and Immunity, University of Texas Medical Branch, Galveston, TX, USA
Correspondence: Nisha J Garg, Email [email protected]
Abstract: Chagas disease (CD) is a major health problem in the Americas and an emerging health problem in Europe and other nonendemic countries. Several studies have documented persistence of the protozoan parasite Trypanosoma cruzi, and oxidative and inflammatory stress are major pathogenic factor. Mural and cardiac thrombi, cardiac arrhythmias, and cardiomyopathy are major clinical features of CD. During T. cruzi infection, parasite-released factors induce endothelial dysfunction along with platelet (PLT) and immune-cell activation. PLTs have a fundamental role in maintaining hemostasis and preventing bleeding after vascular injury. Excessive activation of PLTs and coagulation cascade can result in thrombosis and thromboembolic events, which are recognized to occur in seropositive individuals in early stages of CD when clinically symptomatic heart disease is not apparent. Several host and parasite factors have been identified to signal hypercoagulability and increase the risk of ischemic stroke in early phases of CD. Further, PLT interaction with immune cells and their role in host defense against pathogens and inflammatory processes have only recently been recognized and evolving. In the context of parasitic diseases, PLTs function in directly responding to T. cruzi infection, and PLT interactions with immune cells in shaping the proinflammatory or immunoregulatory function of monocytes, macrophages, and neutrophils remains elusive. How T. cruzi infection alters systemic microenvironment conditions to influence PLT and immune-cell interactions is not understood. In this review, we discuss the current literature, and extrapolate the mechanistic situations to explain how PLT and innate immune cell (especially monocytes and macrophages) interactions might be sustaining hypercoagulability and thromboinflammation in chronic CD.
Keywords: Chagas disease, coagulopathy, inflammation, macrophage activation, platelets, thrombosis
Introduction
Chagas disease (CD) constitutes a leading health problem in the Americas. Though public health records are not up to date, it is estimated that ~8–10 million individuals are infected with Trypanosoma cruzi (Tc) and >110 million people are exposed to a risk of infection in endemic countries in the Americas.1 While direct transmission of the parasite by triatomine vector remains the primary route of infection, consumption of food contaminated with feces of infected triatomines, transfusion of blood from infected donors, and vertical transmission from infected mothers to their newborns are also prominent.2,3 The oral route of transmission is noted to result in a much higher mortality rate of 8%–35% than is generally observed by vectorial transmission, which is <5%–10%.4 Due to the high rate of migration from endemic countries, it is estimated that 68,000–120,000 people with CD are currently living in Europe.5 The Centers for Disease Control and Prevention (CDC) estimates >300,000 immigrants with T. cruzi infection are living in the US, with up to 45,000 cardiomyopathy cases occurring yearly.6 CD is reported to cause >14,000 deaths every year.1 The global economic burden of CD that can be averted by treatment with antiparasite drugs in the early phase of infection and thereby preventing heart failure and death in young adults is estimated to be US$8 billion for 2021–2030.7
The natural course of CD progression is illustrated in Figure 1. CD is initiated with acute parasitemic phase in which an abundance of T. cruzi in circulation, dissemination to tissue, and intracellular replication is noted. With activation of adaptive immunity, infected individuals control the parasitemia and develop to an indeterminate phase in which very low levels of parasites are intermittently detected in circulation and no to mild clinical features are noted.8 Approximately a third of infected individuals progress to a clinically symptomatic chronic phase and are burdened with major complications of the heart. Cardiomyopathy with progressive heart failure, arrhythmias, and cardiac, pulmonary, and cerebral embolism are believed to be the major cause of death in CD patients.8 Up to 20% of infected individuals may also develop gastrointestinal disorders that can result in chronic dysphagia, constipation, and malnutrition.
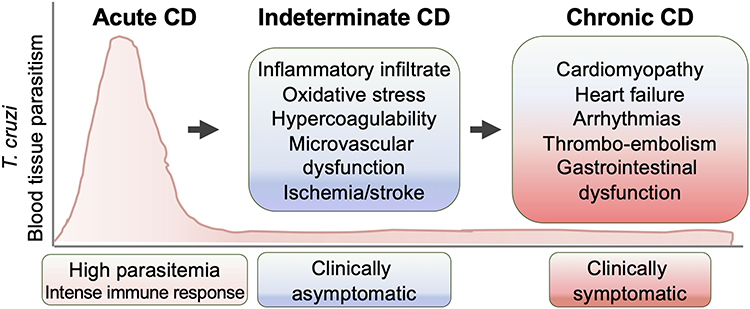 |
Figure 1 Clinical phases of Chagas disease (CD). Upon exposure to Trypanosoma cruzi, the acute phase of CD presents with high blood parasitemia, strong activation of the immune system, and intense inflammatory infiltrates and tissue parasitism. In indeterminate phases of CD, patients exhibit immune control of parasites and are mostly clinically asymptomatic, though pathological processes, including well-recognized inflammatory and oxidative stress, may continue. With progression to chronic-phase CD, tissue parasitism remains low, yet pathological factors lead to evolution of symptomatic heart disease and systemic, cardiac, pulmonary, or cerebral embolism. Some patients may also develop gastrointestinal and neurological disorders. |
Benznidazole and nifurtimox are the currently available antiparasitic drugs that are used as first-line therapy against T. cruzi infection.9 These drugs are recommended for the treatment of young children and acutely infected adults. Infected children (<14 years old) exhibit a cure rate of >85% upon treatment with nifurtimox.10,11 Benznidazole is effective in curing up to 90% of acute or recent cases of infection.12–14 These drugs are contraindicated during pregnancy,9 and their efficacy in treating infected individuals in indeterminate–chronic stages of CD remains controversial.15,16 Several efforts have been employed to identify new antiparasitic drugs, yet no new drug has succeeded in clinical trials so far, likely due to gaps in our knowledge of disease pathogenesis and parasite biology.17
The complexities of CD and disease-management strategies have been discussed in recent reviews.18,19 It is generally accepted that parasite persistence at low levels provides consistent antigens that continue to trigger immunoresponses, cause pathological tissue injury, and subsequently lead to cardiac insufficiency.20 Our work suggests that the myocardium in CD sustains chronic oxidative stress. An increase in macrophage production of superoxide by NADPH oxidase 221 and the mitochondrial generation of reactive oxygen species (ROS) from electron-chain complexes have been noted in the myocardium during CD development.22–26 Proteins, lipids, and DNA oxidative adducts have been detected in the peripheral blood and/or heart tissue of chronically infected rodents and human CD subjects27–34 and exacerbated by inefficient antioxidant capacity.28,31,35–39 Studies demonstrating ROS signaling of cytokine and chemokine production in infected cardiomyocytes and murine hearts40–43 have provided a mechanistic link between ROS and chronic inflammation in CD. Further details regarding the pathogenesis of chronic inflammation in CD can be found in excellent recent reviews.8,44
Parasympathetic and sympathetic dysfunction and microvascular impairments have been described to contribute to chronic CD pathology.8 High incidence of ischemic stroke is frequently noted in CD patients, and prevalence of stroke was higher in CD patients than in the general population of Brazil.45 Cross-sectional studies in the Americas noted higher frequency of stroke in patients with T. cruzi infection and indeterminate or chronic CD than in non-CD individuals (14%–15% vs 2%–6%, p<0.01 in Brazil; 25% vs 2%, p<0.05 in Colombia).46–48 These and other studies infer that ischemic stroke is likely the first clinically relevant sign in indeterminate CD before the clinical symptoms of heart disease become apparent. Cerebral and embolic stroke may occur with the development of chronic CD, involving the left ventricle or left atrium, several types of arrhythmias (right bundle branch block, advanced atrioventricular block, atrial fibrillation, tachycardia), and mural thrombi.8 Beyond the role of structural alterations of the myocardium and conduction disorders, recent research suggests that T. cruzi presence and inflammation conducive to hypercoagulability also contribute to higher risk of clot formation and thromboembolic events in CD. Accordingly, patients with chronic CD are often prescribed oral anticoagulants; however, long-term efficacy of anticoagulants in CD has not been studied.49
In this review, we summarize the current literature on hypercoagulability factors in CD and focus on the potential pathophysiological mechanisms involving platelets (PLTs) and monocytes/macrophages (Mo/Mφ) in sustaining thrombosis and inflammation in CD.
Integration of Platelet Activation and Coagulation Pathway in Hemostasis
Hemostasis reflects a delicate balance between procoagulant and anticoagulant factors for maintaining the blood flow within the vascular compartment and forming blood clots after vascular injury. There are two main components of hemostasis: primary hemostasis refers to PLT aggregation and plug formation, and secondary hemostasis refers to the deposition of insoluble fibrin to further strengthen the PLT plug.50,51 These two processes are mechanically and functionally intertwined. Calcium (Ca2+) ions play a major role in tight regulation of the coagulation cascade that is paramount in the maintenance of hemostasis while preventing pathological thrombosis or bleeding.52
PLTs play a fundamental role in primary hemostasis via adhesion, activation, and aggregation, which are triggered upon vascular tissue injury. First identified by Schultze in 1865, PLTs’ function in hemostasis was elucidated by Bizzazero in 1881,53 and in 1906, Wright showed that PLTs are generated from megakaryocytes. Lacking a nucleus, PLTs do not need a transcriptional machinery, yet they package carefully selected RNA and translational machinery and are equipped with an abundance of genetic information and processing machinery required for a highly efficient rapid response to vascular injury. Briefly, in the event of vascular injury, endothelial cells (ECs) in the blood vessels and circulating PLTs near the injury site produce von Willebrand factor (VWF). ECs also release the contents of their Weibel–Palade bodies, which enhance local concentrations of VWF. PLTs utilize adhesion molecules/ligands/receptors, including GPIa/GPIIa (collagen), GPIb/GPIX/GPV (VWF), and GPIIb/GPIIIa (ICAM1/fibrinogen/VWF) to adhere to the endothelium.54 Adherence and activation of PLTs may also be achieved through agonists (eg, serotonin, thromboxane A2 [TXA2], adenosine diphosphate [ADP], adrenaline, thrombin) that bind to protease-activated receptor 1, GPVI, purinergic receptors (P2Y1/2), and other G-protein-coupled receptors.50 Altogether, these signals lead to PLT aggregation and PLT plug formation at the site of vascular injury (Figure 2).
 |
Figure 2 Platelets: coagulation cascade cross talk in hemostasis. Primary hemostasis refers to platelets’ activation, adhesion, and aggregation at the site of endothelium injury. Adherence of platelets onto the exposed collagen takes place in a multifaceted process, and as a result platelets form a plug at the vascular injury site. Secondary hemostasis refers to the activation of the coagulation cascade. Both the intrinsic and extrinsic pathways converge into a common pathway and involve a series of sequential cleavage and activation of coagulation factors (XII, XI, IX, X, VII, III), ending with zymogen prothrombin (factor II) activation to thrombin (factor IIa). Activated thrombin catalyzes fibrinogen (factor I) cleavage to fibrin (Ia), and polymerization of fibrin monomers into an insoluble fibrin matrix strengthens the clot formation around the platelet plug. Fibrinolysis, through the action of plasmin, prevents unnecessary intravascular fibrin accumulation and enables the removal of thrombi. Protein C (PC), protein S (PS), and antithrombin II (ATII) inhibit the factors Xa/IIa and fibrin (Ia) formation. Tests and biomarkers used in clinical and research settings for examining the activation of platelets and coagulation cascade include, prothrombin fragment F1+2, thrombin time (TT); endogenous thrombin potential (ETP), factor VII, P-selectin, fibrinogen, and tissue factor (TF). Fibrinolysis is measured by plasminogen activator inhibitor 1 (PAI1), plasmin–antiplasmin complex (PAP), and D-dimer tests. Figure created using BioRender. |
Recruitment of PLTs at the vascular injury site stimulates secondary hemostasis, in which several coagulation mediators undergo cleavage to be activated. Stepwise activation of inactive precursors, or zymogens, is necessary for the formation of insoluble fibrin for vascular healing (Figure 2). The intrinsic pathway is dependent on contact with a negatively charged surface (eg, collagen), and involves cleavage-mediated activation of blood coagulation factors XII XI, and IX. The extrinsic pathway is dependent on tissue factor (TF) being exposed to the circulation. TF is synthesized and localized in the cell membrane of ECs, fibroblasts, and Mo/Mφ, among others. The TF-bearing extravascular cells scanning the vascular injury encounter plasma factor VII, which is then rapidly activated and brings VIIa/TF in proximity to PLT surfaces. TF inhibitors control factor VIIa–TF complex activity.54
Both intrinsic and extrinsic pathways converge to a common pathway to produce cross-linked fibrinogen (fibrin) and stabilization of the PLT plug. Herein, activated PLTs release α-granules containing factor V and VWF, and promote the assembly of procoagulant complexes, including tenase (factors VIIIa and IXa) and prothrombinase (factors Xa/Va) to propagate large-scale conversion of prothrombin (factor II) to thrombin (factor IIa) and culminating fibrinogen conversion to insoluble fibrin for the clot formation.54 Naturally occurring anticoagulants, such as protein C (PC), protein S (PS), and antithrombin (AT) keep a check on this process.52 Anticoagulant drugs including oral inhibitors of factor Xa (eg, rivaroxaban, apixaban, edoxaban) and factor IIa (eg, dabigatran) also check excessive activation of coagulation pathway. Finally, fibrinolysis represents the process in which clots are broken down to permit tissue repair.55 Disturbances in procoagulant or anticoagulant factors can delay clot formation or dissolution, cause bleeding disorder, or increase blood clotting and thrombosis.52
Thromboembolism and Stroke in CD
Thromboembolic events have been reported to occur at a high frequency in chronic Chagas heart disease. Samuel et al56 examined 1345 CD patients, of which 44% exhibited cardiac thrombosis and/or thromboembolic events affecting arteries in lungs, kidneys, and brain. Arteaga-Fernandez et al57 recorded 73% of CD patients (81 of 111) had cardiac thrombosis and many of them also showed systemic thrombosis. Further, euglobulin lysis time (measures fibrinolysis) in patients with chronic CD differed significantly from the values obtained in the control group, suggesting an occurrence of active thrombosis with decreased fibrinolysis.56,58 Subsequently, strokes were found to occur at a higher rate in chronic CD patients when compared to patients with heart disease of other etiologies.46 A history of stroke/transient ischemic attack, atrial fibrillation, and CD-positive serology were strongly associated with the occurrence of ischemic stroke in CD.59 Multivariate analysis showed cardioembolic CD patients with apical aneurysm and left ventricular (LV) intracavitary thrombi are highly susceptible to develop into ischemic cerebrovascular events.60 Cardiomyopathy was noted as an independent risk factor for the occurrence of cerebrovascular events in CD patients.46,59 At a population level, up to 18%–20% of CD patients with chronic heart failure exhibit cerebral infarctions, and nearly half of them have an association between cerebral infarction and death.49 It is proposed that progressive cardiac segmental contractile changes and aneurysm induce the formation of paradoxical emboli, ie, thrombi from dyskinetic areas of the cardiac cavities detach and are released in the cardiac vena cava and subsequently into the circulation, from where thrombi can reach the brain or other extremities. Therefore, the American Heart Association scientific statement on current knowledge and management of CD concludes that class III–IV LV dysfunction, cardiac dilation, significant electrocardiogram abnormalities, low ejection fraction, and chronic visceral venous congestion are all risk factors in higher propensity to thromboembolism and ischemic and cerebral stroke in CD patients.19
Role of Hypercoagulation in Induction of Thrombosis in Early Stages of CD
While cardiomyopathy in chronic CD is a known pathological process contributing to thrombosis, several studies have recognized that hypercoagulability, a cause of thrombus formation, occurs in acute-to-indeterminate phases of CD when symptomatic cardiomyopathy is not noted. The presence of procoagulation/prothrombotic phase in early stages of CD has been evidenced by elevated plasma levels of prothrombin fragment F1+2, D-dimer (fibrin degradation product), and the antithrombin enzyme complex in seropositive patients.58,61 However, Melo et al did not find higher prothrombotic states among heart-failure patients with CD compared to that noted in non-CD patients.62 Echeverria et al63 performed a meta-analysis of studies evaluating the pathophysiological aspects of hypercoagulation in CD patients in Argentina, Brazil, and Spain during 1996–2016. Cumulatively, the seven studies included in the meta-analysis enrolled 311 (57%) seropositive CD patients (mostly in the indeterminate phase) and 234 (43%) seronegative, healthy controls. These studies investigated biomarkers of EC activation, ie, sVCAM1, PLT activation (CD41, CD62P, P-selectin after ADP stimulation), thrombosis (F1+2, D-dimer, fibrinogen, FVII), and fibrinolysis (PAI1, PAP). The authors found a significant increase in F1+2 (SMD 5.15, 95% CI 1.92–8.38) and PAI1 (SMD 0.46, 95% CI 0.07–0.89) in CD vs healthy individuals.63 High levels of F1+2, an important marker of thrombin formation in vivo, may reflect endothelium injury by invading parasites or inflammation. PAI1 is a member of the superfamily of serpin-protease inhibitors and the primary inhibitor of tissue and urokinase-type plasminogen activators (tPA and uPA) that promote plasminogen cleavage to plasmin for its essential role in fibrinolysis. Increased PAI1 expression thus indicates excessive fibrin deposition in indeterminate CD.64 Four of the seven studies included in the meta-analysis examined PLT function. One study noted decreased frequency of activated PLTs based on the expression levels of CD41a and CD62P and of P-selectin after ADP stimulation in CD patients.65 Two studies noted significant increases in soluble P-selectin in adults and children with indeterminate CD compared to matched healthy controls.66,67 In a meta-analysis with a random-effect model, P-selectin was significantly increased in the indeterminate phase of CD (SMD 1.8, 95% CI 0.13–3.47).63 P-selectin is the largest member of the selectin family of cell-adhesion receptors, and it provides stability to the PLT aggregates, favoring thicker and more compact thrombi.68 These studies indicate that the high frequency of PLT activation, coagulation disorder, and thrombosis appear in indeterminate CD before the cardiac abnormality of clinical importance is detected in chronic CD patients. Further longitudinal studies will be needed to establish the role of hypercoagulability and prothrombotic status in the development of clinical features of heart failure and cerebral stroke in CD.
Parasite and Host Factors Contributing to Procoagulation State in CD
Parasite Factors
The literature discussed provides some evidence that an active thrombogenesis process takes place during indeterminate CD when cardiomyopathy and heart failure are not apparent. Some studies have indicated the role of T. cruzi factors in hypercoagulation syndrome in early-to-indeterminate CD, and these are discussed next (Figure 3). In 1990s, Tanowitz showed that ECs infected with T. cruzi exhibit increased adherence to PLTs in vitro, and PLTs of Tc-infected (vs control) mice were two–six-fold more sensitive to aggregation induced by ADP and arachidonic acid.69 Tanowitz et al also reported that TXA2 (stimulates PLT activation) is expressed in all developmental forms of T. cruzi, maximal TXA2 is expressed in replicative amastigote form, and Tc TXA2 accounts for up to 90% of the circulating levels of TXA2 in infected mice.70 They concluded that increased production of TXA270 and endothelin 1 (EC-activation marker) in CD71 likely mediate PLT adhesion, activation, and aggregate formation, at least in the acute parasitemic phase of CD. PLT aggregates contributing to transient thrombi have been detected in small epicardial and intramyocardial vessels of T. cruzi–infected animals.72
 |
Figure 3 Trypanosoma cruzi factors contributing to hypercoagulability and thrombosis. T. cruzi (all life-cycle stages) releases thromboxane 2 (TXA2), a known activator of platelets, and trans-sialidase (TS) proteins during differentiation from trypomastigote to amastigote form. Endothelial cells (ECs) were susceptible to TS-induced increases in E selectin, VCAM1, and ICAM1 release. The enzymatically active isoform of TS (transfers sialic acid) induced platelet apoptosis and thrombocytopenia. T. cruzi infection–induced changes in blood rheological properties causing abnormal red blood cell (RBC) morphology can contribute to increased RBC-platelet aggregates and thrombi formation and affect smooth blood flow. Treatment with the antiparasite benznidazole was shown to reduce the biomarkers of platelet activation (P-selectin) and hypercoagulation (fibrinogen, prothrombin fragment F1+2, endogenous thrombin potential (ETP), plasmin antiplasmin complex (PAP)) in individuals exposed to acute T. cruzi infection. Figure created using BioRender. |
T. cruzi expresses a large family of trans-sialidase (TS) surface glycoproteins, a majority of which are enzymatically inactive.73 The enzymatically inactive members of the TS family have been shown to signal the expression of E selectin, VCAM1, and ICAM1, and increase EC susceptibility.74 Others suggested that T. cruzi sheds enzymatically active TS that removes sialic acid from PLT surfaces75 and may contribute to thrombocytopenia in CD.
Blood vessel–wall injury due to circulating parasites and changes in blood viscosity due to parasite-mediated host immuneoresponse may also augment hypercoagulability and facilitate the development of thromboembolic events in early-to-indeterminate CD. Berra et al76 showed increases in hematocrit and mean corpuscular volume, abnormal morphological changes in red blood cells, and increases in plasma viscosity in acutely infected (vs control) rats. The authors concluded that T. cruzi infection induced changes in host blood properties and defensive responses determine the blood hyperviscosity that is an important ischemic risk factor capable of altering microvascular blood flow. The role of abnormally shaped red blood cells in promoting thrombus formation and high viscosity of blood in enhancing the stability of thrombi has also been documented in other diseases.77 Finally, it has been shown that treatment with the antiparasitic benznidazole for 60 days resulted in a significant reduction in plasma levels of P-selectin, F1+2, fibrinogen, PAP, and ETP in acutely infected patients.66,67
Together, these findings imply that parasites and parasite-derived factors can contribute to endothelial dysfunction, PLT activation, and hypercoagulability in CD, and highlight the relevance of treatment with benznidazole in early stages of CD. Whether the benefits of antiparasitic treatment in reducing coagulation factors would translate into protection from stroke and chronic severity of CD remains to be investigated in future studies.
Host Factors
Circulating and tissue parasite burden is very low in the indeterminate-to-chronic phase of CD. Therefore, we postulate that parasite factors may not solely be responsible for hypercoagulability in indeterminate CD. TF stimulation of thrombin is quantified by endogenous thrombin potential that is increased in indeterminate CD.66,67 TF is expressed by several cells, including ECs, neutrophils, and Mo/Mφ, after exposure to inflammatory cytokines, eg, TNFα and IL6, and TF is the primary mediator of interactions between inflammatory and coagulation pathways.78 Patients with indeterminate CD exhibit higher plasma levels of TNFα79 and IL680 than uninfected healthy controls. We have documented a significant increase in peripheral and myocardial levels of CD11b+CD68+TNFα+ Mo/Mφ in CD.81,82 In vitro studies have demonstrated significantly elevated levels of proinflammatory cytokines (IL6, IL1β) in the supernatants of T. cruzi–infected ECs.83 Parasite-infected ECs exhibit NFκB transcriptional activation and increased expression of E-selectin, ICAM1, and VCAM1.83,84 It is thus conceivable that ECs or circulating Mo/Mφ, after encountering T. cruzi, Tc pathogen–associated molecular patterns (PAMPs), or Tc-induced damage associated molecular patterns (DAMPs) and proinflammatory cytokines, begin to express TF that then promotes coagulation cascade and thrombin production in CD (Figure 4). This hypothesis, however, remains to be tested in experimental and clinical studies.
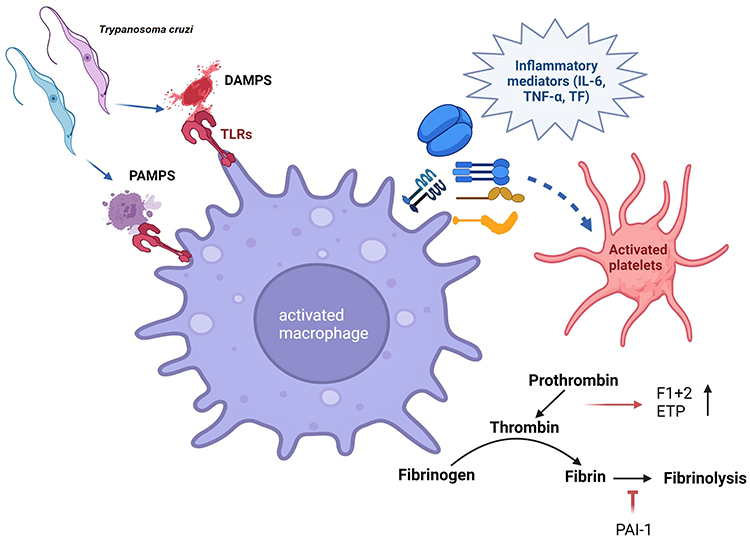 |
Figure 4 Trypanosoma cruzi–macrophage interactions and induction of thrombosis. T. cruzi releases pathogen- associated molecular patterns (PAMPs), and T. cruzi-mediated cellular injury generates damage-associated molecular patterns (DAMPs). Macrophage uptake of PAMPs and DAMPs through Toll-like receptors (TLRs) may signal the release of inflammatory mediators, eg, IL6, TNFα, and tissue factor (TF) to induce the activation of platelets and coagulation cascade. Increase in prothrombin fragment F1+2, endogenous thrombin potential (ETP), and PAI1 is noted in Chagas disease. Figure created using BioRender. |
P-selectin mediates adhesion of PLTs with endothelium and leukocytes, and soluble P-selectin signals recruitment of cells and cell particles containing TF to the injury site.85 Likewise, PAI1 has been identified as a key factor linking fibrinolysis with subclinical (eg, inflammation, atherosclerosis) and clinical (eg, metabolic syndrome) conditions.64 Circulating proinflammatory cytokines can directly activate PLTs and release of P-selectin and PAI1, among others.86,87 An increase in soluble P-selectin and PAI1 associated with an increase in PLT death has been noted in patients with indeterminate CD.63 It is possible that inflammatory responses that continue to persist in all stages of T. cruzi infection and disease progression signal activation and exhaustion of PLTs in CD. If such is the case, these events will contribute to PAI1-mediated increases in fibrin deposition and soluble P-selectin–mediated enhanced stability of PLT–leukocyte aggregates and formation of thicker thrombi. Subsequently, P-selectin may also contribute to apoptosis of activated PLT.
Summarizing, proinflammatory mediators and Tc-activated EC and Mo/Mφ potentially contribute to TF-induced PLT activation and hypercoagulability in acute-to-indeterminate CD. However, mechanistic studies documenting PLT–immune cell interactions and the signals that contribute to disturbances of hemostasis in early-to-indeterminate CD have not yet been conducted.
Platelet Role in Inflammation
A connection between PLTs and inflammation is very well recognized in invertebrate arthropods, in which a single cell type (hemocytes) carries out the function of innate immune cells (eg, phagocytosis, antimicrobial release), as well as wound plugging and repair.88 Considering that PLTs constantly scan the endothelium and vascular injury, it is not surprising that they are well positioned to act as first responders to detect and respond to invading pathogens. We briefly discuss PLT involvement in shaping host immunodefense and inflammatory responses and extrapolate the literature findings in the context of CD here.
PLTs synthesize and package molecules in dense α-granules and selectively release prothrombotic, proinflammatory, or immunoregulatory molecules depending on the microenvironment. Activated PLTs have an arsenal of inflammation inducers, including P-selectin, sCD40L, IL1β, HMGB1, C–C motif chemokine ligand (CCL) family members, such as CCL3 (MIP1α), CCL5 (RANTES), and CCL7 (MIP3), and C–X–C chemokine ligand (CXCL) family members, eg, CXCL1, CXCL4 (PF4), CXCL5, and CXCL7.89 PLTs also release the mediators of tissue repair, such as TGFβ, depending on the microenvironment.90,91 Activated PLTs can rapidly bind to and engulf microbes, and their direct role in the control of pathogens is discussed elsewhere.92
PLT binding shapes the proinflammatory or immunoregulatory activation of immune cells. In general, PLTs utilize the classical PSGL1 axis to adhere to all types of immune cells and employ selective receptors to adhere to specific population of immune cells. Nonclassical pathways also activate PLT–immune cell interactions via engagement of a vast repertoire of pattern-recognition receptors including Toll-like receptors (TLR2, TLR4, TLR9), C-type lectin-like receptors, and TNFα and IL1β receptors.93,94
With reference to influence on Mo/Mφ, PLT interactions with Mo/Mφ through P-selectin–PSGL1 induce the expression of MCP1 and IL8 in Mo/Mφ, while GPIβ–CD11b induces proinflammatory response with increased TNFα production95 (Figure 5). Circulatory PF4/CXCL4 is increased in chronic inflammation of diverse etiologies.96,97 In experimental models, PF4/CXCL4 and P-selectin have been shown to potentiate phagocytosis, respiratory burst, and a three–ten-fold increase in proinflammatory cytokines expression in human monocytes.98,99 PLT release of HMGB1 induced monocyte migration and accumulation in tissue by inhibiting apoptosis in a TLR-dependent manner.100 CD40L is primarily secreted by activated PLTs, which signals a CD40-mediated immunoregulatory phenotype in human monocytes by enhancing IL10 and suppressing TNFα production (Figure 5).101
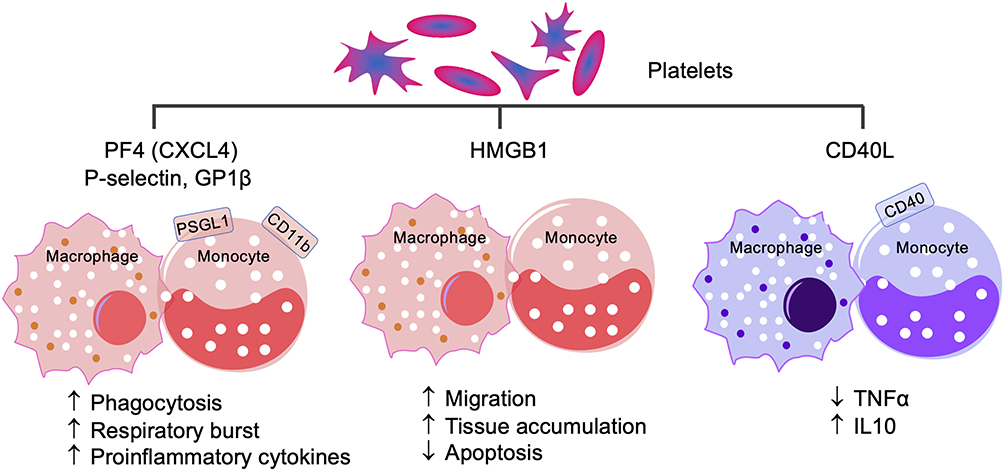 |
Figure 5 Platelet–monocyte/macrophage interactions. PF4, also known as CXCL3, P-selectin, and GPIβ interact with PSGL1 and CD11b receptor–positive monocytes/macrophages to signal proinflammatory activation, while platelet expression and release of HMGB1 signals monocyte/macrophage migration to tissue and cell apoptosis. Conversely, CD40L interacts with CD40 to drive the anti-inflammatory, immunomodulatory monocyte/macrophage response by suppressing proinflammatory cytokine TNFα and increasing anti-inflammatory IL10. |
Regarding PLTs’ role in shaping the neutrophil response, PLT and neutrophil interactions through P-selectin–PSGL1 or GP1β–integrin αMβ2 (Mac1) axes have been shown to modulate neutrophil adhesion to ECs and neutrophil extracellular trap (NET) release (Figure 6A).102 Likewise, PLT release of HMGB1 enhances neutrophil activation and NET formation (Figure 6A).103,104 PLT–neutrophil interactions facilitate NET release, providing protection from poxvirus infection.105 Human PLT release of β-defensin–induced NET formation impairs the growth of clinical strains of Salmonella aureus.106 PLT–neutrophil interactions may also inform the pathogenesis of invasive S. pyogenes infection.107 Conversely, PF4/CXCL4 inhibition decreases vasculature inflammation and preserves heart function by reducing leukocyte recruitment, adhesion, and extracellular nuclear DNA trap release by activated neutrophils.108
 |
Figure 6 Platelet activation of neutrophils and lymphocytes. (A) Platelets signal neutrophils via platelet factor 4 (PF4), GPIβ, or HMGB1 interactions with the macrophage 1 (Mac1) antigen, which enhances neutrophil extracellular trap (NET) release. P-selectin–PSGL1-mediated platelet–neutrophil interaction suppresses neutrophil adhesion to endothelial cell (ECs) and subsequent activation. (B) Platelet–lymphocyte interactions via PF4–PSGL1, CD40L–CD40, or platelet release of TGFβ result in a decline in T-cell proliferation, decreased expression of T helper 1/T helper 17 (Th1/Th17) proinflammatory cytokines and granzyme (GZB) and perforin (PFN) cytotoxic molecules, and upregulation of immunomodulatory Th2 regulatory T-cell response. |
The specific role of PLT-adhesion molecules is, however, context-dependent. For example, PF4 has been shown to inhibit the proinflammatory, cytotoxic activation of CD8+ T cells109 and limit the Th1 and Th17 cell differentiation and cardiac allograft rejection by suppressing RORγ expression (Figure 6B).110 Likewise, elevated CD40L levels induce immunosuppression by mediating the regulatory T cell (Treg) expansion in HIV-infected patients.111 Some studies have shown that PLT expression of TGFβ suppresses lymphocyte function, while IL1α/IL1β plays a crucial role in systemic and cerebrovascular inflammation.112,113 Specifically, PLT TGFβ inhibited the expression of proinflammatory cytokines (IFNγ) and cytotoxic molecules (granzyme B and perforin) in CD8+ T cells, and TGFβ inhibitors improved adoptive T-cell therapy in multiple cancers in mice.114 PLT release of TGFβ also activates the Treg phenotype and suppresses the Th1/Th17 response in human CD4+ T cells115 (Figure 6B). Likewise, PLT–CD4+ T cell aggregates exhibit reduced proliferation and proinflammatory cytokine production in rheumatoid arthritis patients, and higher levels of PLT–CD4+ T-cell aggregates correlate with reduced risk of cardiovascular disease.116
In summary, PLTs can exert dual function, ie, proinflammatory and immunoregulatory on Mo/Mφ, neutrophils, and distinct subsets of T lymphocytes. How the expression of different receptors and ligands on PLTs and immune cells is determined in different conditions, how microenvironmental conditions influence the PLT and immune cells interactions and whether PLT stimulated immune cells migrate to tissue to exert their effects is not completely understood and requires further investigation.
A Perspective on the Role of Platelets’ Partnership with Macrophages in Chronic Inflammation in CD
There is practically no information in literature on PLT-neutrophils partnership in CD and the limited literature on the potential role of neutrophils in CD is summarized recently.117 Herein, we will discuss our perspectives on potential role of PLT – Mφ interactions in chronic inflammation in CD (Figure 7).
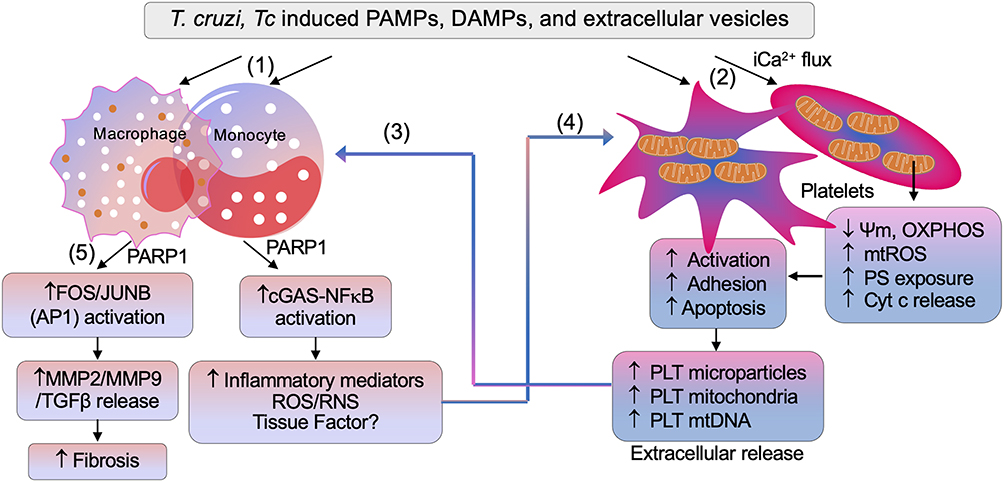 |
Figure 7 Potential cross talk between platelets and macrophages in Chagas disease. (1) Trypanosoma cruzi (Tc), and Tc-induced pathogen associated molecular patterns (PAMPs), damage associated molecular patterns (DAMPs), and extracellular vesicles signal initial proinflammatory activation of macrophages. (2) Platelets encountering T. cruzi–induced molecules undergo intracellular Ca2+ flux–mediated mitochondrial stress presented by a decline in membrane potential (Ψm) and oxidative phosphorylation (OXPHOS) capacity, and an increase in mitochondrial reactive oxygen species (mtROS), Cyt c release, and phosphotidyle serine (PS) exposure. These events signal platelet activation, aggregation, and subsequent apoptosis. (3) Extracellular vesicles, microparticles, mitochondria, and mtDNA released by Tc, Tc-infected cells, and platelets are phagocytized by macrophages, and elicit transcriptional activation of mediators of inflammation and oxidative stress (ie, ROS and reactive nitrogen species [RNS]). (4) Macrophage inflammatory mediators, along with tissue factors, can sustain platelet activation and apoptosis. (5) PARP1–AP1 synergy also signals release of profibrotic molecules, ie, matrix metalloproteinase 2 (MMP2), MMP9, and TGFβ, leading to tissue fibrosis in Chagas disease. Abbreviation: cGAS, cyclic GMP-AMP synthase. |
Proinflammatory Macrophage Response in CD
Studies in human and mouse macrophages show that T. cruzi elicits Ca2+ flux for invasion of immune and non-immune cells, and signals low levels of NADPH oxidase mediated oxidative burst, inducible nitric oxide synthase mediated nitric oxide production and delayed inflammatory cytokines/chemokines response in innate immune cells.118,119 Because of this, Mφ are not able to clear the parasites, and instead may support parasite dissemination to tissue.120,121 With the activation of adaptive B and T cell immunity, parasitemia is controlled, and very low levels of circulating parasites are infrequently detected in the patients progressing from indeterminate to chronic CD. Yet, CD patients present systemic increase in inflammatory markers (N-terminal pro-brain natriuretic peptide, C-reactive protein), inflammatory cytokines (eg, IL1β, IL6, IFNγ, TNFα) and chemokines (eg, CXCL9, CXCL10), and free radical induced oxidative lesions of lipids (lipid hydroperoxides, 4-hydroxynonenal), DNA (8-oxo-7,8-dihydro-2’-deoxyguanosine) and proteins (protein carbonyls, cysteine nitrosylation) during indeterminate-to-chronic CD (reviewed in)122–124. CD patients also display a proinflammatory transcriptomic and proteomic profile in peripheral blood mononuclear cells,125–128 and increased levels of TNFα+ monocytes129–131 in circulation. Tissue distribution of proinflammatory Mo/Mφ is also increased in chronic CD.132 Thus, Mo/Mφ are recognized as major drivers of chronic inflammation in CD.
Extracellular Vesicles Signal Proinflammatory Activation of Macrophages in CD
Recent literature documents that T. cruzi sheds compositionally different membrane vesicles, also referred as extracellular vesicles (EV), depending on their developmental stage.133,134 EV of infective trypomastigote form of T. cruzi were fused to host cell membranes and promoted EV release from THP1 Mφ.135–138 We showed that EV are released in media of cells, eg, human peripheral blood mononuclear cells (PBMC), mouse and human Mφ, and human cardiac myocytes in vitro incubated with T. cruzi and in peripheral blood of chronically infected mice and humans, and these Tc-induced EV signaled the proinflammatory gene expression in primary and cultured Mφ.81,139 Compositional and molecular analysis showed that EV released in plasma of CD patients were of leukocytes and endothelial origin and packaged oxidized DNA (oxDNA) of host and parasite.81,139 Treatment of these EV with DNase abrogated the inflammatory signature, thus indicating that EVoxDNA is necessary for proinflammatory activation of Mφ in CD.81
Poly(ADP-ribose) polymerase 1 (PARP1) is the enzymatically active member of the PARP family of 18 proteins. PARP1 senses DNA oxidative damage to catalyze NAD+ dependent poly-ADP-ribosylation of target histone proteins and functions in DNA repair.140 PARP1 expression and activity were increased in CD;141 however, PARP1 enhanced the mitochondrial ROS production and nuclear factor kappa B (NFκB) transcriptional activation of inflammatory gene expression in cardiac myocytes and Mφ in response to T. cruzi infection.81 PARP1 synergized with cytosolic DNA response element cyclic GMP-AMP synthase (cGAS) in sensing the EVoxDNA shed in plasma of T. cruzi infected animals and humans to transmit the signal for NFκB-dependent inflammatory gene expression in human and mouse Mφ.81 Further, PARP1 signaled the FOS/JUNB (members of the activator protein 1 (AP1) complex) transcriptional activation of matrix metalloproteinases (MMP2, MMP9) and TGFβ expression in Mφ infected with T. cruzi.82 Consequently, PARP1 deficiency by genetic, chemical, or siRNA approaches was beneficial in inhibiting the inflammatory and fibrotic response in Mφ incubated with T. cruzi or Tc-induced EV, decreasing the oxidative and inflammatory stress in infected mice, and improving the mitochondrial and cardiac function in chronic CD mice.81,82,141 These studies conclude that EVoxDNA/PARP1 crosstalk with cGAS-NFκB and AP1 signaling pathways in Mo/Mφ to sustain inflammation and fibrosis with progression of chronic CD. The source of oxidized DNA that triggers chronic proinflammatory/profibrotic Mφ response in CD was not defined in these studies.
Platelet Shedding of Mitochondria and Its Potential Role in Chronic Inflammation in CD
PLTs lack chromosomes but pack 5–8 mitochondria to meet their energy demand. PLT are highly flexible in energy source utilization for their classical function in hemostasis.142 Double knockout of glucose transporters GLUT1 and GLUT3 led to mitochondria reprograming, and reduced thrombosis, PLT activation, and thrombocytopenia.143 Likewise, PLT treated with pharmacological inhibitors of mitochondrial respiratory complexes exhibited reduced thrombosis activity,142 though mechanisms were not discussed.
Recent studies indicate that mitochondria influence the PLT health and activation via their role in regulation of cell survival and apoptosis. Briefly, disturbances in mitochondrial Ca2+ hemostasis can signal mitochondrial membrane permeability transition, increased ROS production, cytochrome c release, and involvement of pro- and anti-apoptotic members of B cell lymphoma 2 (Bcl-2) family to guide the activation of caspase cascade and cell death decision.144,145 B cell lymphoma extra-large (Bcl-xl) protein was shown to govern the lifespan of healthy PLT,146 while iCa2+ flux facilitated the collagen, thrombin, and ADP mediated PLT activation.147 PLT expression of Bcl-2 family proteins and exposure of phosphatidylserine that facilitates recognition and removal of the apoptotic cell by phagocytes were deemed essential for PLT adhesion and apoptosis.146,148 Apoptotic PLT (and their microparticles) induced 50–100-fold higher procoagulant activity than the quiescent, healthy PLTs.149 Overall, these studies indicate that mitochondrial signals drive the thrombotic function and subsequent apoptosis of activated PLTs.
T. cruzi induction of mitochondrial dysfunction is well documented in cardiac myocytes (reviewed in8,26 and briefly discussed in Introduction). Considering that PLT encounter circulating parasites, especially in acute phase, T. cruzi signals Ca2+ flux during invasion, and Tc-released PAMPs and DAMPs signal macrophage proinflammatory response and possibly PLT aggregation and activation, it is conceivable that T. cruzi may also initiate mitochondrial dysfunction in PLTs, and consequently, enhance the PLT apoptosis and PLT mitochondria derived EV release in circulation.
Indeed, cell‐derived mitochondrial components including mtDNA can be found free or enclosed by membrane vesicles in the peripheral blood of healthy subjects and patients with various diseases.150–152 Meddeb et al153 reported that ~50,000 fold more copies of mitochondrial genome than the nuclear genome is present in circulation of healthy individuals, and these further increase in disease conditions. The cell free mtDNA is less fragmented than the nuclear genome,154 and, therefore, circulatory mtDNA has emerged as biomarker of multiple diseases including diabetes, myocardial infarction, cancer, and trauma.150–152 Despite the clinical implications, knowledge regarding the origin and function of cell free mtDNA and how they are protected from degradation in circulation is lacking. Some studies have reported that monocytes and PLT are the primary source of cell free mitochondria and mitochondrial fragments found in circulation, and free and EV-encapsulated mitochondrial fragments and mtDNA released by activated monocytes and PLT serve as inflammatory trigger.155,156 Indeed, mitochondria isolated from lipopolysaccharide (LPS) activated Mo/Mφ, EV released by LPS-activated Mo/Mφ, as well as EV isolated from plasma of human volunteers receiving low-dose LPS-injections induced type I IFN and TNFα response in ECs.157 Activated PLTs released respiratory-competent mitochondria, both within EV and as free organelles, and mitochondrial membranes and mtDNA present in these PLT-derived extracellular mitochondria promoted leukocytes activation, neutrophils adhesion and inflammatory responses.158 The mtDNA encapsulated in circulating EV of chronic CD patients signaled proinflammatory Mφ activation (discussed above), and we postulate that the mtDNA in circulation of CD patients are primarily derived from activated PLT contributing to hypercoagulability. Overall, despite several gaps in our knowledge, we surmise that a crosstalk between PLT and macrophages via circulating mtDNA (oxidized or native) might feed the chronic inflammatory and oxidative stress in progressive CD development.
Concluding Remarks
In this review, we have summarized the current literature describing the general and CD-specific features of PLTs’ role in coagulopathy and inflammation. While inflammation, oxidative stress and hypercoagulability are well-recognized characteristics of CD; the literature discussed here suggests that these pathological events might occur because of the crosstalk between monocytes/macrophages and PLTs. While T. cruzi and Tc-derived PAMPs may initially elicit Mo/Mφ and PLT activation, we propose that a feedback cycle of inflammation and hypercoagulation persists in CD because PLT activation for signaling the coagulation cascade is followed by PLT apoptosis and release of mitochondria, mitochondrial particles and mtDNA in circulation, that then serve as DAMPs in proinflammatory activation of Mo/Mφ in CD. Thus, we envision that new therapies modulating PLT activation and mitochondria dysfunction or reprogramming the Mo/Mφ towards immunomodulatory phenotype will arrest excessive thrombus formation as well as inflammation in chronic CD.
Although some evidence is available regarding the beneficial effect of antiparasitic benznidazole therapy in correcting the hypercoagulable state in acutely infected children and young adults, these findings need to be repeated and verified in larger cohorts. Further, extensive preclinical and clinical research efforts are needed for evaluating the benefits and risks of anti-parasite, anti-PLT, and anticoagulant therapies in arresting the hypercoagulable state and thromboembolic events in indeterminate stage of CD. Longitudinal studies in which seropositive indeterminate CD patients are offered these therapies and followed for several years to monitor the progression to chronic CD would potentially offer new treatments to prevent the clinical manifestation of CD.
Abbreviations
AT, antithrombin; CD, Chagas disease; CCL, C–C motif chemokine ligand family; CXCL, C–X–C chemokine ligand family; EC, endothelial cell; ETP, endogenous thrombin potential; F1+2, prothrombin factor 1 and 2; GP, glycoprotein; Mo/Mφ, monocytes/macrophages; MMP, matrix metalloproteinase; NET, neutrophil extracellular traps; PAP, plasmin–antiplasmin complex; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; SMD, standard mean difference; TF, tissue factor; TXA2, thromboxane 2; VWF, von Willebrand factor.
Acknowledgment
NJG has been supported by grants from the National Institute of Allergy and Infectious Diseases (R21 AI151305, R01AI054578, and R01AI136031) and National Heart Lung, and Blood Institute (R01HL094802) of the National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Chagas disease (also known as American trypanosomiasis) key facts. Geneva, Switzerland: UNDP/World Bank/WHO; 2022. Available from: https://wwwwhoint/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis).
2. World Health Organization. Chagas disease control and elimination. Report of the secretariat. Geneva, Switzerland: UNDP/World Bank/WHO; 2019. Available from: http://apps.who.int/gb/ebwha/pdf_files/WHA63/A63_17-en.pdf.
3. Coura JR. The main sceneries of Chagas disease transmission. The vectors, blood and oral transmissions - A comprehensive review. Mem Inst Oswaldo Cruz. 2015;110:277–282. doi:10.1590/0074-0276140362
4. Antunes D, Marins-Dos-Santos A, Ramos MT, et al. Oral route driven acute Trypanosoma cruzi infection unravels an IL-6 dependent hemostatic derangement. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.01073
5. Antinori S, Galimberti L, Bianco R, Grande R, Galli M, Corbellino M. Chagas disease in Europe: a review for the internist in the globalized world. Eur J Intern Med. 2017;43:6–15. doi:10.1016/j.ejim.2017.05.001
6. Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microbiol Rev. 2011;24:655–681. doi:10.1128/CMR.00005-11
7. Bartsch SM, Avelis CM, Asti L, et al. The economic value of identifying and treating Chagas disease patients earlier and the impact on Trypanosoma cruzi transmission. PLoS Negl Trop Dis. 2018;12:e0006809. doi:10.1371/journal.pntd.0006809
8. Bonney KM, Luthringer DJ, Kim SA, Garg NJ, Engman DM. Pathology and pathogenesis of Chagas heart disease. Annual Rev Pathol. 2020;14:421–447. doi:10.1146/annurev-pathol-020117-043711
9. Meymandi S, Hernandez S, Park S, Sanchez DR, Forsyth C. Treatment of Chagas disease in the United States. Curr Treat Options Infect Dis. 2018;10:373–388. doi:10.1007/s40506-018-0170-z
10. Sales Junior PA, Molina I, Fonseca Murta SM, et al. Experimental and clinical treatment of Chagas disease: a review. Am J Trop Med Hyg. 2017;97:1289–1303. doi:10.4269/ajtmh.16-0761
11. Bianchi F, Cucunuba Z, Guhl F, et al. Follow-up of an asymptomatic Chagas disease population of children after treatment with nifurtimox (Lampit) in a sylvatic endemic transmission area of Colombia. PLoS Negl Trop Dis. 2015;9:e0003465. doi:10.1371/journal.pntd.0003465
12. de Andrade AL, Zicker F, de Oliveira RM, et al. Randomised trial of efficacy of benznidazole in treatment of early Trypanosoma cruzi infection. Lancet. 1996;348:1407–1413. doi:10.1016/S0140-6736(96)04128-1
13. Coura JR. Current prospects of specific treatment of Chagas’ disease. Bol Chil Parasitol. 1996;51:69–75.
14. Bermudez J, Davies C, Simonazzi A, Real JP, Palma S. Current drug therapy and pharmaceutical challenges for Chagas disease. Acta Trop. 2016;156:1–16. doi:10.1016/j.actatropica.2015.12.017
15. Morillo CA, Marin-Neto JA, Avezum A, et al. Randomized trial of benznidazole for chronic Chagas cardiomyopathy. N Engl J Med. 2015;373:1295–1306. doi:10.1056/NEJMoa1507574
16. Mills RM. Chagas disease: epidemiology and barriers to treatment. Am J Med. 2020;133:1262–1265. doi:10.1016/j.amjmed.2020.05.022
17. Kratz JM, Goncalves KR, Romera LM, et al. The translational challenge in Chagas disease drug development. Mem Inst Oswaldo Cruz. 2022;117:e200501. doi:10.1590/0074-02760200501
18. Pinazo MJ, Torrico F, Gascón J. Pathogenesis of Chagas disease in humans. In: Singh SK, editor. Human Emerging and Re‐emerging Infections: Viral and Parasitic Infections. Vol. 1. Wiley Online Library; 2015:349–369.
19. Nunes MCP, Beaton A, Acquatella H, et al. Chagas cardiomyopathy: an update of current clinical knowledge and management: a scientific statement from the American Heart Association. Circulation. 2018;138:e169–e209. doi:10.1161/CIR.0000000000000599
20. Fonseca R, Salgado RM, Borges da Silva H, Nascimento RS, D’Império-Lima MR, Alvarez JM. Programmed cell death protein 1–PDL1 interaction prevents heart damage in chronic Trypanosoma cruzi infection. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.00997
21. Dhiman M, Garg NJ, Vieira LQ. P47phox−/− mice are compromised in expansion and activation of CD8+ T cells and susceptible to Trypanosoma cruzi infection. PLoS Pathog. 2014;10:e1004516. doi:10.1371/journal.ppat.1004516
22. Garg N. Mitochondrial disorders in chagasic cardiomyopathy. Front Biosci. 2005;10:1341–1354. doi:10.2741/1624
23. Zacks MA, Wen JJ, Vyatkina G, Bhatia V, Garg N. An overview of chagasic cardiomyopathy: pathogenic importance of oxidative stress. An Acad Bras Cienc. 2005;77:695–715. doi:10.1590/S0001-37652005000400009
24. Wen JJ, Garg NJ. Mitochondrial generation of reactive oxygen species is enhanced at the Q(o) site of the complex III in the myocardium of Trypanosoma cruzi-infected mice: beneficial effects of an antioxidant. J Bioenerg Biomembr. 2008;40:587–598. doi:10.1007/s10863-008-9184-4
25. Wen JJ, Garg NJ. Mitochondrial complex III defects contribute to inefficient respiration and ATP synthesis in the myocardium of Trypanosoma cruzi-infected mice. Antioxid Redox Signal. 2010;12:27–37. doi:10.1089/ars.2008.2418
26. Lopez M, Tanowitz HB, Garg NJ. Pathogenesis of chronic Chagas disease: macrophages, mitochondria, and oxidative stress. Curr Clin Microbiol Rep. 2018;5:45–54. doi:10.1007/s40588-018-0081-2
27. Wen JJ, Dhiman M, Whorton EB, Garg NJ. Tissue-specific oxidative imbalance and mitochondrial dysfunction during Trypanosoma cruzi infection in mice. Microbes Infect. 2008;10:1201–1209. doi:10.1016/j.micinf.2008.06.013
28. Wen -J-J, Vyatkina G, Garg NJ. Oxidative damage during chagasic cardiomyopathy development: role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radic Biol Med. 2004;37:1821–1833. doi:10.1016/j.freeradbiomed.2004.08.018
29. Dhiman M, Zago MP, Nunez S, Nunez-Burgio F, Garg NJ. Cardiac oxidized antigens are targets of immune recognition by antibodies and potential molecular determinants in Chagas disease pathogenesis. PLoS One. 2012;7:e28449. doi:10.1371/journal.pone.0028449
30. Paiva CN, Medei E, Bozza MT, Gubbels M-J. ROS and Trypanosoma cruzi: fuel to infection, poison to the heart. PLoS Pathog. 2018;14:e1006928. doi:10.1371/journal.ppat.1006928
31. Wen -J-J, Yachelini PC, Sembaj A, Manzur RE, Garg NJ. Increased oxidative stress is correlated with mitochondrial dysfunction in chagasic patients. Free Rad Biol Med. 2006;41:270–276. doi:10.1016/j.freeradbiomed.2006.04.009
32. de Oliveira TB, Pedrosa RC, Filho DW. Oxidative stress in chronic cardiopathy associated with Chagas disease. Int J Cardiol. 2007;116:357–363. doi:10.1016/j.ijcard.2006.04.046
33. Dhiman M, Nakayasu ES, Madaiah YH, et al. Enhanced nitrosative stress during Trypanosoma cruzi infection causes nitrotyrosine modification of host proteins: implications in Chagas’ disease. Am J Pathol. 2008;173:728–740. doi:10.2353/ajpath.2008.080047
34. Dhiman M, Estrada-Franco JG, Pando JM, et al. Increased myeloperoxidase activity and protein nitration are indicators of inflammation in patients with Chagas’ disease. Clin Vaccine Immunol. 2009;16:660–666. doi:10.1128/CVI.00019-09
35. Wen -J-J, Bhatia V, Popov VL, Garg NJ. Phenyl-alpha-tert-butyl nitrone reverses mitochondrial decay in acute Chagas disease. Am J Pathol. 2006;169:1953–1964. doi:10.2353/ajpath.2006.060475
36. Perez-Fuentes R, Guegan JF, Barnabe C, et al. Severity of chronic Chagas disease is associated with cytokine/antioxidant imbalance in chronically infected individuals. Int J Parasitol. 2003;33:293–299. doi:10.1016/S0020-7519(02)00283-7
37. Wen JJ, Porter C, Garg NJ. Inhibition of NFE2L2-Antioxidant Response Element pathway by mitochondrial reactive oxygen species contributes to development of cardiomyopathy and left ventricular dysfunction in Chagas disease. Antioxid Redox Signal. 2017;27:550–566. doi:10.1089/ars.2016.6831
38. Wan X, Wen JJ, Koo SJ, Liang LY, Garg NJ. SIRT1-PGC1alpha-NFkappaB pathway of oxidative and inflammatory stress during Trypanosoma cruzi infection: benefits of SIRT1-targeted therapy in improving heart function in Chagas disease. PLoS Pathog. 2016;12:e1005954. doi:10.1371/journal.ppat.1005954
39. Dhiman M, Wan -X-X, Vargas G, Garg NJ, Garg NJ. MnSOD tg mice control myocardial inflammatory and oxidative stress and remodeling responses elicited in chronic Chagas disease. J Am Heart Assoc. 2013;2:e000302. doi:10.1161/JAHA.113.000302
40. Gupta S, Bhatia V, Wen -J-J, Wu Y, Huang M-H, Garg NJ. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med. 2009;47:1414–1421. doi:10.1016/j.freeradbiomed.2009.08.008
41. Ba X, Gupta S, Davidson M, Garg NJ. Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes. J Biol Chem. 2010;285:11596–11606.
42. Wen -J-J, Gupta S, Guan Z, et al. Phenyl-alpha-tert-butyl-nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic rats. J Am Coll Cardiol. 2010;55:2499–2508. doi:10.1016/j.jacc.2010.02.030
43. Barroso A, Gualdrón-López M, Esper L, et al. The Aryl Hydrocarbon Receptor modulates production of cytokines and reactive oxygen species and development of myocarditis during Trypanosoma cruzi infection. Infect Immun. 2016;84:3071–3082. doi:10.1128/IAI.00575-16
44. Santos ES, Silva DKC, Dos Reis B, et al. Immunomodulation for the treatment of chronic Chagas disease cardiomyopathy: a new approach to an old enemy. Front Cell Infect Microbiol. 2021;11:765879.
45. Pereira AB, Alvarenga H, Pereira RS, Barbosa MT. Stroke prevalence among the elderly in Vassouras, Rio de Janeiro State, Brazil, according to data from the family health program. Cad Saude Publica. 2009;25:1929–1936. doi:10.1590/S0102-311X2009000900007
46. Paixao LC, Ribeiro AL, Valacio RA, Teixeira AL. Chagas disease: independent risk factor for stroke. Stroke. 2009;40:3691–3694. doi:10.1161/STROKEAHA.109.560854
47. Oliveira-Filho J, Viana LC, Vieira-de-Melo RM, et al. Chagas disease is an independent risk factor for stroke: baseline characteristics of a Chagas Disease cohort. Stroke. 2005;36:2015–2017. doi:10.1161/01.STR.0000177866.13451.e4
48. Leon-Sarmiento FE, Mendoza E, Torres-Hillera M, et al. Trypanosoma cruzi-associated cerebrovascular disease: a case-control study in Eastern Colombia. J Neurol Sci. 2004;217:61–64. doi:10.1016/j.jns.2003.08.015
49. Carod-Artal FJ, Gascon J. Chagas disease and stroke. Lancet Neurol. 2010;9:533–542. doi:10.1016/S1474-4422(10)70042-9
50. Rasche H. Hemostasis and thrombosis: an overview. Eur Heart J Supplements. 2001;3:Q3–Q7. doi:10.1016/S1520-765X(01)90034-3
51. George JN. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; 2006.
52. Bonar RA, Lippi G, Favaloro EJ. Overview of Hemostasis and thrombosis and contribution of laboratory testing to diagnosis and management of hemostasis and thrombosis disorders. Methods Mol Biol. 2017;1646:3–27.
53. Bizzozero G. Su di un nuovo elemento morfologico del sangue dei mammiferi e sulla sua importanza nella trombosi e nella coagulazione. [On a new morphological element of mammalian blood and its importance in thrombosis and coagulation]. L’Osservatore Gazz Clin. 1881;17:785–787. Italian.
54. Swieringa F, Spronk HMH, Heemskerk JWM, van der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. 2018;2:450–460. doi:10.1002/rth2.12107
55. Kwaan H, Lisman T, Medcalf RL. Fibrinolysis: biochemistry, clinical aspects, and therapeutic potential. Semin Thromb Hemost. 2017;43:113–114. doi:10.1055/s-0036-1598000
56. Samuel J, Oliveira M, Correa De Araujo RR, Navarro MA, Muccillo G. Cardiac thrombosis and thromboembolism in chronic Chagas’ heart disease. Am J Cardiol. 1983;52:147–151. doi:10.1016/0002-9149(83)90085-1
57. Arteaga Fernández E, Barreto ACP, Ianni BM, et al. Cardiac thrombosis and embolism in patients who died of chronic chagasic cardiopathy/Cardiac thrombosis and embolism in patients who had died with chronic chagasic cardiopathy. Arq Bras Cardiol. 1989;52:189–192.
58. Herrera RN, Diaz E, Perez R, et al. Estado protrombótico en estadios tempranos de la enfermedad de Chagas crónica. [The prothrombotic state in early stages of chronic chagas’ disease]. Rev Esp Cardiol. 2003;56:377–382. Spanish. doi:10.1016/S0300-8932(03)76881-X
59. Cerqueira-Silva T, Goncalves BM, Pereira CB, et al. Chagas disease is an independent predictor of stroke and death in a cohort of heart failure patients. Int J Stroke. 2022;17:180–188. doi:10.1177/17474930211006284
60. Nunes MC, Kreuser LJ, Ribeiro AL, et al. Prevalence and risk factors of embolic cerebrovascular events associated with Chagas heart disease. Glob Heart. 2015;10:151–157. doi:10.1016/j.gheart.2015.07.006
61. Herrera RN, Berman SG, Luciardi HL. Evidence of a prothrombotic state in early stages of chronic Chagas’ disease. Arch Cardiol Mex. 2004;74:259–261.
62. Melo LM, Souza GE, Valim LR, et al. Study of pro-thrombotic and pro-inflammatory factors in Chagas cardiomyopathy. Arq Bras Cardiol. 2010;95:655–662. doi:10.1590/S0066-782X2010005000146
63. Echeverria LE, Rojas LZ, Gomez-Ochoa SA. Coagulation disorders in Chagas disease: a pathophysiological systematic review and meta-analysis. Thromb Res. 2021;201:73–83. doi:10.1016/j.thromres.2021.02.025
64. Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther. 2010;28:e72–91. doi:10.1111/j.1755-5922.2010.00171.x
65. Pengue C, Cesar G, Alvarez MG, et al. Impaired frequencies and function of platelets and tissue remodeling in chronic Chagas disease. PLoS One. 2019;14:e0218260. doi:10.1371/journal.pone.0218260
66. Pinazo MJ, Posada Ede J, Izquierdo L, et al. Altered hypercoagulability factors in patients with chronic Chagas disease: potential biomarkers of therapeutic response. PLoS Negl Trop Dis. 2016;10:e0004269. doi:10.1371/journal.pntd.0004269
67. Laucella SA, Segura EL, Riarte A, Sosa ES. Soluble platelet selectin (sP-selectin) and soluble vascular cell adhesion molecule-1 (sVCAM-1) decrease during therapy with benznidazole in children with indeterminate form of Chagas’ disease. Clin Exp Immunol. 1999;118:423–427. doi:10.1046/j.1365-2249.1999.01070.x
68. Panicker SR, Mehta-D’souza P, Zhang N, Klopocki AG, Shao B, McEver RP. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood. 2017;130:181–191. doi:10.1182/blood-2017-02-770479
69. Tanowitz HB, Burns ER, Sinha AK, et al. Enhanced platelet adherence and aggregation in Chagas’ disease: a potential pathogenic mechanism for cardiomyopathy. Am J Trop Med Hyg. 1990;43:274–281. doi:10.4269/ajtmh.1990.43.274
70. Ashton AW, Mukherjee S, Nagajyothi FN, et al. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. J Exp Med. 2007;204:929–940. doi:10.1084/jem.20062432
71. Tanowitz HB, Huang H, Jelicks LA, et al. Role of endothelin 1 in the pathogenesis of chronic chagasic heart disease. Infect Immun. 2005;73:2496–2503.
72. Woudstra L, Juffermans LJM, van Rossum AC, Niessen HWM, Krijnen PAJ. Infectious myocarditis: the role of the cardiac vasculature. Heart Fail Rev. 2018;23:583–595. doi:10.1007/s10741-018-9688-x
73. Frasch AC. Functional diversity in the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol Today. 2000;16:282–286. doi:10.1016/S0169-4758(00)01698-7
74. Dias WB, Fajardo FD, Graca-Souza AV, et al. Endothelial cell signalling induced by trans-sialidase from Trypanosoma cruzi. Cell Microbiol. 2008;10:88–99. doi:10.1111/j.1462-5822.2007.01017.x
75. Tribulatti MV, Mucci J, Van Rooijen N, Leguizamon MS, Campetella O. The trans-sialidase from Trypanosoma cruzi induces thrombocytopenia during acute Chagas’ disease by reducing the platelet sialic acid contents. Infect Immun. 2005;73:201–207. doi:10.1128/IAI.73.1.201-207.2005
76. Berra HH, Piaggio E, Revelli SS, Luquita A. Blood viscosity changes in experimentally Trypanosoma cruzi-infected rats. Clin Hemorheol Microcirc. 2005;32:175–182.
77. Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. 2017;130:1795–1799. doi:10.1182/blood-2017-03-745349
78. Zelaya H, Rothmeier AS, Ruf W. Tissue factor at the crossroad of coagulation and cell signaling. J Thromb Haemost. 2018;16:1941–1952. doi:10.1111/jth.14246
79. Ferreira RC, Ianni BM, Abel LC, et al. Increased plasma levels of tumor necrosis factor-alpha in asymptomatic/”indeterminate” and Chagas disease cardiomyopathy patients. Mem Inst Oswaldo Cruz. 2003;98:407–411. doi:10.1590/S0074-02762003000300021
80. Lopez L, Arai K, Gimenez E, et al. C-reactive protein and interleukin-6 serum levels increase as Chagas disease progresses towards cardiac failure. Rev Esp Cardiol. 2006;59:50–56.
81. Choudhuri S, Garg NJ, Sacks D. PARP1-cGAS-NF-κB pathway of proinflammatory macrophage activation by extracellular vesicles released during Trypanosoma cruzi infection and Chagas disease. PLoS Pathog. 2020;16:e1008474. doi:10.1371/journal.ppat.1008474
82. Choudhuri S, Garg NJ, Hoft DF, Koshy AA. Trypanosoma cruzi induces the PARP1/AP-1 pathway for upregulation of metalloproteinases and transforming growth factor β in macrophages: role in cardiac fibroblast differentiation and fibrosis in Chagas disease. mBio. 2020;11. doi:10.1128/mBio.01853-20
83. Tanowitz HB, Gumprecht JP, Spurr D, et al. Cytokine gene expression of endothelial cells infected with Trypanosoma cruzi. J Infect Dis. 1992;166:598–603. doi:10.1093/infdis/166.3.598
84. Huang H, Calderon TM, Berman JW, et al. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect Immun. 1999;67:5434–5440. doi:10.1128/IAI.67.10.5434-5440.1999
85. Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost. 2005;3:1590–1596. doi:10.1111/j.1538-7836.2005.01373.x
86. Page MJ, Bester J, Pretorius E. Interleukin-12 and its procoagulant effect on erythrocytes, platelets and fibrin(ogen): the lesser known side of inflammation. Br J Haematol. 2018;180:110–117. doi:10.1111/bjh.15020
87. Bester J, Pretorius E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep. 2016;6:32188. doi:10.1038/srep32188
88. Lavine MD, Strand MR. Insect hemocytes and their role in immunity. Insect Biochem Mol Biol. 2002;32:1295–1309. doi:10.1016/S0965-1748(02)00092-9
89. Ludwig N, Hilger A, Zarbock A, Rossaint J. Platelets at the crossroads of pro-inflammatory and resolution pathways during inflammation. Cells. 2022;11:1957. doi:10.3390/cells11121957
90. Zamora C, Canto E, Vidal S. The dual role of platelets in the cardiovascular risk of chronic inflammation. Front Immunol. 2021;12:625181. doi:10.3389/fimmu.2021.625181
91. Mantovani A, Garlanda C. Platelet-macrophage partnership in innate immunity and inflammation. Nat Immunol. 2013;14:768–770. doi:10.1038/ni.2666
92. Portier I, Campbell RA. Role of platelets in detection and regulation of infection. Arterioscler Thromb Vasc Biol. 2021;41:70–78. doi:10.1161/ATVBAHA.120.314645
93. May F, Hagedorn I, Pleines I, et al. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood. 2009;114:3464–3472.
94. Hally K, Fauteux-Daniel S, Hamzeh-Cognasse H, Larsen P, Cognasse F. Revisiting platelets and toll-like receptors (TLRs): at the interface of vascular immunity and thrombosis. Int J Mol Sci. 2020;21:6150. doi:10.3390/ijms21176150
95. Carestia A, Mena HA, Olexen CM, et al. Platelets Promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep. 2019;28:896–908 e5. doi:10.1016/j.celrep.2019.06.062
96. Patsouras MD, Sikara MP, Grika EP, Moutsopoulos HM, Tzioufas AG, Vlachoyiannopoulos PG. Elevated expression of platelet-derived chemokines in patients with antiphospholipid syndrome. J Autoimmun. 2015;65:30–37. doi:10.1016/j.jaut.2015.08.001
97. Vettori S, Irace R, Riccardi A, et al. Serum CXCL4 increase in primary Sjogren’s syndrome characterizes patients with microvascular involvement and reduced salivary gland infiltration and lymph node involvement. Clin Rheumatol. 2016;35:2591–2596. doi:10.1007/s10067-016-3386-7
98. Kasper B, Brandt E, Brandau S, Petersen F. Platelet factor 4 (CXC chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J Immunol. 2007;179:2584–2591. doi:10.4049/jimmunol.179.4.2584
99. Suzuki J, Hamada E, Shodai T, et al. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int Arch Allergy Immunol. 2013;160:152–160. doi:10.1159/000339857
100. Vogel S, Rath D, Borst O, et al. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem Biophys Res Commun. 2016;478:143–148. doi:10.1016/j.bbrc.2016.07.078
101. Gudbrandsdottir S, Hasselbalch HC, Nielsen CH. Activated platelets enhance IL-10 secretion and reduce TNF-alpha secretion by monocytes. J Immunol. 2013;191:4059–4067. doi:10.4049/jimmunol.1201103
102. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15:384–391. doi:10.1038/nm.1939
103. Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. 2016;7:271. doi:10.3389/fimmu.2016.00271
104. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi:10.1038/nm1565
105. Jenne CN, Wong CH, Zemp FJ, et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. 2013;13:169–180. doi:10.1016/j.chom.2013.01.005
106. Kraemer BF, Campbell RA, Schwertz H, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. 2011;7:e1002355. doi:10.1371/journal.ppat.1002355
107. Hurley SM, Lutay N, Holmqvist B, Shannon O. The dynamics of platelet activation during the progression of Streptococcal sepsis. PLoS One. 2016;11:e0163531. doi:10.1371/journal.pone.0163531
108. Vajen T, Koenen RR, Werner I, et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci Rep. 2018;8:10647. doi:10.1038/s41598-018-29026-0
109. Mulet M, Zamora C, Porcel JM, et al. Platelet factor 4 regulates T cell effector functions in malignant pleural effusions. Cancer Lett. 2020;491:78–86. doi:10.1016/j.canlet.2020.06.014
110. Shi G, Field DJ, Ko KA, et al. Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J Clin Invest. 2014;124:543–552. doi:10.1172/JCI71858
111. Jenabian MA, Patel M, Kema I, et al. Soluble CD40-ligand (sCD40L, sCD154) plays an immunosuppressive role via regulatory T cell expansion in HIV infection. Clin Exp Immunol. 2014;178:102–111. doi:10.1111/cei.12396
112. Burzynski LC, Humphry M, Pyrillou K, et al. The coagulation and immune systems are directly linked through the activation of interleukin-1alpha by thrombin. Immunity. 2019;50:1033–42 e6. doi:10.1016/j.immuni.2019.03.003
113. Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood. 2010;115:3632–3639. doi:10.1182/blood-2009-11-252643
114. Rachidi S, Metelli A, Riesenberg B, et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol. 2017;2. doi:10.1126/sciimmunol.aai7911
115. Zhu L, Huang Z, Stalesen R, Hansson GK, Li N. Platelets provoke distinct dynamics of immune responses by differentially regulating CD4+ T-cell proliferation. J Thromb Haemost. 2014;12:1156–1165. doi:10.1111/jth.12612
116. Zamora C, Canto E, Nieto JC, et al. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol. 2013;94:521–529. doi:10.1189/jlb.0213074
117. de Andrade MF, de Almeida VD, de Souza LMS, Paiva DCC, Andrade CM, De Medeiros Fernandes TAA. Involvement of neutrophils in Chagas disease pathology. Parasite Immunol. 2018;40:e12593. doi:10.1111/pim.12593
118. Koo SJ, Chowdhury IH, Szczesny B, Wan X, Garg NJ. Macrophages promote oxidative metabolism to drive nitric oxide generation in response to Trypanosoma cruzi. Infect Immun. 2016;84:3527–3541. doi:10.1128/IAI.00809-16
119. Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose phosphate shunt modulates reactive oxygen species and nitric oxide production controlling Trypanosoma cruzi in macrophages. Front Immunol. 2018;9:202. doi:10.3389/fimmu.2018.00202
120. Koo SJ, Garg NJ. Metabolic programming of macrophage functions and pathogens control. Redox Biol. 2019;24:101198. doi:10.1016/j.redox.2019.101198
121. Choudhuri S, Chowdhury IH, Garg NJ. Mitochondrial regulation of macrophage response against pathogens. Front Immunol. 2020;11:622602. doi:10.3389/fimmu.2020.622602
122. Sanchez-Villamil JP, Bautista-Nino PK, Serrano NC, Rincon MY, Garg NJ. Potential role of antioxidants as adjunctive therapy in Chagas disease. Oxid Med Cell Longev. 2020;2020:9081813. doi:10.1155/2020/9081813
123. Maldonado E, Rojas DA, Urbina F, Solari A. The oxidative stress and chronic inflammatory process in Chagas disease: role of exosomes and contributing genetic factors. Oxid Med Cell Longev. 2021;2021:4993452. doi:10.1155/2021/4993452
124. Wan X, Garg NJ. Sirtuin Control of mitochondrial dysfunction, oxidative stress, and inflammation in Chagas disease models. Front Cell Infect Microbiol. 2021;11:693051. doi:10.3389/fcimb.2021.693051
125. Keating SM, Deng X, Fernandes F, et al. Inflammatory and cardiac biomarkers are differentially expressed in clinical stages of Chagas disease. Int J Cardiol. 2015;199:451–459. doi:10.1016/j.ijcard.2015.07.040
126. Cunha-Neto E, Teixeira PC, Fonseca SG, Bilate AM, Kalil J. Myocardial gene and protein expression profiles after autoimmune injury in Chagas’ disease cardiomyopathy. Autoimmun Rev. 2011;10:163–165. doi:10.1016/j.autrev.2010.09.019
127. Garg NJ, Soman KV, Zago MP, et al. Changes in Proteome profile of peripheral blood mononuclear cells in chronic Chagas disease. PLoS Negl Trop Dis. 2016;10:e0004490. doi:10.1371/journal.pntd.0004490
128. Ferreira LR, Ferreira FM, Nakaya HI, et al. Blood Gene signatures of Chagas disease cardiomyopathy with or without ventricular dysfunction. J Infect Dis. 2016;214:161–165. doi:10.1093/infdis/jiw095
129. Souza PE, Rocha MO, Menezes CA, et al. Trypanosoma cruzi infection induces differential modulation of costimulatory molecules and cytokines by monocytes and T cells from patients with indeterminate and cardiac Chagas’ disease. Infect Immun. 2007;75:1886–1894. doi:10.1128/IAI.01931-06
130. Souza PE, Rocha MO, Rocha-Vieira E, et al. Monocytes from patients with indeterminate and cardiac forms of Chagas’ disease display distinct phenotypic and functional characteristics associated with morbidity. Infect Immun. 2004;72:5283–5291. doi:10.1128/IAI.72.9.5283-5291.2004
131. Machado FS, Dutra WO, Esper L, et al. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin Immunopathol. 2012;34:753–770. doi:10.1007/s00281-012-0351-7
132. Wan X, Chowdhury IH, Jie Z, Choudhuri S, Garg NJ. Origin of monocytes/macrophages contributing to chronic inflammation in Chagas disease: SIRT1 inhibition of FAK-NFκB-dependent proliferation and proinflammatory activation of macrophages. Cells. 2019;9:80. doi:10.3390/cells9010080
133. Nogueira PM, Ribeiro K, Silveira AC, et al. Vesicles from different Trypanosoma cruzi strains trigger differential innate and chronic immune responses. J Extracell Vesicles. 2015;4:28734. doi:10.3402/jev.v4.28734
134. Ribeiro KS, Vasconcellos CI, Soares RP, et al. Proteomic analysis reveals different composition of extracellular vesicles released by two Trypanosoma cruzi strains associated with their distinct interaction with host cells. J Extracell Vesicles. 2018;7:1463779. doi:10.1080/20013078.2018.1463779
135. Yanez-Mo M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066. doi:10.3402/jev.v4.27066
136. Takasugi M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell. 2018;17:e12734. doi:10.1111/acel.12734
137. Ramirez MI, Deolindo P, de Messias-Reason IJ, et al. Dynamic flux of microvesicles modulate parasite-host cell interaction of Trypanosoma cruzi in eukaryotic cells. Cell Microbiol. 2017;19:e12672. doi:10.1111/cmi.12672
138. Cestari I, Ansa-Addo E, Deolindo P, Inal JM, Ramirez MI. Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J Immunol. 2012;188:1942–1952. doi:10.4049/jimmunol.1102053
139. Chowdhury I, Koo S, Gupta S, et al. Gene expression profiling and functional characterization of macrophages in response to circulatory microparticles produced during Trypanosoma cruzi infection and Chagas disease. J Innate Immunity. 2017;9:203–216. doi:10.1159/000451055
140. Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014;329:18–25. doi:10.1016/j.yexcr.2014.07.003
141. Wen JJ, Yin YW, Garg NJ. PARP1 depletion improves mitochondrial and heart function in Chagas disease: effects on POLG dependent mtDNA maintenance. PLoS Pathog. 2018;14:e1007065. doi:10.1371/journal.ppat.1007065
142. Wang L, Wu Q, Fan Z, Xie R, Wang Z, Lu Y. Platelet mitochondrial dysfunction and the correlation with human diseases. Biochem Soc Trans. 2017;45:1213–1223. doi:10.1042/BST20170291
143. Fidler TP, Campbell RA, Funari T, et al. Deletion of GLUT1 and GLUT3 reveals multiple roles for glucose metabolism in platelet and megakaryocyte function. Cell Rep. 2017;20:881–894. doi:10.1016/j.celrep.2017.06.083
144. Granville DJ, Gottlieb RA. Mitochondria: regulators of cell death and survival. Scientific World J. 2002;2:1569–1578. doi:10.1100/tsw.2002.809
145. Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim Biophys Acta. 2011;1813:1316–1322. doi:10.1016/j.bbamcr.2011.01.031
146. Melchinger H, Jain K, Tyagi T, Hwa J. Role of platelet mitochondria: life in a nucleus-free zone. Front Cardiovasc Med. 2019;6:153. doi:10.3389/fcvm.2019.00153
147. Garcia-Souza LF, Oliveira MF. Mitochondria: biological roles in platelet physiology and pathology. Int J Biochem Cell Biol. 2014;50:156–160. doi:10.1016/j.biocel.2014.02.015
148. Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2012;32:2946–2955. doi:10.1161/ATVBAHA.112.300433
149. Sinauridze EI, Kireev DA, Popenko NY, et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. 2007;97:425–434. doi:10.1160/TH06-06-0313
150. Kohler C, Radpour R, Barekati Z, et al. Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol Cancer. 2009;8:105. doi:10.1186/1476-4598-8-105
151. Sudakov NP, Popkova TP, Katyshev AI, et al. Level of blood cell-free circulating mitochondrial DNA as a novel biomarker of acute myocardial ischemia. Biochemistry. 2015;80:1387–1392. doi:10.1134/S000629791510020X
152. Malik AN, Parsade CK, Ajaz S, et al. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Res Clin Pract. 2015;110:257–265. doi:10.1016/j.diabres.2015.10.006
153. Meddeb R, Dache ZAA, Thezenas S, et al. Quantifying circulating cell-free DNA in humans. Sci Rep. 2019;9:5220. doi:10.1038/s41598-019-41593-4
154. Al Amir Dache Z, Otandault A, Tanos R, et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020;34:3616–3630. doi:10.1096/fj.201901917RR
155. Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. 2018;9:832. doi:10.3389/fimmu.2018.00832
156. Rodriguez-Nuevo A, Zorzano A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress. 2019;3:195–207. doi:10.15698/cst2019.06.190
157. Puhm F, Afonyushkin T, Resch U, et al. Mitochondria Are a subset of extracellular vesicles released by activated monocytes and induce type I IFN and TNF responses in endothelial cells. Circ Res. 2019;125:43–52. doi:10.1161/CIRCRESAHA.118.314601
158. Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–2183. doi:10.1182/blood-2014-05-573543
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
An Exploration of Anti-Inflammatory Therapy in Acute/Subacute Severe Cerebral Venous Thrombosis with Hereditary Protein C/S Deficiency: Case Series
Hao W, Gu Y, Hu S, Ji X, Wang Y, Duan J
Journal of Inflammation Research 2023, 16:5403-5415
Published Date: 21 November 2023
A Microflow Chip Technique for Monitoring Platelets in Late Pregnancy: A Possible Risk Factor for Thrombosis
He C, Ma H, Zhang T, Liu Y, Zhang C, Deng S
Journal of Blood Medicine 2025, 16:15-25
Published Date: 8 January 2025
The Association of Systemic Immune Inflammation Index (SII) and Platelet-to-Lymphocyte Ratio (PLR) on Coagulopathy and Prognosis in Patients with Traumatic Brain Injury
Chen J, Fu J, Liu J, Lu Y, Han D, Zeng J, Zou Z, Li Q, Zhang K, Wei X, Li L, Gu Z
Journal of Inflammation Research 2025, 18:5637-5653
Published Date: 25 April 2025
Clear Cell Ovarian Carcinoma and Its Distinct Coagulopathy Profile: Molecular Drivers and Clinical Implications
Obeagu EI
Cancer Management and Research 2025, 17:2459-2467
Published Date: 27 October 2025
Pulmonary Exposure to Copper Oxide Nanoparticles Induces Systemic Inflammation, Oxidative Stress, and Prothrombotic Responses in BALB/c Mice
Ferdous Z, Beegam S, Zaaba NE, Elzaki O, Greish YE, Nemmar A
International Journal of Nanomedicine 2026, 21:579331
Published Date: 16 June 2026