Back to Journals » Journal of Inflammation Research » Volume 19
Peripheral Blood Mononuclear Cells in Sepsis: Immune Trajectories, Monocyte Dysfunction, and Translational Biomarkers
Authors Qian Z ![]()
Received 23 March 2026
Accepted for publication 12 June 2026
Published 26 June 2026 Volume 2026:19 611267
DOI https://doi.org/10.2147/JIR.S611267
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Xin Du
Zhiheng Qian
Jinhua Central Hospital, Jinhua, People’s Republic of China
Correspondence: Zhiheng Qian, Email [email protected]
Abstract: Sepsis is characterized by a dynamic and heterogeneous immune response in which hyperinflammation and immunosuppression often coexist and change over time. Peripheral blood mononuclear cells (PBMCs), including monocytes and lymphocyte subsets, provide an accessible window into this evolving immune trajectory and can be repeatedly assessed during disease progression. In this narrative review, we synthesize recent evidence from single-cell and spatial omics, mechanistic studies, and PBMC-based biomarker research, with a focus on three connected themes: time-dependent PBMC remodeling, monocyte/macrophage dysfunction, and translational immune biomarkers. Current evidence indicates that PBMC remodeling in sepsis is marked by monocyte state transitions, HLA-DR downregulation, lymphocyte apoptosis and exhaustion, regulated cell death, and immunometabolic reprogramming. These changes help explain why some patients recover after the acute inflammatory phase, whereas others progress toward persistent immunoparalysis, secondary infection, prolonged critical illness, or poor outcomes. PBMC-derived biomarkers, including transcriptomic signatures, monocyte phenotypes, lymphocyte exhaustion markers, metabolic indicators, and functional immune assays, may improve immune phenotyping and risk stratification. However, clinical translation remains limited by cohort heterogeneity, age-related differences, timing-dependent immune states, assay standardization, and insufficient prospective validation. Future studies should prioritize longitudinal, age-stratified, and biomarker-guided designs to determine whether PBMC-based immune monitoring can support individualized immunomodulatory strategies in sepsis.
Keywords: sepsis, peripheral blood mononuclear cells, PBMCs, immunopathogenesis, biomarkers, single-cell omics
Introduction
Sepsis is a life-threatening syndrome caused by a dysregulated host response to infection, leading to organ dysfunction and substantial mortality despite advances in antimicrobial and supportive care.1 Its immune pathophysiology is not a simple linear transition from cytokine storm to immunosuppression. Instead, hyperinflammation and immune paralysis often coexist within the same patient and change over time, influencing tissue injury, pathogen clearance, secondary infection risk, and long-term outcomes.2,3 Although clinical tools such as SOFA scoring, lactate, and procalcitonin remain useful for diagnosis and risk assessment, they do not directly capture the patient’s evolving immune state.4–6 This limitation has increased interest in immune-cell-based approaches that can reflect the biological heterogeneity of sepsis more directly.
Peripheral blood mononuclear cells (PBMCs)—including monocytes, T cells, B cells, natural killer cells, and related subsets—are well suited for this purpose. They are accessible through minimally invasive blood sampling and can be repeatedly assessed during disease progression, making them a practical window into systemic immune dynamics.7,8 Among PBMCs, the monocyte-macrophage lineage is particularly important because it connects pathogen sensing, inflammatory mediator production, antigen presentation, metabolic adaptation, and later immune dysfunction. Key features such as reduced monocyte HLA-DR expression, altered monocyte subsets, lymphocyte exhaustion, and metabolic rewiring provide a framework for understanding why some patients recover after the acute phase whereas others develop persistent immunoparalysis, secondary infections, or chronic critical illness.2,3 Although neutrophils and platelets also contribute to sepsis pathogenesis, comprehensive discussion of these cell types is beyond the scope of this review; they are considered here only where they interact with PBMC-centered immune mechanisms.9–13 This review focuses on PBMC-centered immune remodeling in sepsis, with particular emphasis on monocyte/macrophage dysfunction, time-dependent immune trajectories, and translational biomarkers. We integrate evidence from single-cell and spatial omics, mechanistic studies of inflammatory and immunometabolic regulation, and clinical biomarker investigations. Rather than providing a broad overview of all sepsis-related immune pathways, we aim to clarify how PBMC analysis may improve understanding of sepsis immunopathogenesis, support immune phenotyping, and inform future strategies for diagnosis, risk stratification, and individualized immunomodulatory therapy.
Literature Search Strategy
Given the broad scope of sepsis immunology and the narrative nature of this review, we employed a hybrid search strategy combining systematic keyword search with purposive citation tracking.
Phase 1 - Core Identification: We conducted a PubMed search (2020–2025) using high-specificity keywords: (“sepsis”[MeSH]) AND (“PBMC” OR “single-cell RNA-seq” OR “immunometabolism” OR “HLA-DR”), restricted to high-impact (IF > 5) journals (eg, Nature Medicine, Immunity, Intensive Care Medicine, Critical Care). This yielded 84 highly relevant records.
Phase 2 - Snowballing: From seminal 2024–2025 publications identified in Phase 1 (eg, Scicluna et al’s consensus transcriptomic framework; Hua et al’s CD38high monocyte study; Monneret et al’s 20-year cohort), we performed forward and backward citation tracking to capture mechanistic foundations (eg, Döcke et al, 1997) and latest derivatives.
Phase 3 - Targeted Updates: We conducted targeted searches for specific emerging concepts (eg, “TRAM sepsis”, “ZIP8 ferroptosis”, “4-OI immunometabolism”) to ensure inclusion of cutting-edge mechanisms discussed in this review.
Phase 4 – Eligibility refinement and age-related evidence check. To address age-related differences, we performed targeted searches combining “sepsis” and “PBMC” with age-related terms including “neonate,” “newborn,” “preterm,” “pediatric,” “children,” “older adults,” “elderly,” and “immunosenescence,” together with “HLA-DR” and “immunoparalysis.” Candidate records were then refined according to relevance to PBMC-centered mechanisms, support for the revised article scope, evidence type, and contribution to the central narrative. Studies focused mainly on non-PBMC mechanisms or broad sepsis management without direct relevance to the review focus were excluded or used only for contextual discussion.
Overall, Phase 1 identified 84 records and Phases 2–3 identified 78 additional records, yielding 162 candidate records. After Phase 4 screening, deduplication, and focus-based refinement, 127 references were retained in the final article. The final cited literature included original clinical, mechanistic, translational, biomarker, and interventional studies, with selected reviews, meta-analyses, and consensus papers used for background and synthesis.
The composition of the final cited literature is summarized in Supplementary Table S2 by article type, evidence type, and age-related focus. Because some studies combined clinical, translational, mechanistic, biomarker, or interventional components, categories were not mutually exclusive. The age-related evidence check showed that most PBMC-related evidence came from adult or mixed ICU cohorts or from studies without explicit age stratification, whereas neonatal, pediatric, and older-adult evidence remained limited. These findings informed our discussion of age-related generalizability and evidence gaps.
PBMC-Centered Immune Trajectories in Sepsis
Sepsis is often described as a transition from early hyperinflammation to later immunosuppression. However, these states are not strictly sequential or mutually exclusive. In many patients, inflammatory activation and immune paralysis coexist from the early phase and change in relative dominance over time.2,3,14,15 This dynamic coexistence is central to understanding PBMC remodeling in sepsis. Rather than reflecting a single immune phenotype, PBMCs capture a moving spectrum of immune states that includes inflammatory monocyte activation, impaired antigen presentation, lymphocyte depletion, cellular exhaustion, and metabolic dysfunction.
Within PBMCs, the monocyte/macrophage lineage represents a major axis of sepsis-associated immune dysregulation. During the early response to infection, monocytes and macrophages contribute to pathogen recognition and inflammatory mediator production through NF-κB-dependent and related innate immune pathways.16,17 As sepsis progresses, the same lineage may undergo functional reprogramming characterized by sustained HLA-DR downregulation, impaired antigen presentation, reduced cytokine-production capacity, and diminished antimicrobial competence.2,14 Monneret et al demonstrated in a large cohort of patients with septic shock that decreased monocyte HLA-DR expression is a robust marker of immunoparalysis and is associated with secondary infections and mortality.14 These findings support the view that monocyte dysfunction is not a peripheral phenomenon, but a central PBMC feature linking acute immune activation to later immune failure.
Adaptive immune dysfunction develops in parallel with myeloid reprogramming. Sepsis induces extensive lymphocyte apoptosis, and surviving effector T cells may acquire exhaustion phenotypes marked by checkpoint receptors such as PD-1 and TIGIT.18–21 Preclinical studies suggest that blockade of exhaustion-associated pathways, including TIGIT, can restore aspects of T-cell function in polymicrobial sepsis models.22 At the same time, immunosuppressive populations such as regulatory T cells and myeloid-derived suppressor cell-like populations may accumulate and further suppress lymphocyte proliferation and effector responses.23–25 Together, these changes create a PBMC environment in which impaired antigen presentation, lymphocyte loss, and suppressive cellular states reinforce one another.
This PBMC-centered trajectory may help explain why some patients recover after the acute phase whereas others progress to prolonged immunoparalysis, secondary infection, or chronic critical illness. In patients with uncomplicated recovery, early immune suppression may be transient and resolve as pathogen burden and inflammatory stress decline. In contrast, patients with prolonged hospitalization may experience sustained antigenic stimulation, persistent tissue injury, metabolic exhaustion, lymphocyte depletion, and impaired monocyte antigen presentation. These overlapping processes can prevent restoration of immune homeostasis and contribute to persistent inflammation, immunosuppression, and catabolism syndrome. Thus, the clinically important question is not only whether inflammation or immunosuppression is present, but when each state dominates and whether PBMC function is recovering or deteriorating.
Pathogen type can further modify PBMC immune signatures, but it should be interpreted within this PBMC-centered framework rather than as a separate topic. Viral infections tend to induce interferon-stimulated gene programs through RIG-I-like receptors and endosomal Toll-like receptors, whereas bacterial sepsis more often shows prominent myeloid activation and inflammatory cytokine signaling through pathways such as TLR4–MyD88–NF-κB.26–29 Single-cell and transcriptomic studies indicate that these pathogen-specific responses can generate distinguishable PBMC transcriptional patterns.28 However, mixed infections and treatment exposure may blur these distinctions. Therefore, pathogen class should be considered as an important modifier of PBMC biomarkers and immune phenotypes, rather than as an independent organizing axis for this review.
Age is another major modifier of PBMC responses in sepsis. Preterm neonates may exhibit immature monocyte function and reduced HLA-DR expression, whereas pediatric patients can show rapid progression toward immunoparalysis with lymphocyte apoptosis and T-cell exhaustion.30–32 Adult patients display substantial heterogeneity because of differences in comorbidities, infection source, and treatment exposure.33 Older adults may have immunosenescence, thymic involution, reduced naive T-cell reserves, and baseline low-grade inflammation, all of which can alter the balance between inflammation and immune suppression.34,35 These differences suggest that key PBMC features such as HLA-DR downregulation, lymphocyte exhaustion, metabolic rewiring, and immunoparalysis should not be assumed to occur uniformly across age groups.
Overall, PBMCs provide a practical and biologically informative window into the evolving immune trajectory of sepsis. The central issue is not whether individual inflammatory or suppressive pathways are present, but how PBMC states shift over time and across patient groups. The following section therefore examines the mechanisms that generate these PBMC immune-state transitions, including single-cell remodeling, upstream signaling circuits, regulated cell death, and immunometabolic reprogramming.
PBMC Immune-State Transitions and Mechanistic Reprogramming
The central mechanistic question is not simply which inflammatory pathways are activated during sepsis, but how these pathways reshape PBMC states over time. PBMCs, especially the monocyte/macrophage lineage, undergo dynamic transitions from early inflammatory activation to impaired antigen presentation, metabolic dysfunction, immune exhaustion, and late immunosuppression. Therefore, this section organizes mechanistic evidence around a temporal and functional sequence rather than a list of isolated pathways. We first summarize single-cell evidence for PBMC heterogeneity and immune-state transitions, then discuss upstream signaling circuits, downstream immune failure and cell death, and immunometabolic reprogramming as interconnected mechanisms that define PBMC-centered sepsis progression.
Single-Cell Evidence for Dynamic PBMC Remodeling
Single-cell RNA sequencing has substantially changed the understanding of sepsis-associated PBMC remodeling. Rather than representing a homogeneous immune compartment, PBMCs in sepsis contain shifting populations of inflammatory, regulatory, exhausted, and metabolically altered cells. Zhou et al used scRNA-seq to profile PBMCs from septic patients and healthy controls and identified seven major immune cell types and twelve monocyte subpopulations, including two sepsis-associated monocyte subsets that were markedly expanded in septic patients.17,36 These findings support the view that sepsis is not defined by a single immune-cell abnormality, but by redistribution and functional remodeling of multiple PBMC subsets.
Among the newly identified myeloid populations, CD38high monocytes are particularly relevant because they link cellular phenotype with metabolic dysfunction. This subset shows increased expression of CD38, an NAD+-metabolizing ectoenzyme, and expands during early sepsis.37 Because NAD+ availability influences immune-cell bioenergetics and inflammatory function, CD38high monocytes may reflect both early myeloid activation and metabolic stress. This population also has potential translational relevance as a diagnostic marker and as a possible therapeutic target, although its causal role in sepsis progression remains to be fully established.37
The value of single-cell analysis extends beyond the identification of discrete subpopulations. When combined with pseudotemporal trajectory analysis, single-cell data can reconstruct immune-state transitions during sepsis. Liu et al integrated transcriptomic and epigenetic information from a large single-cell dataset and proposed an “immunological clock” model of sepsis progression.38 In this model, monocytes begin to transition toward macrophage-like states within the first 16–24 hours, CD8+ T-cell exhaustion becomes more prominent at approximately 36–48 hours, and HLA-DRlow immunosuppressive myeloid populations expand after 72 hours.38 The TOX-associated exhaustion program and its epigenetic features are summarized in Figure 1, bottom panel, illustrating why the timing of checkpoint-directed immunomodulation may be critical. This temporal framework suggests that PBMC dysfunction evolves through recognizable transition points rather than through a simple binary shift from hyperinflammation to immunosuppression.
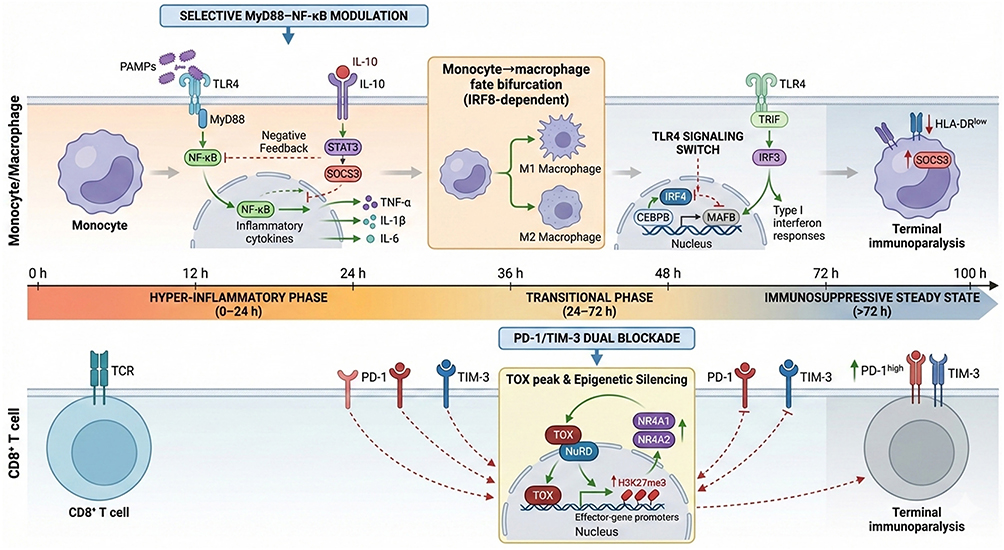 |
Figure 1 Time-resolved model of sepsis immune reprogramming. The timeline depicts three distinct phases: (Left) Early hyper-inflammatory phase (0–24 h): PAMPs engage TLR4, triggering MyD88-dependent NF-κB activation and cytokine production (TNF-α, IL-1β, IL-6), counterbalanced by IL-10–STAT3–SOCS3 negative feedback. (Center) Transitional phase (24–72 h): TLR4 signaling switches to TRIF-dependence, activating IRF3 and type I interferon responses; monocytes undergo IRF8-dependent fate bifurcation into M1/M2 macrophages; CD8 T cells initiate TOX-mediated epigenetic silencing (NuRD complex recruitment, H3K27me3 deposition) and upregulate PD-1/TIM-3. (Right) Late immunosuppressive steady state (>72 h): Predominance of HLA-DR SOCS3 exhausted monocytes and PD-1/TIM-3 T cells with terminal epigenetic silencing. Therapeutic interventions are phase-specific: selective MyD88–NF-κB modulation in early phase; PD-1/TIM-3 dual blockade in late phase. M2-like macrophages (anti-inflammatory) and exhausted macrophages (HLA-DR, functionally impaired) represent distinct late-phase states. |
This trajectory-based interpretation is important for therapeutic timing. Interventions aimed at reducing excessive inflammatory signaling may be most relevant during early hyperactivation, whereas strategies designed to restore antigen presentation or reverse T-cell exhaustion are more likely to be useful after immunosuppressive features emerge. However, these pseudotemporal findings should be interpreted with caution. Many high-dimensional datasets are derived mainly from adult or mixed ICU cohorts, and age- or sex-stratified effects are not consistently reported. Therefore, future studies should clarify whether the same PBMC trajectories apply equally to neonates, children, adults, and older adults.
Myeloid-derived suppressor cells (MDSCs) should also be interpreted within this broader continuum of myeloid dysfunction. In sepsis, MDSC-like populations expand during emergency myelopoiesis and suppress T-cell responses through arginase-1 activity, reactive oxygen species, nitric oxide, IL-10, and TGF-β.39–41 These cells are clinically relevant because their expansion has been associated with disease severity, secondary infection risk, and mortality. However, the phenotypic boundaries between MDSCs and dysfunctional monocytes remain uncertain. Monocytic MDSCs, commonly defined as CD14+HLA-DRlow cells, overlap substantially with sepsis-associated or exhausted monocyte states identified in single-cell studies.42–45 This overlap raises an important unresolved question: whether MDSCs in human sepsis represent a distinct suppressor lineage or a heterogeneous collection of dysfunctional myeloid states.
This distinction matters for therapy. If these cells are immature suppressor cells, differentiation-inducing strategies such as all-trans retinoic acid or vitamin D may help restore myeloid function,46 If they instead represent terminally exhausted monocytes, differentiation therapy may be insufficient, and metabolic or epigenetic reprogramming may be required. Current uncertainty is compounded by the lack of standardized gating strategies and variable use of markers such as CD33, CD11b, and HLA-DR.43,44,47 Single-cell multi-omics integrating transcriptomic and surface-protein information may help determine whether MDSCs are a discrete therapeutic target or part of a broader monocyte dysfunction spectrum.48
Taken together, single-cell and spatial omics support a trajectory-based model of PBMC remodeling in sepsis. Early inflammatory monocyte states may contribute to pathogen recognition and cytokine production, whereas persistent HLA-DRlow, metabolically impaired, or suppressive myeloid states may mark progression toward immunoparalysis, secondary infection risk, and prolonged critical illness. This interpretation provides the conceptual basis for linking cellular heterogeneity with upstream signaling, downstream immune failure, and therapeutic windows.
Upstream Sensing and Cytokine-Feedback Circuits in Monocyte Dysfunction
Monocyte dysfunction in sepsis begins with altered pathogen sensing and inflammatory signal transduction. During early infection, pathogen-associated molecular patterns and damage-associated molecular patterns activate Toll-like receptors and other pattern-recognition receptors on monocytes and macrophages. TLR engagement rapidly activates MyD88-dependent NF-κB signaling, promoting transcription of TNF-α, IL-1β, IL-6, and other inflammatory mediators16,49 (Figure 2, upper left panel). This early NF-κB-dependent response, which is positioned within the early hyperinflammatory phase in Figure 1, top left, is necessary for pathogen containment, but excessive or sustained activation can amplify systemic inflammation, endothelial injury, and organ dysfunction.
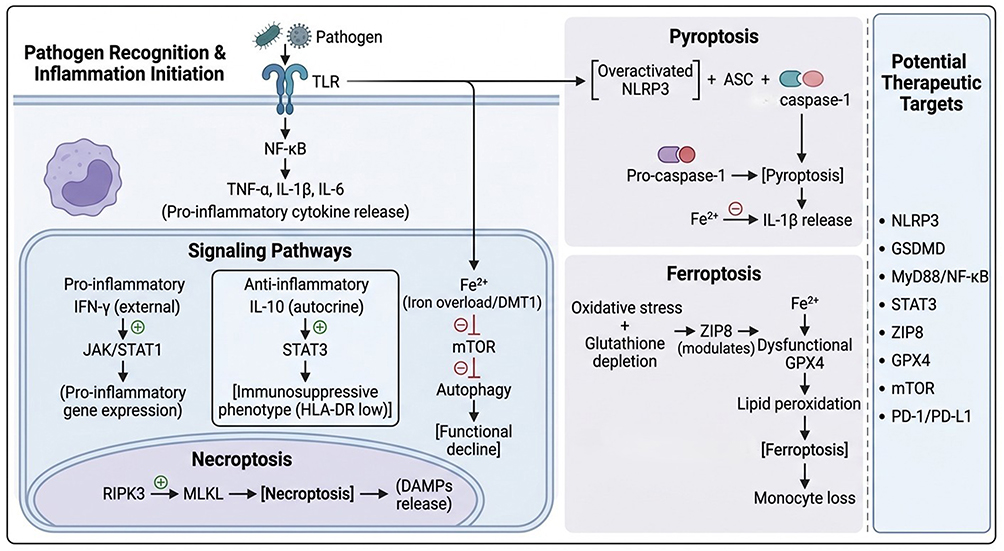 |
Figure 2 Key pathways driving monocyte dysfunction in sepsis. Schematic integration of pattern recognition signaling (upper left), dual JAK/STAT regulation (center), programmed cell death modalities (pyroptosis, necroptosis, ferroptosis), and metabolic perturbations (iron overload/mTOR inhibition). Right panel lists corresponding therapeutic targets discussed in the text. Upon pathogen recognition via TLRs, NF-κB is activated to produce pro-inflammatory cytokines (TNF-α, IL-1β, IL-6). Exogenous IFN-γ activates the JAK/STAT1 pathway (pro-inflammatory). In parallel, autocrine IL-10 activates STAT3 to induce an immunosuppressive phenotype characterized by low HLA-DR expression. Iron overload (Fe2⁺) inhibits mTOR, thereby promoting autophagy and subsequent functional decline. The RIPK3-MLKL axis drives necroptosis with DAMPs release. Overactivated NLRP3 inflammasome triggers pyroptosis via caspase-1 activation (promoted by Fe2⁺), releasing IL-1β. GPX4 dysfunction combined with lipid peroxidation induces ferroptosis, leading to monocyte loss. Potential therapeutic targets are listed. |
The biological meaning of NF-κB activation depends strongly on timing. In the early phase of sepsis, excessive NF-κB activation contributes to cytokine storm and tissue injury. In later stages, however, many patients show impaired monocyte responsiveness and reduced cytokine-production capacity, indicating functional exhaustion rather than persistent activation.50,51 Therefore, NF-κB should not be viewed simply as a harmful inflammatory pathway. Instead, it represents a phase-dependent regulatory node: excessive early activation may require restraint, whereas late immune paralysis may require restoration of monocyte responsiveness.
TLR4 adaptor specificity further shapes this transition. MyD88-dependent signaling mainly supports early NF-κB-driven inflammation, whereas TRAM-TRIF signaling links TLR4 activation to IRF3 activation and type I interferon responses. Recent studies have identified TRAM, encoded by TICAM2, as an important regulator of sepsis-associated monocyte exhaustion.52,53 TRAM-deficient monocytes preserve HLA-DR expression, maintain cytokine-production capacity, and show improved bacterial clearance in experimental models.52 Similarly, TICAM2 ablation facilitates post-septic monocyte recovery during the resolution phase.53 These findings suggest that TLR adaptor usage may influence whether monocytes remain inflammatory, recover immune competence, or progress toward exhaustion.
Cytokine-feedback pathways provide another layer of regulation. JAK/STAT signaling mediates cytokine responses and exerts dichotomous effects during sepsis, as summarized in Figure 2, center panel.21 IFN-γ-mediated STAT1 activation supports M1-like macrophage activation and enhances microbicidal function, whereas IL-10-mediated STAT3 activation promotes an immunosuppressive phenotype and is associated with reduced monocyte HLA-DR expression.54–56 Thus, the JAK/STAT pathway functions as both an immune accelerator and an immune brake. This dual role helps explain why uniform pathway inhibition or stimulation may fail in unselected septic populations.
Negative-feedback circuits are especially relevant to the transition from acute inflammation to immunoparalysis. IL-10–STAT3–SOCS3 signaling can limit excessive inflammation, but persistent activation may impair antigen presentation, reduce inflammatory responsiveness, and promote HLA-DR downregulation.57–59 In this context, immunosuppression is not simply the absence of inflammation; rather, it reflects an actively regulated state shaped by cytokine feedback, adaptor signaling, and metabolic constraints. These mechanisms provide a bridge between early inflammatory activation and later monocyte dysfunction.
Downstream Consequences: Antigen-Presentation Failure, Immune Exhaustion, and Regulated Cell Death
The downstream consequence of dysregulated signaling is not only cytokine release, but progressive loss of immune competence. One of the most clinically relevant features of this process is monocyte HLA-DR downregulation. HLA-DR is required for antigen presentation to CD4+ T cells, and sustained reduction of monocyte HLA-DR reflects impaired antigen-presenting capacity. A large 20-year cohort study including 1023 patients with septic shock confirmed that reduced monocyte HLA-DR expression is a robust marker of immunoparalysis and is associated with secondary infections and mortality.14 HLA-DRlow monocytes therefore represent a functional state that links innate immune dysfunction to impaired adaptive immune activation. IRF8, a master transcription factor for myeloid differentiation, undergoes progressive downregulation during this transitional phase. Loss of IRF8 impairs monocyte-to-dendritic cell differentiation and reduces antigen-presentation capacity, contributing to the HLA-DRlow immunosuppressive state depicted in Figure 1, transitional phase.
Adaptive immune exhaustion develops in parallel with myeloid dysfunction. During sepsis, extensive lymphocyte apoptosis reduces the pool of functional T and B cells, while surviving effector T cells often express exhaustion-associated checkpoint receptors such as PD-1 and TIGIT.18–21 Experimental studies suggest that TIGIT blockade can restore CD4+ T-cell function in polymicrobial sepsis models.22 However, the reversibility of T-cell exhaustion likely depends on timing. Once exhaustion-associated transcriptional and epigenetic programs become established, checkpoint blockade may be less effective. At the chromatin level, this involves NuRD complex recruitment to immune-activation gene loci and concurrent H3K27me3 deposition, which establish a repressive epigenetic state that constrains T-cell responsiveness (Figure 1, transitional phase). This epigenetic constraint helps explain why checkpoint blockade may be insufficient once exhaustion programs are fully consolidated. This reinforces the need to interpret lymphocyte exhaustion as a time-dependent immune state rather than a static therapeutic target.
Regulated cell death pathways contribute to both inflammatory injury and immune-cell loss. NLRP3 inflammasome activation promotes caspase-1-mediated GSDMD cleavage, pore formation, pyroptosis, and release of IL-1β and IL-1860–62 (Figure 2, upper right panel). During early infection, this pathway can support pathogen clearance and amplify innate immune activation. When excessive, however, NLRP3-driven pyroptosis contributes to cytokine release, monocyte/macrophage loss, and tissue injury. Preclinical studies show that inhibition of NLRP3 or its downstream effector GSDMD can attenuate inflammatory responses and improve survival in sepsis models.61,63–66 These findings support the NLRP3–pyroptosis axis as a potential therapeutic target, but its phase-dependent role must be considered.
Other forms of regulated cell death also shape PBMC dysfunction. Apoptosis contributes to lymphocyte depletion and adaptive immunosuppression, whereas necroptosis, mediated by RIPK3–MLKL signaling, promotes DAMP release and inflammatory tissue injury.67,68 PANoptosis integrates pyroptotic, apoptotic, and necroptotic programs and may provide a broader framework for understanding inflammatory cell death during severe infection.67 These overlapping death pathways suggest that immune-cell loss in sepsis is not caused by a single mechanism, but by convergent inflammatory, metabolic, and death-signaling stress.
Ferroptosis has recently emerged as another relevant mechanism, particularly in monocytes and macrophages. Sepsis-associated oxidative stress, glutathione depletion, and GPX4 dysfunction can increase lipid peroxidation and promote ferroptotic cell death.67,69 ZIP8 has been identified as an upstream regulator of monocytic ferroptosis, and ZIP8 overexpression may exacerbate iron influx, lipid peroxidation, monocyte loss, and organ injury.65 In experimental models, ZIP8 suppression or ferroptosis inhibition using reduced glutathione or ferrostatin-1 can attenuate monocyte loss and improve survival.65 Beyond ferroptosis, elevated intracellular Fe2⁺ may also promote caspase-1 activation, linking iron dysregulation to pyroptotic cell death, and has been proposed to inhibit mTOR signaling, contributing to autophagy dysregulation in septic monocytes (Figure 2).65
Despite these promising findings, ferroptosis-targeted therapy requires caution. Ferroptosis-related iron restriction may contribute to host defense against intracellular pathogens, and systemic ferroptosis suppression could theoretically create an iron-replete environment that favors microbial persistence.70 In addition, ferroptosis may have cell type–specific effects: monocyte preservation may be beneficial, whereas endothelial ferroptosis can contribute to capillary leak and organ dysfunction.71 Therefore, regulated cell death should be understood as a downstream consequence of dysregulated inflammation and metabolic stress, not as a set of isolated targets. Effective therapeutic modulation will likely require phase-specific and cell type–specific strategies.
Immunometabolic Reprogramming in Sepsis
Metabolic rewiring provides a mechanistic bridge between early inflammation and later immunoparalysis. During acute sepsis, monocytes and macrophages shift from oxidative phosphorylation toward aerobic glycolysis to rapidly generate ATP and biosynthetic intermediates needed for inflammatory activation57,72 (Figure 3, early phase). This Warburg-like response supports cytokine production and antimicrobial activity. PKM2 (pyruvate kinase M2) is a central driver of this metabolic shift: its nuclear translocation promotes HIF-1α-dependent transcription of glycolytic enzymes and pro-inflammatory mediators including HMGB1, directly coupling metabolic reprogramming to inflammatory gene expression (Figure 3, early phase).57,72 Metabolites such as lactate and succinate can further shape inflammatory transcriptional programs; succinate, for example, stabilizes HIF-1α and sustains inflammatory gene expression.58
 |
Figure 3 Temporal dynamics of immunometabolic remodeling in sepsis progression. Schematic illustration of metabolic reprogramming and functional transitions in immune cells across three distinct phases of sepsis. (Left) Early Phase (0–24h): Innate immune cells (monocytes/macrophages) undergo a Warburg-like metabolic switch characterized by PKM2-driven aerobic glycolysis, resulting in lactate accumulation and inflammatory gene expression. Mitochondrial succinate accumulates (despite low oxygen tension), stabilizing HIF-1α and promoting pro-inflammatory responses. Concurrently, CD4⁺ T cells exhibit PIM1 overexpression and ABCG1 downregulation, leading to intracellular cholesterol accumulation that favors Th1/Th17 polarization over regulatory T cell (Treg) differentiation. Itaconate (and its derivative 4-OI) provides negative feedback by inhibiting SDH and the NLRP3 inflammasome. (Middle) Transition Phase (24–72h): Bifurcation of macrophage fates occurs. (Upper track) Repair-oriented M2-like macrophages maintain intact OXPHOS and enhanced fatty acid oxidation (FAO), supporting tissue repair. (Lower track) Exhausted macrophages exhibit “double failure” of both glycolysis and OXPHOS, accompanied by mitochondrial dysfunction and SOCS3-mediated epigenetic silencing, marking the onset of metabolic paralysis. (Right) Late Phase (Immunoparalysis): Profound ATP depletion and ROS accumulation drive metabolic and immune paralysis. Innate immune cells display downregulated HLA-DR expression (a clinical marker of immunosuppression) and disrupted membrane lipid rafts due to cholesterol depletion, impairing TLR signaling efficiency. Adaptive immune cells enter an exhausted state with altered metabolic dependencies. Dashed arrows indicate therapeutic intervention points or functional consequences; blunt-ended red lines indicate inhibitory actions. |
However, prolonged inflammatory stress can drive immune cells from metabolic activation into metabolic paralysis. In this state, both glycolysis and oxidative phosphorylation become impaired, mitochondrial function declines, ATP production falls, and monocytes lose the bioenergetic capacity required for phagocytosis, cytokine production, and antigen presentation.2,59,72,73 This process helps explain why some patients progress from early inflammatory activation to persistent immunoparalysis, secondary infection risk, and chronic critical illness. Metabolic failure is therefore not merely a consequence of severe sepsis; it is a central mechanism linking PBMC dysfunction with clinical deterioration.
Mitochondrial dysfunction is particularly important in this transition. Damaged mitochondria impair ATP-dependent immune functions and increase reactive oxygen species production.74 In monocytes, mitochondrial dysfunction may contribute directly to HLA-DR downregulation and defective antigen presentation.35 It may also affect the surrounding immune environment by altering metabolite availability and limiting the ability of effector T cells to sustain activation.75 Thus, PBMC mitochondrial dysfunction can suppress immunity through both cell-intrinsic and cell-extrinsic mechanisms.
It is useful to distinguish repair-oriented M2-like macrophage states from terminally exhausted macrophage states. M2-like macrophages rely more on fatty acid oxidation and retain oxidative phosphorylation capacity, supporting tissue repair and resolution of inflammation.76 In contrast, exhausted macrophages show simultaneous glycolytic and mitochondrial failure, ATP depletion, reduced HLA-DR expression, and impaired functional recovery.59,73 This distinction has therapeutic implications. M2-like macrophages may retain enough metabolic plasticity to be repolarized by immunostimulatory signals such as IFN-γ, whereas terminally exhausted macrophages may require deeper metabolic or epigenetic reprogramming.77,78 This metabolic bifurcation between repair-oriented M2-like macrophages and metabolically exhausted macrophages is summarized in Figure 3, transition phase.
mTOR and autophagy also regulate PBMC metabolic fitness. mTOR integrates nutrient availability, growth signals, and immune activation, while autophagy helps remove damaged organelles and maintain cellular homeostasis.79,80 In sepsis, excessive mTOR activity can suppress autophagy and contribute to immune-cell dysfunction. This mechanism may be particularly relevant in older patients, in whom defective autophagy–lysosomal function and mTOR hyperactivation in CD4+ T cells have been linked to impaired immunity and poor outcomes.81 Therefore, mTOR-autophagy balance may influence whether PBMCs recover function or remain metabolically impaired.
Lipid metabolism further modifies immune-cell function. Hypocholesterolemia is common in sepsis and has been associated with poor prognosis.82,83 At the cellular level, cholesterol affects membrane architecture, lipid raft formation, and TLR signaling. Both cholesterol depletion and overload can disrupt immune signaling, indicating that cholesterol homeostasis is required for balanced monocyte activation.83 In T cells, the PIM1–ABCG1 axis regulates cholesterol efflux and influences Th1/Th17 and regulatory T-cell balance19 (Figure 3, late phase). These findings suggest that lipid metabolism may shape both innate and adaptive PBMC responses during sepsis.
Amino-acid metabolism also contributes to PBMC dysfunction and biomarker development. Glutamine supports immune-cell proliferation and biosynthesis, and altered glutamine metabolism has been linked to sepsis susceptibility and immunoparalysis-associated gene signatures, including SRSF7, RAB13, E2F2, and S100A8.84–86 Arginine metabolism regulates nitric oxide production, vascular tone, and antimicrobial function; dysregulated arginase activity in monocytes and macrophages may contribute to impaired pathogen clearance and immunosuppression.87,88
Tryptophan metabolism, including kynurenine-related pathways and CYP1B1 activity, may further influence monocyte inflammation and migration.85
Finally, metabolites can act as direct immunoregulatory signals. Succinate promotes HIF-1α-mediated inflammation, whereas itaconate provides negative feedback by limiting inflammatory activation. The itaconate derivative 4-octyl itaconate inhibits succinate dehydrogenase, suppresses NLRP3 activation, and improves survival in experimental sepsis models89 (Figure 3, late phase). These findings support the concept that metabolic interventions may modulate immune function without globally suppressing host defense. However, clinical translation will require careful patient selection, timing, and validation of immune-state biomarkers.
Mechanistic Synthesis: PBMC States Define Therapeutic Windows
The mechanisms discussed above converge on a trajectory-based model of PBMC dysfunction in sepsis. Early monocyte activation is driven by pathogen sensing, TLR–NF-κB signaling, cytokine production, and glycolytic reprogramming. As inflammation persists, cytokine-feedback pathways, TRAM-TRIF signaling, mitochondrial dysfunction, impaired autophagy, and metabolic exhaustion contribute to HLA-DR downregulation and antigen-presentation failure. In parallel, lymphocyte apoptosis, checkpoint-associated T-cell exhaustion, and regulated cell death pathways further weaken host immune competence.
This model has two important implications. First, PBMC mechanisms must be interpreted temporally. The same pathway may be protective in one phase but harmful in another. Early NF-κB or NLRP3 activation may support pathogen clearance, whereas sustained activation may contribute to tissue injury and immune-cell loss. Conversely, immune stimulation may be harmful during uncontrolled hyperinflammation but beneficial when immunoparalysis dominates. Second, PBMC-based biomarkers should be viewed not as isolated diagnostic markers, but as readouts of immune-state transitions. HLA-DR expression, monocyte subsets, exhaustion markers, metabolic signatures, and functional immune assays may help identify where a patient lies along the sepsis immune trajectory.
This mechanistic framework provides the rationale for the following translational section. If PBMC states define disease phase and immune competence, then clinically useful biomarkers should be evaluated according to their timing, target population, biological meaning, and ability to guide immunomodulatory therapy.
PBMC-Based Biomarkers, Therapeutic Windows, and Clinical Translation
PBMC-based biomarkers provide the translational link between mechanistic immune trajectories and clinical decision-making. Because sepsis immune states change over time, PBMC-derived biomarkers should not be evaluated only as static diagnostic indicators. Instead, they should be interpreted according to three clinically relevant questions: what immune state they reflect, when they are measured, and whether they can guide treatment decisions. In this section, we organize PBMC-derived biomarkers by clinical purpose—early diagnosis, prognostic stratification, functional immune assessment, and therapeutic guidance—rather than by molecular platform alone. This organization links the mechanistic transitions described in PBMC Immune-State Transitions and Mechanistic Reprogramming to clinically usable immune phenotypes.
Conventional screening tools such as SOFA, qSOFA, NEWS, lactate, C-reactive protein, and procalcitonin remain useful for early recognition and risk assessment, but they do not directly define the patient’s immune state.4–6,90 PBMC-based biomarkers may fill this gap by capturing dynamic changes in monocyte activation, antigen-presentation capacity, lymphocyte exhaustion, metabolic stress, and functional immune responsiveness. These biomarkers are summarized in Table 1 according to their biological meaning, clinical use, sampling window, study population, predictive performance, and major limitations.
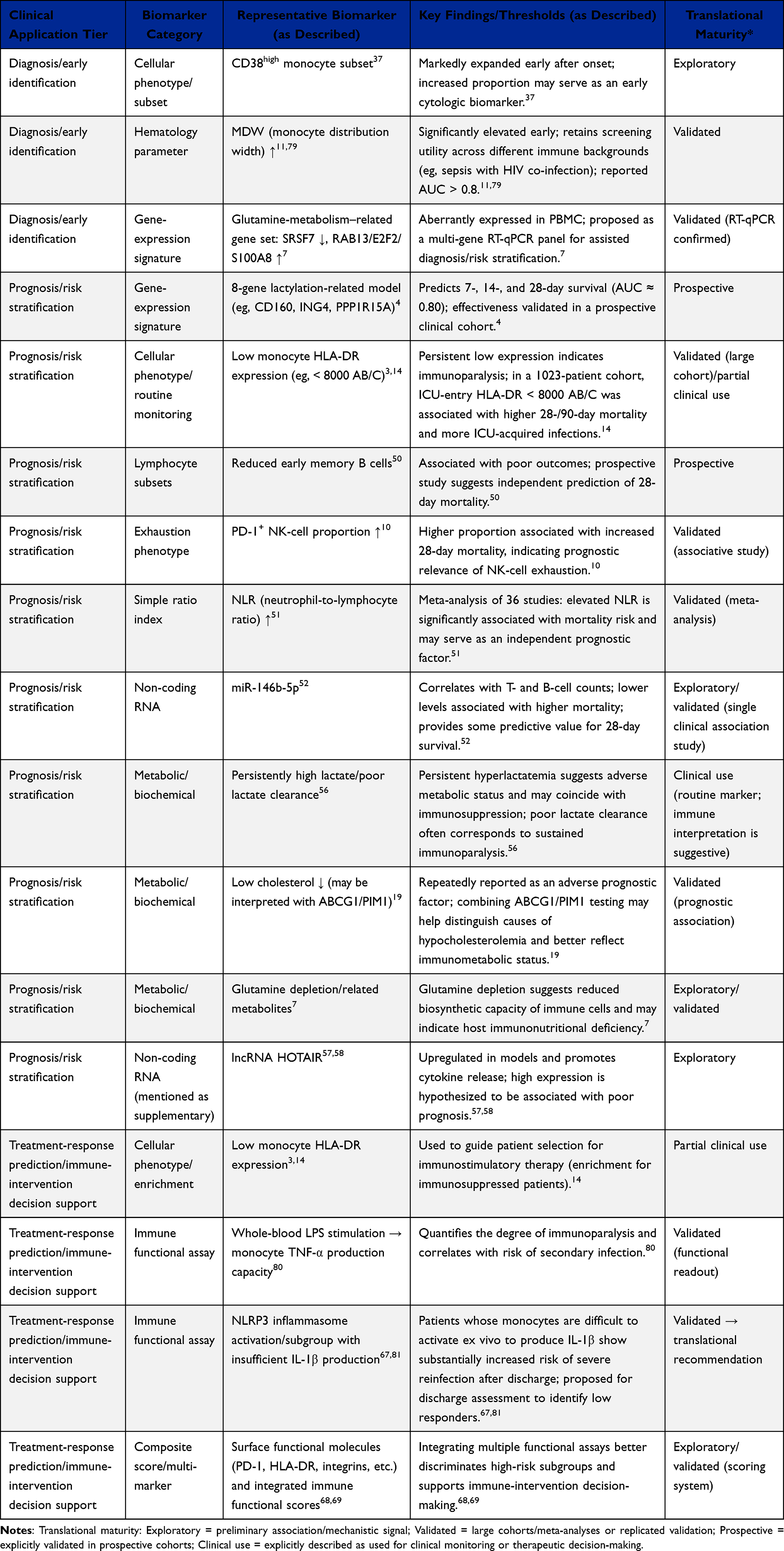 |
Table 1 Overview of PBMC-Related Biomarkers in Sepsis |
From PBMC Immune Trajectories to Biomarker Categories
The biomarker value of PBMCs arises from their ability to reflect different stages of the sepsis immune trajectory. Early in sepsis, markers of myeloid activation and cellular heterogeneity may support diagnosis and early risk stratification. During the transition phase, markers of HLA-DR downregulation, lymphocyte exhaustion, and metabolic dysfunction may identify patients at risk of persistent immunoparalysis. In later or clinically stable phases, functional assays may be more informative because they directly test whether immune cells can respond to stimulation.
This temporal logic is important because the same biomarker may have different meanings depending on when it is measured. For example, early monocyte activation may indicate an appropriate antimicrobial response, whereas persistent HLA-DR downregulation at 48–72 hours may indicate impaired antigen presentation and increased risk of secondary infection. Similarly, metabolic indicators such as lactate, glutamine-related signatures, or cholesterol disturbance should be interpreted in relation to the immunometabolic transitions summarized in Figure 3, rather than as isolated biochemical abnormalities. Therefore, PBMC-based biomarker translation requires integration of biological mechanism, sampling time, and clinical context.
Multi-omics technologies have expanded the range of candidate PBMC biomarkers. Transcriptomics captures dynamic gene-expression programs and is useful for mechanistic discovery and prognostic stratification.91–94 Proteomics provides information closer to functional protein abundance and druggable pathways.95,96 Metabolomics offers immediate functional readouts of immune-cell metabolic status and may support subphenotyping when combined with machine learning.97,98 However, each platform has limitations in turnaround time, cost, analytical complexity, and reproducibility. For near-term clinical use, the most promising approach may combine a small number of robust cellular, molecular, metabolic, and functional indicators rather than rely on a single high-dimensional assay.
Diagnostic Biomarkers of Early PBMC Activation
Diagnostic PBMC biomarkers are most useful when they identify early immune activation before clinical deterioration becomes irreversible or before treatment obscures immune signatures. Among cellular biomarkers, CD38high monocytes are particularly relevant because they connect early myeloid activation with NAD+-linked metabolic stress. Hua et al reported that this subset expands rapidly during early sepsis, with samples collected within 0–24 hours after onset and before empirical antibiotic administration.37 The diagnostic performance of CD38high monocytes was stronger than that of conventional inflammatory markers in this cohort, suggesting that monocyte-state markers may improve early discrimination between sepsis and non-septic inflammation.37 This early activation state is consistent with the temporal PBMC trajectory summarized in Figure 1.
Monocyte distribution width (MDW) represents another clinically attractive diagnostic marker because it can be derived from routine complete blood count analysis. MDW reflects heterogeneity in monocyte cell volume, which may increase during early inflammatory activation. Studies have shown that MDW is elevated in early sepsis compared with non-septic infection and healthy controls.99 Sun et al further validated MDW in HIV-infected patients with sepsis co-infection, reporting favorable diagnostic performance when samples were obtained immediately upon emergency department admission.100 Because MDW is inexpensive, rapid, and widely available, it may be useful as a first-line screening biomarker, particularly when combined with more specific PBMC phenotyping.
Transcriptomic signatures also show diagnostic and prognostic potential. High-throughput transcriptomics combined with machine learning has identified gene-expression models that stratify sepsis risk and outcome. For example, Li et al developed a prognostic model based on lactylation-related genes, including CD160, ING4, and PPP1R15A, using whole-blood RNA samples obtained within 6 hours of ICU admission.75 This model showed good discrimination for 14-day and 28-day mortality and illustrates how metabolic dysregulation can be translated into RNA-based risk stratification.75 Similarly, glutamine metabolism–associated genes, including SRSF7, RAB13, E2F2, and S100A8, have been identified in septic PBMCs and may be developed into targeted qPCR panels for diagnostic or prognostic use.86
Despite their promise, early diagnostic biomarkers face several limitations. First, early sepsis is biologically heterogeneous, and diagnostic signatures may differ by infection source, pathogen class, age group, comorbidity, and treatment exposure. Second, many transcriptomic assays remain too slow or technically demanding for urgent bedside decisions. Third, most biomarkers require prospective validation in diverse cohorts before they can be incorporated into clinical workflows. Therefore, early diagnostic PBMC biomarkers should be viewed as complementary tools that refine, rather than replace, clinical assessment and conventional sepsis screening.
Prognostic Biomarkers of Immunoparalysis and Adverse Outcomes
Prognostic PBMC biomarkers are most valuable when they identify persistent or worsening immune dysfunction rather than a single inflammatory snapshot. Monocyte HLA-DR remains the most established marker in this category. Because HLA-DR is required for antigen presentation, sustained reduction of monocyte HLA-DR reflects impaired antigen-presenting capacity and is commonly interpreted as a marker of immunoparalysis. A large cohort study by Monneret et al including 1023 patients with septic shock showed that low monocyte HLA-DR expression was associated with mortality and ICU-acquired infections.14 This supports the use of HLA-DR as both a prognostic biomarker and an enrichment marker for immunostimulatory strategies.
The timing of HLA-DR measurement is critical. A single early low value may reflect transient immune suppression during acute stress, whereas persistent reduction at 48–72 hours or beyond may identify patients at higher risk of secondary infection and prolonged critical illness. This distinction helps explain why PBMC monitoring should be dynamic. Repeated HLA-DR assessment may better capture whether a patient is recovering immune competence or progressing toward persistent immunoparalysis.
Lymphocyte-based biomarkers provide complementary prognostic information. Sepsis is associated with lymphocyte apoptosis, T-cell exhaustion, and altered B-cell and NK-cell compartments. Sun et al reported that reduced memory B-cell proportions on ICU day 1 were associated with increased 28-day mortality.101 Tang et al showed that elevated PD-1+ NK-cell proportions at 48–72 hours predicted 28-day mortality, supporting the relevance of NK-cell exhaustion as a prognostic indicator.102 These markers suggest that adverse outcomes are not driven solely by myeloid dysfunction, but also by impaired adaptive and innate lymphocyte responses.
Simple blood-count-derived indices may also contribute to prognostic stratification. The neutrophil-to-lymphocyte ratio (NLR) is not PBMC-specific, but it reflects the balance between innate inflammatory activation and lymphocyte depletion. A recent meta-analysis showed that elevated NLR within 48 hours of admission was associated with mortality, and dynamic increases in NLR improved predictive performance compared with single-point measurement.103 Because NLR is inexpensive and widely available, it may be useful as a low-cost screening marker when interpreted alongside PBMC-specific indicators such as HLA-DR and lymphocyte exhaustion markers.
Non-coding RNAs may further refine prognostic assessment. miR-146b-5p has been associated with lymphocyte subset counts and 28-day mortality in sepsis.104 Mechanistically, the miR-146 family is involved in negative feedback regulation of TLR/NF-κB signaling, suggesting that circulating miRNA levels may reflect the balance between inflammatory activation and immune regulation.105 Long non-coding RNAs such as HOTAIR have also been implicated in inflammatory regulation in experimental sepsis models.106,107 However, non-coding RNA biomarkers remain less mature than cellular markers such as HLA-DR, and their clinical utility will require further standardization and validation.
Overall, prognostic PBMC biomarkers are strongest when interpreted as dynamic indicators of immune trajectory. Persistent HLA-DR downregulation, lymphocyte exhaustion, memory B-cell depletion, elevated PD-1+ NK-cell proportions, and worsening NLR may together identify patients who are failing to restore immune competence. These biomarkers may be particularly important for identifying patients at risk of secondary infection, prolonged ICU stay, and chronic critical illness.
Functional Biomarkers of Immune Competence
Functional biomarkers may be closer to clinical decision-making than static molecular measurements because they test whether PBMCs can still respond to immune stimulation. Static markers such as HLA-DR expression or gene-expression signatures infer immune status, whereas functional assays directly measure immune-cell capacity. This distinction is important because two patients with similar biomarker concentrations may differ substantially in their ability to produce cytokines, activate inflammasomes, or respond to immunostimulatory therapy.
Whole-blood lipopolysaccharide (LPS) stimulation assays are among the most widely discussed functional tests. These assays measure monocyte TNF-α release after ex vivo stimulation and are typically performed after initial hemodynamic stabilization, often around ICU days 3–5.108 Reduced TNF-α production suggests impaired monocyte responsiveness and may identify patients with clinically relevant immunoparalysis. This type of assay is particularly useful because it evaluates immune reserve rather than baseline inflammation alone.
Inflammasome function provides another functional readout. PBMC NLRP3 activation assays measure IL-1β release after ex vivo stimulation and may identify patients with impaired inflammasome responsiveness before discharge or during recovery.109,110 Patients with low IL-1β production after stimulation have been reported to be at increased risk of reinfection and readmission.109,110 The mechanistic basis of this assay is linked to the NLRP3–caspase-1–GSDMD pyroptosis axis summarized in Figure 2. Therefore, NLRP3 functional testing may help identify a “cryptic immunosuppression” phenotype that is not captured by conventional inflammatory biomarkers.
Other functional or semi-functional indicators include checkpoint receptor expression, integrin expression, cytokine-production capacity, and composite immune-function scores.111,112 These multidimensional scores may outperform single biomarkers because they integrate myeloid function, lymphocyte exhaustion, and inflammatory responsiveness. However, clinical implementation remains challenging. Functional assays require standardized stimulation protocols, rapid processing, defined thresholds, and prospective evidence that assay-guided interventions improve outcomes.
A practical translational strategy may involve stepwise immune assessment. Rapid and inexpensive markers such as MDW and NLR could be used for initial screening. Flow-cytometric markers such as monocyte HLA-DR, CD38high monocytes, and exhausted lymphocyte subsets could then refine immune phenotyping. Finally, functional assays such as LPS-induced TNF-α release or NLRP3-induced IL-1β production could identify patients who may benefit from immunostimulatory or immune-restorative therapies. This layered approach may be more feasible than routine comprehensive omics for all patients.
Biomarker-Guided Therapeutic Windows and Interventional Evidence
The major clinical implication of PBMC-based immune monitoring is that immunomodulatory therapy should be matched to immune state and timing. These biomarker categories should be interpreted within the temporal immune trajectory summarized in Figure 1. Early inflammatory activation may justify selective suppression of tissue-damaging pathways, whereas persistent HLA-DR downregulation, lymphocyte exhaustion, or impaired functional responses may support immunostimulatory approaches. This temporal principle explains why non-stratified sepsis immunotherapy has often failed.
Several failed trials illustrate the problem of treating sepsis as a uniform syndrome. Trials of recombinant human activated protein C, eritoran, and broad anti-inflammatory interventions failed to demonstrate consistent benefit in unselected sepsis populations.113 Large phase 3 trials of anakinra in the 1990s also failed to show overall survival benefit when patients were not selected by immune phenotype.114,115 These negative results do not necessarily invalidate immunomodulation; rather, they show that the direction and timing of immune intervention matter. A therapy designed to suppress inflammation may be harmful or ineffective in patients who are already immunoparalyzed, whereas immunostimulation may be inappropriate during uncontrolled hyperinflammation.
More recent phenotype-guided studies support a different model. Post-hoc analyses of anakinra trials suggested that IL-1 blockade may benefit patients with macrophage activation syndrome features, including marked hyperferritinemia.115 The PROVIDE trial further supported phenotype-guided anti-inflammatory therapy by using ferritin-based enrichment to identify patients with a macrophage activation phenotype.116 In contrast, the ImmunoSep trial used immune markers such as monocyte HLA-DR and ferritin to guide immunostimulatory therapy; recombinant IFN-γ improved organ function in patients with immunoparalysis characterized by low monocyte HLA-DR and lower ferritin levels.117 These findings support the concept that PBMC-related biomarkers can identify not only prognosis, but also treatment direction.
IL-7 provides another example of immune-state-specific intervention. The IRIS-7 trial showed that IL-7 restored lymphocyte counts in septic patients with profound lymphopenia, inducing sustained increases in CD4+ and CD8+ T cells.118 This approach is biologically distinct from anti-inflammatory therapy: it targets immune restoration rather than inflammatory suppression. Therefore, IL-7-like strategies should be considered only in patients with evidence of lymphocyte depletion or adaptive immune failure, not as universal sepsis therapy.
Mechanism-guided interventions emerging from PBMC biology also require biomarker-based selection. NLRP3 or GSDMD inhibition may be most relevant in patients with excessive inflammasome activation or pyroptosis, whereas IFN-γ, GM-CSF, or IL-7 may be more appropriate in patients with persistent HLA-DR downregulation, impaired cytokine production, or lymphopenia. Metabolic interventions, including approaches targeting succinate, itaconate pathways, mitochondrial dysfunction, or ferroptosis, should be aligned with immunometabolic states summarized in Figure 3. Without such stratification, pathway-targeted therapies risk reproducing the failures of earlier broad sepsis trials.
To address why most immunomodulatory trials in sepsis have failed and how immune phenotyping could improve this, we have compiled Supplementary Table S1, which systematically summarizes PBMC-relevant interventions by target, intended immune state, timing, evidence type, current status, and major limitation. The table includes both failed non-stratified approaches (eg, eritoran/TLR4 antagonism) and emerging biomarker-guided strategies (eg, IFN-γ, GM-CSF, IL-7, checkpoint modulation, NLRP3/GSDMD inhibition, metabolic and ferroptosis-related interventions), because negative studies are essential for understanding the necessity of immune phenotyping.
Future clinical trials should therefore move beyond single-marker enrollment and adopt adaptive immune-phenotyping designs. Such trials should incorporate repeated PBMC monitoring, predefined immune-state thresholds, and dynamic adjustment of therapy according to biomarker response. Platform trial structures may allow parallel testing of anti-inflammatory, immunostimulatory, and metabolic interventions across different immune endotypes. The success of adaptive trial designs in other critical-care contexts, such as REMAP-CAP during the COVID-19 pandemic, supports the feasibility of this approach.119
Overall, PBMC-based biomarkers are most useful when interpreted as dynamic readouts of immune-state transitions rather than isolated laboratory values. Diagnostic markers such as CD38high monocytes and MDW may identify early myeloid activation, whereas prognostic markers such as persistent HLA-DR downregulation, exhausted lymphocyte subsets, and impaired functional responses identify patients at risk of immunoparalysis and secondary infection. The major translational challenge is no longer simply biomarker discovery, but validation of time-specific, population-specific, and treatment-relevant biomarker panels. Future interventional trials should combine patient enrichment, temporal immune monitoring, and adaptive trial designs to test whether PBMC-guided therapy can improve outcomes.
Controversies, Evidence Limitations, and Future Directions
Although PBMC-based immune profiling provides a promising framework for sepsis stratification, several limitations prevent immediate clinical implementation. Current evidence remains heterogeneous with respect to patient age, infection source, comorbidity burden, sampling time, assay platform, and treatment exposure. In addition, many mechanistic findings are derived from preclinical models, in vitro systems, or single-center observational cohorts and have not been prospectively validated in broad clinical populations. Therefore, the main challenge is no longer only to identify additional PBMC biomarkers or pathways, but to determine which findings are reproducible, clinically actionable, and applicable to specific patient populations.
Population Heterogeneity and Age-Related Evidence Gaps
A major limitation of the current evidence base is population heterogeneity. Sepsis is not a single biological entity, and PBMC responses may vary according to infection source, pathogen type, baseline immune status, comorbidities, treatment exposure, and genetic background. Many PBMC transcriptomic and single-cell studies have been performed in adult or mixed ICU cohorts, whereas neonatal, pediatric, and older adult populations remain less consistently represented. This creates uncertainty about whether the same PBMC immune trajectories apply across the lifespan.
Age is particularly important because baseline immune architecture differs substantially between neonates, children, adults, and older adults. Neonates and young children may show developmental immaturity of innate and adaptive immune coordination, whereas older adults may exhibit immunosenescence, thymic involution, reduced naive T-cell pools, and chronic low-grade inflammation. These differences may alter the timing and severity of HLA-DR downregulation, lymphocyte apoptosis, metabolic rewiring, and immunoparalysis. Therefore, PBMC mechanisms identified in adult cohorts should not be assumed to apply directly to pediatric or elderly patients without age-stratified validation.
The temporal immune trajectory summarized in Figure 1 should therefore be validated separately across age groups and clinical trajectories, particularly in patients who progress from acute sepsis to prolonged critical illness. Future studies should report age distribution, sex, infection source, comorbidities, immune-modulating treatments, and sampling time in sufficient detail. Without such information, it remains difficult to determine whether observed PBMC signatures reflect universal sepsis biology or cohort-specific immune states.
Timing Dependence and Reversibility of PBMC Dysfunction
The interpretation of PBMC biomarkers and mechanisms depends strongly on sampling time. Early immune activation, transient immune suppression, and persistent immunoparalysis may produce superficially similar biomarker patterns but have different biological meanings. For example, reduced monocyte HLA-DR expression at admission may reflect acute stress and early immune dysregulation, whereas persistent HLA-DR downregulation at 48–72 hours or beyond more strongly suggests impaired antigen presentation and clinically relevant immunoparalysis. Similarly, early inflammatory monocyte activation may support pathogen clearance, whereas prolonged activation may contribute to tissue injury, metabolic exhaustion, and immune-cell loss.
This timing dependence is especially relevant to patients who survive the acute phase but develop prolonged hospitalization, secondary infection, or persistent inflammation, immunosuppression, and catabolism syndrome. These later phenotypes may emerge because sustained antigenic stimulation, ongoing tissue injury, metabolic stress, lymphocyte depletion, and impaired antigen presentation prevent restoration of immune homeostasis. Thus, the absence of overt immunoparalysis in early survivors does not mean that the same mechanisms are irrelevant; rather, they may become clinically visible only when immune dysfunction persists beyond the early phase.
The reversibility of immune exhaustion is another unresolved issue. Checkpoint-associated T-cell exhaustion and monocyte dysfunction may be reversible during early or intermediate stages, but late-stage exhaustion may become constrained by epigenetic remodeling and terminal differentiation. Recent evidence suggests that by later time points, CD8+ T cells may acquire TOX-associated transcriptional and epigenetic programs that reduce responsiveness to checkpoint blockade.120,121 This “point-of-no-return” concept remains debated, but it has important implications for trial design: immunostimulatory therapies may fail if they are administered after immune dysfunction has become biologically fixed.
Metabolic dysfunction shows similar timing complexity. Early sepsis is often associated with Warburg-like glycolytic activation, which supports inflammatory cytokine production. In prolonged sepsis, however, the metabolic state of PBMCs remains controversial. Some studies describe persistent glycolytic activation or a “glycolytic trap,” whereas others report complete metabolic paralysis with failure of both glycolysis and oxidative phosphorylation.122,123 The immunometabolic transitions summarized in Figure 3 should therefore be interpreted cautiously, because late sepsis may involve different metabolic states depending on cohort characteristics, sampling time, disease duration, and assay platform.
Discordance Between Single-Cell, Bulk, and Functional Immune Assays
Another limitation is the incomplete agreement between different analytical platforms. Single-cell RNA sequencing can identify rare or transient PBMC subsets, such as CD38high monocytes or HLA-DRlow myeloid states, that may be missed by bulk transcriptomic approaches. Conversely, bulk transcriptomics may better capture dominant systemic expression patterns but cannot resolve whether changes arise from altered cell composition, altered gene expression within a subset, or both. Therefore, results from single-cell and bulk studies should not be interpreted as interchangeable.
The clinical significance of rare PBMC subsets also remains uncertain. Some subsets identified by scRNA-seq may represent causal drivers of sepsis pathobiology, whereas others may be epiphenomena of severe inflammation or treatment exposure. For example, rare monocyte states may have disproportionate functional effects if they produce large amounts of cytokines or suppress T-cell responses, but this requires direct functional validation. Without paired transcriptomic, surface-protein, and stimulation-assay data, it is difficult to determine whether a transcriptionally defined subset is clinically actionable.
Functional assays add another layer of complexity. A patient may show a transcriptomic signature of inflammation but have poor ex vivo cytokine-production capacity, or may show low baseline cytokine levels but retain strong inducible responses. As summarized in Table 1, PBMC biomarkers differ substantially in sampling window, clinical purpose, platform requirements, and validation status. Future studies should therefore integrate cell-frequency data, single-cell transcriptomics, surface immunophenotyping, metabolomics, and functional stimulation assays in the same patients. Such integrated designs are needed to distinguish immune-cell abundance, immune-cell state, and immune-cell capacity.
Barriers to Clinical Implementation
Even when PBMC biomarkers show strong biological plausibility, several practical barriers limit clinical translation. High-dimensional technologies such as scRNA-seq, ATAC-seq, mass cytometry, proteomics, and untargeted metabolomics remain expensive, technically demanding, and difficult to implement within the time-sensitive decision windows of sepsis care. Turnaround time is a major limitation: many omics workflows require specialized processing and analysis that may exceed the period during which immunomodulatory decisions are most relevant.124–127
Preanalytical and analytical variability further complicate implementation. Blood collection method, anticoagulant, sample handling time, cell isolation procedure, storage conditions, sequencing depth, gating strategy, and batch effects can all influence PBMC measurements. This is particularly relevant for transcriptomic and metabolomic assays, which may be sensitive to processing delays and inter-laboratory variation. Even for more established markers such as monocyte HLA-DR, differences in antibody panels, calibration methods, and reporting units can limit comparability across studies.
Functional assays face similar standardization problems. LPS-induced TNF-α release, NLRP3-induced IL-1β production, and composite immune-function scores require standardized stimulation conditions, incubation times, readout methods, and clinically validated thresholds. Without harmonized protocols, it is difficult to define which patients truly have immunoparalysis, excessive inflammasome suppression, or preserved immune reserve. Therefore, assay standardization is as important as biomarker discovery.
Another barrier is the lack of prospective evidence that biomarker-guided decisions improve outcomes. Many PBMC biomarkers predict mortality, secondary infection, or immune dysfunction, but prediction alone does not establish clinical utility. To change practice, biomarkers must show that they can guide an intervention, change management, or improve patient-centered outcomes. The intervention landscape summarized in Supplementary Table S1 illustrates that treatment failure often reflects insufficient immune-state enrichment rather than the absence of biologically plausible targets. Future trials must therefore test biomarker-guided treatment algorithms rather than isolated biomarkers.
Future Directions for PBMC-Guided Sepsis Research
Future studies should prioritize longitudinal, age-stratified, and biomarker-guided designs. Longitudinal sampling is essential because PBMC states evolve rapidly during sepsis. Single time-point measurements cannot reliably distinguish transient immune activation from persistent immune paralysis. Repeated sampling at predefined windows—such as admission, 24 hours, 48–72 hours, days 5–7, and recovery or discharge—would help define when specific immune states emerge, persist, or resolve.
Age-stratified studies are also needed. Neonatal, pediatric, adult, and older adult cohorts should be analyzed separately before conclusions are generalized across the lifespan. Such studies should evaluate whether key mechanisms, including HLA-DR downregulation, T-cell exhaustion, metabolic paralysis, and functional immune suppression, follow similar trajectories across age groups. This is particularly important because therapeutic windows and safety profiles may differ between children, adults, and older adults.
Integrated immune phenotyping should become a central design principle. Rather than relying on one marker, future studies should combine cellular phenotyping, transcriptomic or targeted gene-expression signatures, metabolic indicators, and functional assays. A practical clinical panel may include monocyte HLA-DR, selected monocyte and lymphocyte subsets, one or more transcriptomic or metabolic markers, and a functional stimulation assay. Such panels should be evaluated against clinically meaningful outcomes, including secondary infection, organ dysfunction trajectory, ICU length of stay, readmission, and mortality.
Clinical trials should also use immune-state enrichment. Instead of applying uniform immunomodulatory therapy to all septic patients, future trials should allocate therapy according to immune phenotype. Patients with hyperinflammatory features may be considered for selective anti-inflammatory approaches, whereas patients with persistent HLA-DR downregulation, lymphopenia, or impaired cytokine-production capacity may be better candidates for immunostimulatory therapy. The immune-state rationale summarized in Supplementary Table S1 can serve as a framework for matching intervention type to patient biology.
Finally, PBMC-guided sepsis research should move toward adaptive and platform trial designs. Such designs can incorporate repeated immune monitoring, allow treatment allocation to change over time, and test multiple interventions across immune endotypes. This approach is better aligned with the dynamic biology of sepsis than fixed, one-size-fits-all trial designs. Only through standardized, longitudinal, and temporally informed approaches can PBMC-based immune profiling move from mechanistic discovery toward clinically useful precision immunology in sepsis.
Conclusion
PBMCs provide an accessible and clinically relevant window into the dynamic immune response of sepsis. Rather than following a simple linear progression from hyperinflammation to immunosuppression, sepsis involves overlapping and time-dependent immune states that differ across patients and disease stages. Evidence from single-cell profiling, transcriptomics, mechanistic studies, and functional immune assays indicates that PBMC remodeling—particularly monocyte/macrophage dysfunction, HLA-DR downregulation, lymphocyte exhaustion, regulated cell death, and immunometabolic reprogramming—plays a central role in this evolving immune trajectory.
This PBMC-centered perspective has important translational implications. PBMC-derived biomarkers may help move sepsis assessment beyond static inflammatory markers toward dynamic immune phenotyping. Early markers such as CD38high monocytes and monocyte distribution width may support early diagnosis, whereas persistent monocyte HLA-DR downregulation, exhausted lymphocyte subsets, metabolic signatures, and functional stimulation assays may identify patients at risk of immunoparalysis, secondary infection, and prolonged critical illness. These markers may also help define therapeutic windows, allowing anti-inflammatory, immunostimulatory, or metabolic interventions to be matched more closely to the patient’s immune state.
However, PBMC-guided precision immunology in sepsis remains an emerging field. Current evidence is limited by cohort heterogeneity, age-related differences, variable sampling times, assay standardization challenges, and insufficient prospective validation. Future studies should therefore prioritize longitudinal and age-stratified immune monitoring, standardized PBMC assays, integrated biomarker panels, and biomarker-guided interventional trials. With these advances, PBMC-based immune profiling may become a practical tool for improving risk stratification and guiding individualized immunomodulatory strategies in sepsis.
Data Sharing Statement
No new datasets were generated or analyzed in this review. Further information is available from the corresponding author upon reasonable request.
Author Contributions
Zhiheng Qian: Conceptualization, Methodology, Investigation, Literature Search, Data Curation, Formal Analysis, Visualization, Writing – Original Draft, and Writing – Review & Editing. The author has read and approved the final article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zhu L, Hu M, Xu H, et al. The key players of inflammasomes and pyroptosis in sepsis-induced pathogenesis and organ dysfunction. Front Pharmacol. 2025;16:1586364. doi:10.3389/fphar.2025.1586364
2. Ovali MA, Percin S. Sepsis-associated immunosuppression: mechanistic insights, biomarkers, and therapeutic perspectives. Mol Biol Rep. 2025;53:148. doi:10.1007/s11033-025-11312-6
3. Yang Y, Zhang Y, Wu J, Liu Y, Lei X. Decoding immune low-response states in sepsis: single-cell and 3D spatial transcriptomic insights into immunoparalysis. Front Immunol. 2025;16:1696914. doi:10.3389/fimmu.2025.1696914
4. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395:200–23. doi:10.1016/S0140-6736(19)32989-7
5. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–810. doi:10.1001/jama.2016.0287
6. Ni FH. Biomarker-based diagnosis of ventilator-associated pneumonia using serum and bronchoalveolar lavage fluid levels of presepsin, procalcitonin, and lipopolysaccharide-binding protein. Front Cell Infect Microbiol. 2026;16:1747971. doi:10.3389/fcimb.2026.1747971
7. Qiu X, Li J, Bonenfant J, et al. Dynamic changes in human single-cell transcriptional signatures during fatal sepsis. J Leukoc Biol. 2021;110:1253–1268. doi:10.1002/JLB.5MA0721-825R
8. Sun L, Zhang P, Zhang H, et al. Single-Cell transcriptomic profiles of peripheral blood immune cells reveal early monocyte and platelet activation in the transition from high-risk states to clinical sepsis. Sci Rep. 2025;15:32879. doi:10.1038/s41598-025-17078-y
9. Chen S, Wu A, Shen X, Kong J, Huang Y. Disrupting the dangerous alliance: dual anti-inflammatory and anticoagulant strategy targets platelet-neutrophil crosstalk in sepsis. J Control Release. 2025;379:814–831. doi:10.1016/j.jconrel.2025.01.053
10. Qian W, Liu Y, Zhao X, Dong Y, Zhou J, Shou S. Single-cell transcriptomic and m6A methylation analyses reveal platelet-mediated immune regulatory mechanisms in sepsis. Front Immunol. 2025;16:1607732. doi:10.3389/fimmu.2025.1607732
11. Babuta M, Morel C, de Carvalho Ribeiro M, et al. Neutrophil extracellular traps activate hepatic stellate cells and monocytes via NLRP3 sensing in alcohol-induced acceleration of MASH fibrosis. Gut. 2024;73:1854–1869. doi:10.1136/gutjnl-2023-331447
12. Liu Q, Chen R, Zhang Z, Sha Z, Wu H. Mechanisms and immune crosstalk of neutrophil extracellular traps in response to infection. mBio. 2025;16:e0018925. doi:10.1128/mbio.00189-25
13. Yang X, Song J, Ma H, et al. The crucial roles of platelets as immune mediators in sepsis. J Inflamm Res. 2025;18:12825–12845. doi:10.2147/JIR.S535701
14. Monneret G, Lafon T, Gossez M, et al. Publisher correction: monocyte HLA-DR expression in septic shock patients: insights from a 20-year real-world cohort of 1023 cases. Intensive Care Med. 2025;51:2196–2198. doi:10.1007/s00134-025-08161-z
15. Shi J, Zhu L, Sui X, et al. CXCR5-engineered mesenchymal stromal cells home to spleen and mitigate post-sepsis syndrome by preventing secondary infection. Stem Cell Res Ther. 2025;16:616. doi:10.1186/s13287-025-04751-2
16. Yu Z, Xiao C, Liu R, et al. The protective effect of apolipoprotein H in paediatric sepsis. Crit Care. 2024;28:36. doi:10.1186/s13054-024-04809-2
17. Li Y, Shi J, Yu T, et al. FPR1-dependent Pro-inflammatory Ccl4(high) monocytes/macrophages drive and predict sepsis-induced acute lung injury. Respir Res. 2025;26:308. doi:10.1186/s12931-025-03385-5
18. Stegnjaić G, Mićanović D, Saksida T, et al. Sepsis prevents the development of experimental type 1 diabetes. Front Immunol. 2025;16:1658960. doi:10.3389/fimmu.2025.1658960
19. Liu M, Wang JQ, Wang JN, et al. PIM1 orchestrates sepsis-associated inflammatory imbalance in CD4(+) T cell subsets via cholesterol metabolism. mBio. 2025;16:e0168025. doi:10.1128/mbio.01680-25
20. Yang Y, Sun Z, Ge Y. Upadacitinib in the treatment of SAPHO syndrome: a case report. Front Immunol. 2025;16:1662675. doi:10.3389/fimmu.2025.1662675
21. Liu X, Chen L, Peng W, et al. Th17/Treg balance: the bloom and wane in the pathophysiology of sepsis. Front Immunol. 2024;15:1356869. doi:10.3389/fimmu.2024.1356869
22. Zhong X, Xie H, Wang S, et al. TIGIT regulates CD4(+) T cell immunity against polymicrobial sepsis. Front Immunol. 2024;15:1290564. doi:10.3389/fimmu.2024.1290564
23. Jiang Y, Gao S, Li X, et al. Myeloperoxidase-anchored ENO1 mediates neutrophil extracellular trap DNA to enhance Treg differentiation via IFITM2 during sepsis. J Clin Invest. 2025;135. doi:10.1172/JCI183541
24. Wang Y, Guo T, Xing X, et al. The accumulation of myeloid-derived suppressor cells participates in abdominal infection-induced tumor progression through the PD-L1/PD-1 axis. Mol Oncol. 2025;19:1532–1545. doi:10.1002/1878-0261.13767
25. Wang L, Liu Q, Wu C, Hou M, Li Z. Establishment and validation of the prediction model based on lymphocyte subsets for acute kidney injury in sepsis patients. Front Immunol. 2025;16:1674673. doi:10.3389/fimmu.2025.1674673
26. Perschinka F, Lehner GF, Mayerhöfer T, et al. Differences in the phenotype of bacterial and viral sepsis–a prospective, multicenter, observational study. Viruses. 2025;17.
27. Han L, Zhuang MW, Deng J, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol. 2021;93:5376–5389. doi:10.1002/jmv.27050
28. Reyes M, Filbin MR, Bhattacharyya RP, et al. An immune-cell signature of bacterial sepsis. Nat Med. 2020;26:333–340. doi:10.1038/s41591-020-0752-4
29. Tang J, Xu L, Zeng Y, Gong F. Effect of gut microbiota on LPS-induced acute lung injury by regulating the TLR4/NF-kB signaling pathway. Int Immunopharmacol. 2021;91:107272. doi:10.1016/j.intimp.2020.107272
30. De Biasi S, Neroni A, Nasi M, et al. Healthy preterm newborns: altered innate immunity and impaired monocyte function. Eur J Immunol. 2023;53:e2250224. doi:10.1002/eji.202250224
31. Weiss SL, Zhang D, Bush J, et al. Mitochondrial dysfunction is associated with an immune paralysis phenotype in pediatric sepsis. Shock. 2020;54:285–293. doi:10.1097/SHK.0000000000001486
32. Martin MD, Badovinac VP, Griffith TS. CD4 T cell responses and the sepsis-induced immunoparalysis state. Front Immunol. 2020;11:1364. doi:10.3389/fimmu.2020.01364
33. van Amstel RBE, Kennedy JN, Scicluna BP, et al. Uncovering heterogeneity in sepsis: a comparative analysis of subphenotypes. Intensive Care Med. 2023;49:1360–1369. doi:10.1007/s00134-023-07239-w
34. Thomas R, Wang W, Su DM. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun Ageing. 2020;17:2. doi:10.1186/s12979-020-0173-8
35. Wang Y, Zhang H, Miao C. Unraveling immunosenescence in sepsis: from cellular mechanisms to therapeutics. Cell Death Dis. 2025;16:393.
36. Zhou Y, Sun W, Zhou J, et al. Bu Zhong Yi Qi Tang targets key monocyte-associated genes to enhance sepsis therapy. J Ethnopharmacol. 2026;355:120610. doi:10.1016/j.jep.2025.120610
37. Hua N, Kong L, Fan L, et al. Identification of CD38(high) monocyte as a candidate diagnostic biomarker and therapeutic target for sepsis. Adv Sci. 2025;12:e2500457. doi:10.1002/advs.202500457
38. Liu H, Liang Q. Single-cell multi-omics-based immune temporal network resolution in sepsis: unravelling molecular mechanisms and precise therapeutic targets. Front Immunol. 2025;16:1616794. doi:10.3389/fimmu.2025.1616794
39. Liu D, Fu W, Zhang T, et al. Eliminating myeloid-derived suppressor cells alleviates immunosuppression and reduces susceptibility to secondary infections in a two-hit sepsis model. Cytokine. 2025;191:156955. doi:10.1016/j.cyto.2025.156955
40. Yao M, Cao Y, He J, et al. Single-cell transcriptomic analysis reveals heterogeneous features of myeloid-derived suppressor cells in newborns. Front Immunol. 2024;15.
41. Luo K, Xu Y, Chen J, et al. Noncoding RNA-mediated regulation of myeloid-derived suppressor cells in cancer. Cancer Manag Res. 2025;17:2567–2587. doi:10.2147/CMAR.S550896
42. Joshi I, Carney WP, Rock EP. Utility of monocyte HLA-DR and rationale for therapeutic GM-CSF in sepsis immunoparalysis. Front Immunol. 2023;14.
43. Zhang T, Liu H, Tie YF, Meng TW, Liang Q. Potential of interleukin-7 in sepsis as a biomarker and therapeutic agent: a narrative review. Front Med. 2025;12:1649049.
44. Shankar-Hari M, Francois B, Remy KE, et al. A randomized, double-blind, placebo-controlled trial of IL-7 in critically ill patients with COVID-19. JCI Insight. 2025;10. doi:10.1172/jci.insight.189150
45. Fang Z, Gao X, Duan L, et al. Expression and clinical significance of S100A8/9 in adults with secondary phagocytic lymphohistiocytosis. Front Mol Biosci. 2025;12:1607352. doi:10.3389/fmolb.2025.1607352
46. Zhao Y, Du J, Shen X. Targeting myeloid-derived suppressor cells in tumor immunotherapy: current, future and beyond. Front Immunol. 2023;14.
47. Halstead ES, McKeone DJ, Samuelsen AM, Zhou S, Bonavia AS. Functional immune profiling of hyper- and hypo-inflammatory subphenotypes of critical illness: a secondary analysis. Front Immunol. 2025;16:1520848. doi:10.3389/fimmu.2025.1520848
48. Saavedra-Torres JS, Pinzón-Fernández MV, Nati-Castillo HA, et al. Immunodynamic Disruption in Sepsis: mechanisms and Strategies for Personalized Immunomodulation. Biomedicines. 2025;13:2139.
49. Kawata K, Hatano S, Baba A, Imabayashi K, Baba Y. Bruton’s tyrosine kinase inhibition limits endotoxic shock by suppressing IL-6 production by marginal zone B cells in mice. Front Immunol. 2024;15:1388947.
50. Tullie S, Nicholson T, Bishop JRB, et al. Severe thermal and major traumatic injury results in elevated plasma concentrations of total heme that are associated with poor clinical outcomes and systemic immune suppression. Front Immunol. 2024;15:1416820.
51. Leite GGF, de Brabander J, Michels EHA, et al. Monocyte state 1 (MS1) cells in critically ill patients with sepsis or non-infectious conditions: association with disease course and host response. Crit Care. 2024;28:88. doi:10.1186/s13054-024-04868-5
52. Wang J, Wu Y, Lin R, Zhang Y, Li L. TRAM deletion attenuates monocyte exhaustion and alleviates sepsis severity. Front Immunol. 2023;14:1297329. doi:10.3389/fimmu.2023.1297329
53. Caldwell BA, Ie S, Lucas A, Li L. Ticam2 ablation facilitates monocyte exhaustion recovery after sepsis. Sci Rep. 2025;15:2059. doi:10.1038/s41598-025-86103-x
54. Chen S, Saeed A, Liu Q, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8:207.
55. O’Farrell AM, Liu Y, Moore KW, Mui AL. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO j. 1998;17:1006–1018.
56. Bah I, Kumbhare A, Nguyen L, McCall CE, El Gazzar M. IL-10 induces an immune repressor pathway in sepsis by promoting S100A9 nuclear localization and MDSC development. Cell Immunol. 2018;332:32–38.
57. Yang L, Xie M, Yang M, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436.
58. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi:10.1038/nature11986
59. Maheshwari D, Kumar D, Jagdish RK, et al. Bioenergetic failure drives functional exhaustion of monocytes in acute-on-chronic liver failure. Front Immunol. 2022;13:856587. doi:10.3389/fimmu.2022.856587
60. Mao JY, Xie YW, Lei XL, Zhang JH, Cheng W, Cui N. Effects of neutrophil granule proteins on sepsis-associated lymphopenia and their relationship with CD4(+) T-cell pyroptosis. Front Immunol. 2025;16:1507800. doi:10.3389/fimmu.2025.1507800
61. Dai W, Zheng P, Wu J, et al. Integrated analysis of single-cell RNA-seq and chipset data unravels PANoptosis-related genes in sepsis. Front Immunol. 2023;14:1247131. doi:10.3389/fimmu.2023.1247131
62. Cornelius DC, Travis OK, Tramel RW, et al. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS One. 2020;15:e0234039. doi:10.1371/journal.pone.0234039
63. Su E, Song X, Wei L, et al. Endothelial GSDMD underlies LPS-induced systemic vascular injury and lethality. JCI Insight. 2025;10. doi:10.1172/jci.insight.182398
64. Mallarpu CS, Chelluri SI, Katragadda TK, Singarapu M, Chelluri LK, Madiraju C. Programmed cell death markers in COVID-19 survivors with and without sepsis. Front Immunol. 2025;16:1535938.
65. Zhang T, Wang S, Hua D, et al. Identification of ZIP8-induced ferroptosis as a major type of cell death in monocytes under sepsis conditions. Redox Biol. 2024;69:102985.
66. Mei Y, Chen X, Shi S, et al. GI-Y2, a novel gasdermin D inhibitor, attenuates sepsis-induced myocardial dysfunction by inhibiting gasdermin D-mediated pyroptosis in macrophages. Br J Pharmacol. 2025;182:3503–3521. doi:10.1111/bph.70040
67. Yang Z, Gao Y, Zhao L, Lv X, Du Y. Molecular mechanisms of Sepsis attacking the immune system and solid organs. Front Med. 2024;11:1429370.
68. Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–2605. doi:10.1001/jama.2011.1829
69. Ye Z, Xie B, Tao Y, Xiao D. Mechanism of ferroptosis and its role in disease development. Int J Biol Sci. 2025;21:5328–5360. doi:10.7150/ijbs.102859
70. Arcos M, Liu Z, Villareal LB, et al. Myeloid NCOA4 sequesters KEAP1 to reduce ferroptosis for protection against salmonellosis in mice. Res Sq. 2024.
71. Fang P, Li S, Lu ZQ, et al. Histone lactylation exacerbates acute lung injury in septic mice by promoting ferroptosis in pulmonary microvascular endothelial cells. Burns Trauma. 2025;13:tkaf056. doi:10.1093/burnst/tkaf056
72. Wasyluk W, Zwolak A. Metabolic Alterations in Sepsis. J Clin Med. 2021;10.
73. Tang J, Yan M, Li M, Hu Z, Zhou K. From “metabolic storm” to “immune paralysis”: the dynamic evolution of macrophages and metabolism reprogramming in ARDS. Front Immunol. 2025;16:1738713. doi:10.3389/fimmu.2025.1738713
74. Rahmel T, Marko B, Nowak H, et al. Mitochondrial dysfunction in sepsis is associated with diminished intramitochondrial TFAM despite its increased cellular expression. Sci Rep. 2020;10:21029. doi:10.1038/s41598-020-78195-4
75. Li C, He M, Shi P, et al. A novel, rapid, and practical prognostic model for sepsis patients based on dysregulated immune cell lactylation. Front Immunol. 2025;16:1625311. doi:10.3389/fimmu.2025.1625311
76. Appiah MG, Park EJ, Akama Y, et al. Cellular and exosomal regulations of sepsis-induced metabolic alterations. Int J Mol Sci. 2021;22:8295. doi:10.3390/ijms22158295
77. Davis MJ, Tsang TM, Qiu Y, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio. 2013;4:e00264–13. doi:10.1128/mBio.00264-13
78. Ghamangiz S, Jafari A, Maleki-Kakelar H, Azimi H, Mazloomi E. Reprogram to heal: macrophage phenotypes as living therapeutics. Life Sci. 2025;371:123601. doi:10.1016/j.lfs.2025.123601
79. Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–311. doi:10.1016/j.immuni.2010.09.002
80. Wang H, Bai G, Cui N, Han W, Long Y. T-cell-specific mTOR deletion in mice ameliorated CD4(+) T-cell survival in lethal sepsis induced by severe invasive candidiasis. Virulence. 2019;10:892–901. doi:10.1080/21505594.2019.1685151
81. Cheng W, Zhang J, Li D, et al. The endoplasmic reticulum stress status of CD4+ T lymphocytes and its association with mTOR-mediated autophagic-lysosomal disorder in elderly sepsis patients. Front Immunol. 2025;16:1648075. doi:10.3389/fimmu.2025.1648075
82. Cai J, Li H, Cui W, et al. Herpesvirus entry mediator on effector memory CD8+ T cells mediates the effects of sterol ester (27:1/18:1) levels on critical sepsis. Immunol Lett. 2026;277:107103. doi:10.1016/j.imlet.2025.107103
83. Amunugama K, Pike DP, Ford DA. The lipid biology of sepsis. J Lipid Res. 2021;62:100090. doi:10.1016/j.jlr.2021.100090
84. Gao X, Cai S, Li X, Wu G. Sepsis-induced immunosuppression: mechanisms, biomarkers and immunotherapy. Front Immunol. 2025;16:1577105. doi:10.3389/fimmu.2025.1577105
85. Chen LH, Lin WR, Hu HR, Chen ZY, Wang YH. Identification and validation of the important role of tryptophan-related gene CYP1B1 in the development and progression of sepsis. PLoS One. 2025;20:e0335944. doi:10.1371/journal.pone.0335944
86. Shi Z, Wang F, Yang L, et al. Identification of sepsis biomarkers through glutamine metabolism-mediated immune regulation: a comprehensive analysis employing mendelian randomization, multi-omics integration, and machine learning. Front Immunol. 2025;16:1640425. doi:10.3389/fimmu.2025.1640425
87. Heuser SK, Li J, Pudewell S, LoBue A, Li Z, Cortese-Krott MM. Biochemistry, pharmacology, and in vivo function of arginases. Pharmacol Rev. 2025;77:100015. doi:10.1124/pharmrev.124.001271
88. Long Q, Song S, Li J, et al. Pharmacological and genetic inhibition of ARG2 in CXCR2(Hi) myeloid-derived suppressor cells combats sepsis-induced lymphopenia. Theranostics. 2025;15:7990–8011. doi:10.7150/thno.112339
89. Hooftman A, Angiari S, Hester S, et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. 2020;32:468–478.e7. doi:10.1016/j.cmet.2020.07.016
90. Mignot-Evers L, Raaijmakers V, Buunk G, et al. Comparison of SIRS criteria and qSOFA score for identifying culture-positive sepsis in the emergency department: a prospective cross-sectional multicentre study. BMJ Open. 2021;11:e041024. doi:10.1136/bmjopen-2020-041024
91. Garduno A, Cusack R, Leone M, Einav S, Martin-Loeches I. Multi-omics endotypes in ICU sepsis-induced immunosuppression. Microorganisms. 2023;11:1119. doi:10.3390/microorganisms11051119
92. Lu J, Zhang W, He Y, et al. Multi-omics decodes host-specific and environmental microbiome interactions in sepsis. Front Microbiol. 2025;16:1618177. doi:10.3389/fmicb.2025.1618177
93. Scicluna BP, Cano-Gamez K, Burnham KL, et al. A consensus blood transcriptomic framework for sepsis. Nat Med. 2025;31:4119–4130. doi:10.1038/s41591-025-03964-5
94. Sendak MP, Ratliff W, Sarro D, et al. Real-world integration of a sepsis deep learning technology into routine clinical care: implementation study. JMIR Med Inform. 2020;8:e15182. doi:10.2196/15182
95. Mi Y, Burnham KL, Charles PD, et al. High-throughput mass spectrometry maps the sepsis plasma proteome and differences in patient response. Sci Transl Med. 2024;16:eadh0185. doi:10.1126/scitranslmed.adh0185
96. Zhou W, Lin Z, Deng H, et al. Identification of potential therapeutic drug targets for sepsis by combining the human plasma proteome and genome: a Mendelian randomization study. J Thorac Dis. 2025;17:2576–2593. doi:10.21037/jtd-2025-590
97. Chang Y, Yoo HJ, Kim SJ, et al. A targeted metabolomics approach for sepsis-induced ARDS and its subphenotypes. Crit Care. 2023;27:263. doi:10.1186/s13054-023-04552-0
98. She H, Du Y, Du Y, et al. Metabolomics and machine learning approaches for diagnostic and prognostic biomarkers screening in sepsis. BMC Anesthesiol. 2023;23:367. doi:10.1186/s12871-023-02317-4
99. Ligi D, Della Franca C, Pelloso M, et al. Comparative analysis of monocyte distribution width alterations in Escherichia coli sepsis: insights from in vivo and ex vivo models. Clin Chem Lab Med. 2026;64:237–249. doi:10.1515/cclm-2025-0487
100. Sun J, Shao Y, Jiang R, et al. Monocyte distribution width (MDW) as a reliable diagnostic biomarker for sepsis in patients with HIV. Emerg Microbes Infect. 2025;14:2479634. doi:10.1080/22221751.2025.2479634
101. Sun Y, Lu Y, Pan X, Zhang C, Wang L, Zhang L. Early B lymphocyte subsets in blood predict prognosis in sepsis. Front Immunol. 2024;15:1437864. doi:10.3389/fimmu.2024.1437864
102. Tang J, Shang C, Chang Y, et al. Peripheral PD-1(+)NK cells could predict the 28-day mortality in sepsis patients. Front Immunol. 2024;15:1426064. doi:10.3389/fimmu.2024.1426064
103. Wu H, Cao T, Ji T, Luo Y, Huang J, Ma K. Predictive value of the neutrophil-to-lymphocyte ratio in the prognosis and risk of death for adult sepsis patients: a meta-analysis. Front Immunol. 2024;15:1336456. doi:10.3389/fimmu.2024.1336456
104. Cheng S, Wang J, Wang P, et al. Relationship between peripheral blood miR-146b-5p levels and lymphocyte subsets in patients with sepsis and its predictive value for prognosis: a single-center study. Front Immunol. 2025;16:1608576. doi:10.3389/fimmu.2025.1608576
105. Antonakos N, Gilbert C, Théroude C, Schrijver IT, Roger T. Modes of action and diagnostic value of miRNAs in sepsis. Front Immunol. 2022;13:951798. doi:10.3389/fimmu.2022.951798
106. Ho J, Chan H, Wong SH, et al. The involvement of regulatory non-coding RNAs in sepsis: a systematic review. Crit Care. 2016;20:383. doi:10.1186/s13054-016-1555-3
107. Wu H, Liu J, Li W, Liu G, Li Z. LncRNA-HOTAIR promotes TNF-α production in cardiomyocytes of LPS-induced sepsis mice by activating NF-κB pathway. Biochem Biophys Res Commun. 2016;471:240–246. doi:10.1016/j.bbrc.2016.01.117
108. Drewry AM, Ablordeppey EA, Murray ET, et al. Comparison of monocyte human leukocyte antigen-DR expression and stimulated tumor necrosis factor alpha production as outcome predictors in severe sepsis: a prospective observational study. Crit Care. 2016;20:334. doi:10.1186/s13054-016-1505-0
109. Martínez-García JJ, Martínez-Banaclocha H, Angosto-Bazarra D, et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat Commun. 2019;10:2711. doi:10.1038/s41467-019-10626-x
110. Coudereau R, Monneret G, Lukaszewicz AC, et al. Altered ex vivo NLRP3 inflammasome activation is associated with 28-day mortality in septic patients. Viruses. 2023;15:2419. doi:10.3390/v15122419
111. Li M, Huang H, Ke C, et al. Identification of a novel four-gene diagnostic signature for patients with sepsis by integrating weighted gene co-expression network analysis and support vector machine algorithm. Hereditas. 2022;159:14. doi:10.1186/s41065-021-00215-8
112. Hernández-Jiménez E, Plata-Menchaca EP, Berbel D, et al. Assessing sepsis-induced immunosuppression to predict positive blood cultures. Front Immunol. 2024;15:1447523. doi:10.3389/fimmu.2024.1447523
113. Slim MA, van Mourik N, Bakkerus L, et al. Towards personalized medicine: a scoping review of immunotherapy in sepsis. Crit Care. 2024;28:183. doi:10.1186/s13054-024-04964-6
114. Fisher CJ, Dhainaut J-FA, Opal SM, et al. Recombinant human Interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome: results from a randomized, double-blind, Placebo-Controlled Trial. JAMA. 1994;271:1836–1843. doi:10.1001/jama.1994.03510470040032
115. Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III Trial. Crit Care Med. 2016;44:275–281. doi:10.1097/CCM.0000000000001402
116. Leventogiannis K, Kyriazopoulou E, Antonakos N, et al. Toward personalized immunotherapy in sepsis: the PROVIDE randomized clinical trial. Cell Rep Med. 2022;3:100817. doi:10.1016/j.xcrm.2022.100817
117. Giamarellos-Bourboulis EJ, Kotsaki A, Kotsamidi I, et al. Precision immunotherapy to improve sepsis outcomes: the ImmunoSep randomized clinical trial. JAMA. 2026;335:775–786. doi:10.1001/jama.2025.24175
118. Francois B, Jeannet R, Daix T, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. 2018;3. doi:10.1172/jci.insight.98960
119. Writing Committee for the REMAP-CAP Investigators. Long-term (180-Day) outcomes in critically ill patients with COVID-19 in the REMAP-CAP randomized clinical trial. JAMA. 2023;329:39–51. doi:10.1001/jama.2022.23257
120. Li C, Yuan Y, Jiang X, Wang Q. Epigenetic regulation of CD8(+) T cell exhaustion: recent advances and update. Front Immunol. 2025;16:1700039. doi:10.3389/fimmu.2025.1700039
121. Niu B, Zhou F, Su Y, et al. Different expression characteristics of LAG3 and PD-1 in sepsis and their synergistic effect on T cell exhaustion: a new strategy for immune checkpoint blockade. Front Immunol. 2019;10:1888. doi:10.3389/fimmu.2019.01888
122. Wu F, Chen Y, Chen L, Wei X, Zhang L, Shen Y. Immunometabolic reprogramming in experimental sepsis: a driver of multiple organ dysfunction syndrome. J Inflamm Res. 2026;19:577143. doi:10.2147/JIR.S577143
123. Paul SN, Nessel I, Puthucheary Z, Henson SM. Sepsis and the immunometabolic inflammatory response. NPJ Metab Health Dis. 2026;4:4. doi:10.1038/s44324-025-00095-w
124. Rashid A, Al-Obeidat F, Kanthimathinathan HK, et al. Advancing sepsis clinical research: harnessing transcriptomics for an omics-based strategy - a comprehensive scoping review. Inf Med Unlocked. 2024;44:101419. doi:10.1016/j.imu.2023.101419
125. Malic L, Zhang PGY, Plant PJ, et al. Publisher correction: a machine learning and centrifugal microfluidics platform for bedside prediction of sepsis. Nat Commun. 2025;16:5330. doi:10.1038/s41467-025-61096-3
126. Jin X, Shen H, Zhou P, et al. Research progress on sepsis diagnosis and monitoring based on Omics Technologies: a review. Diagnostics. 2025;15:2887. doi:10.3390/diagnostics15222887
127. Bouttell J, Heggie R, Oien K, et al. Economic evaluation of genomic/genetic tests: a review and future directions. Int J Technol Assess Health Care. 2022;38:e67. doi:10.1017/S0266462322000484
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Endothelial-Related Biomarkers in Evaluation of Vascular Function During Progression of Sepsis After Severe Trauma: New Potential Diagnostic Tools in Sepsis
Yang B, Wang X, Liu Z, Lu Z, Fang G, Xue X, Luo T
Journal of Inflammation Research 2023, 16:2773-2782
Published Date: 6 July 2023
Comprehensive Sepsis Risk Prediction in Leukemia Using a Random Forest Model and Restricted Cubic Spline Analysis
Kou Y, Tian Y, Ha Y, Wang S, Sun X, Lv S, Luo B, Yang Y, Qin L
Journal of Inflammation Research 2025, 18:1013-1032
Published Date: 22 January 2025
Single-Cell Multi-Omics Deciphers Core Gene Networks and Immune Interaction Collapse in Sepsis-Associated T Cell Dysfunction

Li X, Chen Z, Yao Y, Chen M, Hu Y
Infection and Drug Resistance 2025, 18:4863-4885
Published Date: 13 September 2025
A Reproducible Ceramide Phenotype of Sepsis Across Aetiologies – A Monocenter Cohort Study
Pavel V, Mester P, Höring M, Krautbauer S, Liebisch G, Schmid S, Müller M, Buechler C
Journal of Inflammation Research 2026, 19:571836
Published Date: 20 February 2026
Bioinformatic Analysis and Experimental Verification Reveal the Protective Role of Baicalein Against LPS-Induced Endothelial Cell Dysfunction
Luo F, Ao J, Chen Y, Cui Y, Gan X, Li M
Journal of Inflammation Research 2026, 19:555260
Published Date: 27 February 2026