Back to Journals » International Journal of General Medicine » Volume 19
Phosphatidylethanolamine Alleviates Osteoarthritis Progression by Inhibiting Oxidative Stress-Induced Chondrocyte Ferroptosis in a Lysosomal-Dependent Manner
Authors Zheng H ![]() , Zhu Z, Zhu J, Chen D, Ruan R, Ning R
, Zhu Z, Zhu J, Chen D, Ruan R, Ning R
Received 29 November 2025
Accepted for publication 30 March 2026
Published 14 April 2026 Volume 2026:19 581128
DOI https://doi.org/10.2147/IJGM.S581128
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Vinay Kumar
Hanlin Zheng,1,2,* Zhengming Zhu,1,2,* Jiawang Zhu,3,* Desheng Chen,3 Renzhi Ruan,1,2 Rende Ning1,2
1Department of Orthopedics, The Third Affiliated Hospital of Anhui Medical University, Hefei, 230061, People’s Republic of China; 2Department of Orthopedics, The First People’s Hospital of Hefei, Hefei, 230061, People’s Republic of China; 3Department of Sports Medicine and Arthroscopy, Tianjin Hospital of Tianjin University, Tianjin, 300210, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Rende Ning, Department of Orthopedics, The Third Affiliated Hospital of Anhui Medical University, Huaihe Road 390#, Hefei, 230061, People’s Republic of China, Email [email protected]
Purpose: Lysosomal dysfunction and chondrocyte ferroptosis are pivotal drivers of osteoarthritis (OA) pathogenesis, yet their interlinked molecular mechanisms remain poorly defined. This study investigates the associations between lysosomal dysfunctions and ferroptosis in OA chondrocytes, aiming to identify actionable therapeutic targets.
Patients and Methods: Human OA cartilage samples were categorized into intact and damaged groups based on structural integrity. Lysosomal fractions were isolated from both groups for comparative lipidomic profiling and functional assays. A rat OA model was established via anterior cruciate ligament transection, followed by histopathological evaluation using hematoxylin-eosin (HE) staining, Safranin-O Fast Green scoring, and immunohistochemical analysis to quantify cartilage repair and degeneration.
Results: The damaged groups displayed significantly increased lysosomal membrane permeability (LMP) and ferroptosis activation compared to intact groups. Lysosomal lipidomics revealed oxidative stress-induced down-regulation of phosphatidylethanolamine (PE), a key membrane-stabilizing phospholipid, in chondrocytes. Functional studies demonstrated that PE supplementation rescued chondrocyte viability (CCK-8 assay) and attenuated LMP-driven ferroptosis by restoring lysosomal integrity and suppressing lipid peroxidation. In vivo, intra-articular PE administration markedly reduced OA progression, as evidenced by improved cartilage histology scores, and downregulated ferroptosis markers.
Conclusion: PE supplementation restores lysosomal PE levels, reduces LMP, and alleviates ferroptotic phenotypes in preclinical models, suggesting therapeutic potential. These findings significantly increase our understanding of the pathogenesis of OA and reveal potential therapeutic targets for its management.
Keywords: osteoarthritis, LMP, ferroptosis, PE, lipidomics
Background
Osteoarthritis (OA), a degenerative joint disease, represents a substantial threat to the quality of life of middle-aged and elderly individuals. Chronic pain, joint stiffness, and reduced mobility significantly impair daily activities, often leading to physical decline, social isolation, and depression.1 Over 30% of adults aged 65 and older experience symptomatic OA, with the knee and hip joints being the most frequently affected areas.2 This condition not only exacerbates age-related comorbidities such as obesity and cardiovascular risk but also diminishes independence in performing essential tasks.3 The therapeutic landscape remains challenging owing to the limited availability of disease-modifying treatments. Current treatment strategies predominantly focus on symptom management via the use of non-steroidal antiinflammatory drug (NSAIDs), which poses risks of gastrointestinal and cardiovascular complications with prolonged use, or invasive joint replacement surgeries, which involve surgical risks and considerable financial burdens.4 The contrast between therapeutic challenges and the increasing prevalence of OA in aging populations underscores the pressing need for innovative treatment strategies that address both the biological and psychosocial aspects of this debilitating condition.
Ferroptosis, a form of iron-dependent regulated cell death characterized by lipid peroxidation and inactivation of glutathione peroxidase 4 (GPX4), has emerged as a critical factor in the pathogenesis of OA.5,6 In OA, chondrocytes undergo metabolic disturbances and oxidative stress, which create an environment conducive to ferroptotic processes. The accumulation of excess iron in articular cartilage disrupts redox homeostasis, promoting the generation of reactive oxygen species (ROS) and driving lipid peroxidation.7,8 Meanwhile, mechanical stress and inflammation elevate ROS levels in chondrocytes, surpassing the capacity of GPX4 and facilitating chondrocyte ferroptosis. Ferroptosis-induced chondrocyte death accelerates extracellular matrix degradation through the upregulation of matrix metalloproteinases and aggrecanases while concurrently inhibiting collagen synthesis. Previous studies have demonstrated that ferroptosis inhibitors or iron chelators can mitigate cartilage degeneration and improve joint function in OA models.9,10 Therefore, elucidating the regulatory mechanisms of ferroptosis may offer potential strategies for OA management by preserving chondrocyte viability and maintaining cartilage integrity.
Emerging evidence underscores the intricate interplay between lysosomal dysfunction and the pathogenesis of OA.11,12 Lysosomes, which are essential for maintaining cellular homeostasis through macromolecule degradation and autophagy, are impaired in OA chondrocytes.13 This impairment results in the accumulation of damaged organelles and toxic metabolites. In addition, the destabilization of lysosomes disrupts autophagy flux, thereby exacerbating oxidative stress and mitochondrial dysfunction.14 Excessive iron accumulation within chondrocytes catalyzes Fenton reactions, leading to the generation of ROS that overwhelm antioxidant defenses, ultimately triggering lipid peroxidation and membrane rupture.15 Lysosomal damage may synergize with ferroptosis by releasing stored iron and proteolytic enzymes, further destabilizing cellular membranes and promoting the release of inflammatory cytokines. Although ferroptosis inhibitors show promise in preclinical studies, they lack lysosomal targeting. Targeting lysosomal repair mechanisms may have therapeutic potential for mitigating the progression of OA.
We observed a significant decrease in the abundance of phosphatidylethanolamine (PE) through lysosomal lipidomic analysis. As an essential component of lysosomal membrane lipids, the conical shape of PE promotes lysosomal formation, membrane curvature, and fusion, which is critical for lysosomal stability.16,17 Notably, alterations in phospholipid composition have been previously reported in joint tissues and synovial fluid from OA patients, further supporting the translational relevance of targeting PE in this context.18–20 However, the known biological functions of PE in regulating membrane repair, antioxidant activity, and preventing ferroptosis have not been sufficiently addressed. This study aimed to investigate the protective effects of PE on human chondrocytes against oxidative stress-induced ferroptosis in OA.
Materials and Methods
Human Samples Collection and Cell Culture
The studies were approved by the Ethics Committee of the Third Affiliated Hospital of Anhui Medical University (2024-241-01). All research activities adhered strictly to the Declaration of Helsinki. Human articular cartilage samples were collected from patients diagnosed with knee OA (aged 67 to 82 years, Kellgren-Lawrence grade 4) who underwent total knee arthroplasty. The samples were categorized into intact and damaged regions according to the degree of degeneration.21 Human chondrocytes were isolated and cultured in accordance with a previously established protocol.22 The samples were obtained from the articular cartilage surface during knee joint replacement. After washing away the blood with normal saline, a bone rongeur was used to crush the cartilage, which usually had a diameter of about 3 to 5 millimeters. These pieces were first treated with 0.25% trypsin for 30 minutes, then subjected to digestion with 0.25% type II collagenase for 6 hours. The isolated primary chondrocytes were subsequently resuspended and maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution, under a humidified environment containing 5% CO2 at 37°C. COL2A1 was utilized for chondrocyte identification via immunofluorescence assay.23 For experimentation, chondrocytes from the second passage were utilized.
Subcellular Fractionation
Chondrocytes were isolated and used to obtain cytosolic and lysosome-enriched fractions using the Lysosome Enrichment Kit (Solarbio, China). The cell pellet was resuspended in PBS and homogenized on ice using a Dounce homogenizer with approximately 20 strokes. Subsequently, differential centrifugation was performed in accordance with the manufacturer’s instructions to isolate crude lysosomes, while the remaining supernatant was collected as the cytosolic fraction. WB analysis of LAMP1 was used to validate the purity and normalize the content. The prepared subcellular fractions were stored at −80°C for further analysis.24
Proteomics and Lipidomics
The proteomics and lipidomics analysis was performed at OUYI Corporation (Shanghai, China) in accordance with established methodologies.18,25,26 Proteins with a P value ≤ 0.05 and a fold change ≥ 1.2 or ≤ 0.83 were considered upregulated and downregulated, respectively. The criteria for identifying significantly differentially abundant metabolites were a variable importance in the projection (VIP) value >1 and a P value < 0.05.
Western Blot (WB) Analysis
Proteins were isolated from the samples using RIPA buffer (Beyotime, China), and the concentration of the extracted proteins was measured via the Bradford method. Following this, the proteins were electrophoretically transferred onto PVDF membranes (Merck Millipore, USA). To prevent non-specific binding, the membranes were incubated in a blocking solution containing 5% non-fat dry milk for one hour at ambient temperature. They were then exposed to primary antibodies at 4°C for 12 hours. The primary antibodies applied in this study included: anti-GAPDH (Servicebio, China; 1:2000), anti-LAMP1 (Zenbio, China; 1:1000), anti-CTSD (Proteintech, China; 1:1000), anti-COX2 (Proteintech, China; 1:1000), anti-TF (Proteintech, China; 1:1000), anti-FTH1 (Huabio, China; 1:1000), and anti-GPX4 (Huabio, China; 1:1000). After the primary antibody incubation, the membranes were rinsed three times with TBST, each wash lasting 15 minutes, and subsequently treated with secondary antibodies for a duration of 2 hours at room temperature. GAPDH served as the loading control for data normalization.
Immunofluorescence (IF)
Chondrocytes grown in confocal dishes were first fixed using 4% paraformaldehyde for a duration of 15 minutes, followed by two washes with PBS. Next, the cells were permeabilized with a solution containing 0.1% Triton X-100 for 15 minutes. After permeabilization, the samples were incubated with 2% goat serum for 30 minutes at room temperature to block nonspecific binding. Subsequently, the sections were treated overnight at 4°C with primary antibodies specific to LAMP1 (Zenbio, China; dilution: 1:100) and CTSD (Proteintech, China; dilution: 1:200). The following day, the samples were exposed to Alexa Fluor-labeled secondary antibodies for one hour at room temperature. To visualize nuclei, sections were stained with DAPI for five minutes. Images were taken with Zeiss LSM800 confocal microscope (Zeiss, Germany). The fluorescence intensities quantified using Image J.
Lipid ROS Staining
The levels of lipid ROS were measured through staining with BODIPY-C11 (Invitrogen, Germany). Chondrocytes were treated with 5 μM of BODIPY 581/591 C11 for 30 minutes at 37°C. After the treatment, cells were washed three times with PBS to eliminate residual dye.
Lysosomal Enzyme Assay
The enzymatic functions of CTSD (Proteintech, China) and NAGLU (Sigma-Aldrich, USA) were evaluated following the guidelines outlined by the manufacturers. The activity levels of these enzymes in both lysosomal and cytosolic fractions were determined by analyzing the corresponding changes in optical density.
FerroOrange Staining
FerroOrange (Dojindo, Japan) was utilized to label intracellular ferrous iron by applying a 5 μM solution of FerroOrange. The chondrocytes were then incubated for a period of 30 minutes at 37°C. Following incubation, the cells were washed three times with PBS to eliminate any residual dye. Fluorescence imaging was performed using a confocal microscope (Zeiss, Germany).
Lyso-Tracker Staining
Lysosomal staining was carried out with the aid of Lyso-Tracker Red (Beyotime, China). The cells were exposed to 50 nM Lyso-Tracker Red for a duration of 30 minutes at 37°C, after which they were rinsed thoroughly with PBS three times. Subsequently, the stained cells were examined using a confocal microscopy (Zeiss, Germany).
Animal Experiment
The animal study was reviewed and approved by the Animal Care and Use Committee of the Anhui Medical University (20242502). All animal experiments were conducted following the 3R principle of animal experiment ethics. Male Sprague-Dawley (SD) rats, approximately 6 to 8 weeks old, were randomly divided into three experimental groups: the control group, the degeneration group, and the PE treatment group, with five animals in each group. In the control group, only surgical incisions were made to the skin and joint capsules, which were then immediately closed without further manipulation. For all surgical procedures, rats were first anesthetized using isoflurane and then secured in a stereotactic apparatus under continuous anesthesia. The knee OA model was induced following previously established protocols.21,27 This involved making a medial parapatellar incision to access the right knee joint cavity, followed by the removal of the medial collateral ligament. The knee was flexed to allow for identification and excision of the anterior cruciate ligament, and the medial meniscus was then removed. After surgery, all animals were returned to their cages and allowed free movement. For the PE group, 40 µL of PE (6 µM) was directly injected into the joint twice a week. In contrast, the sham and degeneration groups received intra-articular injections of 40 µL of normal saline on the same schedule. Eight weeks post-surgery, the right knee joints were harvested for further analysis.
Histology and Immunohistochemical (IHC) Staining
Following decalcification, the specimens underwent dehydration and were embedded in paraffin wax. The prepared paraffin blocks were then sectioned into 5 μm thick coronal slices. These sections were subjected to HE staining and safranin O staining for histological analysis. For IHC labeling, the sections were initially deparaffinized and rehydrated. Antigen retrieval was carried out via microwave exposure in a 0.01 mol/L citrate buffer for 15 minutes. Endogenous peroxidase activity was blocked by incubating the sections with goat serum albumin for 30 minutes. Subsequently, the tissue sections were incubated overnight at 4°C with primary antibodies targeting FTH1 (Huabio, China; 1:200 dilution), COX2 (Proteintech, China; 1:200 dilution), TF (Proteintech, China; 1:200 dilution), and GPX4 (Huabio, China; 1:200 dilution). Afterward, the sections were exposed to the corresponding secondary antibodies for 1 hour at room temperature and finally counterstained with hematoxylin.
Statistical Analysis
The data are presented as the mean ± standard deviation (SD). To compare two groups, statistical significance was assessed using Student’s t-test for normally distributed data or the Mann–Whitney U-test for data that were not normally distributed. When comparing multiple groups, one-way analysis of variance (ANOVA) was employed for normally distributed datasets, while the Kruskal–Wallis test was used for those that were not normally distributed. All statistical evaluations were conducted using GraphPad Prism software, with significance levels set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
LMP and Ferroptosis in Chondrocytes Were Observed During OA
To address the association between lysosomal dysfunction and OA, we analyzed the LMP between histologically defined intact OA cartilage and damaged OA cartilage (Figure 1A). Compared with those in the intact samples, significantly greater concentrations of lysosomal enzymes were detected in the cytosolic fractions from the damaged samples. This increase was accompanied by a corresponding decrease in enzyme content within the lysosomal fractions (Figure 1B). Costaining of CTSD and LAMP1 revealed that, compared with those in the intact group, CTSD exhibited a more diffuse distribution and fewer puncta in the damaged group (Figure 1C). LysoTracker staining revealed fewer healthy lysosomes in the damaged group than in the control group (Figure 1D). Furthermore, the levels of ferroptosis markers, including FerroOrange and Lipid ROS, were significantly elevated in damaged samples compared with intact samples (Figure 1E–H). These results revealed that lysosomal dysfunction and ferroptosis occurred during the course of OA.
 |
Figure 1 Lysosomal dysfunction and ferroptosis in chondrocytes during OA course. (A) Intact and damaged articular cartilages from OA patients; (B) Activity of lysosomal CTSD and NAGLU is reduced in lysosomal fractions and elevated in cytosolic fractions in damage group. n=3. (C) IF staining reveals leakage of CTSD (Red) from lysosomes (Green) into the cytosol following in damaged group. Scale bar:10μm. (D) Lyso-tracker staining is utilized to visualize lysosomes in chondrocytes. Scale bar: 10μm. (E and F) FerroOrange staining and quantification for Fe2+ levels in intact and damaged group. Scale bar:30μm. n=3. (G and H) Lipid ROS staining and quantification for lipid peroxidation in intact and damaged group. Scale bar: 30μm. n=3. *P <0.05, **P <0.01, and ***P <0.001. |
Oxidative Stress Leads to Lysosomal Damage and Ferroptosis in Chondrocytes
To investigate the effects of oxidative stress on chondrocytes, we conducted proteomics analysis after treatment with the oxidative stress inducer tert-butyl hydroperoxide (TBHP) at a concentration of 100 µM for 48 hours. The PCA revealed a clear separation between the control and treated groups (Figure 2A). The enriched analysis revealed that the terms “lysosome membrane” and “lysosome” were the most notable (Figure 2B). Upon treatment with TBHP, a significant increase in lysosomal enzyme concentration was observed in the cytosolic fractions. This elevation coincided with a corresponding decrease in enzyme levels within the lysosomal fractions (Figure 2C–F). Costaining of CTSD and LAMP1 revealed that following TBHP treatment, CTSD exhibited a more diffuse distribution with fewer puncta (Figure 2G). Moreover, there was a notable reduction in the number of lysosomes after TBHP treatment (Figure 2H). WB analysis of the FTH1, GPX4, COX2, and TF proteins confirmed that oxidative stress induces ferroptosis in chondrocytes (Figure 2I and J). Additionally, the levels of FerroOrange and lipid ROS were significantly elevated following TBHP treatment (Figure 2K–N).
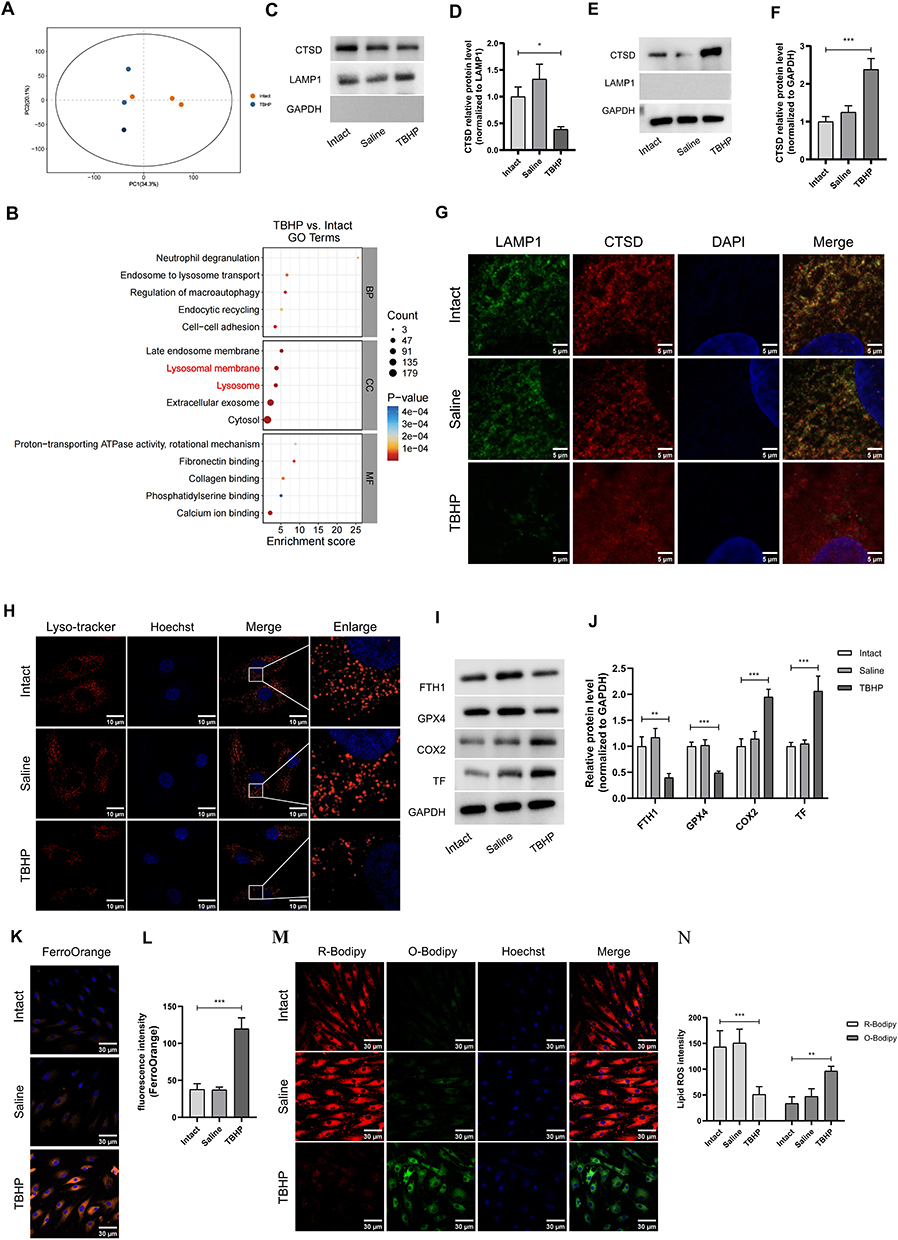 |
Figure 2 Oxidative stress leads to lysosomal damage and ferroptosis in chondrocytes. (A) Proteomics analysis for human chondrocytes after TBHP treatment. (B) GO analysis of the different proteins indicates that “lysosomal membrane” and “lysosome” are the enriched items. (C and D) CTSD in the lysosomes fractions were extracted after oxidative stress overload, and quantification of the protein levels of CTSD in lysosomes fractions. n=3. (E and F) CTSD in the cytoplasm were extracted from chondrocytes, and quantification of the protein levels of CTSD in cytoplasm fractions. n=3. (G) IF staining reveals leakage of CTSD (Red) from lysosomes (Green) into the cytosol following after oxidative stress overload. Scale bar:10μm. (H) Lyso-tracker staining is utilized to visualize lysosomes in chondrocytes after oxidative stress overload. Scale bar: 10μm. (I and J) WB analysis and quantification of COX2, TF, FTH1 and GPX4 protein levels. n=3. (K and L) FerroOrange staining and quantification for Fe2+ levels. Scale bar:30μm. n=3. (M and N) Lipid ROS staining and quantification for lipid peroxidation. Scale bar: 30μm. n=3. *P <0.05, **P <0.01, and ***P <0.001. |
Lipidomics of the Lysosomal Fractions Revealed a Reduction in PE After Oxidative Stress
Given that proteomics sequencing revealed an aberrant lysosomal membrane structure, and considering that the lipid composition of the lysosomal membrane is critically important for its functionality, we prepared lysosome-enriched fractions for lipidomic analysis following oxidative stress overload with TBHP at a concentration of 100 µM for 48 hours. The orthogonal partial least squares discriminant analysis (OPLS-DA) model demonstrated a distinct separation between the two groups (Figure 3A). Volcano plot and heatmap analyses revealed substantial metabolic differences between the intact group and those treated with TBHP (Figure 3B and C). Notably, the abundance of PEs was significantly decreased after oxidative stress (Figure 3D–F). Moreover, the abundance of PEs in the damaged group was significantly lower than that observed in the intact group (Figure 3G). This analysis reveals the process by which PE participates in lysosomal damage under oxidative stress conditions.
 |
Figure 3 Lipidomics analysis for lysosomal fractions. (A) The Orthogonal partial least squares-discriminant analysis (OPLS-DA) model demonstrates a distinct separation between the two groups. (B and C) Volcano plot and heat map analysis reveal substantial metabolic differences between intact lysosome fractions and those under oxidative stress overload. (D–F) The PEs abundance was significantly down-regulated after oxidative stress overload. n=5. (G) The relative of PE in damaged group was significantly decreased when compared to intact group. n=5. *P <0.05, **P <0.01. |
PE Protects Chondrocytes from Oxidative Stress-Induced Ferroptosis and Improves Lysosomal Functionality
Given that the functional role of PE in chondrocytes remains unclear, we subsequently investigated whether PE can also mitigate ferroptosis and lysosomal dysfunction. The PE (Sigma - Aldrich, USA) was dissolved in saline using ultrasonic waves, and the concentration was adjusted according to the experimental requirements. As shown in Figure 4A, PE significantly promoted cell viability of the experimental group after 24 h at concentrations ranging from 2 to 12 μmol. Notably, the most substantial protective effect was observed at a concentration of 6 μmol, leading to its selection for subsequent experiments. PE treatment resulted in decreased levels of COX2 and TF but concurrently increased levels of FTH1 and GPX4 (Figure 4B and C). Additionally, PE treatment led to reductions in both iron and lipid peroxide levels (Figure 4D–G).
 |
Figure 4 PE protects chondrocytes from oxidative stress induced ferroptosis. (A) CCK8 results of human chondrocytes treated with various concentrations of PE. n=3. (B and C) WB analysis and quantification of COX2, TF, FTH1 and GPX4 protein levels after 72h PE treatment. n=3. (D and E) FerroOrange staining and quantification for Fe2+ levels. Scale bar:50μm. n=3. (F and G) Lipid ROS staining and quantification for lipid peroxidation. Scale bar: 50μm. n=3. *P <0.05, **P <0.01, and ***P <0.001. |
PE is a key component of lysosomal membrane lipids. On the basis of these findings, we speculate that PE may repair lysosomal membrane damage in the damaged group. As shown in Figure 5A–D, PE decreased lysosomal content leakage from lysosomes to the cytoplasm. Costaining of CTSD and LAMP1 demonstrated that, following PE treatment, CTSD exhibited a less diffuse distribution with fewer puncta (Figure 5E). LysoTracker staining of lysosomes revealed an increased number of healthy lysosomes after PE treatment (Figure 5F). Lipidomic analysis revealed that the abundance of lysosomal PEs was significantly increased following PE treatment (Figure 5G–J). The total abundance of PEs was significantly up-regulated after PE treatment (Figure 5K). These results indicate that PE protects chondrocytes from ferroptosis and improves lysosomal functionality.
 |
Figure 5 PE normalizes lysosomal functions. (A and B) CTSD in the lysosomes fractions were extracted after oxidative stress overload, and quantification of the protein levels of CTSD in lysosomes fractions. n=3. (C and D) CTSD in the cytoplasm were extracted from chondrocytes, and quantification of the protein levels of CTSD in cytoplasm fractions. n=3. (E) IF staining reveals leakage of CTSD (Red) from lysosomes (Green) into the cytosol was inhibited following PE treatment. Scale bar:10μm. (F) Lyso-tracker staining is utilized to visualize lysosomes in chondrocytes. (G) The Orthogonal partial least squares-discriminant analysis (OPLS-DA) model demonstrates a distinct separation after PE treatment. (H) Volcano plot and heat map analysis reveal substantial metabolic differences between intact lysosome fractions and those under oxidative stress overload. (I–K) The total abundance of PEs was significantly up-regulated after PE treatment. n=3. Scale bar: 10μm. *P <0.05, **P <0.01, and ***P <0.001. |
PE Protects Against Chondrocyte Ferroptosis in a Lysosomal-Dependent Manner
The present study further investigated the potential protective effects of PE on oxidative stress in chondrocytes through the lysosomal pathway. Following PE treatment, the chondrocytes were subsequently exposed to LMP inducer N-dodecylimidazole (NDI, 30μM) for 48 hours. PE significantly attenuated the leakage of lysosomal contents from lysosomes to the cytoplasm, but treatment with NDI blocked the protective effect of PE (Figure 6A–D). Following NDI disturbance, CTSD exhibited a more diffuse distribution with fewer puncta (Figure 6E) and fewer healthy lysosomes (Figure 6F). Moreover, the level of ferroptosis significantly increased after NDI disturbance (Figure 6G–L). These findings suggest that PE protects against chondrocyte ferroptosis in a lysosomal-dependent manner.
 |
Figure 6 PE protects chondrocytes ferroptosis in a lysosomal dependent manner. (A and B) CTSD in the lysosomes fractions were extracted after NDI treatment, and quantification of the protein levels of CTSD in lysosomes fractions. n=3. (C and D) CTSD in the cytoplasm fractions were extracted after NDI treatment, and quantification of the protein levels of CTSD in cytoplasm fractions. n=3. (E) IF staining reveals leakage of CTSD (Red) from lysosomes (Green) into the cytosol following NDI treatment. Scale bar:10μm. (F) Lyso-tracker staining for the lysosomes in human chondrocytes. Scale bar:10μm. (G and H) WB analysis and quantification of COX2, TF, FTH1 and GPX4 protein levels after NDI treatment. n=3. (I and J) FerroOrange staining and quantification for Fe2+ levels. Scale bar:30μm. n=3. (K and L) Lipid ROS staining and quantification for lipid peroxidation. Scale bar: 30μm. n=3. *P <0.05, **P <0.01, and ***P <0.001. |
Therapeutic Effect of PE in an OA Model
To investigate the therapeutic effects of PE in vivo, we utilized a rat model of OA. Figure 7A shows the schematic illustration of the animal experimental procedure. Histological analysis via HE and safranin O staining demonstrated that PE attenuated the progression of OA (Figure 7B and C). IHC staining for COX2, TF, and GPX4 indicated that the OA models exhibited a significant degree of ferroptosis, which was partially reversed following treatment with PE (Figure 7D–I).
 |
Figure 7 Therapeutic effect of PE in a OA model. (A) Schematic illustration of the animal experimental procedure. (B) HE and S-O staining of OA models eight weeks postoperatively. n=5 per group; Scale bar: 50 µm. (C) Osteoarthritis Research Society International (OARSI) grades of the joints. n=5 per group. (D and E) IHC staining and quantification of COX2, and the enlarged area within the red frame. n=5 per group; Scale bar: 50 µm. (F and G) IHC staining and quantification of TF positive cells, and the enlarged area within the red frame. n=5 per group; Scale bar: 50 µm. (H and I) IHC staining and quantification of TF positive cells, and the enlarged area within the red frame. n=5 per group; Scale bar: 50 µm. *P <0.05, **P <0.01, and ***P <0.001. |
Discussion
LMP, characterized by the leakage of lysosomal contents into the cytosol, has emerged as a critical mechanism driving ferroptosis and is implicated in the progression of OA.13 Lysosomes, which house hydrolytic enzymes such as cathepsins and store redox-active iron, play important roles in maintaining cellular homeostasis and mediating stress responses.14 Under pathological conditions such as oxidative stress or inflammation, the lysosomal membrane becomes destabilized, leading to the release of proteolytic enzymes and iron ions.28,29 This leakage triggers a cascade of events: cathepsins disrupt mitochondrial integrity, exacerbating oxidative stress, whereas free iron catalyzes lipid peroxidation via the Fenton reaction, a hallmark of ferroptosis.14 In OA, articular chondrocytes are particularly vulnerable to LMP-induced ferroptosis. The degenerative joint environment, which is characterized by chronic inflammation and excessive ROS, promotes lysosomal fragility and dysfunction.11,30 Our work is consistent with previous studies revealing that chondrocytes in OA patients exhibit elevated levels of lysosomal LMP and ferroptosis. The loss of functional chondrocytes disrupts extracellular matrix synthesis, accelerating cartilage degradation and joint dysfunction. Thus, targeting LMP and ferroptosis pathways may offer novel strategies to mitigate OA progression.
In our study, we also observed that LMP and ferroptosis occurred under conditions of oxidative stress. Lysosomal membranes are uniquely composed of a single phospholipid bilayer enriched with specialized lipids, including cholesterol, phosphatidylcholine, phosphatidylinositol, and PE, which collectively ensure membrane integrity, stability, and functionality.16 Alterations in the lipid composition of lysosomal membranes are critical drivers of lysosomal damage and increased permeability, contributing to cellular dysfunction and death. In a study on random skin samples, Lou et al24 demonstrated that the hydrolysis of phospholipids in the lysosomal membrane leads to increased LMP and subsequent cell necroptosis via the lysosomal pathway. Similarly, Sarkar et al31 found that low levels of phospholipids such as PE and phosphatidylcholine contribute to LMP and lysosomal dysfunction, ultimately resulting in cell death. Under pathological conditions such as oxidative stress, aging, or lipid peroxidation, cholesterol accumulation or imbalances in phospholipid ratios disrupt membrane fluidity and stability.32 For example, oxidative stress in OA oxidizes PE, compromising its ability to maintain membrane structure. These lipid perturbations destabilize the membrane, leading to LMP, leakage of cathepsins into the cytosol, and activation of ferroptosis pathways.33 Therefore, therapeutic strategies targeting lipid homeostasis, such as the use of antioxidants to mitigate lipid peroxidation, small molecules to restore phospholipid balance, or cholesterol-lowering agents, may stabilize lysosomal membranes, preserve autophagic flux, and prevent cell death.
Lipidomics is an emerging and rapidly evolving field within the broader omics domain dedicated to the systematic study of lipid-related pathways in biological systems.26,34 This discipline not only examines the metabolic pathways of endogenous metabolites within organisms, organs, or tissues but also assesses alterations in metabolic profiles caused by both internal and external stress conditions. By thoroughly profiling lipid modifications, lipidomics has facilitated the discovery of various biological processes involved in specific pathophysiological pathways. Integrating changes in metabolic profiles with these pathways holds significant promise for advancing disease diagnosis and treatment strategies. Unlike other omics approaches, lipidomics operates downstream of gene regulation and protein interaction networks, offering a direct reflection of biochemical activities and delivering a precise depiction of an organism’s ultimate phenotypic characteristics. By employing lipidomic analysis, Radulovic et al35 demonstrated that cholesterol transfer mediated by endoplasmic reticulum contacts facilitates lysosomal damage repair and provides lipids essential for membrane restoration. In this context, we conducted a lipidomic analysis to investigate alterations in the metabolite profiles of lysosomal fractions under oxidative stress conditions.
Through this lipidomic approach, we identified significant downregulation of PE under oxidative stress conditions. Furthermore, the abundance of PEs in the damaged group was lower than that in the intact group. PE, a key phospholipid component of lysosomal membranes, plays a pivotal role in maintaining lysosomal membrane integrity and function.16 PE contributes to membrane curvature and stability due to its conical molecular shape, which is critical for lysosomal membrane dynamics, including fusion with autophagosomes or endosomes.17 PE is essential for preserving lysosomal structural integrity, as its deficiency or oxidative modification under OA-related oxidative stress can destabilize lysosomal membranes, leading to enzyme leakage and impaired autophagic flux.24,31 In OA, oxidative stress-induced lipid peroxidation may alter the physicochemical properties of PEs, compromising their lysosomal acidification and degradative capacity. Furthermore, PE interacts with lysosomal membrane proteins and ion channels, regulating lysosomal exocytosis and calcium signaling.36 The downregulation of PE has been linked to lysosomal storage disorder-like phenotypes, exacerbating chondrocyte dysfunction through the accumulation of undegraded macromolecules. Therapeutic strategies targeting PE homeostasis may restore lysosomal functions in OA.
PE plays tissue-specific therapeutic roles in disease modulation, influenced by its unique metabolic pathways and functional demands in different organs. In the liver, PEs play critical roles in lipid metabolism and lipoprotein secretion.37 Inhibition of PE synthesis disrupts very-low-density lipoprotein assembly, leading to hepatic steatosis. Restoring the hepatic phosphatidylcholine-to-PE molar ratio through dietary interventions or pharmacological modulation of the Kennedy pathway may alleviate fatty liver disease and enhance postsurgical liver regeneration.38 In cardiac tissue, mitochondrial PE is essential for maintaining cristae structure and ATP production.39 Deficiencies in PE-plasmalogens are associated with cardiac dysfunction. Supplementation with plasmalogen precursors or antioxidants to prevent PE oxidation may protect against cardiomyopathy and ischemia‒reperfusion injury.40 However, further research is needed to validate the specific functional role of PE in OA.
In our study, we demonstrated the therapeutic potential of PE in the inhibition of LMP. The unique conical shape of PE promotes membrane curvature, facilitating lysosomal vesicle formation and repair. During lysosomal damage, PE externalization serves as a signal for membrane repair systems, such as the endosomal sorting complexes required for transport (ESCRT) complex, which seals membrane ruptures to prevent enzyme leakage.16 PE also interacts with autophagy-related proteins such as LC3, enabling autophagosome‒lysosome fusion, a critical step in clearing damaged organelles or pathogens.41,42 Conversely, PE deficiency disrupts lysosomal acidification and hydrolase activity, exacerbating substrate accumulation and oxidative stress. PE also plays a role in lysosome-dependent cell death. Excessive damage induces PE peroxidation, destabilizes membranes and releases cathepsins, which activate caspase-dependent apoptosis or NLRP3 inflammasome-driven ferroptosis.14,43 Therefore, PE may function as both a structural component and a dynamic signaling center, integrating lysosomal integrity with adaptive cellular mechanisms to prevent oxidative stress-induced chondrocyte ferroptosis in OA. PE depletion could indirectly affect lysosomal stability by impairing autophagic flux or other lipid trafficking pathways that are crucial for lysosomal homeostasis.
As the most prevalent degenerative joint disease, OA has significant socioeconomic impacts and a reduced quality of life. Current pharmacological interventions remain limited in efficacy, necessitating the urgent development of disease-modifying therapies. Through integrated lipidomics and functional validation, we elucidated the regulatory mechanism by which PE modulates chondrocyte ferroptosis, establishing its potential therapeutic value for OA management. As an economical and readily available phospholipid compound, PE has remarkable translational potential. Our findings not only reveal novel molecular targets but also suggest PE as a potential agent for OA therapeutics. PE is a relatively cost-effective and readily available agent. The reported alterations in lysosomal PE are of transitional significance, as they offer novel molecular targets and potential therapeutic drugs for the research on OA treatment. However, the method of administering PE for human OA treatment poses significant concerns. Intra-articular injection can be regarded as a feasible approach; nevertheless, such attempts have only been conducted in animal experiments. The next challenge is to ensure the safe and efficient delivery of PE to the joints.44 PE can be incorporated into the composition of delivery systems, as demonstrated by the clinical development of MM-II (NCT04506463).45 This clinical results from the phase 2b trial demonstrated that a single 3 mL injection of MM-II provided pain relief lasting up to 26 weeks, with improvements in weekly average daily pain and reduced rescue medication use, supporting the therapeutic potential of phospholipid-based formulations. Current research on articular administration remains confined to preclinical models, highlighting the imperative to develop safe and effective joint-specific delivery systems for human applications.
Our study was subject to several limitations. First, the limited cohort size in lipidomic profiling may constrain the statistical power to delineate comprehensive metabolic signatures, potentially introducing bias in capturing systemic lipidomic variations. Secondly, all human samples are from knees of KL grade 4, which is suitable for advanced OA but restricts the generalizability to earlier stages. Third, the precise regulatory mechanisms underlying PE downregulation under oxidative stress remain unclear, particularly concerning epigenetic modifications or posttranslational regulation of lipid metabolic enzymes. This knowledge gap underscores the need for comprehensive mechanistic studies that integrate multiomics approaches and include functional validation to fully elucidate this pathway. Lastly, human OA is a slowly progressive, long-term degenerative joint disorder. By contrast, the anterior ACLT-induced OA model reflects an acute, trauma-driven onset and fails to fully recapitulate the gradual, multifactorial nature of human OA development. Although the in-vitro model cannot fully simulate the etiology and pathological mechanism of OA suitable model, we can be rationally chosen according to the observed phenotypic features or the underlying disease mechanism, offering a promising approach to evaluate the therapeutic potential of candidate targets.
Conclusion
Through lipidomic analysis of lysosomal fractions in chondrocytes, we observed a significant decrease in the abundance of PEs under oxidative stress conditions. As an essential component of lysosomal membrane lipids, PE plays a critical role in maintaining the stability and functionality of the lysosomal membrane. PE supplementation restores lysosomal PE levels, reduces LMP, and inhibits ferroptosis in preclinical models, suggesting therapeutic potential. These findings significantly increase our understanding of the pathogenesis of OA and reveal therapeutic targets for its management.
Data Sharing Statement
The data from this study are available upon request from the corresponding author.
Ethics Statement
The studies involving human participants underwent a rigorous review and were subsequently approved by the Ethics Committee of the Third Affiliated Hospital of Anhui Medical University (2024-241-01). All research activities adhered strictly to the World Medical Association’s Declaration of Helsinki, ensuring compliance with ethical standards for human experimentation. The privacy rights of all human subjects were meticulously safeguarded, and informed consent was obtained from each participant. The animal study was reviewed and approved by the Animal Care and Use Committee of the Anhui Medical University (20242502). All animal experiments were conducted following the 3R principle of animal experiment ethics.
Funding
This project has received funding from the Beijing-Tianjin-Hebei Cooperative Basic Research Program (21JCZXJC00030).
Disclosure
The authors declare that they have no competing interests.
References
1. Stoffer-Marx MA, Klinger M, Luschin S, et al. Functional consultation and exercises improve grip strength in osteoarthritis of the hand - a randomised controlled trial. Arthritis Res Ther. 2018;20(1):253. doi:10.1186/s13075-018-1747-0
2. Jiang Y. Osteoarthritis year in review 2021: biology. Osteoarthritis Cartilage. 2022;30(2):207–16. doi:10.1016/j.joca.2021.11.009
3. Nelligan RK, Hinman RS, Kasza J, et al. Effects of a self-directed web-based strengthening exercise and physical activity program supported by automated text messages for people with knee osteoarthritis: a randomized clinical trial. JAMA Intern Med. 2021;181(6):776–785. doi:10.1001/jamainternmed.2021.0991
4. Dantas LO, Salvini TF, McAlindon TE. Knee osteoarthritis: key treatments and implications for physical therapy. Braz J Phys Ther. 2021;25(2):135–146. doi:10.1016/j.bjpt.2020.08.004
5. Wang S, Li W, Zhang P, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75. doi:10.1016/j.jare.2022.01.004
6. Wang D, Fang Y, Lin L, et al. Upregulating miR-181b promotes ferroptosis in osteoarthritic chondrocytes by inhibiting SLC7A11. BMC Musculoskelet Disord. 2023;24(1):862. doi:10.1186/s12891-023-07003-7
7. Fan F, Yang C, Piao E, Shi J, Zhang J. Mechanisms of chondrocyte regulated cell death in osteoarthritis: focus on ROS-triggered ferroptosis, parthanatos, and oxeiptosis. Biochem Biophys Res Commun. 2024;705:149733. doi:10.1016/j.bbrc.2024.149733
8. Tao L, Yang K, Wang K, Yang Y. NOX1-mediated oxidative stress induces chondrocyte ferroptosis by inhibiting the Nrf2/HO-1 pathway. Sci Rep. 2024;14(1):19877. doi:10.1038/s41598-024-70991-6
9. Zhang X, Hou L, Guo Z, et al. Lipid peroxidation in osteoarthritis: focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 2023;9(1):320. doi:10.1038/s41420-023-01613-9
10. Pan X, Kong X, Feng Z, et al. 4-Octyl itaconate protects chondrocytes against IL-1β-induced oxidative stress and ferroptosis by inhibiting GPX4 methylation in osteoarthritis. Int Immunopharmacol. 2024;137:112531. doi:10.1016/j.intimp.2024.112531
11. Yang H, Wen Y, Zhang M, et al. MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint. Autophagy. 2020;16(2):271–288. doi:10.1080/15548627.2019.1606647
12. Lee JS, Kim YH, Jhun J, et al. Oxidized LDL accelerates cartilage destruction and inflammatory chondrocyte death in osteoarthritis by disrupting the TFEB-regulated autophagy-lysosome pathway. Immune Netw. 2024;24(3):e15. doi:10.4110/in.2024.24.e15
13. Ye T, Wang C, Yan J, et al. Lysosomal destabilization: a missing link between pathological calcification and osteoarthritis. Bioact Mater. 2023;34:37–50. doi:10.1016/j.bioactmat.2023.12.001
14. Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. doi:10.1111/tra.12613
15. An F, Zhang J, Gao P, et al. New insight of the pathogenesis in osteoarthritis: the intricate interplay of ferroptosis and autophagy mediated by mitophagy/chaperone-mediated autophagy. Front Cell Dev Biol. 2023;11:1297024. doi:10.3389/fcell.2023.1297024
16. Casares D, Escribá PV, Rosselló CA. Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int J Mol Sci. 2019;20(9):2167. doi:10.3390/ijms20092167
17. Rockenfeller P, Koska M, Pietrocola F, et al. Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ. 2015;22(3):499–508. doi:10.1038/cdd.2014.219
18. Jónasdóttir HS, Brouwers H, Kwekkeboom JC, et al. Targeted lipidomics reveals activation of resolution pathways in knee osteoarthritis in humans. Osteoarthritis Cartilage. 2017;25(7):1150–1160. doi:10.1016/j.joca.2017.01.018
19. Kosinska MK, Liebisch G, Lochnit G, et al. A lipidomic study of phospholipid classes and species in human synovial fluid. Arthritis Rheum. 2013;65(9):2323–2333. doi:10.1002/art.38053
20. Sluzalska KD, Liebisch G, Lochnit G, et al. Interleukin-1β affects the phospholipid biosynthesis of fibroblast-like synoviocytes from human osteoarthritic knee joints. Osteoarthritis Cartilage. 2017;25(11):1890–1899. doi:10.1016/j.joca.2017.07.011
21. Lv Z, Han J, Li J, et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. EBioMedicine. 2022;84:104258. doi:10.1016/j.ebiom.2022.104258
22. Tang S, Nie X, Ruan J, et al. Circular RNA circNFKB1 promotes osteoarthritis progression through interacting with ENO1 and sustaining NF-κB signaling. Cell Death Dis. 2022;13(8):695. doi:10.1038/s41419-022-05148-2
23. Huang H, Liu K, Ou H, et al. Phgdh serves a protective role in Il‑1β induced chondrocyte inflammation and oxidative‑stress damage. Mol Med Rep. 2021;23(6):419. doi:10.3892/mmr.2021.12058
24. Lou J, Wang X, Zhang H, et al. Inhibition of PLA2G4E/cPLA2 promotes survival of random skin flaps by alleviating Lysosomal membrane permeabilization-Induced necroptosis. Autophagy. 2022;18(8):1841–1863. doi:10.1080/15548627.2021.2002109
25. Chen X, Chen K, Hu J, et al. Multiomics analysis reveals the potential of LPCAT1-PC axis as a therapeutic target for human intervertebral disc degeneration. Int J Biol Macromol. 2024;276(Pt 1):133779. doi:10.1016/j.ijbiomac.2024.133779
26. Tu S, Dong Y, Li C, et al. Phosphatidylcholine ameliorates palmitic acid-induced lipotoxicity by facilitating endoplasmic reticulum and mitochondria contacts in intervertebral disc degeneration. JOR Spine. 2025;8(2):e70062. doi:10.1002/jsp2.70062
27. Yuan Z, Yang L, Li Y, et al. FTH1 protects against osteoarthritis by MAPK pathway inhibition of extracellular matrix degradation. BMC Musculoskelet Disord. 2024;25(1):282. doi:10.1186/s12891-024-07411-3
28. Jeong E, Willett R, Rissone A, et al. TMEM55B links autophagy flux, lysosomal repair, and TFE3 activation in response to oxidative stress. Nat Commun. 2024;15(1):93. doi:10.1038/s41467-023-44316-6
29. Choi C, Jeong YL, Park KM, et al. TM4SF19-mediated control of lysosomal activity in macrophages contributes to obesity-induced inflammation and metabolic dysfunction. Nat Commun. 2024;15(1):2779. doi:10.1038/s41467-024-47108-8
30. Ansari MY, Ahmad N, Haqqi TM. Oxidative stress and inflammation in osteoarthritis pathogenesis: role of polyphenols. Biomed Pharmacother. 2020;129:110452. doi:10.1016/j.biopha.2020.110452
31. Sarkar C, Jones JW, Hegdekar N, et al. PLA2G4A/cPLA2-mediated lysosomal membrane damage leads to inhibition of autophagy and neurodegeneration after brain trauma. Autophagy. 2020;16(3):466–485. doi:10.1080/15548627.2019.1628538
32. Nagata K, Hishikawa D, Sagara H, et al. Lysophosphatidylcholine acyltransferase 1 controls mitochondrial reactive oxygen species generation and survival of retinal photoreceptor cells. J Biol Chem. 2022;298(6):101958. doi:10.1016/j.jbc.2022.101958
33. Santos M, Melo T, Maurício T, et al. The non-enzymatic oxidation of phosphatidylethanolamine and phosphatidylserine and their intriguing roles in inflammation dynamics and diseases. FEBS Lett. 2024;598(17):2174–2189. doi:10.1002/1873-3468.14992
34. Zhou Q, Ghorasaini M, Cornelis FMF, et al. Lipidomics unravels lipid changes in osteoarthritis articular cartilage. Ann Rheum Dis. 2025;84(7):1264–1276. doi:10.1016/j.ard.2025.01.009
35. Radulovic M, Wenzel EM, Gilani S, et al. Cholesterol transfer via endoplasmic reticulum contacts mediates lysosome damage repair. EMBO J. 2022;41(24):e112677. doi:10.15252/embj.2022112677
36. Thekkinghat AA, Yadav KK, Rangarajan PN. Apolipoprotein L9 interacts with LC3/GABARAP and is a microtubule-associated protein with a widespread subcellular distribution. Biol Open. 2019;8(9):bio045930. doi:10.1242/bio.045930
37. Elmihi KA, Leonard KA, Nelson R, et al. The emerging role of ethanolamine phosphate phospholyase in regulating hepatic phosphatidylethanolamine and plasma lipoprotein metabolism in mice. FASEB J. 2024;38(18):e70063. doi:10.1096/fj.202401321R
38. Ling J, Chaba T, Zhu LF, et al. Hepatic ratio of phosphatidylcholine to phosphatidylethanolamine predicts survival after partial hepatectomy in mice. Hepatology. 2012;55(4):1094–1102. doi:10.1002/hep.24782
39. Xu G, Xiao W, Sun P, et al. Lysophosphatidylethanolamine improves diastolic dysfunction by alleviating mitochondrial injury in the aging heart. J Lipid Res. 2025;66(1):100713. doi:10.1016/j.jlr.2024.100713
40. Drgová A, Likavcanová K, Dobrota D. Changes of phospholipid composition and superoxide dismutase activity during global brain ischemia and reperfusion in rats. Gen Physiol Biophys. 2004;23(3):337–346.
41. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020;21(8):439–458. doi:10.1038/s41580-020-0241-0
42. Yang A, Pantoom S, Wu YW. Distinct Mechanisms for Processing Autophagy Protein LC3-PE by RavZ and ATG4B. Chembiochem. 2020;21(23):3377–3382. doi:10.1002/cbic.202000359
43. Li W, Luo LX, Zhou QQ, et al. Phospholipid peroxidation inhibits autophagy via stimulating the delipidation of oxidized LC3-PE. Redox Biol. 2022;55:102421. doi:10.1016/j.redox.2022.102421
44. Maudens P, Jordan O, Allémann E. Recent advances in intra-articular drug delivery systems for osteoarthritis therapy. Drug Discov Today. 2018;23(10):1761–1775. doi:10.1016/j.drudis.2018.05.023
45. Schnitzer TJ, Chevalier X, Rovsing H, et al. Intra-articular MM-II for the treatment of knee osteoarthritis pain: efficacy and safety results from a 26-week, phase 2b, placebo-controlled, double-blind, randomized dose-ranging trial. Osteoarthritis Cartilage. 2025;33(7):897–906. doi:10.1016/j.joca.2025.04.006
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Bioinformatics and Integrative Experimental Method to Identifying and Validating Co-Expressed Ferroptosis-Related Genes in OA Articular Cartilage and Synovium
Ma J, Yu P, Ma S, Li J, Wang Z, Hu K, Su X, Zhang B, Cheng S, Wang S
Journal of Inflammation Research 2024, 17:957-980
Published Date: 12 February 2024
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024
Ferroptosis in Osteoarthritis: Current Understanding
Liu Y, Zhang Z, Fang Y, Liu C, Zhang H
Journal of Inflammation Research 2024, 17:8471-8486
Published Date: 7 November 2024
Molecular Mechanisms of Immunoinflammatory Infiltration and Ferroptosis in Arthritis Revealed by a Combination of Bioinformatics and Single-Cell Analysis
Song C, Song W, Liu Y, Zhou D, Cai W, Mei Y, Liu F, Jiang F, Chen F, Liu Z
Journal of Inflammation Research 2025, 18:2409-2432
Published Date: 18 February 2025
MXene-Mediated Nanocarrier Delivery Enhances the Chondroprotective Effects of Quercetin in Experimental Osteoarthritis
Gan K, Li J, Zhang X, Feng Z, Liu J, Zhao F
International Journal of Nanomedicine 2025, 20:11553-11567
Published Date: 20 September 2025