Back to Journals » OncoTargets and Therapy » Volume 13
Physiological and Pathophysiological Roles of Ion Transporter-Mediated Metabolism in the Thyroid Gland and in Thyroid Cancer
Authors Wang H, Ma Z, Cheng X, Tuo B, Liu X, Li T
Received 7 September 2020
Accepted for publication 17 November 2020
Published 3 December 2020 Volume 2020:13 Pages 12427—12441
DOI https://doi.org/10.2147/OTT.S280797
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Hu Wang,1 Zhiyuan Ma,1 Xiaoming Cheng,1 Biguang Tuo,2,3 Xuemei Liu,2,3 Taolang Li1
1Department of Thyroid and Breast Surgery, Affiliated Hospital of Zunyi Medical University, Zunyi, People’s Republic of China; 2Department of Gastroenterology, Affiliated Hospital of Zunyi Medical University, Zunyi, People’s Republic of China; 3Digestive Disease Institute of Guizhou Province, Zunyi, People’s Republic of China
Correspondence: Taolang Li
Department of Thyroid and Breast Surgery, Affiliated Hospital, Zunyi Medical University, Zunyi 563003, People’s Republic of China
Email [email protected]
Xuemei Liu
Department of Gastroenterology, Affiliated Hospital, Zunyi Medical University, Zunyi 563003, People’s Republic of China
Email [email protected]
Abstract: Thyroid cancer is the most common type of endocrine tumor and has shown an increasing annual incidence, especially among women. Patients with thyroid cancer have a good prognosis, with a high five-year survival rate; however, the recurrence rate and disease status of thyroid cancer remain a burden for patients, which compels us to further elucidate the pathogenesis of this disease. Recently, ion transporters have gradually become a hot topic in the field of thyroid gland biology and cancer research. Additionally, alterations in the metabolic state of tumor cells and protein molecules have gradually become the focus of scientific research. This review focuses on the progress in understanding the physiological and pathophysiological roles of ion transporter-mediated metabolism in both the thyroid gland and thyroid cancer. We also hope to shed light on new targets for the treatment and prognosis of thyroid cancer.
Keywords: thyroid cancer, metabolism, ion transporters, physiology and pathophysiology, regulation factors
Introduction
Globally, the incidence of thyroid cancer (TC) is increasing annually according to the latest global statistics on the epidemiology of this malignancy published by the International Agency for Research on Cancer. TC ranks ninth among malignancies, killing more than 40,000 people annually, most of whom are women.1 Therefore, clarification of its etiology and pathogenesis is important for identifying effective therapeutic targets for early diagnosis and prevention.
It is well known an adequate energy supply is required for the growth and survival of cells, including tumor cells, which provides a good entry point for tumor research. The metabolic status of tumor cells has been studied for nearly a hundred years, and as research has progressed, researchers have found that metabolic reprogramming is ubiquitous among tumor cells.2–4 Healthy cells use carbohydrates, fats, amino acids, and other substances to produce energy in the form of adenosine triphosphate (ATP) as well as biomacromolecules to maintain normal cell function. This complex process involves glycolysis, oxidative phosphorylation (OXPHOS), gluconeogenesis, and the tricarboxylic acid (TCA) cycle and requires a stable internal environment and an adequate oxygen supply within the cell. For tumor cells, with their high metabolic needs and proliferative activities, the above processes are adjusted accordingly, which means they need rapid ATP production to maintain their energy consumption and increased biosynthesis of macromolecules. Thus, the carbohydrate, lipid, protein, and nucleic acid requirements necessary for cell maintenance are increased. Decades ago, Professor Otto Warburg described a metabolic phenotype observed in cancer cells in which the cells relied on glycolysis rather than the OXPHOS pathway to produce ATP, even when oxygen concentrations were sufficient.2 This phenomenon, later known as the “Warburg effect”, greatly aided subsequent studies of tumor metabolism and led to the accepted theory of aerobic glycolysis. Glycolysis pathways are less efficient at producing ATP than is OXPHOS, so as the demand for glucose in tumor cells increases, the glycolytic production of pyruvates and lactates also increases. Furthermore, increased glucose consumption by tumors has been confirmed to correlate with poor tumor prognosis.5 Glutamine is an amino acid that is essential for cell survival and is used as a precursor for the biosynthesis of proteins, nucleotides, and amino sugars; furthermore, its carbon skeleton structure can be used in the production process of the mitochondrial TCA cycle.6 Studies have confirmed that in fast-growing tumors, cells are more likely to use glutamine for energy production.7–10 Lactate produced by glycolysis in tumor cells also plays a very important role in tumor development. The levels of lactate from glycolysis in tumor cells and surrounding cells have been associated with tumor invasion and progression.11–14 In addition to the three molecules mentioned above, there are changes in the tumor microenvironment, metabolism-related organelle functions and ions in the metabolic process of tumor cells. TC, as a rapidly growing solid tumor, unavoidably undergoes the above metabolic changes, and alterations such as these have an impact on the growth, proliferation, metastasis and treatment resistance of TC.15
Ion transport proteins, which are widely distributed in cells, dominate the transport of cellular metabolites. Studies have reported that different ion transporters in various tumors play an essential role in tumor cell proliferation, metastasis, invasion, and apoptosis.16–20 The sodium-iodide symporter and thyroid hormone (TH) transporter in the thyroid gland, which are responsible for iodine uptake and TH secretion, respectively, are critical proteins that are thought to play an important role in TC.21–23 The expression and activity of ion transporters associated with tumor metabolism, such as glucose metabolism-related glucose transporters, lactate metabolism-related monocarboxylate transporters, amino acid metabolism-related amino acid transporters, and L-type amino acid transporters, are also changed.24–28 Several studies have confirmed that these ion transporters control the prognosis of patients with TC.
TC undergoes extensive metabolic changes related to the growth, proliferation, metastasis, and invasion of tumor cells, and ion transporters mediate the transmission of substances required for metabolism; therefore, the regulation of metabolism by ion transporters is vital for tumor cell survival. The main purpose of this review is to illuminate how some ion transporters in TC regulate metabolism and thus affect the development and progression of TC.
Sodium-Iodide Symporter (NIS) and Iodide Metabolism in TC
Physiological Role of NIS in the Thyroid Gland
NIS is an integral plasma membrane glycoprotein that is encoded by the SLC5A5 gene and is widely expressed in different organs in the human body.22 In the thyroid gland, NIS is expressed on the basolateral membrane of follicular cells and can absorb I–into the cells for the synthesis of THs and the maintenance of iodine homeostasis in humans.21 This I− uptake process is passive; after a Na+ concentration gradient is produced by the Na+/K+ ATPase, I− enters the cell together with two Na+ ions.29 Based on the iodine uptake function of the thyroid gland mediated by NIS, the application of radioiodide in the diagnosis and treatment of primary TC and its metastatic lesions has become an important clinical approach.30 In the healthy thyroid gland, the expression of NIS is mainly controlled by thyroid-stimulating hormone (TSH), which can regulate the expression and distribution of NIS in thyroid follicular cells via cyclic adenosine 3ʹ,5ʹ-monophosphate (cAMP).21,31,32 Additionally, studies have demonstrated that adenosine monophosphate-activated protein kinase (AMPK) can regulate the expression of NIS in thyroid cells.33,34 Organisms can control NIS expression via these methods to maintain metabolic iodine homeostasis in the human body. Based on these descriptions, we know that NIS is involved in the maintenance of iodine homeostasis in vivo and plays a certain role in the treatment of thyroid diseases. It is also regulated by various factors, which impact thyroid function.
Aberrant NIS Expression Regulates I− Metabolism at the Onset of TC
Radioactive iodine (RAI) has been used for decades to treat TC, especially micrometastases after thyroidectomy.35,36 The basis for this treatment is that NIS is expressed on the membrane of the thyroid follicles and can transport 131I into the nidus. However, several studies have demonstrated that NIS in TC has abnormalities relative to that in normal thyroid cells, such as altered expression, different intracellular localization, and loss of function.21,22,36–41 More than 20 years ago, Professor Sebastiano Filetti demonstrated abnormalities in the function and expression of NIS in various TC samples and indicated that abnormal changes in NIS function would reduce iodine uptake.42 These changes in NIS interfere with the treatment of some TCs, which can lead to poor prognosis and relapse; moreover, abnormal iodine metabolism in TC also affects the process of tumor proliferation.43 Researchers have conducted relevant studies to identify the underlying mechanism and pointed out that these changes in NIS expression and function may be directly or indirectly related to mutations in tumor suppressor genes or oncogenes, activation of signaling pathways, modifications in the metabolic status, changes in the intracellular environment and other factors.33,34,44–49 It is obvious that the change in NIS iodine uptake is not caused by a single factor but by several interlaced factors, which finally leads to the alteration of the NIS functional state. Studies have elucidated that the BRAF-V600E mutation and mutant RAS genes, which are the most commonly mutated genes in TC, cause the activation of signaling pathways such as the mitogen-activated protein kinases (MAPK), phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT), AMPK and mammalian target of rapamycin (mTOR) pathways. These alterations affect the expression of NIS and change I metabolism.34,46,49,50 Previous studies have shown that the BRAF-V600E mutation can cause downregulation of NIS expression and is associated with the MAPK pathway.50 PI3K and its downstream molecule mTOR also exert an impact on the function of NIS and reduce I− absorption.33,51 Andrade’s group demonstrated that AMPK affects the uptake of iodine and glucose in rat thyroid cells, which ultimately display decreased iodine uptake, and further confirmed that AMPK is upregulated in TC.34,52–54 Moreover, studies have shown that AMPK can downregulate NIS expression in TC.54,55 Therefore, we can see that the upregulation and activation of AMPK in TC can downregulate the expression of NIS and thus reduce the uptake of I−. Additionally, NIS expression is regulated by microRNAs. Studies have shown that microRNA-339-5p can regulate NIS expression at the RNA level in TC, thereby affecting iodine uptake.56 Last year, one study identified two key factors that interact with NIS, and blocking these factors allowed NIS to resume normal function in TC cells, thus improving the prognosis of patients.57 Furthermore, EI Mokh’s group demonstrated that by inhibiting NIS-related regulators (BRAF-V600E, MAPK, PI3K), iodine uptake levels in TC could be effectively promoted.58 To date, preclinical and clinical studies on NIS as a therapeutic target have been conducted, some of which have been selected and are presented in Table 1. It can be concluded that NIS plays a significant role in TC; its aberrant expression in TC is caused by many factors and leads to abnormal iodine metabolism, meaning that radioactive iodide ions cannot be effectively taken up, thus affecting the prognosis of TC. Interestingly, abnormal iodide metabolism is associated with the proliferation of TC.
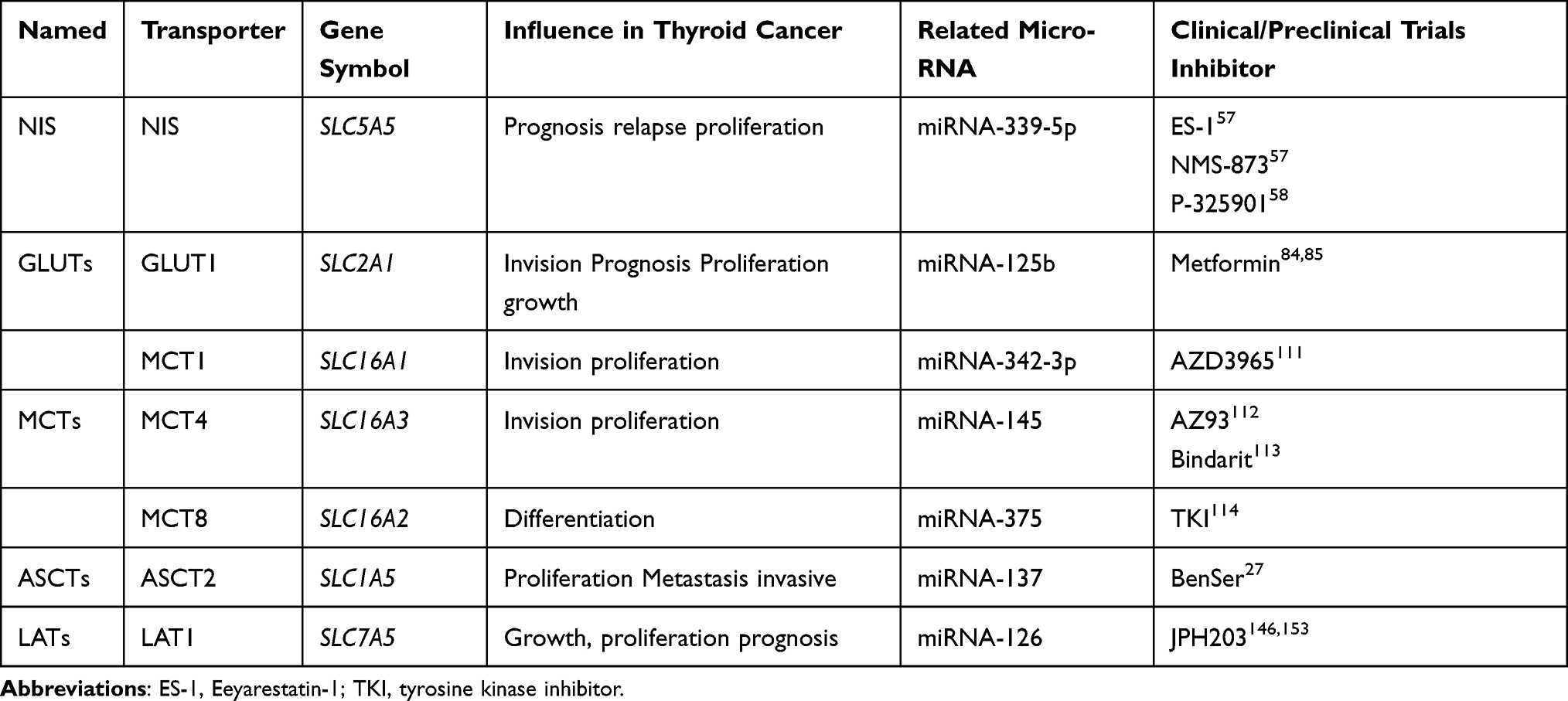 |
Table 1 Expression, Influence, Related MicroRNA, Clinical/Preclinical Trials of Ion Transporters in Thyroid Cancer |
Glucose Transporters (GLUTs) and Energy Metabolism in TC
Physiological Role of GLUTs in the Thyroid Gland
The GLUT family comprises transmembrane proteins encoded by SLC2 genes, which are widely expressed on the plasma membrane of various eukaryotic cells, and the main function of these proteins is to mediate the transport of carbohydrates into cells.59 In the human body, 14 members of the GLUT family are distributed on different cells.60 The main function of GLUTs in human cells is to absorb glucose from the extracellular environment and blood circulation into cells for energy metabolism.18 Studies have confirmed that GLUTs regulate glucose uptake and energy metabolism in thyroid cells.61–65 GLUT1, GLUT3, GLUT4, and GLUT10 are expressed in rat thyroid cells, with GLUT1 being the main metabolism-related subtype.61,62,65 Matsuzu’s team summarized previous studies and verified the expression of GLUTs in the thyroid gland; they also found that GLUT1 was of great significance to thyroid gland function.61 GLUT1 is encoded by the SLC2A1 gene and is expressed on the plasma membrane.59 However, due to technical limitations, the only certainty is that GLUT1 is expressed on the plasma membrane, and any specific localization is unclear.61 The primary substrate for GLUT1 is glucose, but it is also involved in the transport of mannose, glucosamine, galactose, and reduced ascorbate.66 GLUT1 can transport glucose into thyroid cells, which is then converted into ATP via biosynthesis processes such as glycolysis and OXPHOS to provide energy for cell survival. GLUT1 expression in the thyroid gland is regulated by TSH and AMPK.52,61 From the above observations, we can conclude that as a member of the GLUT family GLUT1 is necessary for the energy metabolism of the thyroid gland.
GLUT1 Regulates Metabolism at the Onset of TC
GLUT1 has been extensively studied. Researchers have found that GLUT1 expression is upregulated in many different types of tumor cells and is closely linked to tumor progression.67–69 Based on the “Warburg effect”, we know that tumor cells are significantly more dependent on ATP than are normal cells and that this dependence depends on activation of aerobic glycolysis.2–4,70,71 As a transporter directly related to the intracellular energy source, GLUT1 is bound to be closely related to cellular metabolism. Studies have reported a substantial increase in GLUT1 expression in TC cells.18,25,26 By analyzing the immunohistochemical data of more than 500 patients with TC, Nahm et al25 found that GLUT1 expression was increased in TC and that its activity was enhanced in cells with increased glycolysis. They also noted that increased GLUT1 expression is associated with invasion and poor prognosis of TC.25 TC, as a tumor with high proliferative and metabolic activity, has a high energy demand and rapidly consumes ATP, which mainly depends on the glucose transport function of GLUT1. Therefore, only increased expression and enhanced function of GLUT1 can meet the energy needs of TC. The study also confirmed that glucose uptake was significantly increased with upregulated GLUT1 expression in TC cells.72 At this point, we can conclude that when GLUT1 expression is upregulated, the amount of glucose entering the cells and the subsequent levels of substrates involved in aerobic glycolysis, OXPHOS and gluconeogenesis increase, which provides hospitable conditions for the growth and proliferation of tumor cells. The expression of GLUT1 in TC is regulated by many factors. Studies have demonstrated that the PI3K pathway can upregulate the expression of GLUT1 in RAS-mutated TC cells.73 The transcription factor hypoxia-inducible factor 1 α (HIF-1α), whose expression is induced in hypoxic tumor environments, has also been linked to GLUT1 expression in TC. Jóźwiak’s group demonstrated that in the TC cell lines FTC-133 and 8305c, HIF-1α can upregulate the expression of GLUT1 and promote glucose uptake of cancer cells.74 In addition, after applying siRNA to knockdown GLUT1 expression in TC cells, they found that the cells’ ability to uptake glucose was reduced and that their ability to proliferate was also diminished.74 HIF-1α is an important factor in the metabolic changes in tumor cells and can activate the glycolytic pathway and inhibit OXPHOS in mitochondria.75,76 Studies have shown that HIF-1α can activate many transporters associated with cellular aerobic glycolysis, including GLUT1, and enhance their expression in TC.24–26,77 There have been many well-executed experiments on the regulation of GLUT1 expression in TC and other cancers. In addition to the two factors mentioned above, oncogenes and tumor suppressor factors such as Raf, Myc, Src, p53 and PTEN have also been proven to regulate GLUT1 expression in TC and promote glucose uptake.78–82 Moreover, there exists a regulatory microRNA that targets GLUT1. Zhang et al confirmed that microRNA-125b can reduce glucose uptake in TC cells by downregulating GLUT1 expression, thus affecting the development and progression of cancer.83 Due to these changes in TC, GLUT1 expression increases to compensate for the metabolic reprogramming so that enough glucose can be taken up to fulfill the needs of TC cells with high metabolism and elevated proliferative activities, and such changes in TC are associated with its growth, proliferation and progression. In summary, we can conclude that after the occurrence of TC, many factors in TC can promote GLUT1 expression, which further regulates the uptake of glucose through its own changes in expression and then affects the energy metabolism of cells to confront the needs of TC cells to maintain their growth, proliferation and progression. GLUT1 has been used as an effective target for tumor therapy in many studies. In TC, a preclinical study by Shen et al confirmed that metformin could effectively reduce GLUT1 expression and thus inhibit the progression of TC.84 Other studies have identified GLUT1 as an important therapeutic target in treating tumors, and these studies have been well summarized in an excellent review by Zambrano.85
Monocarboxylate Transporters (MCTs) and Metabolism in TC
Physiological Role of MCTs in the Thyroid Gland
The MCT family, encoded by the SLC16 gene, is a proton-linked membrane transport protein located in the cell membrane.86 It consists of 14 members; however, only MCT (1–4) are involved in monocarboxylate transport in human cells.86 Their main function is to transport intracellular monocarboxylic acid substances, such as lactate, pyruvate and ketone bodies.86 These substances are necessary for the energy metabolism and material synthesis of organelles. When the oxygen supply fails to meet the metabolic needs of the cell, the cell relies on glycolysis to produce energy, which increases the production of lactate and pyruvate and consequently leading to the accumulation of these products in the cell. MCTs can transport these redundant substances out of the cell or into other cells for further metabolism, thus maintaining cellular homeostasis. In healthy thyroid tissues, MCT (1–4) have not been precisely defined, but under pathological conditions, MCT1 and MCT4 are clearly expressed in the thyroid and are closely related to the energy metabolism of cells.25,87 MCT1, which is encoded by the SLC16A1 gene, is a bidirectional transporter located on the plasma membrane whose main function is to transport lactate into the cell.16,88 The lactate that enters the cell can be reversibly converted to pyruvate by lactate dehydrogenase (LDH) or to other energy materials by gluconeogenesis.16 The factors that regulate MCT1 expression in normal tissues have not been clearly elucidated, but studies have found that it is associated with metabolism and AMPK.88 MCT4, encoded by SLC16A3, is also distributed across the plasma membrane.16 Compared with MCT1, MCT4 is mainly involved in glycolysis metabolism in cells and has a different functional structure.16,88 It has a low affinity for lactate, and its primary function is to transport intracellular lactate out of the cell.89 Studies have reported that MCT4 expression is mainly regulated by cell metabolism and the hypoxic environment.90 In addition to these two transporters, MCT8, a member of the MCT family, is related to TH transport and is also distributed in the thyroid gland.91 MCT8 is encoded by the SLC16A2 gene and is widely distributed in the basolateral membrane of thyroid follicular epithelial cells.91 Its main function is to transport THs, especially 3.3′,5-triiodothyronine (T3).91 TH is synthesized in thyroid follicular epithelial cells and then transported by MCT8 to cells throughout the body to act on the corresponding cell receptors, thereby causing a series of biological reactions.92 In healthy thyroid cells, MCT8 expression is mainly regulated by cAMP and TSH.23 In summary, MCT1, MCT4 and MCT8 play a significant role in the energy metabolism of and transport of materials between cells.
MCTs Regulate Metabolism in the Onset of TC
MCT1 and MCT4 are essential in the metabolic process of TC cells. As noted above, most tumor cells rely on anaerobic glycolysis to provide energy for their growth and survival; this process eventually produce a large amount of lactate, which plays an important role in cancer metabolism.93,94 During aerobic glycolysis in tumor cells, a large amount of pyruvate is produced when excessive glucose is consumed to produce ATP; then, pyruvate is converted to lactate via LDH. This process is reversible, which means that lactate can also be transformed into pyruvate.3,95,96 In addition, there is another source of lactate in TC cells: the surrounding fibroblasts, immune cells, epithelial cells and so on constitute the tumor microenvironment. Studies have shown that cancer-associated fibroblasts (CAFs), also called stromal fibroblasts, play an important role in tumor metabolism and clarified that these cells are definitely present in TC.97,98 CAFs can produce lactate via aerobic glycolysis and release lactate into the surrounding environment; then, tumor cells absorb this lactate and convert it into pyruvate to produce energy in mitochondria.13 This process, known as the “Reverse Warburg Effect”, allows CAFs to provide some lactate that is absorbed by highly metabolic tumor cells to further participate in the TCA cycle to produce energy. The transportation of lactate is dependent on MCT1 and MCT4. Studies have confirmed that the expression levels of MCT1 and MCT4 are increased to varying degrees in different types of TC.89,99,100 Pioneering researchers conducted an immunohistochemical analysis of orthotopic xenograft tumors and clinicopathological specimens and found that MCT1 was highly expressed relative to MCT4 in anaplastic TC (ATC).87 Nahm et al conducted a similar experiment with the clinicopathological specimens of 566 patients with TC and immunohistochemically analyzed the expression of proteins related to tumor glycolysis.25 They finally concluded that MCT4 expression increased, mainly in ATCs.25 Other researchers have found that MCT4 is associated with the invasion and proliferation of different tumors.99,101 Therefore, as a highly invasive and metastatic tumor, the incidence of ATC may be associated with MCT4. Studies also stated that cellular hypoxia can induce upregulated MCT4 expression through the action of HIF-1α interacting with the promoter of MCT4 to adapt tumor cells to hypoxia, maintain intracellular acid-base balance and prevent intracellular lactic acid accumulation.90,102 Currently, studies on the regulation of microRNA-mediated MCT1/4 expression in TC are lacking, but there relevant reports in other tumors. Studies have confirmed that microRNA-342-3p can target MCT1 in breast cancer and then change the metabolic state of tumor cells.103 Regarding MCT4, studies have confirmed that microRNA-145 can act on MCT4 in hepatocellular carcinoma, thereby changing the homeostasis of tumor cells.104 The above two microRNAs can serve as potential therapeutic targets for further study in TC, a tumor with high levels of proliferation and metabolic characteristics. The expression of MCT1 and MCT4 in TC is increased through various factors and accelerates the transport of lactate and other energy substances to satisfy the energy requirements of the cells. In summary, MCT1 and MCT4 play an essential role in the metabolism of TC because they regulate the uptake and release of metabolic compounds to adjust to the changing metabolic needs of TC via changes in their expression levels, thus promoting the growth, proliferation and invasion of TC. MCT8 is a member of the MCT family, and its functional role in TC is quite different from that of MCT1 and MCT4. Bidziong et al demonstrated that MCT8 expression in TC tissues was significantly lower than that in healthy thyroid tissues; therefore, MCT8 could be regarded as a biomarker of TC differentiation.23 It is worth mentioning that, as a TH transporter, MCT8 (namely, downregulation of its expression) is of great significance in TC. Studies have confirmed that TH can promote the proliferation, metastasis and development of TC through the MAPK and PI3K signaling pathways.105 Therefore, downregulation of MCT8 expression in TC can effectively reduce TH secretion and thus lead to the accumulation of TH in TC tissue, which ultimately promotes the progression of TC. Currently, there are few studies on MCT8 in TC. Smith et al proved that pituitary tumor transforming gene-binding factor (PBF) could regulate the expression and function of MCT8 in TC.106 At the microRNA level, MCT8 has been shown to be a target of microRNA-375 in TC.107 In summary, we have highlighted the role of MCTs in the development of TC and their regulatory factors in TC. Currently, there are many preclinical trials regarding them as tumor therapeutic targets; however, there have been few preclinical trials investigating MCTs as a druggable target in TC.108–110 Polanski et al demonstrated that the inhibitor AZD3965 can block MCT1 function in small cell lung cancer, thereby stunting tumor development.111 This inhibitor is currently in clinical trials, and we believe that its effects on TC will be reported in the near future. For MCT4, AstraZeneca has developed AZ93, a specific inhibitor of MCT4, but it has not yet entered preclinical trials.112 Futagi et al confirmed that bindarit effectively inhibits MCT4 in human cells and could be used in antitumor research.113 MCT8 is regulated by TSH, but no direct inhibitor has been reported in TC. Krajewska summarized the study of MCT8 as a therapeutic target and found that tyrosine kinase inhibitors can affect the transport function of MCT8.114 In summary, MCTs play an important role in TC and may be a potential therapeutic target, but further research is needed.
Neutral Amino Acid Transporters (ASCTs) and Energy Metabolism in TC
Physiological Role of ASCTs in the Thyroid Gland
ASCTs, encoded by the SLC1 gene, belong to the amino acid transporter family.115 In humans, the amino acid transport family consists of seven members, five of which mostly transport glutamate and the other two mainly transport neutral amino acids.115 ASCTs, including ASCT1 and ASCT2, facilitate the transport of neutral amino acids; these proteins are encoded by SLC1A4 and SLC1A5, respectively.115 In the thyroid, we focused on the functional role of ASCT2. ASCT2 is located in the cell plasma membrane and is widely expressed in various tissues of the human body.115,116 The name ASCT2 comes from its ability to transport alanine, serine, cysteine, and threonine.117,118 In fact, ASCT2 can transport glutamine as well as these neutral amino acids with high affinity.119 Glutamine is used as a precursor for the biosynthesis of many proteins, nucleotides, and amino sugars, and its carbon skeleton structure can be used in the production process of the mitochondrial TCA cycle.6 When cells are in a proliferative state, all biosynthetic and metabolic requirements increase, and the requirements of glutamine also rise; these changes rely on the function of transporters, ASCT2 among them.120,121 Glutamine is transported into cells by ASCT2 and then into mitochondria, where it is converted to glutamate by phosphate-dependent glutaminase. Furthermore, glutamate is converted into α-ketoglutarate (α-KG) for use in the TCA cycle of mitochondria and participates in energy metabolism.122 At present, studies on factors influencing the regulation of ASCT2 expression in normal tissues are not clear. Most of the studies are in tumors, which will be elaborated in the next section. In conclusion, ASCT2 plays an important role in the energy supply and material transfer of cells.
ASCT2 Regulates Metabolism in the Onset of TC
As previously mentioned, to meet the needs of growth and proliferation, tumor cells change their metabolic state, mainly producing energy by replacing OXPHOS with aerobic glycolysis, and some studies suggest that mitochondrial function in tumor cells is impaired.123 However, after much research and debate, researchers found that these claims are not rigorous. Researchers have found that in tumor cells, mitochondria still function normally.122,124,125 In addition to using glucose for energy, cancer cells also uptake fats, proteins, and amino acids to survive and proliferate, and mitochondria play an important role in the metabolism of these molecules. In a study on the metabolism of tumor cells, researchers discovered another important substance – glutamine.6 Studies have confirmed that in fast-growing tumors, tumor cells are more likely to use glutamine for energy production.9,10 Aerobic glycolysis cooperates with glutamine metabolism to maintain cell proliferation, while glutamine can also maintain mitochondrial function and participate in the synthesis of nonessential amino acids and nucleotides (purines and pyrimidines) in mitochondria.9,10 Some researchers have realized that these changes in energy metabolism in tumor cells cause the cells to produce an energy stress response, which is associated with tumor proliferation and metastasis.126 Above, we stated the importance of glutamine in the energy metabolism of tumor cells. After glutamine enters the cell, it is first catalyzed by glutaminase to produce glutamate, which is transported into the mitochondria and then converted into α-KG, alanine, aspartate and other substances by metabolism-related enzymes to participate in intracellular metabolic functions.127 Therefore, ASCT2, as a transporter of glutamine, occupies a significant position in the regulation of cancer metabolism. Kim’s group used immunohistochemistry to stain for glutamine metabolism-related proteins in a TMA comprising 557 TC samples and found that ASCT2 was expressed in all TC tissues; however, the expression level was dissimilar in different types of TC samples.27 They analyzed the statistical correlations between these staining results and eventually discovered that the expression of glutamine metabolism-related proteins was highest in ATC and BRAF-V600E-mutated papillary TC (PTC) and that ASCT2 expression was higher in medullary TC (MTC) than in other types of TC.27 Several studies have reported that proteins involved in glutamine metabolism, such as ASCT2, are associated with tumor aggressiveness.128,129 Among the subtypes of TC, PTC with the BRAF-V600E mutation and MTC are both highly invasive and prone to metastatic behavior.36,44,130,131 Therefore, ASCT2 may be associated with the invasive characteristics and poor prognosis of TC. ASCT2 expression in TC is affected by a variety of factors, among which include gene mutations that cause changes in signaling pathways involved in ASCT2 expression. In TC, in addition to the common BRAF and RAS gene mutations, MYC gene mutations also exist, which mainly occur in MTC.132,133 MYC can stimulate ASCT2 expression and promote the utilization of glutamine.134 In addition, ASCT2 is regulated by microRNA-137.135 In summary, we can conclude that TC, as a tumor with high metabolic needs, has increased energy demands, so utilizing glutamine as a source of energy is necessary, and that ASCT2, as a transporter of glutamine into cells, cannot be neglected. Abnormal expression of ASCT2 regulates glutamine metabolism to meet the energy metabolism needs of TC and is related to tumor growth, proliferation, metastasis, invasion, and poor prognosis. Researchers have demonstrated that ASCT2 can be a therapeutic target for tumors and that blocking ASCT2 can prevent tumor growth and progression.136 Wang et al demonstrated that BenSer, an inhibitor of ASCT2, can significantly attenuate tumor proliferation in malignant melanoma.27 However, this needs to be further verified in TC.
L-Type Amino Acid Transporters (LATs) and Energy Metabolism in TC
Physiological Role of LATs in the Thyroid Gland
The SLC7 solute carrier family consists of two subfamilies: LATs and cationic amino acid transporters (CATs).137 The LAT subfamily comprises four members, LAT1-4, which are widely distributed on the plasma membrane of various specific cells in the human body, and their key function is to participate in the transport of essential amino acids (EAAs) throughout the human body.138 Furthermore, due to differences in their function and structure, the four members of the LATs are divided into two groups. LAT1 and LAT2, which have a high affinity for EAAs, must combine with the 4F2 antigen heavy chain (4F2hc) to constitute a heterodimer.139,140 However, LAT3 and LAT4 do not need to form a heterodimer; they can directly participate in EAA transport, but with low affinity.141,142 Amino acids provide a nitrogen source for the synthesis of nucleotides, amino sugars and proteins in cells; meanwhile, the carbon skeleton of amino acids can be used for OXPHOS to produce ATP, which is also involved in lipid synthesis in cells.143 Therefore, as carriers of human amino acids, LATs play a very important role in human metabolism. Of the four members of the LAT family, LAT1, which is encoded by the SLC7A5 gene, has been the most extensively studied, so it has existing research value in thyroid physiology and pathology. In polarized epithelial cells, LAT1 is mainly localized to the basolateral membrane.137,144 On the cell membrane, LAT1 and 4F2hc constitute a heterodimer that transports EAAs, such as leucine and phenylalanine, and at the same time participate in the exchange of EAAs and glutamine in a Na+-independent manner.139,145 Through this transport function of LAT1, cells can meet their growth and proliferation needs. Unfortunately, however, the expression of LAT1 protein was not detected in healthy thyroid tissue, but it was clearly expressed in pathological tissue.146 The regulatory factors of LAT1 expression in healthy tissues have not been elaborated in detail, as most are regulatory studies in pathological conditions, which will be elaborated in the next part. In summary, LAT1 plays a specific role in human metabolism, and abnormalities in its expression can cause changes in human cell metabolism, which can be used in tumor research.
LAT1 Regulates Metabolism in the Onset of TC
Tumor cells are characterized by high proliferation and metabolism, which depend on the consumption of nutrients such as glucose, glutamine and EAAs. By consuming these nutrients, cells can synthesize proteins and produce ATP.11,147,148 More than 60 years ago, after conducting experiments with HeLa cells, Harry Eagle discovered that EAAs are needed for cell proliferation.149 Studies have reported a higher uptake of EAAs in a variety of tumors than that in healthy tissue, suggesting that EAAs are necessary for cell proliferation.143,150,151 Of the eight EAAs needed by the human body, leucine is worth mentioning because it is an effective activator of mechanistic target of rapamycin kinase complex 1 (mTORC1), and this signaling pathway can promote the growth, proliferation and apoptosis resistance of tumor cells.152 In addition, leucine is an allosteric agent of glutamate dehydrogenase, which can regulate the activity of glutamate dehydrogenase in mitochondria and thus affect glutamate metabolism.147 Therefore, as a transporter of EAAs (including leucine), LAT1 plays an important role in the metabolic regulation of amino acids in tumor cells. Professor Enomoto proved through experiments that LAT1 and 4F2hc are overexpressed in ATC tissues and were expressed in ATC cell lines (8505C, OCUT-2, and OCUT-6); moreover, these two proteins are closely related to the growth and proliferation of tumor cells.146 Furthermore, the experiment verified that the uptake of EAAs decreased and that the mTOR signaling pathway was stalled after inhibition with LAT1.146 Other researchers conducted similar experiments in ATC, PTC and their cell lines (8505c, LNCaP, SW1736, Hth104, KTC1 and TPC-1) and observed that LAT1 was overexpressed in both of these tumor tissues. These studies concluded that this growth was related to the growth, proliferation and prognosis of tumor cells.153 At the same time, they also confirmed that inhibition of LAT1 would affect the mTORC1 pathway.153 From these studies, we can see that LAT1 plays a role in the amino acid metabolism of TC. The regulatory factors of LAT1 expression have not been thoroughly studied in TC, but some other tumor studies have shown that the expression of LAT1 is affected by many factors. Studies have shown that LAT1 expression is regulated by vascular endothelial growth factor and that this process is related to the hypoxic environment of tumor cells.154–156 Studies have also shown that the overexpression of LAT1 is related to the amplification of the MYC gene.157 In addition, one study confirmed that LAT1 expression was related to the Ras-MEK-ERK signaling pathway in a mouse thyroid tumor model.153 Furthermore, studies have confirmed that LAT1 is the target of microRNA-126.158,159 In summary, we can conclude that LAT1 plays an important role in the amino acid metabolism of TC. Various factors in TC caused alterations in LAT1 expression, and LAT1 transported more leucine through these changes to meet the metabolic needs of TC, thus promoting the development of TC. The existence of LAT1 is also associated with the prognosis of TC. At present, there are relevant studies on LAT1 as a therapeutic target in TC. Hafliger et al demonstrated that JPH203, an inhibitor of LAT1, could effectively block LAT1 function and thus inhibit TC proliferation in a mouse model.153 Furthermore, Enomoto et al demonstrated that JPH203 could inhibit the progression of TC via LAT1.146
Conclusions and Perspectives
The prevalence of TC is increasing every year. Although the prognosis may be acceptable, it (along with the recurrence rate) of TC with a high degree of malignancy still cannot be ignored. Alterations in intracellular metabolic status and associated ion transporters are markers of tumorigenesis. However, the relationship among TC, cell metabolism, and ion transporters has not been thoroughly elucidated. We attempted to clarify the physiological and pathophysiological connections (Figure 1). In this paper, the status of NIS in TC related to the treatment of TC and its relationship with metabolism are described, providing both a reference for the treatment of patients resistant to radiotherapy and profound ideas for TC researchers. We also discussed GLUT1, MCT1/4/8, ASCT2, and LAT1 plasma membrane transporters and their association with energy metabolism in TC while also confirming their association with TC and metabolism in previous studies. In TC, these ion transporters can regulate the metabolism of corresponding substances through both their own function and expression changes to meet the needs of TC cells. Although many studies have investigated the abovementioned ion transporters, few effective drugs have been applied in clinical practice, and the incidence of TC and prognosis of patients have not been significantly improved. This paper summarizes the metabolism-related ion transporters, their regulatory factors, and relevant preclinical and clinical trials in TC (Table 1) with the goal of providing hope for TC patients and researchers with in-depth research directions.
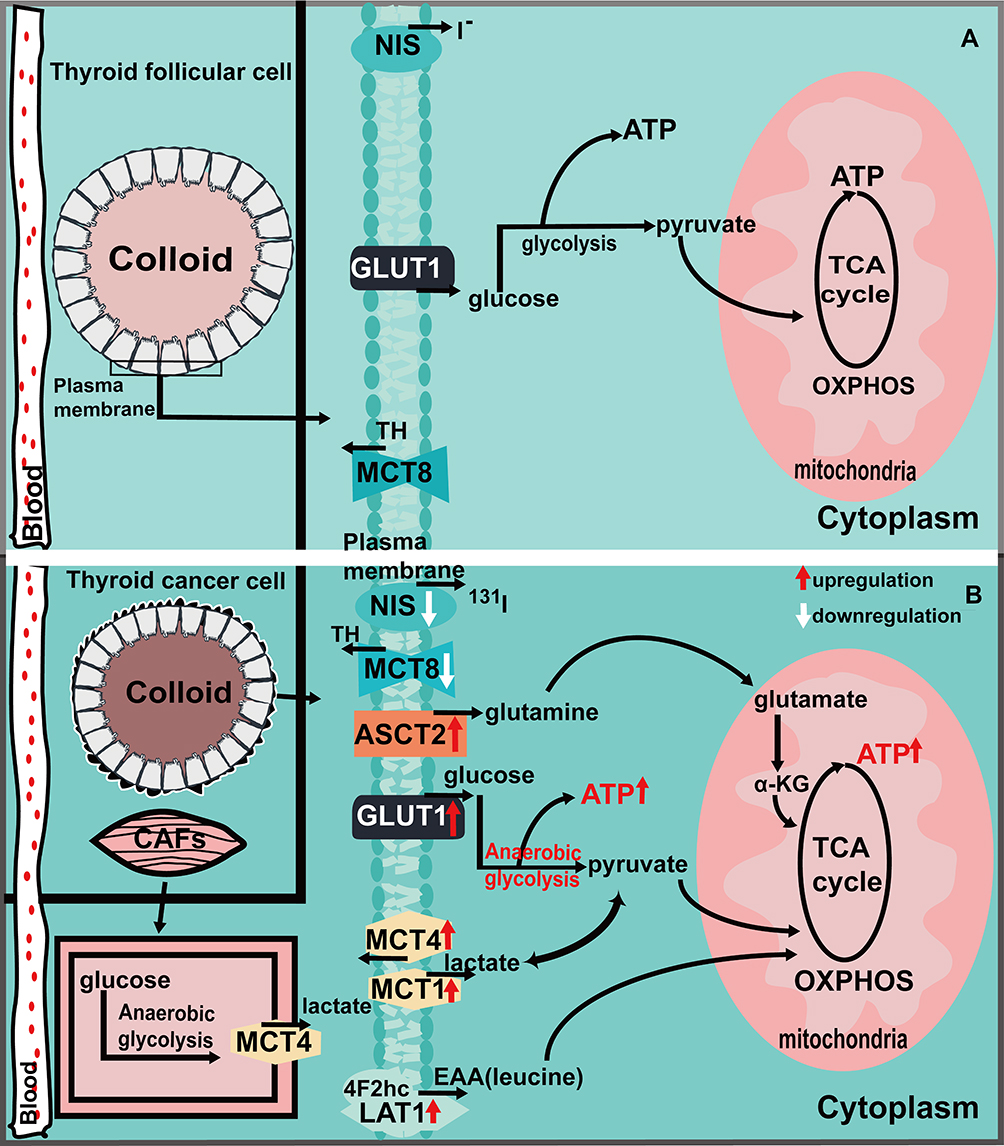 |
Figure 1 Ion transporters and the metabolic situation in normal thyroid cells (A) and cancer cells (B). Figure (A) shows that normal thyroid cells rely on GLUT1 transport of glucose to provide energy through glycolysis and oxidative phosphorylation, NIS is involved in intracellular iodide metabolism, and MCT8 is involved in the transportion of TH. The expression of MCT1/4, ASCT2, and LAT1 has not been elucidated. Figure (B) shows the upregulation of GLUT1, ASCT2, LAT1 and MCT1/4 in thyroid cancer, which regulates the metabolism of tumor cells to meet their growth and proliferation needs through the transport of corresponding metabolic substrates. The downregulated expression of NIS leads to the decreased function of iodine intake, affects the treatment of radioactive iodine, and promotes the poor prognosis of the tumor. The downregulation of MCT8 expression affects the transport of TH, thus affecting the progression of thyroid cancer. |
Abbreviations
α-KG, α-ketoglutarate; AMPK, adenosine monophosphate-activated protein kinase; ATP, adenosine triphosphate; ASCTs, amino acid transporters; ATC, anaplastic thyroid cancer; CAFs, cancer-associated fibroblasts; cAMP, cyclic adenosine 3ʹ,5ʹ-monophosphate; HIF-1α, hypoxia-inducible factor 1 alpha; EAA, essential amino acids; GLUTs, glucose transporters; LATs, L-neutral amino acid transporters; LDH, lactate dehydrogenase; MAPK, mitogen-activated protein kinases; MCTs, monocarboxylate transporters; MTC, medullary thyroid cancer; mTOR, mammalian target-of-rapamycin; mTORC1, mechanistic target of rapamycin kinase complex 1; NIS, sodium-iodide symporter; OXPHOS, oxidative phosphorylation; PTC, papillary thyroid cancer; PI3K/AKT, phosphatidylinositol 3 kinase/protein kinase B; RAI, radioactive iodine; TC, thyroid cancer; TCA, tricarboxylic acid; TH, thyroid hormone; TSH, thyroid-stimulating hormone; 4F2hc, 4F2 antigen heavy chain; T3, 3, 3ʹ, 5-triiodothyronine.
Data Sharing Statement
Not applicable.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Acknowledgments
We thank JXZ, GRW, HJ and JXA, who gave us the suggestions for the paper and support for the daily experiments.
Funding
This research was supported by the National Natural Science Foundation of China (81660098 to T.L.L., 81860103 and 82070536 to X.M.L. and 81572438 to B.G.T.), the Outstanding Scientific Youth Fund of Guizhou Province (2017-5608 to X.M.L.), and the 15851 Talent Projects of Zunyi City (to T.L.L).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. doi:10.1085/jgp.8.6.519
3. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13(6):472–482. doi:10.1016/j.ccr.2008.05.005
4. Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274(6):1393–1418. doi:10.1111/j.1742-4658.2007.05686.x
5. Som P, Atkins HL, Bandoypadhyay D, et al. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med. 1980;21(7):670–675.
6. DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29(3):313–324. doi:10.1038/onc.2009.358
7. Pedersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res. 1978;22:190–274. doi:10.1159/000401202
8. Rodríguez-Enríquez S, Torres-Márquez ME, Moreno-Sánchez R. Substrate oxidation and ATP supply in AS-30D hepatoma cells. Arch Biochem Biophys. 2000;375(1):21–30. doi:10.1006/abbi.1999.1582
9. Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254(8):2669–2676.
10. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Ann Review Biomed Eng. 2017;19(1):163–194. doi:10.1146/annurev-bioeng-071516-044546
11. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi:10.1126/science.1160809
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
13. Pavlides S, Whitaker-Menezes D, Castello-Cros R, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. doi:10.4161/cc.8.23.10238
14. Xu XD, Shao SX, Jiang HP, et al. Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol Res Treat. 2015;38(3):117–122. doi:10.1159/000375435
15. Coelho RG, Fortunato RS, Carvalho DP. Metabolic reprogramming in thyroid carcinoma. Front Oncol. 2018;8:82. doi:10.3389/fonc.2018.00082
16. Halestrap AP, Wilson MC. The monocarboxylate transporter family–role and regulation. IUBMB Life. 2012;64(2):109–119. doi:10.1002/iub.572
17. Prevarskaya N, Skryma R, Shuba Y. Ion channels and the hallmarks of cancer. Trends Mol Med. 2010;16(3):107–121. doi:10.1016/j.molmed.2010.01.005
18. Medina RA, Owen GI. Glucose transporters: expression, regulation and cancer. Biol Res. 2002;35(1):9–26. doi:10.4067/s0716-97602002000100004
19. Becchetti A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am J Physiol Cell Physiol. 2011;301(2):C255–C265. doi:10.1152/ajpcell.00047.2011
20. Litan A, Langhans SA. Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front Cell Neurosci. 2015;9:86. doi:10.3389/fncel.2015.00086
21. Bizhanova A, Kopp P. Minireview: the sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology. 2009;150(3):1084–1090. doi:10.1210/en.2008-1437
22. Smanik PA, Ryu KY, Theil KS, Mazzaferri EL, Jhiang SM. Expression, exon-intron organization, and chromosome mapping of the human sodium iodide symporter. Endocrinology. 1997;138(8):3555–3558. doi:10.1210/endo.138.8.5262
23. Badziong J, Ting S, Synoracki S, et al. Differential regulation of monocarboxylate transporter 8 expression in thyroid cancer and hyperthyroidism. Eur J Endocrinol. 2017;177(3):243–250. doi:10.1530/eje-17-0279
24. Chen M, Shen M, Li Y, et al. GC-MS-based metabolomic analysis of human papillary thyroid carcinoma tissue. Int J Mol Med. 2015;36(6):1607–1614. doi:10.3892/ijmm.2015.2368
25. Nahm JH, Kim HM, Koo JS. Glycolysis-related protein expression in thyroid cancer. Tumour Biol. 2017;39(3):1010428317695922. doi:10.1177/1010428317695922
26. Grabellus F, Nagarajah J, Bockisch A, Schmid KW, Sheu S-Y. Glucose transporter 1 expression, tumor proliferation, and iodine/glucose uptake in thyroid cancer with emphasis on poorly differentiated thyroid carcinoma. Clin Nucl Med. 2012;37(2):121–127. doi:10.1097/RLU.0b013e3182393599
27. Kim HM, Lee YK, Koo JS. Expression of glutamine metabolism-related proteins in thyroid cancer. Oncotarget. 2016;7(33):53628–53641. doi:10.18632/oncotarget.10682
28. Häfliger P, Charles R-P. The L-type amino acid transporter LAT1-an emerging target in cancer. Int J Mol Sci. 2019;20(10):E2428. doi:10.3390/ijms20102428
29. Portulano C, Paroder-Belenitsky M, Carrasco N. The Na+/I- symporter (NIS): mechanism and medical impact. Endocr Rev. 2014;35(1):106–149. doi:10.1210/er.2012-1036
30. Carvalho DP, Ferreira ACF. The importance of sodium/iodide symporter (NIS) for thyroid cancer management. Arq Bras Endocrinol Metabol. 2007;51(5):672–682. doi:10.1590/s0004-27302007000500004
31. Riedel C, Levy O, Carrasco N. Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem. 2001;276(24):21458–21463. doi:10.1074/jbc.M100561200
32. Kaminsky SM, Levy O, Salvador C, Dai G, Carrasco N. Na(+)-I- symport activity is present in membrane vesicles from thyrotropin-deprived non-I(-)-transporting cultured thyroid cells. Proc Natl Acad Sci USA. 1994;91(9):3789–3793. doi:10.1073/pnas.91.9.3789
33. de Souza ECL, Padrón AS, Braga WMO, et al. MTOR downregulates iodide uptake in thyrocytes. J Endocrinol. 2010;206(1):113–120. doi:10.1677/JOE-09-0436
34. Andrade BM, Araujo RL, Perry RLS, et al. A novel role for AMP-kinase in the regulation of the Na+/I–symporter and iodide uptake in the rat thyroid gland. Am J Physiol Cell Physiol. 2011;300(6):C1291–C1297. doi:10.1152/ajpcell.00136.2010
35. Levy O, Carrasco N. Structure and function of the thyroid iodide transporter and its implications for thyroid disease. Curr Opin Endocrinol Diabetes Obes. 1997;4(5):364–370.
36. Haddad RI, Nasr C, Bischoff L, et al. NCCN guidelines insights: thyroid carcinoma, version 2.2018. J Natl Compr Canc Netw. 2018;16(12):1429–1440. doi:10.6004/jnccn.2018.0089
37. Mian C, Barollo S, Pennelli G, et al. Molecular characteristics in papillary thyroid cancers (PTCs) with no 131I uptake. Clin Endocrinol (Oxf). 2008;68(1):108–116. doi:10.1111/j.1365-2265.2007.03008.x
38. Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature. 1996;379(6564):458–460. doi:10.1038/379458a0
39. Ajjan RA, Kamaruddin NA, Crisp M, Watson PF, Ludgate M, Weetman AP. Regulation and tissue distribution of the human sodium iodide symporter gene. Clin Endocrinol (Oxf). 1998;49(4):517–523. doi:10.1046/j.1365-2265.1998.00570.x
40. Lazar V, Bidart JM, Caillou B, et al. Expression of the Na+/I- symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab. 1999;84(9):3228–3234. doi:10.1210/jcem.84.9.5996
41. Dohán O, De la Vieja A, Paroder V, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24(1):48–77. doi:10.1210/er.2001-0029
42. Filetti S, Bidart JM, Arturi F, Caillou B, Russo D, Schlumberger M. Sodium/iodide symporter: a key transport system in thyroid cancer cell metabolism. Eur J Endocrinol. 1999;141(5):443–457. doi:10.1530/eje.0.1410443
43. Filetti S, Rapoport B. Autoregulation by iodine of thyroid protein synthesis: influence of iodine on amino acid transport in cultured thyroid cells. Endocrinology. 1984;114(4):1379–1385. doi:10.1210/endo-114-4-1379
44. Ricarte-Filho JC, Ryder M, Chitale DA, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69(11):4885–4893. doi:10.1158/0008-5472.CAN-09-0727
45. Chakravarty D, Santos E, Ryder M, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest. 2011;121(12):4700–4711. doi:10.1172/JCI46382
46. Beyer S, Lakshmanan A, Liu YY, et al. KT5823 differentially modulates sodium iodide symporter expression, activity, and glycosylation between thyroid and breast cancer cells. Endocrinology. 2011;152(3):782–792. doi:10.1210/en.2010-0782
47. Abdulrahman RM, Boon MR, Sips HCM, et al. Impact of Metformin and compound C on NIS expression and iodine uptake in vitro and in vivo: a role for CRE in AMPK modulation of thyroid function. Thyroid. 2014;24(1):78–87. doi:10.1089/thy.2013.0041
48. Schmutzler C, Köhrle J. Implications of the molecular characterization of the sodium-iodide symporter (NIS). Exp Clin Endocrinol Diabetes. 1998;106(Suppl 3):S1–10. doi:10.1055/s-0029-1212036
49. Lakshmanan A, Scarberry D, Shen DH, Jhiang SM. Modulation of sodium iodide symporter in thyroid cancer. Horm Cancer. 2014;5(6):363–373. doi:10.1007/s12672-014-0203-0
50. Riesco-Eizaguirre G, Rodríguez I, De la Vieja A, et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 2009;69(21):8317–8325. doi:10.1158/0008-5472.CAN-09-1248
51. Kogai T, Sajid-Crockett S, Newmarch LS, Liu -Y-Y, Brent GA. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J Endocrinol. 2008;199(2):243–252. doi:10.1677/JOE-08-0333
52. Andrade BM, Cazarin J, Zancan P, Carvalho DP. AMP-activated protein kinase upregulates glucose uptake in thyroid PCCL3 cells independent of thyrotropin. Thyroid. 2012;22(10):1063–1068. doi:10.1089/thy.2012.0041
53. Vidal AP, Andrade BM, Vaisman F, et al. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur J Endocrinol. 2013;169(4):521–528. doi:10.1530/EJE-13-0284
54. Andrade BM, de Carvalho DP. Perspectives of the AMP-activated kinase (AMPK) signalling pathway in thyroid cancer. Biosci Rep. 2014;34(2). doi:10.1042/BSR20130134
55. Cazarin JM, Andrade BM, Carvalho DP. AMP-activated protein kinase activation leads to lysome-mediated NA(+)/I(-)-symporter protein degradation in rat thyroid cells. Hormone Metabol Res. 2014;46(5):313–317. doi:10.1055/s-0034-1371803
56. Lakshmanan A, Wojcicka A, Kotlarek M, Zhang X, Jazdzewski K, Jhiang SM. microRNA-339-5p modulates Na+/I- symporter-mediated radioiodide uptake. Endocr Relat Cancer. 2015;22(1):11–21. doi:10.1530/erc-14-0439
57. Fletcher A, Read ML, Thornton CEM, et al. Targeting novel sodium iodide symporter interactors ADP-ribosylation factor 4 and valosin-containing protein enhances radioiodine uptake. Cancer Res. 2020;80(1):102–115. doi:10.1158/0008-5472.can-19-1957
58. ElMokh O, Taelman V, Radojewski P, et al. MEK inhibition induces therapeutic iodine uptake in a murine model of anaplastic thyroid cancer. J Nucl Med. 2019;60(7):917–923. doi:10.2967/jnumed.118.216721
59. Augustin R. The protein family of glucose transport facilitators: it’s not only about glucose after all. IUBMB Life. 2010;62(5):315–333. doi:10.1002/iub.315
60. Mueckler M, Caruso C, Baldwin SA, et al. Sequence and structure of a human glucose transporter. Science. 1985;229(4717):941–945. doi:10.1126/science.3839598
61. Matsuzu K, Segade F, Wong M, Clark OH, Perrier ND, Bowden DW. Glucose Transporters in the Thyroid. Thyroid. 2005;15(6):545–550. doi:10.1089/thy.2005.15.545
62. Samih N, Hovsepian S, Aouani A, Lombardo D, Fayet G. Glut-1 translocation in FRTL-5 thyroid cells: role of phosphatidylinositol 3-kinase and N-glycosylation. Endocrinology. 2000;141(11):4146–4155. doi:10.1210/endo.141.11.7793
63. Filetti S, Vetri M, Damante G, Belfiore A. Thyroid autoregulation: effect of iodine on glucose transport in cultured thyroid cells. Endocrinology. 1986;118(4):1395–1400. doi:10.1210/endo-118-4-1395
64. Filetti S, Damante G, Foti D. Thyrotropin stimulates glucose transport in cultured rat thyroid cells. Endocrinology. 1987;120(6):2576–2581. doi:10.1210/endo-120-6-2576
65. Hosaka Y, Tawata M, Kurihara A, Ohtaka M, Endo T, Onaya T. The regulation of two distinct glucose transporter (GLUT1 and GLUT4) gene expressions in cultured rat thyroid cells by thyrotropin. Endocrinology. 1992;131(1):159–165. doi:10.1210/endo.131.1.1319316
66. Carruthers A, DeZutter J, Ganguly A, Devaskar SU. Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab. 2009;297(4):E836–E848. doi:10.1152/ajpendo.00496.2009
67. Yamamoto T, Seino Y, Fukumoto H, et al. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170(1):223–230. doi:10.1016/0006-291x(90)91263-r
68. Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J. Wide expression of the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Res. 1996;56(5):1164–1167.
69. Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89(1):3–9. doi:10.1079/BJN2002763
70. Ziegler A, von Kienlin M, Décorps M, Rémy C. High glycolytic activity in rat glioma demonstrated in vivo by correlation peak 1H magnetic resonance imaging. Cancer Res. 2001;61(14):5595–5600.
71. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi:10.1038/nrc1478
72. Heydarzadeh S, Moshtaghie AA, Daneshpoor M, Hedayati M. Regulators of glucose uptake in thyroid cancer cell lines. Cell Commun Signal. 2020;18(1). doi:10.1186/s12964-020-00586-x
73. Prante O, Maschauer S, Fremont V, et al. Regulation of uptake of 18F-FDG by a follicular human thyroid cancer cell line with mutation-activated K-ras. J Nucl Med. 2009;50(8):1364–1370. doi:10.2967/jnumed.109.062331
74. Jóźwiak P, Krześlak A, Bryś M, Lipińska A. Glucose-dependent glucose transporter 1 expression and its impact on viability of thyroid cancer cells. Oncol Rep. 2015;33(2):913–920. doi:10.3892/or.2014.3673
75. Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi:10.1074/jbc.M800102200
76. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–713. doi:10.1038/nrc2468
77. Haber RS, Weiser KR, Pritsker A, Reder I, Burstein DE. GLUT1 glucose transporter expression in benign and malignant thyroid nodules. Thyroid. 1997;7(3):363–367. doi:10.1089/thy.1997.7.363
78. Yun J, Rago C, Cheong I, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325(5947):1555–1559. doi:10.1126/science.1174229
79. Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987;235(4795):1492–1495. doi:10.1126/science.3103217
80. Osthus RC, Shim H, Kim S, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275(29):21797–21800. doi:10.1074/jbc.C000023200
81. Zhang C, Liu J, Liang Y, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi:10.1038/ncomms3935
82. Morani F, Phadngam S, Follo C, et al. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J Mol Endocrinol. 2014;53(2):247–258. doi:10.1530/JME-14-0118
83. Zhang G, Zhou S, Yang Q, Liu F. MicroRNA-125b reduces glucose uptake in papillary thyroid carcinoma cells. Oncol Lett. 2020;20(3):2806–2810. doi:10.3892/ol.2020.11832
84. Shen CT, Wei WJ, Qiu ZL, et al. Metformin reduces glycometabolism of papillary thyroid carcinoma in vitro and in vivo. J Mol Endocrinol. 2017;58(1):15–23. doi:10.1530/jme-16-0134
85. Zambrano A, Molt M, Uribe E, Salas M. Glut 1 in cancer cells and the inhibitory action of resveratrol as a potential therapeutic strategy. Int J Mol Sci. 2019;20(13). doi:10.3390/ijms20133374
86. Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447(5):619–628. doi:10.1007/s00424-003-1067-2
87. Johnson JM, Lai SY, Cotzia P, et al. Mitochondrial metabolism as a treatment target in anaplastic thyroid cancer. Semin Oncol. 2015;42(6):915–922. doi:10.1053/j.seminoncol.2015.09.025
88. Halestrap AP. The monocarboxylate transporter family–Structure and functional characterization. IUBMB Life. 2012;64(1):1–9. doi:10.1002/iub.573
89. Gill KS, Tassone P, Hamilton J, et al. Thyroid cancer metabolism: a review. J Thyroid Disord Ther. 2016;5(1):200. doi:10.4172/2167-7948.1000200
90. Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism. J Biol Chem. 2006;281(14):9030–9037. doi:10.1074/jbc.M511397200
91. Heuer H, Visser TJ. Pathophysiological importance of thyroid hormone transporters. Endocrinology. 2009;150(3):1078–1083. doi:10.1210/en.2008-1518
92. Visser WE, Friesema ECH, Jansen J, Visser TJ. Thyroid hormone transport in and out of cells. Trends Endocrinol Metabol. 2008;19(2):50–56. doi:10.1016/j.tem.2007.11.003
93. Alfarouk KO, Shayoub MEA, Muddathir AK, Elhassan GO, Bashir AHH. Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (dismantling of multicellularity). Cancers (Basel). 2011;3(3):3002–3017. doi:10.3390/cancers3033002
94. Mathupala SP, Colen CB, Parajuli P, Sloan AE. Lactate and malignant tumors: a therapeutic target at the end stage of glycolysis. J Bioenerg Biomembr. 2007;39(1):73–77. doi:10.1007/s10863-006-9062-x
95. Weinhouse S, Warburg O, Burk D, Schade AL. On respiratory impairment in cancer cells. Science. 1956;124(3215):267–272. doi:10.1126/science.124.3215.267
96. Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. 2008;134(5):703–707. doi:10.1016/j.cell.2008.08.021
97. Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5(15):1597–1601. doi:10.4161/cc.5.15.3112
98. Sun W-Y, Jung W-H, Koo JS. Expression of cancer-associated fibroblast-related proteins in thyroid papillary carcinoma. Tumour Biol. 2016;37(6):8197–8207. doi:10.1007/s13277-015-4684-4
99. Curry JM, Tuluc M, Whitaker-Menezes D, et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle. 2013;12(9):1371–1384. doi:10.4161/cc.24092
100. Curry JM, Tassone P, Cotzia P, et al. Multicompartment metabolism in papillary thyroid cancer. Laryngoscope. 2016;126(10):2410–2418. doi:10.1002/lary.25799
101. Gotanda Y, Akagi Y, Kawahara A, et al. Expression of monocarboxylate transporter (MCT)-4 in colorectal cancer and its role: MCT4 contributes to the growth of colorectal cancer with vascular endothelial growth factor. Anticancer Res. 2013;33(7):2941–2947.
102. Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, Baltazar F. Role of monocarboxylate transporters in human cancers: state of the art. J Bioenerg Biomembr. 2012;44(1):127–139. doi:10.1007/s10863-012-9428-1
103. Romero-Cordoba SL, Rodriguez-Cuevas S, Bautista-Pina V, et al. Loss of function of miR-342-3p results in MCT1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. Sci Rep. 2018;8(1):12252. doi:10.1038/s41598-018-29708-9
104. Zhao Y, Li W, Li M, et al. Targeted inhibition of MCT4 disrupts intracellular pH homeostasis and confers self-regulated apoptosis on hepatocellular carcinoma. Exp Cell Res. 2019;384(1):111591. doi:10.1016/j.yexcr.2019.111591
105. Liu YC, Yeh CT, Lin KH. Molecular functions of thyroid hormone signaling in regulation of cancer progression and anti-apoptosis. Int J Mol Sci. 2019;20(20). doi:10.3390/ijms20204986
106. Smith VE, Read ML, Turnell AS, et al. PTTG-binding factor (PBF) is a novel regulator of the thyroid hormone transporter MCT8. Endocrinology. 2012;153(7):3526–3536. doi:10.1210/en.2011-2030
107. Hudson J, Duncavage E, Tamburrino A, et al. Overexpression of miR-10a and miR-375 and downregulation of YAP1 in medullary thyroid carcinoma. Exp Mol Pathol. 2013;95(1):62–67. doi:10.1016/j.yexmp.2013.05.001
108. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123(9):3685–3692. doi:10.1172/jci69741
109. Yadav S, Pandey SK, Kumar A, Kujur PK, Singh RP, Singh SM. Antitumor and chemosensitizing action of 3-bromopyruvate: implication of deregulated metabolism. Chem Biol Interact. 2017;270:73–89. doi:10.1016/j.cbi.2017.04.015
110. Puri S, Juvale K. Monocarboxylate transporter 1 and 4 inhibitors as potential therapeutics for treating solid tumours: a review with structure-activity relationship insights. Eur J Med Chem. 2020;199:112393. doi:10.1016/j.ejmech.2020.112393
111. Polański R, Hodgkinson CL, Fusi A, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014;20(4):926–937. doi:10.1158/1078-0432.ccr-13-2270
112. Marchiq I, Pouysségur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berlin, Germany). 2016;94(2):155–171. doi:10.1007/s00109-015-1307-x
113. Futagi Y, Kobayashi M, Narumi K, Furugen A, Iseki K. Identification of a selective inhibitor of human monocarboxylate transporter 4. Biochem Biophys Res Commun. 2018;495(1):427–432. doi:10.1016/j.bbrc.2017.10.025
114. Krajewska J, Jarzab B. Novel therapies for thyroid cancer. Expert Opin Pharmacother. 2014;15(18):2641–2652. doi:10.1517/14656566.2014.969240
115. Kanai Y, Clémençon B, Simonin A, et al. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol Aspects Med. 2013;34(2–3):108–120. doi:10.1016/j.mam.2013.01.001
116. Poffenberger MC, Jones RG. Amino acids fuel T cell-mediated inflammation. Immunity. 2014;40(5):635–637. doi:10.1016/j.immuni.2014.04.017
117. Arriza JL, Kavanaugh MP, Fairman WA, et al. Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J Biol Chem. 1993;268(21):15329–15332.
118. Shafqat S, Tamarappoo BK, Kilberg MS, et al. Cloning and expression of a novel Na(+)-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J Biol Chem. 1993;268(21):15351–15355.
119. Bröer A, Wagner C, Lang F, Bröer S. Neutral amino acid transporter ASCT2 displays substrate-induced Na+ exchange and a substrate-gated anion conductance. Biochem J. 2000;346(Pt 3):705–710.
120. Newsholme P, Procopio J, Lima MMR, Pithon-Curi TC, Curi R. Glutamine and glutamate–their central role in cell metabolism and function. Cell Biochem Funct. 2003;21(1):1–9. doi:10.1002/cbf.1003
121. Pochini L, Scalise M, Galluccio M, Indiveri C. Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Front Chem. 2014;2:61. doi:10.3389/fchem.2014.00061
122. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi:10.1016/j.ccr.2012.02.014
123. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309. doi:10.1126/science.123.3191.309
124. Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. doi:10.1038/nrc3365
125. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2(10):881–898. doi:10.1158/2159-8290.CD-12-0345
126. Yang L, Moss T, Mangala LS, et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol. 2014;10:728. doi:10.1002/msb.20134892
127. Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 2005;15(4):254–266. doi:10.1016/j.semcancer.2005.04.005
128. Huang F, Zhang Q, Ma H, Lv Q, Zhang T. Expression of glutaminase is upregulated in colorectal cancer and of clinical significance. Int J Clin Exp Pathol. 2014;7(3):1093–1100.
129. Kim S, Jung WH, Koo JS. The expression of glutamine-metabolism-related proteins in breast phyllodes tumors. Tumour Biol. 2013;34(5):2683–2689. doi:10.1007/s13277-013-0819-7
130. Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res. 2007;13(4):1341–1349. doi:10.1158/1078-0432.CCR-06-1753
131. Riesco-Eizaguirre G, Gutiérrez-Martínez P, García-Cabezas MA, Nistal M, Santisteban P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I- targeting to the membrane. Endocr Relat Cancer. 2006;13(1):257–269. doi:10.1677/erc.1.01119
132. Wang DG, Liu WH, Johnston CF, Sloan JM, Buchanan KD. Bcl-2 and c-Myc, but not bax and p53, are expressed during human medullary thyroid tumorigenesis. Am J Pathol. 1998;152(6):1407–1413.
133. Boultwood J, Wyllie FS, Williams ED, Wynford-Thomas D. N-myc expression in neoplasia of human thyroid C-cells. Cancer Res. 1988;48(14):4073–4077.
134. Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105(48):18782–18787. doi:10.1073/pnas.0810199105
135. Dong J, Xiao D, Zhao Z, et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis. 2017;6(7):e356. doi:10.1038/oncsis.2017.59
136. Scalise M, Pochini L, Console L, Losso MA, Indiveri C. The human SLC1A5 (ASCT2) amino acid transporter: from function to structure and role in cell biology. Front Cell Dev Biol. 2018;6:96. doi:10.3389/fcell.2018.00096
137. Fotiadis D, Kanai Y, Palacín M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med. 2013;34(2–3):139–158. doi:10.1016/j.mam.2012.10.007
138. Wang Q, Holst J. L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. Am J Cancer Res. 2015;5(4):1281–1294.
139. Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J Biol Chem. 1998;273(37):23629–23632. doi:10.1074/jbc.273.37.23629
140. Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem. 1999;274(28):19745–19751. doi:10.1074/jbc.274.28.19745
141. Babu E, Kanai Y, Chairoungdua A, et al. Identification of a novel system L amino acid transporter structurally distinct from heterodimeric amino acid transporters. J Biol Chem. 2003;278(44):43838–43845. doi:10.1074/jbc.M305221200
142. Bodoy S, Martín L, Zorzano A, Palacín M, Estévez R, Bertran J. Identification of LAT4, a novel amino acid transporter with system L activity. J Biol Chem. 2005;280(12):12002–12011. doi:10.1074/jbc.M408638200
143. Hosios AM, Hecht VC, Danai LV, et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell. 2016;36(5):540–549. doi:10.1016/j.devcel.2016.02.012
144. Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 2004;447(5):532–542. doi:10.1007/s00424-003-1086-z
145. Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136(3):521–534. doi:10.1016/j.cell.2008.11.044
146. Enomoto K, Sato F, Tamagawa S, et al. A novel therapeutic approach for anaplastic thyroid cancer through inhibition of LAT1. Sci Rep. 2019;9(1):14616. doi:10.1038/s41598-019-51144-6
147. Scalise M, Pochini L, Galluccio M, Console L, Indiveri C. Glutamine transport and mitochondrial metabolism in cancer cell growth. Front Oncol. 2017;7:306. doi:10.3389/fonc.2017.00306
148. Bhutia YD, Ganapathy V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta. 2016;1863(10):2531–2539. doi:10.1016/j.bbamcr.2015.12.017
149. Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. 1955;122(3168):501–514. doi:10.1126/science.122.3168.501
150. Qi W, Guan Q, Sun T, Cao Y, Zhang L, Guo Y. Improving detection sensitivity of amino acids in thyroid tissues by using phthalic acid as a mobile phase additive in hydrophilic interaction chromatography-electrospray ionization-tandem mass spectrometry. Anal Chim Acta. 2015;870:75–82. doi:10.1016/j.aca.2015.02.048
151. Wang LB, Shen JG, Zhang SZ, Ding KF, Zheng S. Amino acid uptake in arterio-venous serum of normal and cancerous colon tissues. World J Gastroenterol. 2004;10(9):1297–1300. doi:10.3748/wjg.v10.i9.1297
152. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–371. doi:10.1016/j.cell.2017.03.035
153. Häfliger P, Graff J, Rubin M, et al. The LAT1 inhibitor JPH203 reduces growth of thyroid carcinoma in a fully immunocompetent mouse model. J Exp Clin Cancer Res. 2018;37(1):234. doi:10.1186/s13046-018-0907-z
154. Kaira K, Oriuchi N, Imai H, et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) and 4F2 heavy chain (CD98) expression in early stage squamous cell carcinoma of the lung. Cancer Sci. 2009;100(2):248–254. doi:10.1111/j.1349-7006.2008.01029.x
155. Kaira K, Sunose Y, Arakawa K, et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br J Cancer. 2012;107(4):632–638. doi:10.1038/bjc.2012.310
156. Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270(22):13333–13340. doi:10.1074/jbc.270.22.13333
157. Hayashi K, Jutabha P, Endou H, Anzai N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol Rep. 2012;28(3):862–866. doi:10.3892/or.2012.1878
158. Cao D, Mikosz AM, Ringsby AJ, et al. MicroRNA-126-3p inhibits angiogenic function of human lung microvascular endothelial cells via LAT1 (L-type amino acid transporter 1)-mediated mTOR (Mammalian Target of Rapamycin) signaling. Arterioscler Thromb Vasc Biol. 2020;40(5):1195–1206. doi:10.1161/atvbaha.119.313800
159. Wang J, Chen X, Su L, et al. MicroRNA-126 inhibits cell proliferation in gastric cancer by targeting LAT-1. Biomed Pharmacother. 2015;72:66–73. doi:10.1016/j.biopha.2015.04.001
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.