Back to Journals » Journal of Inflammation Research » Volume 16
An Exploration of Anti-Inflammatory Therapy in Acute/Subacute Severe Cerebral Venous Thrombosis with Hereditary Protein C/S Deficiency: Case Series
Authors Hao W, Gu Y, Hu S, Ji X, Wang Y, Duan J
Received 6 August 2023
Accepted for publication 18 October 2023
Published 21 November 2023 Volume 2023:16 Pages 5403—5415
DOI https://doi.org/10.2147/JIR.S428589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Wensi Hao,1,2 Yaqin Gu,1,2 Shuyuan Hu,1,2 Xunming Ji,1,2 Yuping Wang,2 Jiangang Duan1,2
1Department of Emergency, Xuanwu Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Jiangang Duan, Department of Emergency, Xuanwu Hospital, Capital Medical University, No. 45, Changchun St, Xicheng District, Beijing, 100053, People’s Republic of China, Email [email protected]
Background: Inflammation was associated with the severity of severe cerebral venous thrombosis (CVT) on admission and poor prognosis at discharge. Hereditary protein C/S deficiency (hereditary PCD/PSD) not only promotes thrombosis but also activates the inflammatory response, further inducing venous thrombosis. However, conventional treatments such as standard anticoagulant/endovascular therapy (EVT) do not seem to improve prognosis. Anti-inflammatory therapy may be a new way to treat the disease.
Methods: We enrolled five patients with acute/subacute severe CVT with hereditary PCD/PSD from January 2020 to July 2022. In addition to standard anticoagulant therapy, all of them were given short-term methylprednisolone pulse therapy. Neurological deficit, increased intracranial pressure, venous recanalization, serum and cerebrospinal fluid (CSF) inflammatory markers and adverse events were retrospectively described before and after treatment and at 6 months after discharge.
Results: Inflammatory indexes of all patients were significantly elevated on admission. After methylprednisolone pulse therapy, serum inflammatory indexes including neutrophil-to-lymphocyte ratio (P=0.043); platelet-to-lymphocyte ratio (P=0.043); systemic immune inflammatory index (P=0.043); interleukin-6 (P=0.043) and hypersensitive C-reactive protein (P=0.022) reduced dramatically compared with baseline. CSF inflammatory indexes had a decreasing trend compared with baseline (P> 0.05). In terms of venous recanalization, one patient achieved complete recanalization, four patients obtained partial recanalization. Compared with baseline on admission, the NIH Stroke Scale (NIHSS), modified Rankin Scale (mRS) and intracranial pressure were all considerably lower at discharge (P=0.029, P=0.041 and P=0.017). At 6-month follow-up, NIHSS and mRS further declined. During hospitalization and 6-month follow-up, none of the five patients experienced severe steroid-related adverse effects such as recurrence of venous thrombosis, spontaneous fracture or osteonecrosis, and gastroduodenal ulcer.
Conclusion: Acute/subacute severe CVT with hereditary PCD/PSD has high levels of inflammation. In addition to conventional anticoagulant therapy, early anti-inflammatory therapy using steroids may be necessary. Nevertheless, substantial randomized controlled trials with larger sample sizes are required for further investigation.
Keywords: severe cerebral venous thrombosis, hereditary protein C deficiency, hereditary protein S deficiency, thrombosis, inflammation, methylprednisolone pulse therapy
Introduction
Severe cerebral venous thrombosis (CVT), which presents as cerebral venous infarction or hemorrhage, seizures, or disturbance of consciousness, has more severe clinical manifestations and a worse prognosis.1,2 Patients with severe CVT who also have hereditary protein C and protein S deficiency (hereditary PCD/PSD) have a substantially worse prognosis and an increased risk of distant seizures.3–5 However, in addition to conventional anticoagulant therapy, endovascular therapy (EVT) consisting of mechanical thrombectomy, and local pharmacological thrombolysis fails to achieve good curative effect for severe CVT.6,7
More and more studies have demonstrated that inflammatory markers such as neutrophil-to-lymphocyte ratio (NLR),8 systemic immune inflammatory index (SII),9 hypersensitive C-reactive protein (hs-CRP)10,11 and interleukin-6 (IL-6)10 were associated with the severity of CVT on admission and poor prognosis at discharge. As risk factors for CVT, hereditary PCD/PSD not only weakened the anticoagulant function, leading to thrombosis, but also decreased the anti-inflammatory effect, resulting in inflammatory reaction.12,13 Inflammation contributes to thrombosis,14 while thrombosis itself stimulates the production of pro-inflammatory mediators.15 Our team’s previous research10,11,16 indicated that inflammatory response is closely related to the pathophysiology of severe CVT, suggesting that anti-inflammatory therapy using steroids, may be an effective treatment for this disease.
Glucocorticoids have the effect of anti-inflammation and reducing edema, which can delay the progression of stroke,17 while the application in CVT has been controversial. The International Study on Cerebral Veins and Dural Sinus Thrombosis (ISCVT) pointed out that glucocorticoids did not improve the prognosis of acute CVT patients and were even detrimental in CVT without parenchymal lesions.18 This conclusion has been cited by definitive guidelines,5,19,20 but is based on low-level evidence, suggesting that more high-level studies are needed to determine whether glucocorticoids are suitable for the management of acute/subacute severe CVT.
We collected five acute/subacute severe CVT patients with hereditary PCD/PSD accompanied by PROC/PROS1 mutations and innovatively treated them with methylprednisolone based on anticoagulant therapy. Changes in clinical symptoms, levels of inflammatory factors, imaging characteristics, neurological deficits and intracranial pressure were described at baseline, after treatment and at the 6-month follow-up, respectively. We initially investigated the effect of glucocorticoid anti-inflammatory therapy combined with anticoagulant therapy in acute/subacute severe CVT with hereditary PCD/PSD.
Materials and Method
Selection of the Study Patients
We enrolled five patients diagnosed with acute/subacute severe CVT with hereditary PCD/PSD at Xuanwu Hospital from January 2020 to July 2022. Informed consent was obtained from all patients, and this study was approved by the Ethics Committee of Xuanwu Hospital Capital Medical University.
Inclusion Criteria
The diagnosis of severe CVT following the guidelines5,19,20 was defined as a CVT patient with seizures, coma, or thrombosis of a straight sinus with one or more parenchymal lesions such as infarction, hemorrhage, or edema.2,6 Acute phase (0 −7 days), subacute phase (8–15 days) and chronic phase (>15 days) of CVT were confirmed based on the interval between symptom onset and admission.5 Furthermore, the presence of thrombi signals on T1 or T2-weighted sequences, contrast-enhanced MRI/MRV and MRBTI indicated that the patient was in the acute/subacute phase.21,22 Nevertheless, early manifestations of CVT might be difficult to detect, and MR images sometimes reveal both acute and subacute thrombotic findings in the same individual. Therefore, this study did not distinguish between patients with acute and subacute CVT.
The diagnosis of hereditary PCD/PSD was determined by clinical biochemical parameters on admission and genetic analysis of the proband and related family members.23 Clinical biochemical parameters include blood routine, coagulation function, plasma PC activity (reference range 70–140%) and PS activity (reference range 75–130%). Patients with PC/PS levels below the usual low values and their family numbers were screened for responsible gene mutations using second-generation whole-exome sequencing.
Exclusion Criteria
Patients will be eliminated if they have the following conditions: (1) malignant tumor; (2) severe liver or kidney failure; (3) decreased protein C/S levels due to warfarin.
Data Collection
Baseline data of all patients were collected on admission including demographic characteristics, the interval from symptom onset to admission, clinical manifestations (eg, coma, headache, vomiting, and visual disturbances, seizure, motor or sensory deficit, and aphasiaetc.), past medical histories (eg pregnancy, puerperium, systemic diseases, infection, medication use, etc.), laboratory tests (eg PC activity, PS activity, etc.) and brain imaging characteristics such as brain CT/MRV/MRI and so on. We collected serum inflammatory markers (eg white blood cell (WBC) count, NLR, platelet-to-lymphocyte ratio (PLR), SII, defined as follows: Platelet (/L) × neutrophils (/L)/lymphocytes (/L), hs-CRP, IL-6, IL-8, etc.) and the levels of IgA, IgM, IgG and interleukin in CSF on admission and at 1 week after steroid therapy. We assessed the severity of clinical symptoms and prognosis on admission, at discharge and at 6-month follow-up using the NIH Stroke Scale (NIHSS) and the modified Rankin Scale (mRS). Intracranial pressure and fundus photographs were also taken on admission, at discharge and at 6-month follow-up, respectively. As an efficacy endpoint, we assessed cerebral venous recanalization, measured by the change from baseline to repeat neuroimaging at the end of the treatment and at 6-month follow-up using the Modified Qureshi Scale,24 which was classified as no, partial, or complete recanalization according to the proposed criteria. In addition, recurrence of venous thrombosis, occurrence of venous thrombosis in other parts, and serious steroid-related complications (eg infection, spontaneous fracture or osteonecrosis, and gastroduodenal ulcer, etc.25) were recorded at discharge and during the 6-month follow-up period.
Genetic Testing Protocol
Peripheral blood (3–5 mL) was obtained from the proband and each family member using an ethylenediaminetetraacetic acid (EDTA) anticoagulant tube, and genomic DNA was extracted following the DNA extraction kit (QIAamp DNA Blood MiniKit, Qiagen). DNA libraries were established based on the Illumina protocol. Sequence capture was performed using a Roche liquid chip (NimbleGen Sequence Capture chip, Roche). Pooled libraries were hybridized for 16–20 h at 47°C, followed by elution and purification. The enriched libraries were then amplified by polymerase chain reaction (PCR). Targeted sequencing by Illumina HiSeq X10 High-throughput sequencer was performed to detect mutations in the PROC and PROS1 genes. Illumina Pipeline software (version 1.3.4) was used to analyze the data, and Hg19 and GRCh37 were used as reference gene banks.26
Treatment Protocol
All patients began receiving standardized anticoagulant therapy immediately after diagnosis according to the guideline:5,20 subcutaneous injection of low molecular weight heparin (LMWH). Four patients (4/5) were treated with glucocorticoids (500 mg methylprednisolone once a day, intravenous drip for 3–5 days, then reduce the 80mg once a day, intravenous drip for 3–5 days, next changed to methylprednisolone/prednisone oral 1 mg/kg body weight, then reduced by 8 mg/10 mg per week until withdrawal). Only 1 patient (1/5) was referred to our ward after 16 days of mechanical thrombectomy in ICU without improvement in symptoms, where strength of the thrombus signal indicated that she was still in the acute/subacute stage of the disease. In our ward admission, she was still lethargic and could not walk independently. We gave her 80mg of methylprednisolone for 5 days and then switched to oral steroid therapy and then gradually reduced dose as previously project. In addition, one patient underwent decompressive craniectomy, and all patients were administered mannitol dehydration/antiepileptic therapy (Details are given in Table 1).
 |
Table 1 Clinical Characteristics of five Patients with Severe CVT During Admission |
Follow-Up and Clinical Outcome
Follow-up visits were performed at 6 months after discharge through clinical outpatient visits. NIHSS and mRS at discharge served as the primary efficacy endpoints. NIHSS and mRS at 6-month follow-up, modified Frisén Grades and venous recanalization were used as secondary efficacy endpoints. Recurrence of CVT or venous thrombosis at other locations during hospitalization and 6-month follow-up were the primary safety endpoints. Severe glucocorticoid complications within 6 months follow-up, such as spontaneous fracture or osteonecrosis, induced or worsened infection, induced or aggravated gastroduodenal ulcer, and so on, were secondary safety endpoints.
Statistical Analysis
Descriptive statistics and summary tables were used to summarize the data. All the data were analyzed with SPSS 25.0 (SPSS Inc., Chicago, IL). The normality of continuous variables was evaluated using Shapiro-Wilk test and the homogeneity of variance was also analyzed. Quantitative variables with a normal distribution were specified as mean±standard deviation, while quantitative variables with an abnormal distribution were presented as medians (interquartile range, IQR), categorical variables were presented as numbers (percentages). When quantitative variables met a normal distribution and homogeneous variance, paired t-test was used to compare whether there was a statistical difference before and after methylprednisolone therapy, otherwise, the Wilcoxon test was used. P < 0.05 was considered to be a statistical difference for all tests.
Results
Clinical Characteristics
Five patients with acute/subacute severe CVT with hereditary PCD/PSD from January 2020 to July 2022 were recruited. Two patients were female, and three patients were male, with a median age of 28 years (24, 34). The mean time from onset of symptoms to steroid therapy was 10.60±8.17 days. On admission, 60% (3/5) had generalized tonic-clonic seizures, 80% (4/5) were in coma. In the previous history, one patient had a history of using oral contraceptives, one patient was pregnant, and one patient had a history of venous sinus thrombosis and intermittent use of warfarin. All patients had cerebral venous infarction or hemorrhage. Superior sagittal sinus was most frequently involved (5/5, 100%), followed by transverse sinus (4/5, 80%), and 2 patients (40%) had straight sinus involvement (Details are given in Table 1).
PC/PS Levels and Sequence Map of PROC/PROS1 Gene Mutation in the Patients and Their Families
Three patients had decreased levels of plasma PC, while all patients had decreased levels of plasma PS. Thrombophilia gene screening results showed that 40% (2/5) have PROC gene mutations and 60% (3/5) possessed PROS1 gene mutations. Four patients’ fathers had heterozygous mutations at the same locus in the same gene, whereas one patient’s mother had the same mutation (Details are given in Table 2).
 |
Table 2 PC/PS Levels and Sequence Map in Patients and Their Families |
Changes of Inflammatory Indexes in Serum and CSF Before and After Steroid Therapy
We collected five patients’ serum and four patients’ CSF inflammatory indexes on admission and at discharge. Serum inflammatory indexes were significantly elevated in all patients. After steroid therapy, serum inflammatory indexes NLR (medians 12.39 vs 3.28, P=0.043); PLR (medians 327.59 vs 81.03, P=0.043); SII (medians 4707.41 vs 768.6, P=0.043); IL-6 (medians 30.64 vs 2.71, P=0.043) and hs-CRP (mean 28.26 vs 0.96, P=0.022) reduced dramatically compared with baseline. CSF inflammatory indexes decreased compared with baseline, but there was no statistical difference (Details are given in Figure 1 and Table 3).
 |
Table 3 Levels of Inflammatory Indexes in CSF Before and After Steroid Therapy |
 |
Figure 1 Levels of inflammatory indexes in serum before and after steroid therapy. (*P<0.05). |
Neurological Impairment and Increased Intracranial Pressure Improved Significantly at Discharge and 6-Month Follow-Up
Compared with baseline, NIHSS (mean 24.60 vs 2.40, P=0.029) and mRS (medians 5 vs 1, P=0.041) of all patients were significantly decreased at discharge. At 6-month follow-up, the neurological impairments of all patients further improved. NIHSS (mean 24.60 vs 0.80, P=0.031) and mRS (medians 5 vs 0, P=0.025) were decreased substantially (Details are given in Figure 2).
 |
Figure 2 Changes in neurological impairment and intracranial pressure at discharge and 6-month follow-up. (*P<0.05). |
Four patients received lumbar punctures on admission and at discharge. Compared with baseline, intracranial pressure (mean 348.75 vs 222.5, P=0.017) at discharge had decreased significantly. At 6-month follow-up, no patient received lumbar puncture to measure intracranial pressure. On admission, four patients underwent fundus examinations with grades 1 (3/4) and 5 (1/4) on the modified Frisén scale, whereas 1 patient with critical condition did not have examinations. Four patients received again fundus examination at discharge with Frisén grades 0 (3/4) and 4 (1/4), indicating that intracranial pressure decreased compared with baseline (Frisén grades, P=0.046). Frisén grade 0 was seen in three patients at 1-month, 3-month and 6-month follow-up, respectively, indicating continued decrease of intracranial pressure (Details are given in Figure 2).
Venous Sinus Vessels Recanalization at Discharge and 6-Month Follow-Up
We compared each patient’s venous sinus recanalization at discharge, as measured by the Modified Qureshi scale. The result showed that four patients obtained partial recanalization (grade I) and one patient achieved complete recanalization (grade II) at discharge. At 3-month follow-up, three patients completed imaging examinations, and imaging data revealed that two patients had complete recanalization and one had partial recanalization compared to pre-steroid therapy. Furthermore, two patients had complete recanalization at 6-month follow-up.
Safety Evaluation
During steroid therapy and 6 months after discharge, none of the five patients experienced thrombosis recurrence or thrombosis in other locations. And no adverse effects (eg infection induced or exacerbated, spontaneous fracture or osteonecrosis, and gastroduodenal ulcer, etc.) associated with steroid therapy were observed.
Typical Imaging Changes
We included the imaging changes of two typical cases: one patient suffered from severe CVT with hereditary PCD (Figure 3) and the other suffered from severe CVT with hereditary PSD (Figure 4). Detailed clinical data are shown in Table 1. On admission, the patients presented with cerebral hemorrhagic infarction and severe CVT imaging changes. Following conventional anticoagulant therapy, there were no significant imaging changes. On this basis, after the addition of steroid anti-inflammatory therapy, the area of cerebral hemorrhage infarction and brain edema were alleviated, and venous sinus thrombosis was also reduced to varying degrees. Furthermore, neither cerebral hemorrhage infarction nor recurrence of venous thrombosis occurred during the patients’ follow-up.
 |
Figure 3 Case 2: (A) Brain CT on admission indicated new cerebral hemorrhagic infarction in the right frontoparietotemporal lobe with obvious local edema and right cerebral hernia. (B) MRBTI on admission displayed superior sagittal sinus thrombosis. (C) After methylprednisolone therapy, compared with A, brain CT demonstrated that the edema and hemorrhage area of the right frontoparietotemporal lobe were significantly reduced and the left shift of midline structure alleviated. (D) After methylprednisolone therapy, compared with (B), MRBTI showed the thrombus in superior sagittal sinus was reduced. (E) At 3-month follow-up, compared with (C), brain CT showed the edema of the right frontoparietotemporal lobe disappeared and only a few previous infarcts remained. (F) At 3-month follow-up, CE-MRV indicated a considerable decrease of thrombus in the superior sagittal sinus without new thrombosis in comparison to (D). (G) At 6-month follow-up, compared with (E), Axial T2-MRI revealed no significant changes in right frontoparietotemporal lobe and no new infarcts were detected. (H) At 6-month follow-up, MRBTI demonstrated a further reduction of superior sagittal sinus thrombus compared to (F), leaving a negligible quantity of residual thrombus. Abbreviations: MRBTI, magnetic resonance black-blood thrombus imaging; MRI, magnetic resonance imaging; CE-MRV, contrast-enhanced magnetic resonance venography; CT, computerized tomography. The red arrows indicate the lesion. |
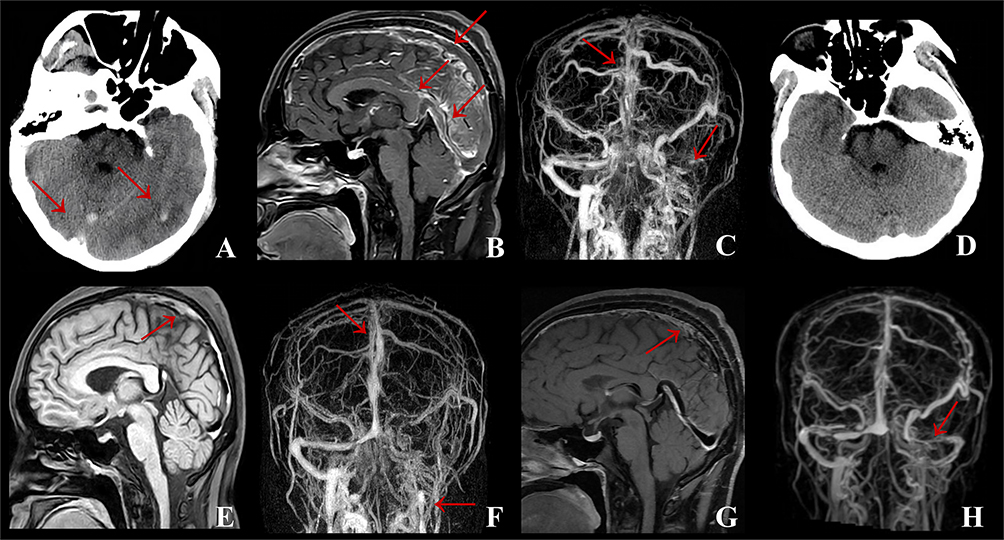 |
Figure 4 Case 4: (A) Brain CT on admission showed the thrombosis of straight sinus and left temporal-occipital lobe cerebral infarction with hemorrhage. (B) CE-MRBTI on admission revealed thrombosis occurred in the vein of Galen, straight sinus, superior sagittal sinus and torcular herophili. (C) CE-MRV on admission displayed that thrombosis was involved in the superior sagittal sinus, torcular herophili, left transverse sinus, sigmoid sinus and internal jugular vein. (D) After methylprednisolone therapy, brain CT demonstrated the straight sinus thrombosis and left temporaloccipital lobe cerebral infarction with hemorrhage disappeared. (E) After methylprednisolone therapy, MRBTI showed a reduction of thrombi in the vein of Galen, straight sinus, superior sagittal sinus and torcular herophili. (F) After methylprednisolone therapy, compared with (C), CE-MRV revealed a considerable decrease of thrombi in the superior sagittal sinus, torcular herophili, left transverse sinus, sigmoid sinus and internal jugular vein. (G) At 6-month follow-up, compared with (E), MRBTI showed thrombi in the vein of Galen, straight sinus and torcular herophili disappeared. (H) At 6-month follow-up, compared with (F), CE-MRV indicated thrombi of left transverse sinus and sigmoid sinus reduced. Abbreviations: MRBTI, magnetic resonance black-blood thrombus imaging; CE-MRV, contrast-enhanced magnetic resonance venography; CT, computerized tomography. The red arrows indicate the lesion. |
Discussion
In our investigation, all patients with severe CVT with hereditary PCD/PSD had high levels of inflammation, which seemed to be proportional to the severity of the disease on admission. Considering the obvious correlation between inflammation and the occurrence and prognosis of severe CVT, in addition to standard anticoagulant therapy, all patients received anti-inflammatory therapy. After methylprednisolone pulse therapy, the inflammation indexes of all patients decreased obviously, venous sinus vessel recanalization, neurological function and increased intracranial pressure also improved dramatically with a good prognosis at discharge and completely recovered during follow-up without new thrombosis and steroid-related adverse effects. It suggested that steroid therapy based on anticoagulant therapy appeared to have a remarkable and distinctive effect in patients with severe CVT combined with hereditary PCD/PSD.
However, until now, there are no systematic and prospective studies on the treatment of CVT with hereditary PCD/PSD.5,20,27,28 For severe CVT, the available evidence does not support thrombolytic therapy in acute phase, and compared with conventional anticoagulant therapy, endovascular treatment of severe CVT does not seem to have a positive effect6,7,20,29. Current guidelines recommend glucocorticoid therapy for autoimmune disease-associated CVT, such as Behcet’s disease, systemic lupus erythematosus and antiphospholipid syndrome.30,31 For non-inflammatory associated severe CVT, steroid therapy is not recommended, but the level of evidence is low.18 Therefore, there are still significant dilemmas in the treatment of severe CVT with hereditary PCD/PSD. Here, we report five severe CVT patients with hereditary PCD/PSD who responded positively to anti-inflammatory therapy, indicating that anti-inflammatory therapy based on anticoagulant therapy may be effective for patients with severe CVT.
Recently, more and more studies have shown that inflammatory response is closely related to the pathophysiology of severe CVT. After the occurrence of CVT, inflammatory cells are activated, which promotes the release of cytokines, chemokines and other inflammatory mediators, resulting in brain injuries,32 including blood-brain barrier (BBB) disruption,33 brain edema and cerebral venous infarction.34 Compared with healthy controls, serum levels of IL-6, hs-CRP10,11 /CRP,35 interleukin-1β (IL-1β),36 tumor necrosis factor-α (TNF-α)37 and claudin-511 were significantly increased in patients with severe CVT. Several recent clinical studies have shown the importance of inflammatory biomarkers in assessing the severity and prognosis of severe CVT. Our previous studies10 showed that baseline serum NLR, CSF IgA, CSF IgM, and CSF IgG levels were positively correlated with NIHSS. Hs-CRP levels were positively correlated with incidence of seizures at the baseline, and baseline IL-6, NLR, and CRP were significantly associated with adverse functional outcomes at 90 days,38 and NLR was the strongest predictor of poor prognosis in CVT. SII,9 a novel cellular immune inflammatory marker reflecting the balance between host immune and inflammatory status, has been positively correlated with CVT severity, poor prognosis, and increased mortality for acute/subacute CVT.8 Therefore, anti-inflammatory therapy is extremely significant as an innovative approach to severe CVT therapy.
PC can be activated to activated protein C (APC) by interacting with the thrombin-thrombomodulin complex and the endothelial cell protein receptor (EPCR) to inactivate factors FV/Va and FVIII/ VIIa, in order to induce anticoagulation.39 APC also promotes fibrinolysis.40 In addition to its anticoagulant function, APC also has strong anti-inflammatory properties. EPCR-bound APC can initiate anti-inflammatory, anti-apoptotic, and barrier protective functions by activating proteinase-activated receptor 1 (PAR-1)41 cleavage on endothelial cells and can inhibit neutrophil and IL-8 chemotaxis42 (The specific mechanism is shown in Figure 5).
 |
Figure 5 Anticoagulant and anti-inflammatory effects of PC/PS and the pathophysiology of hereditary PCD/PSD in severe CVT. PC interacts with the thrombin-TM complex and the EPCR to activate APC, which inhibits the anticoagulant effects of factors Va and VIIIa. PS, sphingolipids, and HDL can all accelerate this process. Furthermore, APC can promote fibrinolysis by inhibiting PAI-1. PS, in addition to being a cofactor of APC, can directly inhibit prothrombin, resulting in an anticoagulant effect. APC can activate PAR-1 in an EPCR-dependent manner, inhibit the transcriptional activation of MMP-9 dependent on NF- κB, and then protect the BBB. In addition, it exerts anti-inflammatory effects through S1P and Rac1-dependent mechanisms. PS inhibits the TLR -induced signaling cascade and inhibits inflammatory cytokines by binding to TAMRs. CRP can promote the formation of PAI-1 and tissue factor, and inhibit fibrinolysis. Endotoxin, IL-1β and TNF-α reduce APC production by down-regulating thrombomodulin and EPCR, thereby inhibiting anticoagulant and anti-inflammatory responses. Consequently, for hereditary PCD/PSD, the inflammatory and thrombosis cascade continues to amplify, resulting in damage to the BBB and nerve cells, thus aggravating the patient’s condition. Blue Arrow: the inhibiting effect, Red Arrow: the activation effect, Black Star and Arrow: the damaging effect. Abbreviations: PC, Protein C; PS, Protein S; Hereditary PCD/PSD, Hereditary Protein C/S Deficiency; TM, Thrombomodulin; EPCR, Endothelial PC Receptor; APC, Activated PC; HDL, High-Density Lipoprotein; PAI-1, Plasminogen Activator Inhibitor 1; BBB, Blood-Brain Barrier; MMP-9, matrix metalloproteinase-9; S1P, Sphingosine 1-Phosphate; TLR, Toll-Like Receptor; TAMRs, TAM receptors; CRP, C-Reactive Protein; IL-1β, Interleukin-1β, TNF-α, Tumor Necrosis Factor-α. |
As a physiological anticoagulant, free PS can enhance APC-induced cleavage of FVa and FVIII 20-fold and 3-fold,43 respectively. In addition to anticoagulant effects, PS is involved in early inflammatory responses. PS synthesized and secreted by other cells, such as megakaryocytes, endothelial cells, and dendritic cells activates a unique family of receptor protein tyrosine kinases (PTK), comprising Tyro3, Axl, and Mer, also known as TAM receptors (TAMRs).44 TAMRs suppress inflammation and inhibit inflammatory cytokines45 (The specific mechanism is shown in Figure 5).
Inflammation amplifies the coagulation process by upregulating tissue factors that lead to coagulation initiation, elevating plasminogen activator inhibitor 1 (PAI-1) to inhibit fibrinolysis, reducing the natural anticoagulant pathway and down-regulating the PC anticoagulant pathway to induce thrombosis.46,47 Thrombosis also leads to inflammation. In the coagulation pathway, pro-inflammatory cytokines and growth factors such as TNF-α, IL-1β and monocyte chemoattractant protein-1 (McP-1) are induced by the TF/FVIIa complex, TF/FVIIa/FXa complex, FXa,48,49 thrombin and fibrin, to aggravate inflammation, resulting in a vicious cycle.
Consequently, hereditary PCD/PSD can not only cause thrombosis but also trigger an inflammatory response, which can further promote thrombosis. Nevertheless, thrombosis can stimulate inflammatory response, resulting in severe CVT and poor prognosis. Therefore, based on anticoagulant therapy, anti-inflammatory therapy is particularly important for severe CVT with hereditary PCD/PSD.
Glucocorticoids, as broad-spectrum anti-inflammatory drugs, inhibiting the expression of various inflammatory factors,50 such as cytokines, enzymes, adhesion molecules, interleukins (IL-1, IL-2, IL-3, IL-6, IL-11, IL-18, TNF-α, IL-5), can not only inhibit the progression of hypoxic-ischemic brain injury in a variety of ways but also stabilize the BBB, reduce brain edema and intracranial pressure so as to delay the progression of stroke.51 In the pathophysiology of CVT, the cascade between inflammation and thrombosis is continuously amplified, thus aggravating the brain injury of CVT, we gave patients methylprednisolone pulse combined with standard anticoagulant therapy and were surprised to find that serum and CSF inflammatory indicators were significantly decreased in patients with severe CVT with hereditary PCD/PSD after treatment. In addition, we also discovered that NIHSS and mRS significantly declined at discharge following steroid therapy and further decreased at 6-month follow-up compared with baseline. In addition to the improvement of the patients’ neurological impairment, intracranial pressure and grades of Frisén also have a downward trend compared with baseline. These phenomena indicate that the decreased inflammatory response is consistent with the improvement of the condition. Therefore, our research suggests that glucocorticoids may reduce the level of inflammation in severe CVT through anti-inflammatory effects, thereby inhibiting the progression of hypoxic-ischemic brain injury, reducing cerebral edema, and enhancing CSF circulation to improve prognosis. In addition, during our 6-month follow-up, none of the five patients with severe CVT experienced thrombosis recurrence or substantial adverse events due to steroid use. We inferred that short-term glucocorticoids combined with anticoagulation may be helpful and safe in the treatment of acute/subacute severe CVT.
Nevertheless, this study has several deficiencies and restrictions, due to the relatively small sample size and the self-controlled study, which could cause bias in the findings, and make it impossible to fully explain the improvement in neurological function after anti-inflammatory therapy. As a result, cohort studies and randomized controlled trials with a larger sample size are needed in further investigations, and we should also adopt logistic regression analysis to analyze the independent variables associated with the severity and outcome of CVT in further study.
Conclusion
Severe CVT with hereditary PCD/PSD has high levels of inflammation, which is associated with poor prognosis of severe CVT. Hereditary PCD/PSD can not only cause thrombosis but also trigger an inflammatory response. Anti-inflammatory therapy, such as methylprednisolone pulse therapy combined with standard anticoagulant therapy, may be effective for severe CVT with hereditary PCD/PSD without significant adverse effects.
Data Sharing Statement
The original contributions presented in the study will be made available by the authors, without undue reservation. Any further inquiries should be directed to the corresponding author/s.
Consent for Publication
Every patient whose data are included in this manuscript has provided their approval for publication.
Consent to Participate
Written informed consent was obtained from the patients.
Funding
This study was supported by the Beijing Natural Science Foundation (7182064) and the Beijing Municipal Science & Technology Commission (Z161100000516088).
Disclosure
The authors declared that there were no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
1. Stam J, Majoie CB, van Delden OM, van Lienden KP, Reekers JA. Endovascular thrombectomy and thrombolysis for severe cerebral sinus thrombosis: a prospective study. Stroke. 2008;39(5):1487–1490. doi:10.1161/STROKEAHA.107.502658
2. Kowoll CM, Kaminski J, Weiss V, et al. Severe cerebral venous and sinus thrombosis: clinical course, imaging correlates, and prognosis. Neurocrit Care. 2016;25(3):392–399. doi:10.1007/s12028-016-0256-8
3. Davoudi VKK, Saadatnia M, Saadatnia M. Risk factors for remote seizure development in patients with cerebral vein and dural sinus thrombosis. Seizure. 2014;23:135–139. doi:10.1016/j.seizure.2013.10.011
4. Appenzeller SZC, Annichino-Bizzachi JM, Annichino-Bizzachi JM, et al. Cerebral venous thrombosis: influence of risk factors and imaging findings on prognosis. Clin Neurol Neurosurg. 2005;107:371–378. doi:10.1016/j.clineuro.2004.10.004
5. Ferro JM, Bousser MG, Canhao P, et al. European stroke organization guideline for the diagnosis and treatment of cerebral venous thrombosis - Endorsed by the European Academy of Neurology. Eur Stroke J. 2017;2(3):195–221. doi:10.1177/2396987317719364
6. Dentali F, Squizzato A, Gianni M, et al. Safety of thrombolysis in cerebral venous thrombosis. A systematic review of the literature. Thromb Haemost. 2010;104(5):1055–1062. doi:10.1160/TH10-05-0311
7. Coutinho JM, Zuurbier SM, Bousser MG, et al. Effect of endovascular treatment with medical management vs standard care on severe cerebral venous thrombosis: the TO-ACT randomized clinical trial. JAMA Neurol. 2020;77(8):966–973. doi:10.1001/jamaneurol.2020.1022
8. Karahan SZ, Gazioglu S, Dilaver I, Boz C. the role of thrombo-inflammatory biomarkers in the prognosis of cerebral venous sinus thrombosis. Curr Neurovasc Res. 2021;18(2):237–243. doi:10.2174/1567202618666210607151518
9. Walzik D, Joisten N, Zacher J, Zimmer P. Transferring clinically established immune inflammation markers into exercise physiology: focus on neutrophil-to-lymphocyte ratio, platelet-to-lymphocyte ratio and systemic immune-inflammation index. Eur J Appl Physiol. 2021;121(7):1803–1814. doi:10.1007/s00421-021-04668-7
10. Wang L, Duan J, Bian T, et al. Inflammation is correlated with severity and outcome of cerebral venous thrombosis. J Neuroinflammation. 2018;15(1):329. doi:10.1186/s12974-018-1369-0
11. Duan J, Leng X, Han Z, et al. Identifying biomarkers associated with venous infarction in acute/subacute cerebral venous thrombosis. Aging Dis. 2021;12(1):93–101. doi:10.14336/AD.2020.0405
12. Suleiman L, Negrier C, Boukerche H. Protein S: a multifunctional anticoagulant vitamin K-dependent protein at the crossroads of coagulation, inflammation, angiogenesis, and cancer. Crit Rev Oncol Hematol. 2013;88(3):637–654. doi:10.1016/j.critrevonc.2013.07.004
13. Dinarvand P, Moser KA. Protein C Deficiency. Arch Pathol Lab Med. 2019;143(10):1281–1285. doi:10.5858/arpa.2017-0403-RS
14. Aksu K, Donmez A, Keser G. Inflammation-induced thrombosis: mechanisms, disease associations and management. Curr Pharm Des. 2012;18(11):1478–1493. doi:10.2174/138161212799504731
15. Margetic S. Inflammation and haemostasis. Biochem Med. 2012;22(1):49–62. doi:10.11613/BM.2012.006
16. Hu S, Lee H, Zhao H, Ding Y, Duan J. Inflammation and severe cerebral venous thrombosis. Front Neurol. 2022;13:873802. doi:10.3389/fneur.2022.873802
17. Singhal AB, Topcuoglu MA. Glucocorticoid-associated worsening in reversible cerebral vasoconstriction syndrome. Neurology. 2017;88(3):228–236. doi:10.1212/WNL.0000000000003510
18. Canhao P, Cortesao A, Cabral M, et al. Are steroids useful to treat cerebral venous thrombosis? Stroke. 2008;39(1):105–110. doi:10.1161/STROKEAHA.107.484089
19. Saposnik G, Barinagarrementeria F, Brown RD, et al. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American heart association/American Stroke Association. Stroke. 2011;42(4):1158–1192. doi:10.1161/STR.0b013e31820a8364
20. Fan Y, Yu J, Chen H, et al. Chinese stroke association guidelines for clinical management of cerebrovascular disorders: executive summary and 2019 update of clinical management of cerebral venous sinus thrombosis. Stroke Vasc Neurol. 2020;5(2):152–158. doi:10.1136/svn-2020-000358
21. Yang Q, Duan J, Fan Z, et al. Early detection and quantification of cerebral venous thrombosis by magnetic resonance black-blood thrombus imaging. Stroke. 2016;47(2):404–409. doi:10.1161/STROKEAHA.115.011369
22. Connor SE, Jarosz JM. Magnetic resonance imaging of cerebral venous sinus thrombosis. Clin Radiol. 2002;57(6):449–461. doi:10.1053/crad.2001.0880
23. Wypasek E, Undas A. Protein C and protein S deficiency - practical diagnostic issues. Adv Clin Exp Med. 2013;22(4):459–467.
24. Qureshi AI, Classification A. Scheme for assessing recanalization and collateral formation following cerebral venous thrombosis. J Vasc Interv Neurol. 2010;3(1):1–2.
25. Stanbury RM, Graham EM. Systemic corticosteroid therapy--side effects and their management. Br J Ophthalmol. 1998;82(6):704–708. doi:10.1136/bjo.82.6.704
26. Fang F, Xu Z, Suo Y, et al. Gene panel for Mendelian strokes. Stroke Vasc Neurol. 2020;5(4):416–421. doi:10.1136/svn-2020-000352
27. Saito K, Ishii K, Furuta K, Kobayashi M, Wada Y, Morishita E. Recurrent cerebral venous thrombosis treated with direct oral anticoagulants in a Japanese man with hereditary protein C deficiency. J Stroke Cerebrovasc Dis. 2021;30(1):105320. doi:10.1016/j.jstrokecerebrovasdis.2020.105320
28. Wang T, Zhao XJ, Zhu HD, Lu M, Wen B, Ma L. Clinical characteristics, genes identification and follow-up study of a patient with central venous thrombosis from a protein S deficiency pedigree. Eur Rev Med Pharmacol Sci. 2021;25(1):353–361. doi:10.26355/eurrev_202101_24402
29. Xu Z, Li X, Feng D, et al. Endovascular therapy versus anticoagulation for treatment of cerebral venous sinus thrombosis: a meta-analysis. Neurologist. 2021;27(2):69–73. doi:10.1097/NRL.0000000000000369
30. Zhang B, Lang Y, Zhang W, Cui L, Deng F. Characteristics and management of autoimmune disease-associated cerebral venous sinus thrombosis. Front Immunol. 2021;12:671101. doi:10.3389/fimmu.2021.671101
31. Ferro JM, Bousser MG, Canhao P, et al. European stroke organization guideline for the diagnosis and treatment of cerebral venous thrombosis - endorsed by the European Academy of Neurology. Eur J Neurol. 2017;24(10):1203–1213. doi:10.1111/ene.13381
32. Vidale S, Consoli A, Arnaboldi M, Consoli D. Postischemic inflammation in acute stroke. J Clin Neurol. 2017;13(1):1–9. doi:10.3988/jcn.2017.13.1.1
33. Piazza G. Cerebral venous thrombosis. Circulation. 2012;125(13):1704–1709. doi:10.1161/CIRCULATIONAHA.111.067835
34. de la Vega Muns G, Quencer R, Ezuddin NS, Saigal G. Utility of Hounsfield unit and hematocrit values in the diagnosis of acute venous sinus thrombosis in unenhanced brain CTs in the pediatric population. Pediatr Radiol. 2019;49(2):234–239. doi:10.1007/s00247-018-4273-y
35. Tekesin A, Tunc A. Inflammatory markers are beneficial in the early stages of cerebral venous thrombosis. Arq Neuropsiquiatr. 2019;77(2):101–105. doi:10.1590/0004-282x20190001
36. Rashad S, Niizuma K, Sato-Maeda M, et al. Early BBB breakdown and subacute inflammasome activation and pyroptosis as a result of cerebral venous thrombosis. Brain Res. 2018;1699:54–68. doi:10.1016/j.brainres.2018.06.029
37. Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory Disequilibrium in Stroke. Circ Res. 2016;119(1):142–158. doi:10.1161/CIRCRESAHA.116.308022
38. Aguiar de Sousa D, Pereira-Santos MC, Serra-Caetano A, et al. Blood biomarkers associated with inflammation predict poor prognosis in cerebral venous thrombosis:: a multicenter prospective observational study. Eur J Neurol. 2021;28(1):202–208. doi:10.1111/ene.14526
39. McDonnell CJ, Soule EE, Walsh PT, O’Donnell JS, Preston RJS. The immunoregulatory activities of activated protein C in inflammatory disease. Semin Thromb Hemost. 2018;44(2):167–175. doi:10.1055/s-0037-1608910
40. Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. 2012;95(4):333–345. doi:10.1007/s12185-012-1059-0
41. Dinarvand P, Hassanian SM, Weiler H, Rezaie AR. Intraperitoneal administration of activated protein C prevents postsurgical adhesion band formation. Blood. 2015;125(8):1339–1348. doi:10.1182/blood-2014-10-609339
42. Galley HF, El Sakka NE, Webster NR, Lowes DA, Cuthbertson BH. Activated protein C inhibits chemotaxis and interleukin-6 release by human neutrophils without affecting other neutrophil functions. Br J Anaesth. 2008;100(6):815–819. doi:10.1093/bja/aen079
43. O’Brien LM, Mastri M, Fay PJ. Regulation of factor VIIIa by human activated protein C and protein S: inactivation of cofactor in the intrinsic factor Xase. Blood. 2000;95(5):1714–1720. doi:10.1182/blood.V95.5.1714.005k40_1714_1720
44. van der Meer JH, van der Poll T, van ‘t Veer C. TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood. 2014;123(16):2460–2469. doi:10.1182/blood-2013-09-528752
45. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–1136. doi:10.1016/j.cell.2007.10.034
46. De Caterina R, D’Ugo E, Libby P. Inflammation and thrombosis - testing the hypothesis with anti-inflammatory drug trials. Thromb Haemost. 2016;116(6):1012–1021. doi:10.1160/TH16-03-0246
47. Vazquez-Garza E, Jerjes-Sanchez C, Navarrete A, Joya-Harrison J, Rodriguez D. Venous thromboembolism: thrombosis, inflammation, and immunothrombosis for clinicians. J Thromb Thrombolysis. 2017;44(3):377–385. doi:10.1007/s11239-017-1528-7
48. van der Poll T, de Jonge E, Levi M. Regulatory role of cytokines in disseminated intravascular coagulation. Semin Thromb Hemost. 2001;27(6):639–651. doi:10.1055/s-2001-18868
49. Peerschke EI, Yin W, Ghebrehiwet B. Platelet mediated complement activation. Adv Exp Med Biol. 2008;632:81–91. doi:10.1007/978-0-387-78952-1_7
50. Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998;94(6):557–572. doi:10.1042/cs0940557
51. Slivka AP, Murphy EJ. High-dose methylprednisolone treatment in experimental focal cerebral ischemia. Exp Neurol. 2001;167(1):166–172. doi:10.1006/exnr.2000.7532
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Platelets, Macrophages, and Thromboinflammation in Chagas Disease
Choudhuri S, Garg NJ
Journal of Inflammation Research 2022, 15:5689-5706
Published Date: 4 October 2022
Pulmonary Exposure to Copper Oxide Nanoparticles Induces Systemic Inflammation, Oxidative Stress, and Prothrombotic Responses in BALB/c Mice
Ferdous Z, Beegam S, Zaaba NE, Elzaki O, Greish YE, Nemmar A
International Journal of Nanomedicine 2026, 21:579331
Published Date: 16 June 2026