Back to Journals » Drug Design, Development and Therapy » Volume 20
Phase I Study Evaluating Pharmacokinetics, Pharmacodynamics, Safety, and Immunogenicity of Recombinant Human Serum Albumin (rHSA) in Healthy Adults
Authors Panuganti VK, Dundigalla MR, Grandhi VR, Khambhampaty S, Madala PK, Mohammad J ![]() , Shaik A, Shaik MB, KSSVV S, Sangeetham SK
, Shaik A, Shaik MB, KSSVV S, Sangeetham SK
Received 25 January 2026
Accepted for publication 4 June 2026
Published 10 July 2026 Volume 2026:20 598274
DOI https://doi.org/10.2147/DDDT.S598274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Veerendra Kumar Panuganti,1 Mamatha Reddy Dundigalla,2 Venkata Ramalingayya Grandhi,2 Sridevi Khambhampaty,3 Pavan Kumar Madala,4 Javeed Mohammad,1 Althaf Shaik,1 Mahaboob Basha Shaik,1 Sanyasirao KSSVV,1 Sai Kumar Sangeetham1
1Clinical Affairs, Shilpa Medicare Limited, Hyderabad, Telangana, 500076, India; 2Medical Affairs Department, Shilpa Medicare Limited, Hyderabad, Telangana, 500076, India; 3Shilpa Biologicals Private Limited, Dharwad, Karnataka, 580011, India; 4Bioanalytical Laboratory, Shilpa Medicare Ltd, Hyderabad, Telangana, 500076, India
Correspondence: Veerendra Kumar Panuganti, Clinical Affairs Division, Shilpa Medicare Limited, Unit VII, Plot No. 79, Survey No 125, IDA, Mallapur, Nacharam, Hyderabad, Telangana, PIN-500076, India, Email [email protected]
Purpose: To compare the safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and immunogenicity of recombinant human serum albumin (rHSA) with plasma-derived human serum albumin (pdHSA) following intravenous administration to healthy adults.
Patients and Methods: This Phase I study enrolled 62 healthy adults aged 18– 45 years in two sequential parts. Part 1 (n=12) evaluated safety and tolerability of rHSA using a single ascending dose design (10 g, 20 g and 40 g) with 3 cohorts. Based on these outcomes, Part 2 (n=50) was conducted as a randomized (1:1), active-controlled, parallel-group study comparing rHSA with pdHSA (Flexbumin® 20%) administered at escalating doses (10 g, 20 g, and 40 g) with 3-week intervals. Primary PK endpoints were Cmax (1st dose) and AUC∞ (3rd dose). Secondary endpoints included PK parameters: Tmax (Time to peak concentration), t½ (Elimination half-life), Elimination rate constant (Kel) and CL (Clearance). PD markers: Colloid Osmotic Pressure (COP) and Hematocrit Ratio (HCT), Safety, Tolerability, and Immunogenicity.
Results: In Part 1, rHSA was tolerated up to 40 g as single dose, and at cumulative dose of 70 g in part 2, with no dose-limiting toxicities. PK and PD profiles were comparable between test rHSA and pdHSA, with geometric mean ratios for Cmax and AUCt (baseline uncorrected) falling within the accepted 80– 125% bioequivalence range. COP and HCT ratios were consistent across doses, indicating equivalent pharmacodynamic activity. No severe adverse events (SAEs) occurred during the study. All reported adverse events (AE) were mild-to-moderate in severity and resolved without sequelae. Low-titre IgG antibodies were detected in a few participants, without clinical relevance, with no IgE or anti-host cell protein (HCP) antibodies detected.
Conclusion: The test rHSA was well tolerated at cumulative doses of 70 g and demonstrated comparable PK, PD, safety and immunogenicity profiles to pdHSA, supporting its potential as a viable alternative.
Keywords: albumin, recombinant proteins, Pichia pastoris, pharmacokinetics, pharmacodynamics, immunogenicity, bioequivalence, safety, phase i
Introduction
Human serum albumin (HSA) is the most abundant protein in the plasma (~ 43 gL−1; 0.6 mM), accounting for approximately 60% of the total plasma proteins.1 Plasma-derived human serum albumin (pdHSA) is a single, non-glycosylated polypeptide comprising 585 amino acids that forms a heart-shaped three-domain structure with a molecular weight of 66.5 kDa. Synthesized in the liver, human albumin contributes approximately 80% of colloid osmotic pressure (COP; 25–33 mmHg) and plays a pivotal role in maintaining blood pH.2 In addition to its oncotic function, albumin serves as a versatile carrier molecule, binding and transporting a broad spectrum of endogenous and exogenous ligands, including proteins, peptides, fatty acids, amino acids, hormones, nutrients, drugs and metal ions.3,4 These multifunctional binding properties, combined with its low toxicity and non-immunogenic nature, render albumin an ideal component in various pharmaceutical and biotechnological applications, serving as stabilizer in vaccines and therapeutic protein drugs, biocompatible coating for medical devices, and as a component in drug delivery and imaging systems.2,5,6
Clinically, pdHSA is widely employed to manage several conditions, including hemorrhagic shock, burns resuscitation, hypovolemia, hypoproteinemia, hypoalbuminemia, trauma, and liver diseases such as decompensated cirrhosis, acute-on-chronic liver failure, hepato-renal syndrome (HRS), sepsis, haemolytic disease of the new-born and erythroblastosis fetalis.4,7–11 Traditionally, pdHSA is obtained through fractionating human plasma from voluntary donors and remains one of the most extensively used biopharmaceuticals, with global production reaching several hundred metric tons annually.12,13 However, plasma-derived products carry inherent limitations, including risk of transmitting blood-borne and emerging pathogens such as HIV, HBV, HCV, HAV, HEV, SARS-CoV, West Nile virus, and variant Creutzfeldt-Jacob disease.14 Dependence on plasma donations exposes supply fluctuations related to seasonal, economic, and global disruptions, as observed during the COVID-19 pandemic.15 In addition, viral inactivation and removal processes increases the manufacturing complexity and cost, especially in resource-limited settings.16
To address these challenges, rHSA has been developed as a promising pathogen-free and sustainable alternative to mitigate supply related disruptions. Recombinant DNA (rDNA) technology enables large-scale production of albumin using prokaryotic and eukaryotic hosts, such as Escherichia coli, Saccharomyces cerevisiae, Kluyveromyces lactis, Pichia pastoris, transgenic plants, animals, and suspension cell cultures.17 Among these, Pichia pastoris (Komagataella phaffii) is preferred because of its ability to achieve high cell density, secrete properly folded proteins, and perform post-translational modifications similar to those in higher eukaryotes.18
Although rHSA products are approved for non-therapeutic use as excipients and/or carrier molecules, no rHSA products are currently approved for direct clinical/therapeutic use by the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA). Preclinical and previously established studies have demonstrated close similarity between various rHSA products and pdHSA with respect to molecular weight, tertiary structure, charge heterogeneity, and absence of glycosylation.19–26 A previous Phase I study conducted on a potential rHSA product showed favourable pharmacokinetics, pharmacodynamics, safety, and immunogenicity profiles to pdHSA, with similar increases in serum albumin concentration and COP.26 Several ongoing/completed Phase II/III trials are currently evaluating various rHSA products in participants with decompensated liver cirrhosis; however, the results are not yet publicly available, and recent sources mention that these studies have met their primary endpoint. (NCT06553456, NCT05858853, NCT06355479, NCT04835480, NCT06411743).27 Although Phase II/III studies are ongoing in patient populations, controlled evaluation in healthy volunteers remains essential to establish baseline pharmacokinetics, pharmacodynamics, and safety without disease-related variability, which is critical for dose optimization and regulatory acceptance.
This recombinant approach offers significant advantages including improved viral safety, batch-to-batch consistency, high purity, and scalable production, making it a safer and more reliable alternative to pdHSA.28 The test-rHSA (20% w/v) used in this study was expressed in Pichia pastoris and extensively characterised for its structural and physicochemical equivalence to native pdHSA. Analytical assessments, including molecular weight determination, size exclusion, ion-exchange chromatography, isoelectric focusing, glycosylation analysis, and purity evaluation confirmed a high degree of structural similarity, providing a mechanistic rationale for its expected pharmacodynamic behaviour.
This Phase I study evaluated the safety, tolerability, pharmacokinetics, pharmacodynamics, and immunogenicity of test-rHSA compared with pdHSA in healthy volunteers, with the objective of establishing PK/PD equivalence and confirming the functional integrity of rHSA prior to patient-based Phase III clinical studies.
Materials and Methods
Study Design
This Phase I open-label, randomized, single-centre study was designed to investigate the safety, tolerability, PK/PD, and immunogenicity of rHSA in comparison with pdHSA in healthy adult participants (CTRI/2023/04/051487). This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice (GCP) guidelines, and applicable regulatory requirements. Written informed consent was obtained from all participants prior to study initiation. The study was conducted in two sequential parts. Part 1 (n=12) used a standard 3+3 dose-escalation design to evaluate safety and tolerability of intravenous rHSA with escalating doses at 10 g, 20 g and 40 g administered in three individual cohorts, which is consistent with standard first-in-human study designs.29 Based on the outcomes, Part 2 (n=50) was conducted as a 1:1 randomized, parallel-arm comparative Phase of rHSA and pdHSA to assess the pharmacokinetics (PK), pharmacodynamics (PD), safety, and immunogenicity with escalating doses, ie. 10 g, 20 g and 40 g with a dosing interval of 3-weeks in the same cohort. The dose selection was based on prior clinical evidence, and established safety profiles of human serum albumin in the relevant studies.24,26,30–32 Furthermore, a placebo group was not included in this Phase 1 study due to highly varying endogenous nature of albumin and in accordance with regulatory guidance for biosimilars, which recommends direct PK comparability between test and reference products to establish bioequivalence.33 The primary objective was to characterize the PK profile of test rHSA in comparison to pdHSA in healthy volunteers, while secondary objectives included evaluation of safety, tolerability, PD parameters (COP and HCT ratio), and immunogenicity. The overall study duration per participant in Part 1 was approximately 4 days, and 95 days for Part 2, encompassing screening, dosing, and post-treatment follow-up. The study protocol and all amendments were approved by the Institutional Ethics Committee (IEC) and conducted in compliance with ICH guidelines under the supervision of the Independent Data Monitoring Committee (IDMC).
Study Participants
This study enrolled 62 healthy adults aged 18–45 years with a body mass index (BMI) between 18.5 and 30.0 kg/m2, who were determined to be in good health based on pre-study physical examination, vital signs, and clinical laboratory assessments. Healthy male participants were included in Part 1 (n=12), whereas both healthy male participants and nonpregnant, nonlactating female participants were eligible for Part 2 (n=50) of the study. However, no female participants were enrolled in the study. All participants provided written informed consent prior to enrolment and met pre-specified eligibility criteria. The inclusion criteria required participants to be free from any clinically significant medical history or active disease, as determined by comprehensive screening assessments that included detailed medical history, physical examination, clinical laboratory tests, chest radiography (performed within 11 months prior to dosing), and a 12-lead electrocardiogram (ECG). Additional gynaecologic evaluations with acceptable methods of contraception were planned for female participants, but were not conducted because no female participants were enrolled. All participants were required to effectively communicate with study personnel, be available for the entire study duration, and have no history of drug or alcohol abuse or dependence.
Exclusion criteria included known hypersensitivity to human plasma proteins or yeast-derived products (Pichia pastoris, Saccharomyces cerevisiae), or any contraindication to plasma protein administration. Participants with significant cardiovascular, respiratory, hepatic, renal, haematologic, gastrointestinal, endocrine, immunologic, dermatologic, musculoskeletal, neurological, or psychiatric disorders or any clinically relevant illness within four weeks before dosing were excluded. Additional exclusion criteria included a history of severe allergic or anaphylactoid reactions (eg. asthma and urticaria) following drug exposure, recent (within four weeks) treatment with corticosteroids or human plasma derivatives, or recent or planned surgical procedures within 20 days of the study drug administration. Participants who tested positive for alcohol or drugs of abuse prior to check-in, or who were unwilling to abstain from tobacco, xanthine-containing beverages, alcohol, grapefruit, cranberry juice, or any prescription, over-the-counter, or recreational drugs (including marijuana, amphetamines, barbiturates, cocaine, benzodiazepines, and opioids) during the study period were excluded. Individuals with recent or ongoing hormone replacement therapy or an unwillingness to abstain from androgens or anabolic steroids during the study period were also excluded.
Sample Size Calculation
The sample size of 12 participants in Part 1 was deemed adequate for standard 3+3 dose-escalation design to evaluate safety and tolerability. The sample size in Part 2 was determined based on statistical considerations for bioequivalence, rather than recruitment limitations, using statistical assumptions adapted from Bosse et al24 considering a test-to-reference (T/R) ratio of 90.00%–111.11%, an inter-subject coefficient of variation (CV) of approximately 15%, a two-sided significance level of 5%, and a statistical power of 80%, with bioequivalence acceptance limits of 80.00%–125.00%. Based on these assumptions, a minimum of 42 participants were required to provide adequate power to demonstrate the bioequivalence between test rHSA with pdHSA. Although a minimum of 42 participants was estimated to achieve 80% power, the sample size was increased to 50 participants (25 per treatment arm) to account for an anticipated dropout rate (~20%) and variability associated with endogenous albumin, ensuring sufficient evaluable data for robust bioequivalence analysis. The assumed intra-subject coefficient of variation (ISCV) of approximately 15% for Cmax further supported this estimation, ensuring at least 80% power to confirm equivalence at an α level of 0.05.
Study Procedures
Part 1: Safety Cohorts
A 3 + 3 dose escalation design was employed in Part 1 to evaluate the safety and tolerability of the test rHSA during first-in-human exposure. Participants in each safety cohort received the test rHSA as an intravenous (IV) infusion via an infusion pump at a constant rate of 2 mL/min on day 1. This adaptive design minimizes the potential risk while enabling the efficient identification of a safer dose. The participants in Part 1 were sequentially assigned to three dose cohorts for the rHSA test: 10 g (cohort 1, n = 3), 20 g (cohort 2, n = 3), and 40 g (cohort 3, n = 3 + 3), based on the absence of any adverse events or dose-limiting toxicities (DLTs). All participants were monitored continuously for 48 h post infusion. DLTs were defined as adverse events that were deemed certainly, probably, or possibly related to the investigational product (IP) as per the World Health Organization (WHO) causality assessment criteria, and considered unacceptable due to their severity or irreversibility, thereby precluding further dose escalation or continued administration at that level.
Part 2: PK/PD and Immunogenicity Cohorts
The participants in Part 2 received three consecutive doses of the assigned IP at 3-week intervals via intravenous infusion at a controlled rate of 2 mL/min. The dosing regimen followed was: Dose 1–10 g (50 mL) infused over 25 min on Day 1; Dose 2–20 g (100 mL) infused over 50 min on day 22; and Dose 3–40 g (200 mL) infused over 100 min on day 43. The maximum cumulative dose administered to each participant during the study was 70 g. The participants were closely monitored during and for at least one-hour after each infusion for infusion-related reactions or adverse events. Baseline correction was used to adjust endogenous levels and better treatment-related exposure.
Study Outcomes
Primary endpoints: The primary pharmacokinetic (PK) endpoints included Cmax (Maximum observed serum concentration following the 1st dose), representing peak systemic exposure, and AUC∞ (Area under the concentration-time curve extrapolated to infinity following the 3rd dose), reflecting total systemic exposure. Secondary endpoints: Pharmacodynamic (PD) measures included COP (Colloid osmotic pressure), reflecting oncotic activity of albumin, and HCT (Hematocrit) ratio, indicating hemodilution following infusion. Secondary PK parameters included AUCt, (Area under the concentration-time curve up to the last measurable concentration), AUC% (Area under the concentration extrapolated), representing the proportion of total exposure estimated beyond the last measurable time point and indicating reliability of AUC∞), Kel (Terminal elimination rate constant), Tmax (Time to reach peak concentration), t1/2 (Terminal elimination half-life, indicating the persistence of albumin in circulation) Vd (Volume of distribution), reflecting the extent of tissue distribution), and CL (Clearance represents the rate of systemic elimination). Safety endpoints included assessment of adverse events (AEs) and serious adverse events (SAEs), and immunogenicity assessment with presence of anti-drug antibodies (ADAs) and anti-HCP antibodies.
Bioanalytical Method
Serum albumin concentrations were quantified using a validated competitive enzyme-linked immunosorbent assay (ELISA) capable of detecting both rHSA and pdHSA. The bioanalytical assay was validated in accordance with ICH M10 bioanalytical method validation guidelines and demonstrated acceptable accuracy, precision, selectivity, and linearity across the full calibration range. Accuracy and precision were within ±15% for quality control samples and within ± 20% for the lower and upper limits of quantification (LLOQ and ULOQ, respectively), confirming stable assay performance at low concentrations. The validated range spanned 1.030 µg/mL and 25.750 µg/mL, using serum as the biological matrix.
Pharmacokinetic Assessments
In Part 2, a total of 49 blood samples (4.0 mL each) were collected from each participant for pharmacokinetic analysis. This included 4 pre-infusion samples (−72.0 h, −48.0 h, −24.0 h, and pre-dose (0.0)), followed by 7 samples on Day 1 (Post-infusion samples were collected at 0.083 h (5 min), 0.25 h (15 min), 0.5 h (30 min); and 1.0 h, 2.0 h, 6.0 h, 12.0 h), and subsequent samples at 24.0 h (Day 2), 72.0 h (Day 4), 144.0 h (Day 7), and 192.0 h (Day 9) for the 10 g dose. The same schedule was repeated for 20 g dose (Day 22). Likewise, for 40 g dose (Day 43), additional samples were collected at 360.0 h (Day 58), 528.0 h (Day 65), 768.0 h (Day 75), and 1152.0 h (Day 91), resulting in a comprehensive characterization of the concentration-time profile. The pharmacokinetic parameters for rHSA and pdHSA were calculated using both baseline-uncorrected and baseline-corrected serum albumin concentrations, with correction performed by subtracting the mean pre-dose value. Because albumin is endogenous, the baseline correction method can potentially affect late-time serum concentrations. A minimum of three quantifiable terminal-phase concentrations were required to estimate the terminal elimination rate constant (Kel); participants not meeting this criterion were excluded from the AUC0-∞ estimation. PK parameters were estimated using non-compartmental analysis (Phoenix® WinNonlin® v8.1.1).
Handling of BLQ Data
Concentrations below the lower limit of quantification (BLQ; < 1.000 µg/mL) were handled in accordance with ICH M10 regulatory guidance. The BLQ values prior to the first quantifiable concentration were treated as zero to avoid inflating the early exposure estimates. BLQ values following the last quantifiable concentration were excluded from the non-compartmental calculations of the AUC and Terminal elimination rate constant (Kel) to prevent bias in the terminal phase estimation. Interpolation was not applied to the BLQ data. This approach ensured the consistency, transparency, and robustness of PK parameter estimation across all participants and dose levels. Both baseline-corrected and uncorrected albumin concentrations were analyzed to distinguish endogenous albumin from exogenous albumin.
Pharmacodynamic Assessments
PD evaluations (COP and HCT ratio) included the collection of 40 blood samples (2.0 mL each) per participant at time points aligned with the PK sampling schedule. These PD samples were collected separately and were not derived from residual volumes of the PK samples. In contrast, PK assessments involved 49 blood samples (4.0 mL each), collected independently for serum albumin concentration analysis. PD Samples were planned after each dose in Part 2 at the time points aligned with PK sampling. HCT was determined by microcentrifugation and expressed as the pre- and post-dose value ratio. COP was measured using a mathematical formula under standardized conditions in accordance with the manufacturer’s instructions. The Area Under Effective Curve (AUEC) was computed using the linear trapezoidal method.
Pharmacokinetic and Pharmacodynamic Analysis Plan
PK/PD parameters were derived from plasma concentration-time profiles by non-compartmental analysis (NCA) performed using Phoenix® WinNonlin® professional software (Version 8.1.1; Pharsight Corporation, USA). The linear-up/log-down trapezoidal rule was used to calculate the AUC. The terminal elimination rate constant (Kel) was estimated using at least three data points within the terminal log-linear phase, with an adjusted R2 > 0.80 as the acceptance criterion. Pharmacokinetic parameters were analyzed based on the bioavailability ratio (test/reference-T/R) of the geometric least square means (LSMs) for the natural log-transformed parameters Cmax (1st dose) and AUC∞ (3rd dose). The 90% confidence intervals (CIs) of the T/R ratios were required to fall within the standard bioequivalence range of 80.00% to 125.00%. Log-transformed Cmax (1st dose) and AUC∞ (3rd dose) were calculated using ANOVA method with PROC GLM in SAS®. Differences due to treatment were considered as fixed effects.
The geometric least-squares mean ratios (test/reference) and the corresponding 90% confidence intervals were calculated to compare the test and reference treatments. The pharmacodynamic parameters were analyzed using T/R ratios with 90% confidence intervals (CIs) for the comparability of PD markers, that is, COP and HCT ratio, following the third dose (40 g dose).
Baseline correction was performed to account for endogenous albumin concentrations and to better isolate exogenous drug exposure. However, given the inherent variability associated with endogenous proteins, both baseline-corrected and uncorrected analyses were evaluated. Importantly, in Phase I biosimilar PK studies, standard non-compartmental bioequivalence analysis is preferred over population-based PK modeling due to regulatory acceptance, simplicity and suitability for dense data, with popPK used only as supportive, which may further improve robustness and will be considered in future studies.
Safety Assessments
All participants who received the investigational products (IPs) were evaluated for safety through comprehensive physical examinations, including vital signs, 12-lead electrocardiogram (ECG), and clinical laboratory investigations (Laboratory assessments were performed 48 h post-infusion in Part 1 and on Day 91 [± 3 days] in Part 2).
Immunogenicity Assessments
Immunogenicity was assessed by the presence of ADAs; Immunoglobulin E (IgE), immunoglobulin G (IgG), and antibodies against host cell proteins (anti-HCPs), present in test and reference products, using a bridging-format enzyme-linked immunosorbent assay (validated two-tier ELISA). In Part 1, two samples (4.0 mL each) were collected, one prior to the infusion of test rHSA or pdHSA, and another post-infusion at 48 h. In Part 2, immunogenicity was evaluated using a predefined sampling schedule comprising collection of 12 blood samples (4.0 mL each) per participant. Samples were collected at pre-dose, post-dose at 24.0 h, 192.0 h, and following each dose, with additional follow-up assessments conducted after each infusion at 61, 81, and 91 days. This sampling framework was uniformly applied to all enrolled participants (N=50), resulting in 600 total samples for immunogenicity analysis. Positive samples were further evaluated for titres and IgG/IgE subclasses.
Statistical Analysis
All continuous data are presented as the mean ± standard deviation (SD), along with the minimum and maximum values (range). Categorical variables were summarised as counts and percentages, as appropriate. PK parameters were log-transformed and analysed using analysis of variance (ANOVA). Bioequivalence was determined, if the 90% confidence intervals (CIs) for the geometric mean ratios of the primary PK parameters fell within the acceptance range of 80–125%. Statistical analyses were performed using the SAS® software (Version 9.4, SAS Institute Inc., Cary, NC, USA).
Results
Demographic Characteristics
A total of 87 participants were screened, of whom 25 were excluded for not meeting the eligibility criteria. A total of 62 participants were enrolled and allocated to Part 1 (n=12) and Part 2 (n=50). Four participants discontinued across both parts: 2 participants in Part 1 and 2 participants in Part 2. CONSORT flow diagram (Figure 1). All participants were Asian and identified as non-Hispanic/Latino males. Healthy volunteers were selected to minimize confounding factors and reduce variability, thereby enabling a more sensitive assessment of pharmacokinetic and pharmacodynamic differences between treatments. The mean (± SD) age ranged from 32.3 ± 6.66 to 35.30 ± 4.73 years across the study parts. The mean BMI values were comparable across Part 2, ranging from 22.86 ± 2.89 kg/m2 to 26.57 ± 2.78 kg/m2. The demographic characteristics are summarized in Table 1.
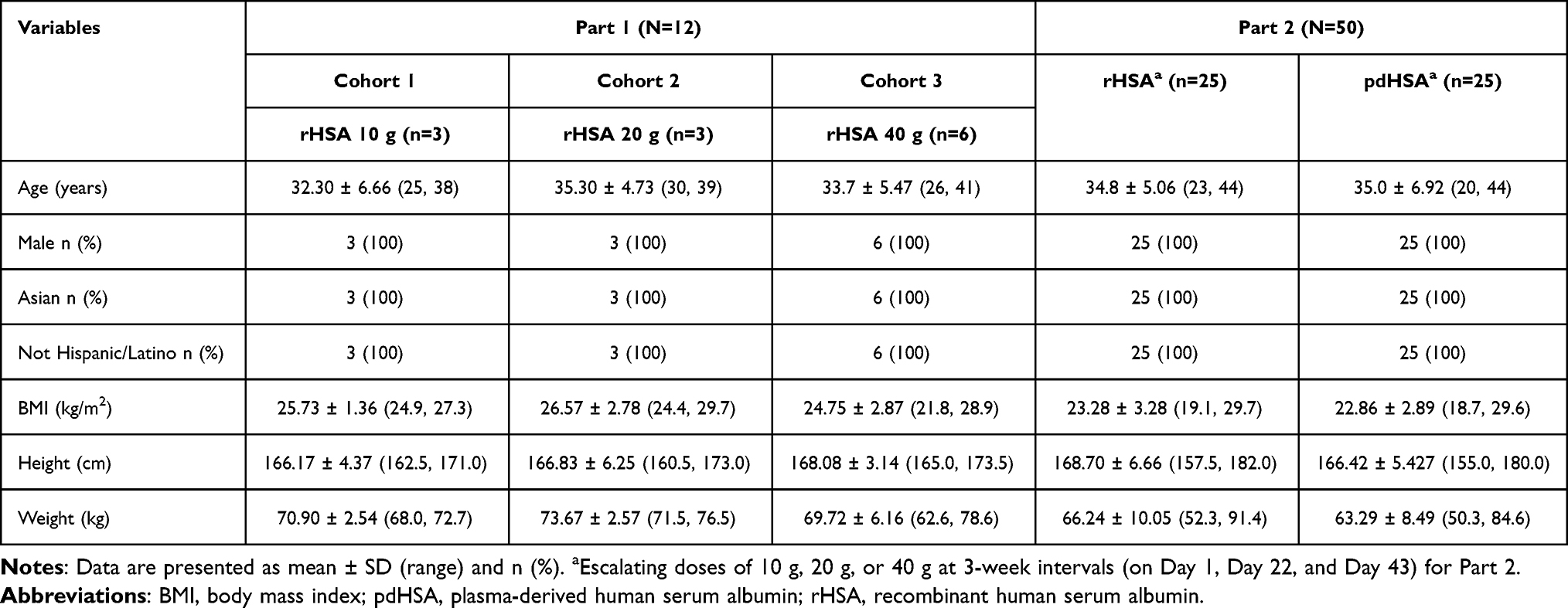 |
Table 1 Demographic Characteristics |
 |
Figure 1 CONSORT flow diagram. |
Pharmacokinetic Parameters
Serum albumin concentrations increased markedly following each dose of rHSA, with peak levels observed after the third dose (40 g). The pattern of changes in serum albumin concentration-time profiles was comparable between the test rHSA and pdHSA in Part 2, with elevated levels maintained and minimal variability observed up to day 91 for both baseline albumin-corrected and-uncorrected data (Figure 2A and B). To facilitate interpretation of variability, the baseline-corrected profiles demonstrate treatment-attributable changes but exhibit increased variability, particularly at later timepoints. In contrast, uncorrected profiles show more stable trends within the physiological range, providing a clinically relevant representation of albumin levels. In both analyses, rHSA and pdHSA exhibited comparable profiles across all dose levels. In the baseline albumin-corrected data, test-to-reference (T/R) ratio of the geometric means of Cmax for rHSA and pdHSA following 1st dose (10 g) was 131.15%, with 90% CIs of 80.03%–214.93%, exceeding the predefined bioequivalence limits of 80–125%. Baseline correction reduced post-dose concentrations (most of the patients had no change in albumin levels after baseline correction) and amplified inter-individual variability owing to endogenous albumin fluctuations. Consequently, only one participant met the criteria for reliable terminal slope estimation, precluding the robust estimation of AUC0–∞ in most participants. However, in three-five participants, the baseline uncorrected total albumin profiles demonstrated comparatively better post-dose signals and yielded stable slope estimates. Moreover, the Cmax and AUCt parameters were evaluated in more participants (baseline-corrected 11 vs. baseline-uncorrected up to n=25), and these parameters were within the accepted bioequivalence limits, supporting reliable PK comparability between rHSA and pdHSA.
 |
Figure 2 (A) Linear scale of mean serum albumin concentration (+ SEM) (g/dL) vs time profiles of recombinant human serum albumin (rHSA) and plasma derived human serum albumin (pdHSA) up to 91 days following escalating doses of 10 g, 20 g, and 40 g with baseline albumin corrected data. Reference, pdHSA and Test, rHSA. (B) Linear scale of mean serum albumin concentration (+ SEM) (g/dL) vs time profiles of test recombinant human serum albumin (rHSA) and plasma derived human serum albumin (pdHSA) up to 91 days following escalating doses of 10 g, 20 g, and 40 g with baseline albumin uncorrected data. Reference, pdHSA and Test, rHSA. |
Furthermore, following the 3rd dose, the baseline-corrected T/R ratio for AUCt was 189.73% with a 90% CI (92.68% - 388.40%), which was outside the bioequivalence limits. However, analyses based on the baseline-uncorrected data demonstrated PK comparability between rHSA and pdHSA. Specifically, the T/R ratio of Cmax after 1st dose was 99.12% and AUCt after 3rd dose was 97.36%, with corresponding 90% CIs (Cmax of 1st dose: 88.97–110.42%; AUCt of third dose: 86.41–109.70%) falling within the accepted bioequivalence range of 80–125% (Table 2).
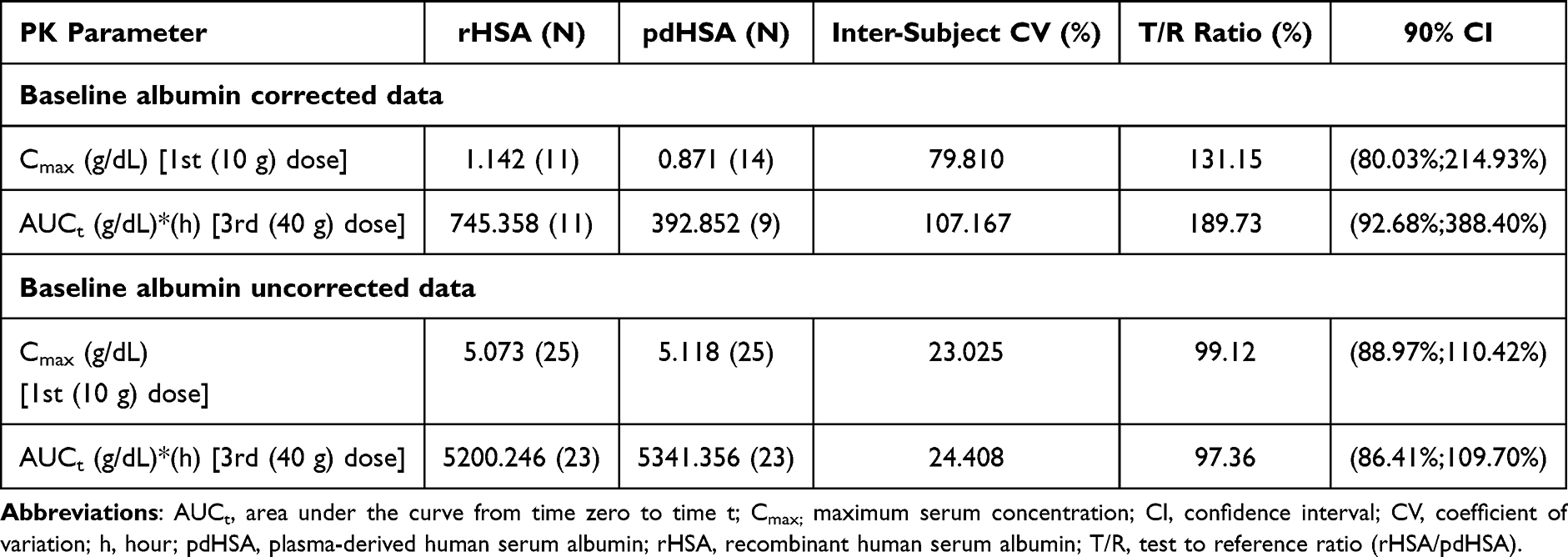 |
Table 2 Comparison of Geometric Means of Primary Pharmacokinetic Parameters for Baseline Albumin Corrected/Uncorrected Data Between Test Recombinant Human Serum Albumin (rHSA) and Plasma-Derived Human Serum Albumin (pdHSA) in Healthy Volunteers |
Pharmacodynamic Parameters
Colloid Osmotic Pressure (COP)
In Part 2, the increase in COP observed in the rHSA-treated group was comparable to that in the pdHSA group, with peak values occurring one day after administration of the second ascending dose (20 g) on day 23 in both groups. A similar peak was observed on Day 44 following the administration of the third ascending dose (40 g), corresponding to the trends in serum albumin levels (Figure 3). The AUECt for COP following the final dose demonstrated bioequivalence between rHSA and pdHSA, with a test-to-reference (T/R) ratio of 103.43% and 90% CIs (100.23%; 106.74%), falling within the predefined bioequivalence range (Table 3).
 |
Table 3 Comparison of Pharmacodynamic Parameters (COP and HCT Ratio) Between Test Recombinant Human Serum Albumin (rHSA) and Plasma-Derived Human Serum Albumin (pdHSA) in Healthy Volunteers Following Third (40 g) Dose |
 |
Figure 3 Linear scale of mean colloid osmotic pressure (mmHg) (±SEM) vs time profiles of test recombinant human serum albumin (rHSA) and plasma derived human serum albumin (pdHSA) up to 91 days following escalating doses of 10 g, 20 g, and 40 g. Reference, pdHSA and Test, rHSA. |
Hematocrit (HCT) Ratio
Maximum haemodilution was observed following administration of the 40 g dose in both rHSA- and pdHSA-treated groups in Part 2. The AUECt for the HCT ratio following the administration of a 40 g dose demonstrated bioequivalence between test rHSA and pdHSA with a T/R ratio of 95.43% with 90% CIs (91.89%; 99.10%) within the predefined bioequivalence limits (Table 3).
Safety
No SAEs occurred in either Part 1 and Part 2 during the study. Notably, in Part 1, no DLTs were observed within the first 48 h following rHSA infusion at doses up to 40 g. In Part 2, 4 AEs, including vomiting (1), dizziness (1), and urticaria (2), were reported in the rHSA group. All reported AEs were mild-to-moderate in severity and resolved without sequelae. Furthermore, 11 clinically insignificant laboratory abnormalities were observed during the study and were considered unlikely to be related to the investigational products (test-rHSA/reference-pdHSA). These include increased alanine aminotransferase (1), increased blood glucose (1), decreased blood phosphorus (2), increased blood triglycerides (1), decreased lymphocyte percentage (1), increased neutrophil percentage (1), and increased white blood cell count (3) in the test rHSA group. In the pdHSA group, a single event of increased blood bilirubin level was reported (Table 4).
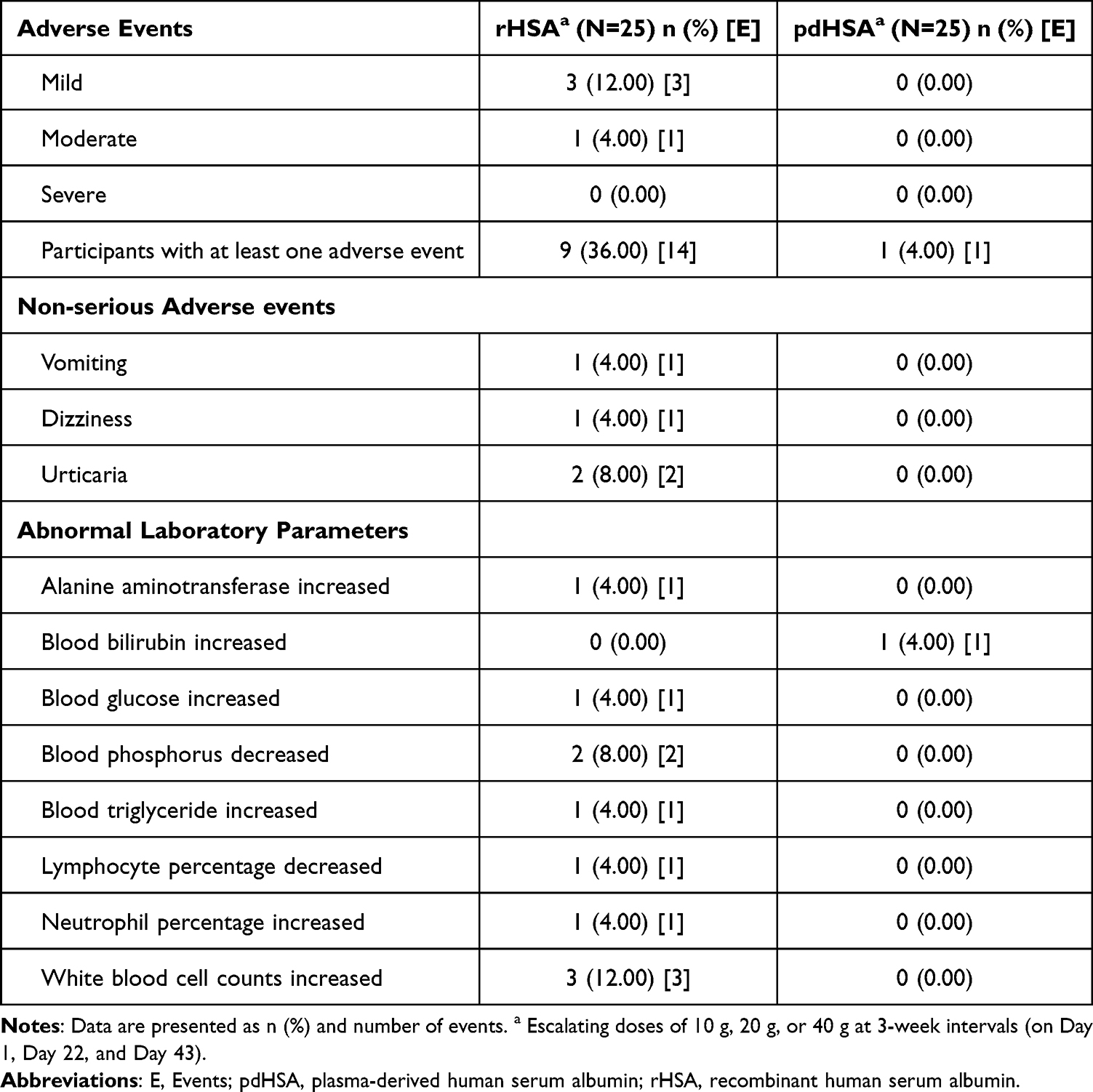 |
Table 4 Summary of Adverse Events (AEs) |
Immunogenicity (ADAs-IgG and IgE, and Anti- HCPs)
All participants tested negative for IgE and anti-HCP antibodies across all doses and treatment groups. In Part 1, no immunogenic response was observed in any participant. In Part 2, none of the participants in the test rHSA group developed IgG antibodies after the first two doses. One participant in the pdHSA group had pre-existing IgG antibodies that remained positive throughout the study period (up to day 90). Following the administration of the third dose, IgG antibodies were detected on day 51 (192 h) in four (16.0%) participants receiving the test rHSA. In the pdHSA group, IgG antibodies were detected in one (4.0%) participant on days 43 and 44 and subsequently in three (12.0%) participants on day 51 (192 h). One participant in the rHSA group developed transient ADAs that tested positive on day 61 and negative thereafter. Most IgG-positive participants in both groups remained IgG-positive at the follow-up visit on day 91 (EOS). No statistically significant differences in immunogenic responses were observed between both the groups (Table 5).
 |
Table 5 Participants with Positive IgG (Confirmatory) in Test Recombinant Human Serum Albumin (rHSA) and Plasma-Derived Human Serum Albumin (pdHSA)-Treated Group |
Discussion
The present study evaluated the safety, tolerability, PK/PD, and immunogenicity of a newly developed rHSA test product compared with those of pdHSA in healthy volunteers. The rationale for the selected dose range (10 g, 20 g, and 40 g) was based on previous reports demonstrating the safety of albumin administration up to 50 g in healthy individuals.21–26,34 The absence of DLTs across all dose levels supports the favourable safety profile of the test rHSA. The study drug was safely administered at up to 40 g with no DLTs during Part 1, which also served as the highest dose evaluated in Part 2 of the study. A three-week dosing interval (Days 1, 22, and 43) was implemented in Part 2 for administering the escalating doses, ie. 10 g, 20 g and 40 g to reflect the long physiological turnover period of albumin (approximately 25 days, reported by Levitt and Levitt). Albumin also has a long pharmacokinetic half-life in healthy adults (~19–21 days), and this dosing interval was selected to reduce the likelihood of drug accumulation.4,35
Pharmacokinetic comparability between test rHSA and pdHSA was demonstrated using baseline-uncorrected albumin concentrations. In contrast, the baseline-corrected and uncorrected PK outcomes showed discrepancies, reflecting the known challenges in estimating PK for endogenous proteins.36 Baseline correction amplified physiological fluctuations and increased the influence of low-level variability at late sampling points, destabilizing the terminal elimination rate constant (Kel) and inflating the extrapolated portion of AUC0–∞. Nonetheless, these effects did not reflect assay limitations, sampling issues, or true differences in disposition between rHSA and pdHSA.
The uncorrected total-albumin profiles showed an orderly terminal decline and yielded reproducible PK estimates. The geometric mean (T/R) ratios observed for Cmax (1st dose) and AUCt (3rd dose) were within the accepted bioequivalence range, indicating comparable systemic exposure and elimination kinetics. The lack of equivalence observed with baseline-corrected data most likely reflects the endogenous nature of albumin, its internal redistribution (eg. vascular-to-interstitial shifts), the healthy-participant study population, and increased inter-individual variability. In the uncorrected dataset, albumin concentrations remained consistently quantifiable at late time points for most participants, whereas an apparent decline emerged only after baseline subtraction, indicating that baseline correction rather than assay performance contributed to the observed instability. Albumin is subjected to tight homeostatic regulation, and AUC0–∞ could not be reliably estimated in most participants, and was therefore assessed descriptively in a limited subset, in alignment with previously reported studies on recombinant albumin formulations.24,26 These observations highlight the known limitations of baseline correction for endogenous proteins, where physiological variability may influence parameter estimation. Advanced modeling approaches, such as mixed-effects models, may be warranted to provide improved characterization of PK profiles in future study investigations. Furthermore, the wide confidence intervals observed in baseline-corrected PK parameter analyses further highlight the variability and reduced reliability of these estimates.
The originally planned parameter AUC∞ could not be reliably estimated because of the absence of a consistent terminal log-linear phase, limited quantifiable post-Tmax concentrations, and presence of highly extrapolated fractions (> 20%). This is in-line with similar challenges that have been noticed in earlier bioequivalence assessment studies of albumin,16,24 emphasizing the practical limitations of deriving AUC∞ for large endogenous proteins. Minor differences in clearance (CL) and volume of distribution (Vd) between formulations were observed after baseline correction, with the test rHSA exhibiting slightly lower CL values, suggesting slower systemic elimination. The Vd values were comparable across certain doses, indicating similar tissue distribution characteristics between the two formulations.
A direct comparison of the elimination half-life (t½) between test rHSA and pdHSA was not performed, as reliable estimation of the terminal phase was limited by the highly variable endogenous nature of albumin, baseline correction, and low post-dose signals in several participants. However, in the baseline-uncorrected analysis, (CL) and (Vd) values were closely aligned between treatments across all dose levels, suggesting similar elimination kinetics for rHSA and pdHSA. These findings highlight that endogenous and exogenous albumin are handled similarly by physiological regulatory mechanisms, with dynamic redistribution between intravascular and extravascular compartments to maintain systemic homeostasis. These observed PK differences may be influenced by intrinsic molecular- and production-related factors. The rHSA was expressed in recombinant systems (such as Pichia pastoris), which can introduce minor charge heterogeneity or glycosylation patterns. Nonetheless, the observed high inter-subject variability was within the expected physiological range and did not indicate any clinically meaningful differences.
Pharmacodynamic outcomes further supported the PK findings in a direct manner. Administration of rHSA and pdHSA produced comparable increases in COP, with corresponding reductions in the HCT ratio, indicating equivalent oncotic activity. Albumin contributes approximately 80% of plasma COP and its synthesis is primarily regulated by oncotic pressure; thus, these findings were consistent with the classic work of Tayek and Blackburn et al, who demonstrated that circulating albumin concentration directly influences plasma oncotic pressure and HCT responses in healthy individuals.24,37 The observed PD similarity supports functional equivalence between test rHSA and pdHSA and reinforces the clinical relevance of the pharmacokinetic comparability findings. Although no point-by-point statistical comparison of concentration time profiles was not performed, the observed similarity between treatments is supported by demonstrated bioequivalence of key PK parameters. Additionally, exploratory correlation was not performed, the PK exposures directly correlated with the PD (COP and HCT) as discussed above and also the immunogenicity endpoints were not correlated and warrant further investigation in larger studies.
The test rHSA 20% solution was well tolerated across all evaluated doses (10, 20, and 40 g), with cumulative exposure up to 70 g. No serious or severe adverse events occurred in the study. Reported adverse events, including urticaria, vomiting, and dizziness, were mild-to-moderate, transient, and self-limiting. These findings are consistent with previous reports on recombinant albumin safety in humans.26
Immunogenicity assessments demonstrated low and comparable rates of IgG positivity between the treatment groups. Low-titre IgG antibodies were detected in a small proportion of participants following repeated dosing; however, no IgE-mediated responses or anti-host cell protein antibodies were observed. Although these IgG responses were low-titre and not associated with any clinical or pharmacological impact, their potential long-term implications cannot be fully excluded and warrant further evaluation in larger and longer-term studies. This minimal immunogenic potential reflects the high degree of molecular similarity between rHSA and its plasma-derived counterpart. The absence of hypersensitivity reactions, lack of impact on pharmacokinetic profiles, and comparable incidence between treatment groups suggest minimal clinical relevance of immunogenic potential. The expression of rHSA in Pichia pastoris and stringent purification processes effectively reduce residual impurities and mitigate the risk of immune-mediated responses. Additionally, some antibody responses were transient in nature. These findings are consistent with the low immunogenic potential expected for a highly purified recombinant protein with structural similarity to endogenous albumin. Nevertheless, long-term studies in patient populations are warranted to fully evaluate the clinical implications of repeated exposure.
The findings of this study are consistent with the predefined objectives and demonstrate comparable pharmacokinetic, pharmacodynamic, and safety profiles between recombinant and plasma-derived human serum albumin. These results align with previous Phase I studies, which have similarly reported no significant differences in PK/PD behavior or safety outcomes between recombinant and plasma-derived albumin formulations.26 Furthermore, Phase II/III clinical trials in patients with liver cirrhosis and ascites have reported that rHSA produces comparable increases in serum albumin levels and maintains a favourable safety and immunogenicity profile relative to human serum albumin.29
Overall, this study demonstrated that rHSA is pharmacokinetically, pharmacodynamically, and immunogenically comparable to pdHSA with excellent safety and tolerability across clinically relevant doses. These findings confirm that rHSA can be considered a safe, tolerable, and pathogen-free alternative to pdHSA for clinical and biopharmaceutical applications. Further studies in patient populations and under long-term treatment conditions are warranted to confirm the therapeutic potential of rHSA.
Limitations
The limited demographic diversity of the study population, including the absence of female participants and restricted ethnic representation, should be considered when interpreting the generalizability of the findings. This was a single-centre study of healthy volunteers with a modest sample size (but a generally employed sample size for Phase I Studies) and without a placebo control. The absence of arm was due to the endogenous nature of albumin and regulatory guidance for Phase I biosimilars, instead an active comparator was used. Conducting the study in healthy volunteers minimized the disease-related variability and ensured internal validity. Given that albumin is an endogenous protein and both treatments are active comparators, inclusion of a placebo arm was not considered scientifically or ethically necessary. However, the absence of a placebo arm limits the ability to fully characterize natural fluctuations in endogenous albumin. In addition, the interstitial albumin levels could not be assessed. Additionally, the limited number of evaluable participants for certain PK parameters (Estimation of AUC∞) may reduce confidence in these estimates, which are further constrained by variability in terminal-phase extrapolation across most profiles; however, no historical data have been reported owing to the challenges involved. Furthermore, the wide confidence intervals observed for certain baseline-corrected PK parameters indicate variability and imprecision, likely due to sample size and inter-individual variability, limiting their interpretability. Nonetheless, these findings should be interpreted cautiously and confirmed in patients with hypoalbuminaemia or liver disease, where the albumin kinetics may differ. The long-term clinical significance of observed IgG immunogenic responses remains to be established.
Conclusion
In conclusion, rHSA demonstrated favourable safety and tolerability, and was pharmacologically comparable to pdHSA following intravenous administration in healthy adults. Comparable pharmacokinetic and pharmacodynamic profiles, together with the absence of clinically relevant immunogenic responses, indicate that rHSA is a safe and pathogen-free alternative to pdHSA. Test rHSA was safely administered at cumulative single doses of up to 40 g, with AUCt serving as the key exploratory pharmacokinetic parameter confirming the similarity between the two products. These findings warrant further clinical evaluation of rHSA in patients requiring albumin therapy, including those with liver cirrhosis, septic shock, or HRS. A randomised Phase III clinical trial comparing the efficacy, safety, and immunogenicity of rHSA and pdHSA in participants with decompensated liver cirrhosis with ascites is currently under progress.
CTRI Trial Registration
This study followed CONSORT reporting standards, and the trial was registered with the Clinical Trials Registry of India (CTRI/2023/04/051487).
Data Sharing Statement
The datasets generated and/or analyzed during the current study are not publicly available due to confidentiality considerations but are available from the corresponding author upon reasonable request via Email at [email protected].
Ethics Approval and Consent to Participate
This study was conducted in accordance with the Declaration of Helsinki. The study protocol number C1B01642 (approval number), received approval from the Sangini Hospital Ethics Committee, Ahmedabad, Gujarat, India. All participants provided written informed consent prior to enrolment after being fully informed about the study’s purpose, procedures, and potential risks, and were made aware of their right to withdraw at any time.
Acknowledgments
We thank Cliantha Research Limited, Ahmedabad, India, for successful execution and completion of the Phase I clinical study on recombinant human serum albumin (rHSA). The authors thank all trial participants, their families, the investigators and site staff, and the members of the safety-monitoring committees for their valuable contributions. The authors also acknowledge Dr. Sandeep K, and Dr. Karunakar T for their scientific writing support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. All the authors have read and approved the final manuscript.
Funding
Financial support was provided by Shilpa Biologicals Pvt. Limited, Belur, Dharwad, Karnataka, India.
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Adamczyk Z, Pomorska A, Nattich-Rak M, et al. Protein adsorption mechanisms at rough surfaces: serum albumin at a gold substrate. J Colloid Interface Sci. 2018;530:631–16. doi:10.1016/j.jcis.2018.06.063
2. Peters Jr T. All About Albumin. Biochemistry, Genetics, and Medical Applications. San Diego, California: Academic Press, Inc; 1996.
3. Ghuman J, Zunszain PA, Petitpas I, et al. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol. 2005;353(1):38–52. doi:10.1016/j.jmb.2005.07.075
4. Fanali G, Di Masi A, Trezza V, et al. Human serum albumin: from bench to bedside. Mol Aspects Med. 2012;33(3):209–290. doi:10.1016/j.mam.2011.12.002
5. Emerson Jr TE. Unique features of albumin: a brief review. Crit Care Med. 1989;17(7):690–694. doi:10.1097/00003246-198907000-00020
6. Chuang VT, Kragh-Hansen U, Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharm Res. 2002;19(5):569–577. doi:10.1023/A:1015396825274
7. Lei J, Guan B, Li B, et al. Expression, purification and characterization of recombinant human interleukin-2-serum albumin (rhIL-2-HSA) fusion protein in Pichia pastoris. Protein Expr Purif. 2012;84(1):154–160. doi:10.1016/j.pep.2012.05.003
8. Trebicka J. Role of albumin in the treatment of decompensated liver cirrhosis. Curr Opin Gastroenterol. 2022;38(3):200–205. doi:10.1097/MOG.0000000000000838
9. Gagliardi R, Zeni N, Piano S. Intravenous albumin in cirrhosis: updated clinical uses and novel perspectives. Ann Hepatol. 2023;28(6):101150. doi:10.1016/j.aohep.2023.101150
10. Tufoni M, Zaccherini G, Caraceni P, et al. Albumin: indications in chronic liver disease. United Eur Gastroenterol J. 2020;8(5):528–535. doi:10.1177/2050640620910339
11. Abedi F, Zarei B, Elyasi S. Albumin: a comprehensive review and practical guideline for clinical use. Eur J Clin Pharmacol. 2024;80(8):1151–1169. doi:10.1007/s00228-024-03664-y
12. IMARC Group. (2024). Global albumin market expected to reach USD 10.5 billion by 2033. Available from: https://www.imarcgroup.com/albumin-market-statistics.
13. Matejtschuk P, Dash CH, Gascoigne EW. Production of human albumin solution: a continually developing colloid. Br J Anaesth. 2000;85(6):887–895. doi:10.1093/bja/85.6.887
14. Simon T, Schumann P, Bieri M, et al. Hyperoncotic human albumin solutions for intravenous fluid therapy: effectiveness of pathogen safety and purification methods, and clinical safety. Biosaf Health. 2022;5(1):21–29. doi:10.1016/j.bsheal.2022.12.004
15. Turecek PL, Hibbett D, Kreil TR. Plasma procurement and plasma product safety in light of the COVID-19 pandemic from the perspective of the plasma industry. Vox Sang. 2022;117(6):780–788. doi:10.1111/vox.13267
16. Ohnishi K, Kawaguchi A, Nakajima S, et al. A comparative pharmacokinetic study of recombinant human serum albumin with plasma-derived human serum albumin in patients with liver cirrhosis. J Clin Pharmacol. 2008;48(2):203–208. doi:10.1177/0091270007310549
17. Chen Z, He Y, Shi B, et al. Human serum albumin from recombinant DNA technology: challenges and strategies. Biochim Biophys Acta. 2013;1830(12):5515–5525. doi:10.1016/j.bbagen.2013.04.037
18. Pan Y, Yang J, Wu J, et al. Current advances of Pichia pastoris as cell factories for production of recombinant proteins. Front Microbiol. 2022;13:1059777. doi:10.3389/fmicb.2022.1059777
19. Ohtani W, Masaki A, Ikeda Y, et al. Structure of recombinant human serum albumin from Pichia pastoris. Yakugaku Zasshi. 1997;117(4):220–232. doi:10.1248/yakushi1947.117.4_220. Japanese. Erratum in: Yakugaku Zasshi 1997;117(12):1033
20. Okano K. Metabolic fate of recombinant human serum albumin (rHSA)(2). Jpn Pharmacol Ther. 1987;25:2007–2018.
21. Chuang VT, Otagiri M. Recombinant human serum albumin. Drugs Today. 2007;43(8):547–561. doi:10.1358/dot.2007.43.8.1067343
22. Kasahara A, Kita K, Tomita E, et al. Repeated administration of recombinant human serum albumin caused no serious allergic reactions in patients with liver cirrhosis: a multicenter clinical study. J Gastroenterol. 2008;43(6):464–472. doi:10.1007/s00535-008-2178-5
23. Margarson MP, Soni NC. Changes in serum albumin concentration and volume expanding effects following a bolus of albumin 20% in septic patients. Br J Anaesth. 2004;92(6):821–826. doi:10.1093/bja/aeh111
24. Bosse D, Praus M, Kiessling P, et al. Phase I comparability of recombinant human albumin and human serum albumin. J Clin Pharmacol. 2005;45(1):57–67. doi:10.1177/0091270004269646
25. Bihari S, Wiersema UF, Perry R, et al. Efficacy and safety of 20% albumin fluid loading in healthy subjects: a comparison of four resuscitation fluids. J Appl Physiol. 2019;126(6):1646–1660. doi:10.1152/japplphysiol.01058.2018
26. Li C, Xiang W, Wu M, et al. A randomized dose-escalation study on the safety, tolerability, immunogenicity, pharmacokinetics and pharmacodynamics of a novel recombinant human albumin in healthy subjects. Eur J Pharm Sci. 2021;165:105923. doi:10.1016/j.ejps.2021.105923
27. Niu J, Gao Y, Wang G, et al. Rice-derived recombinant human serum albumin as an alternative to human plasma for patients with decompensated liver cirrhosis: a randomised, double-blind, positive-controlled and non-inferiority trial. Gut. 2025;74(9):1476–1485. doi:10.1136/gutjnl-2025-335577
28. Tarelli E, Mire-Sluis A, Tivnann HA, et al. Recombinant human albumin as a stabilizer for biological materials and for the preparation of international reference reagents. Biologicals. 1998;26(4):331–346. doi:10.1006/biol.1998.0163
29. Wang X, Li W, Kong F, et al. A randomized, phase Ib trial of recombinant human serum albumin in cirrhotic patients with ascites. Hepatol Int. 2026;20(1):91–101. doi:10.1007/s12072-025-10871-x
30. Rubio-Baines I, Camporota L, González-Delgado D, et al. Use of human serum albumin in critically ill patients: a narrative review. J Clin Med. 2026;15(5):1981. doi:10.3390/jcm15051981
31. Infusino I, Panteghini M. Serum albumin: accuracy and clinical use. Clin Chim Acta. 2013;419:15–18. doi:10.1016/j.cca.2013.01.005
32. Farrugia A. Concerning Chapter 5 - Human albumin; in cross-sectional guidelines for therapy with blood components and plasma derivatives. Transfus Med Hemother. 2009;36(6):399–407.
33. U.S. Food and Drug Administration. Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. Silver Spring, MD: FDA; 2014.
34. Li W, Wang X, Gao SY, et al. Study of the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of recombinant human albumin in patients with ascites due to cirrhosis. Hepatology. 2021;2021:1245.
35. Levitt DG, Levitt MD. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J Gen Med. 2016;9:229–255. doi:10.2147/IJGM.S102819
36. Chan SF, Macaskill P, Irwig L, Walter SD. Adjustment for baseline measurement error in randomized controlled trials induces bias. Control Clin Trials. 2004;25(4):408–416. doi:10.1016/j.cct.2004.06.001
37. Tayek JA, Blackburn GL. Goals of nutritional support in acute infections. Am J Med. 1984;76(5A):81–90. doi:10.1016/0002-9343(84)90248-1
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of Pharmacokinetics and Safety with Bioequivalence of Ibuprofen Sustained-Release Capsules of Two Formulations, in Chinese Healthy Volunteers: Bioequivalence Study
Huang C, Yin Z, Yang Y, Mo N, Yang H, Wang Y
Drug Design, Development and Therapy 2023, 17:1881-1888
Published Date: 23 June 2023
Comparative Pharmacokinetics and Bioequivalence Evaluation of Two Formulations of Pramipexole Dihydrochloride Extended-Release Tablets in Healthy Chinese Subjects Under Fasted and Fed States: A Randomized, Open-Label, Single-Dose, Two-Period Crossover Clinical Trial
Yang L, Zhang L, Luo Z
Drug Design, Development and Therapy 2023, 17:2369-2381
Published Date: 15 August 2023
Evaluation of Olaparib Tablet Safety and Pharmacokinetics in Healthy Chinese Male Subjects
Dong R, Chen J, Guo N, Yang Y, Wu J, Wang X, Song Y, Zhang X
Drug Design, Development and Therapy 2024, 18:5529-5539
Published Date: 3 December 2024
Pharmacokinetics and Bioequivalence of Two Fixed-Dose Combination Tablets of Valsartan/Amlodipine (80/5 Mg) in Healthy Chinese Subjects
Tian M, Huang J, Chen Y, Jin Q, Jiang H, Shi C, Mei J, Xu M, Yu X, Yang S
Drug Design, Development and Therapy 2025, 19:11-22
Published Date: 3 January 2025
Pharmacokinetics, Pharmacodynamic, Safety and Tolerability of Fazamorexant, a Novel Dual Orexin Receptor Antagonist: Report of the First-in-Human Study
Ni J, Jin L, Zhao D, Zhang W, Li B, Huang X, Hao X
Drug Design, Development and Therapy 2025, 19:5271-5282
Published Date: 19 June 2025