Back to Journals » Infection and Drug Resistance » Volume 19
Pathogenic Characteristics and Molecular Epidemiological Analysis of Klebsiella pneumoniae in Clinical Urinary Tract Infections in Beijing, China
Authors Zhao Z, Cao X, Li J, Wang K, Li P, Zhang W, Liu J, Sun T, Yang X, Wang Y, Peng M, Li P
Received 8 December 2025
Accepted for publication 24 February 2026
Published 19 March 2026 Volume 2026:19 581687
DOI https://doi.org/10.2147/IDR.S581687
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hazrat Bilal
Zihan Zhao,1,2,* Xueping Cao,3,* Jinhui Li,2,* Kaiying Wang,2,* Peng Li,2 Wei Zhang,3 Jia Liu,3 Tao Sun,3 Xiaoli Yang,3 Yufei Wang,3 Mengling Peng,1 Peihan Li2
1College of Veterinary Medicine, Anhui Agricultural University, Anhui, 230036, People’s Republic of China; 2Department of Biosecurity, The Chinese PLA Center for Disease Control and Prevention, Beijing, 100071, People’s Republic of China; 3Department of Clinical Laboratory, The Third MedicalCenter, PLA General Hospital, Beijing, 100039, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Mengling Peng, College of Veterinary Medicine, Anhui Agricultural University, Anhui, 230036, People’s Republic of China, Email [email protected] Peihan Li, Department of Biosecurity, The Chinese PLA Center for Disease Control and Prevention, Beijing, 100071, People’s Republic of China, Email [email protected]
Introduction: Klebsiella pneumoniae (KP), a Gram-negative bacterium of the Enterobacteriaceae family, is a major opportunistic pathogen responsible for severe infections, particularly urinary tract infections (UTIs). The increasing incidence of KP infections poses a significant challenge to global healthcare systems.
Methods: In this study, we isolated KP strains from UTI patients and performed antimicrobial susceptibility testing, whole-genome sequencing (WGS), and comprehensive genomic analyses to delineate their molecular characteristics.
Results: Multilocus sequence typing (MLST) identified 41 sequence types (STs), with ST11 being the most prevalent. Phylogenetic analysis revealed that most local strains clustered closely with the globally disseminated KP clonal group 258 (CG258), suggesting a potential origin from this epidemic lineage. All isolates carried virulence and antibiotic resistance genes, with ST11 strains exhibiting the highest resistance gene burden, classifying them as multidrug-resistant (MDR). We further characterized the plasmid pNDM-MAR and identified biosynthetic gene clusters (BGCs) for redox-cofactors, azole-containing RiPPs, terpene precursors, NRP-metallophores, type I polyketide synthases (T1PKS), RiPP-like compounds, and non-ribosomal peptide synthetase-independent siderophores (NI-siderophores).
Discussion: These findings underscore the convergence of hypervirulence and multidrug resistance in KP, highlighting the need for continuous genomic surveillance to inform infection control strategies and antimicrobial stewardship.
Keywords: Klebsiella pneumoniae, urinary tract infection, phylogenetic analysis, epidemiological characteristics, secondary metabolite genes, plasmid
Introduction
Klebsiella pneumoniae (KP) is a significant opportunistic pathogen associated with a range of infections, including pneumonia, urinary tract infections (UTIs), and bloodstream infections.1,2 Since the mid-1980s, hypervirulent KP strains have emerged as a cause of severe disseminated infections.3,4 The World Health Organization (WHO) has designated KP as a priority pathogen due to its escalating antimicrobial resistance (AMR); notably, carbapenem-resistant KP was recently classified as a critical priority in the 2024 WHO bacterial pathogen priority list.5,6 KP accounts for approximately 10% of all hospital-acquired infections (HAIs) and is the second most common Gram-negative pathogen in hospital-acquired pneumonia.7 UTIs represent one of the most frequent infectious diseases globally, contributing to 25% of HAIs and posing substantial clinical burdens.8 Annually, over 150 million UTI cases are reported worldwide, with KP responsible for 15–20% of these infections, often transmitted via cross-contamination in healthcare settings.9,10
Patients in intensive care medicine wards (ICMWs) are particularly vulnerable to KP infections due to critical illness, immune compromise, prolonged hospitalization, and extensive antibiotic exposure.11 In particular, the incidence of drug-resistant and hypervirulent KP strains in this ward is notably higher than in other departments.12 Results from a systematic review indicate that the mortality rate of ICMW patients infected with KP reaches as high as 48.9%.13 The ICMW carries a 3- to 10-fold greater risk of HAIs compared to general wards,14 underscoring the importance of understanding local epidemiology to guide effective infection control.
In this study, we employed genomic analysis to conduct whole-genome comparative research on KP strains from different departments and with different clinical phenotypes, thereby accurately deciphering the genetic variations underlying differences in strain pathogenicity, diversity of drug resistance profiles, and epidemiological characteristics. This research can provide theoretical support for formulating strategies to prevent and control pathogenic infections and blocking the nosocomial transmission and community spread of drug-resistant strains. It holds practical significance in improving the diagnosis and treatment of clinical infectious diseases and enhancing the construction of public health prevention and control systems.
Materials and Methods
Isolation and Identification of Bacterial Strains
From January 2023 to February 2025, a total of 102 bacterial isolates were collected from urine specimens of KP-positive patients admitted to various wards, The Third Medical Center, PLA General Hospital. In accordance with the standard operating procedures,15 midstream morning urine samples were collected from patients using sterile urinary catheters at the aforementioned hospital. After thorough mixing, 5 μL of each sample was inoculated onto blood agar plates and MacConkey agar plates under sterile conditions in the clinical laboratory of the hospital, followed by aerobic incubation at 37°C for 24–48 hours. This study was conducted in accordance with the Declaration of Helsinki (revised 2013).
Antimicrobial Susceptibility Testing
Strain identification and antimicrobial susceptibility testing were conducted using the VITEK 2 COMPACT system (bioMérieux, Marcy-l’Étoile, France) with the AST-GN identification card, as well as the compatible AST-GN13 and AST-GN334 antimicrobial susceptibility cards. The tested antimicrobial agents included ciprofloxacin, levofloxacin, piperacillin/tazobactam, ceftazidime, cefepime, meropenem, imipenem, amikacin, trimethoprim-sulfamethoxazole, and nitrofurantoin. The criteria for determining antimicrobial susceptibility were referenced to the guidelines of the Clinical and Laboratory Standards Institute (CLSI, document M100).16
Bioinformatics Analysis
Whole-Genome Sequencing
Genomic DNA was extracted from the bacterial cultures using the TIANamp Bacteria DNA Kit (Cat. No. DP802, TIANGEN BIOTECH, Beijing, China). Negative controls were included to monitor contamination during the experimental process. Library construction for sequencing was performed using the NEBNext® UltraTM II DNA Library Prep Kit (New England Biolabs, USA) according to the manufacturer’s instructions. Whole-genome sequencing was performed on the Illumina NovaSeq X Plus platform (Illumina, San Diego, CA, USA) with a 2×150 bp paired-end configuration by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China); the sequencing was conducted with the quality metrics preset at Q20 > 90% and Q30 > 85% and the sequencing depth ranged primarily from 34.5× to 42.9×. Subsequent genome assembly was performed using SPAdes v3.15.5 with six specified k-mer sizes: 21, 33, 55, 77, 99, and 127.
Phylogenetic Analysis
The major clones of KP worldwide were identified through a literature survey.17 Genomic sequences were retrieved from the Pasteur Institute KP Database according to the clone types, and five genomes were randomly downloaded for each clonal complex. These downloaded genomes were combined with 102 locally isolated KP genomes to construct an intermediate phylogenetic tree, based on which the majority of the local strains were determined to belong to the CG258 clone. Subsequently, additional genomes of the CG258 clone were downloaded, and together with five genomes of other clones that served as the outgroup, a final dataset comprising 76 strains was established. Using the KP genome (GCF_000240185.1) as the reference, Snippy v4.6.0 was employed for single nucleotide polymorphism (SNP) calling,18 followed by phylogenetic tree construction with the IQ-TREE v2.4.0 tool, in which the ultrafast bootstrap approximation was selected as the validation method and the GTR substitution model was employed. The constructed, test criterion-compliant phylogenetic tree was visualized, processed, and refined using the Interactive Tree Of Life (iTOL) online platform to generate the final phylogenetic tree.19
Correlation Analysis Between Drug Resistance, Virulence, and Strains
Batch analyses were performed using Kleborate v3.1.3, a dedicated KP analysis tool developed by the Holt Laboratory, yielding genomic annotation results.20 These results were transformed and integrated, followed by correlation analysis of strains with their virulence, and drug resistance profiles using Python.
Prediction of Secondary Metabolites
The online tool antiSMASH was employed to predict secondary metabolites in KP, aiming to detect the presence of biosynthetic gene clusters (BGCs) for secondary metabolites within the genomic sequence of KP.
Plasmid Analysis
We performed assembly based on the second-generation sequencing data. Concurrently, plasmid sequence files of KP were downloaded from the NCBI database to construct a local plasmid database. The BLAST v2.16.0 tool was used to align the genomic sequences of all bacterial strains against this database, with the homology threshold set to 85% and the coverage threshold set to 50%. Based on the alignment results, plasmids with relatively high identity (NZ_CP186632.1, NZ_CP159675.1) were selected as representative plasmids. Gene prediction and functional annotation of these plasmids were performed using the online RAST platform.21 Circular maps of the plasmids were generated with CGview.22
Results
Microbiological Characteristics
Among the 102 collected cases of UTIs, a total of 82 cases were patients aged 60 years and above, accounting for 80.4% of the total cases. The case collection covered 21 departments (Table 1). Thirty-five cases were collected from the Department of Urology, where patients developed UTIs due to urinary system lesions; 15 cases were collected from the Intensive Care Medicine Ward, where patients were mostly in critical condition with impaired immune function, making them susceptible to bacterial invasion. The results of antimicrobial susceptibility testing showed that these strains were susceptible to most antimicrobial agents, but exhibited high resistance to ceftazidime and levofloxacin (Figure 1A). Among them, strains K52 and K54 were resistant to all tested antimicrobial agents (Figure 1B).
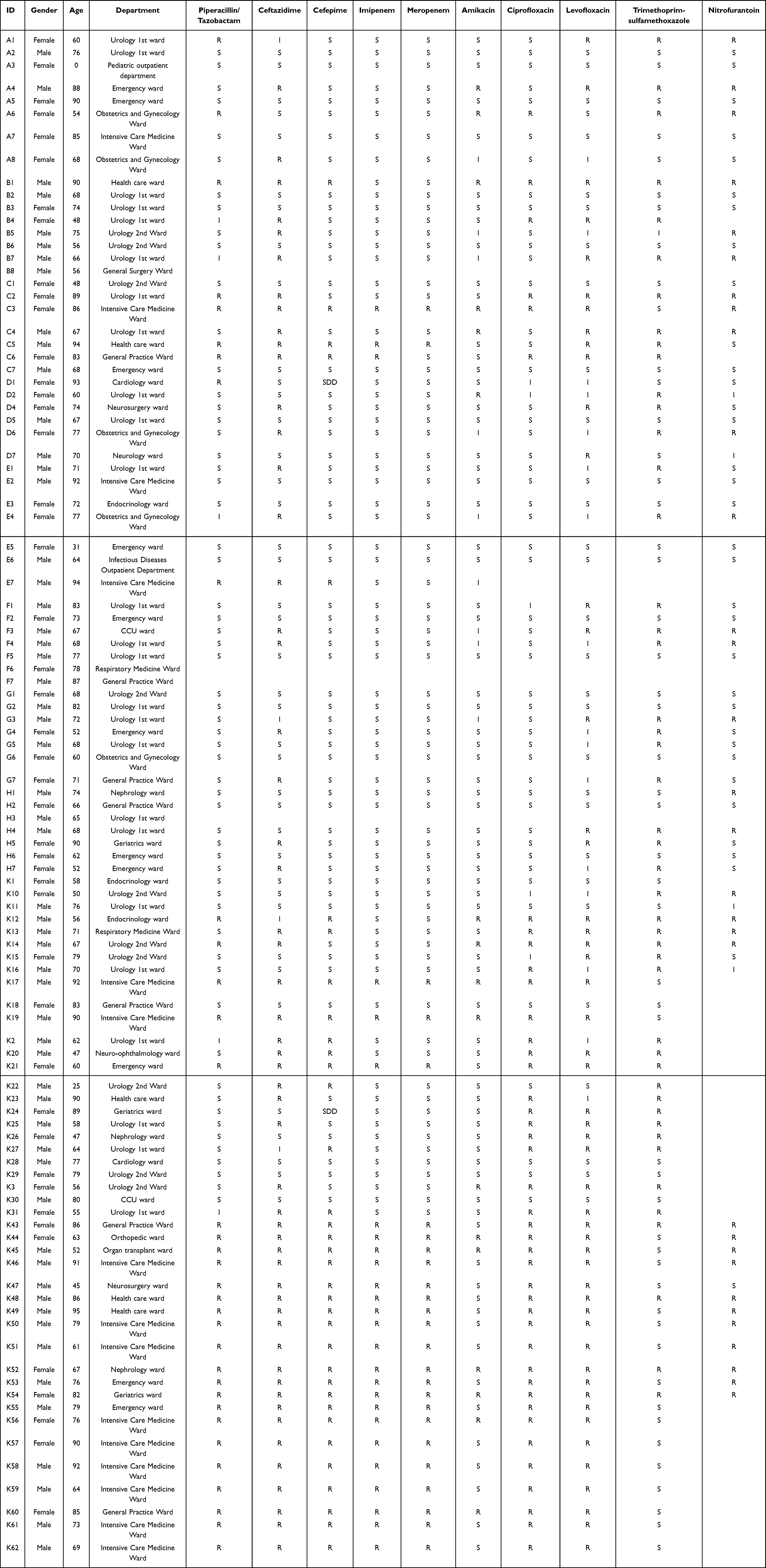 |
Table 1 Clinical Information Table of Strain Collection |
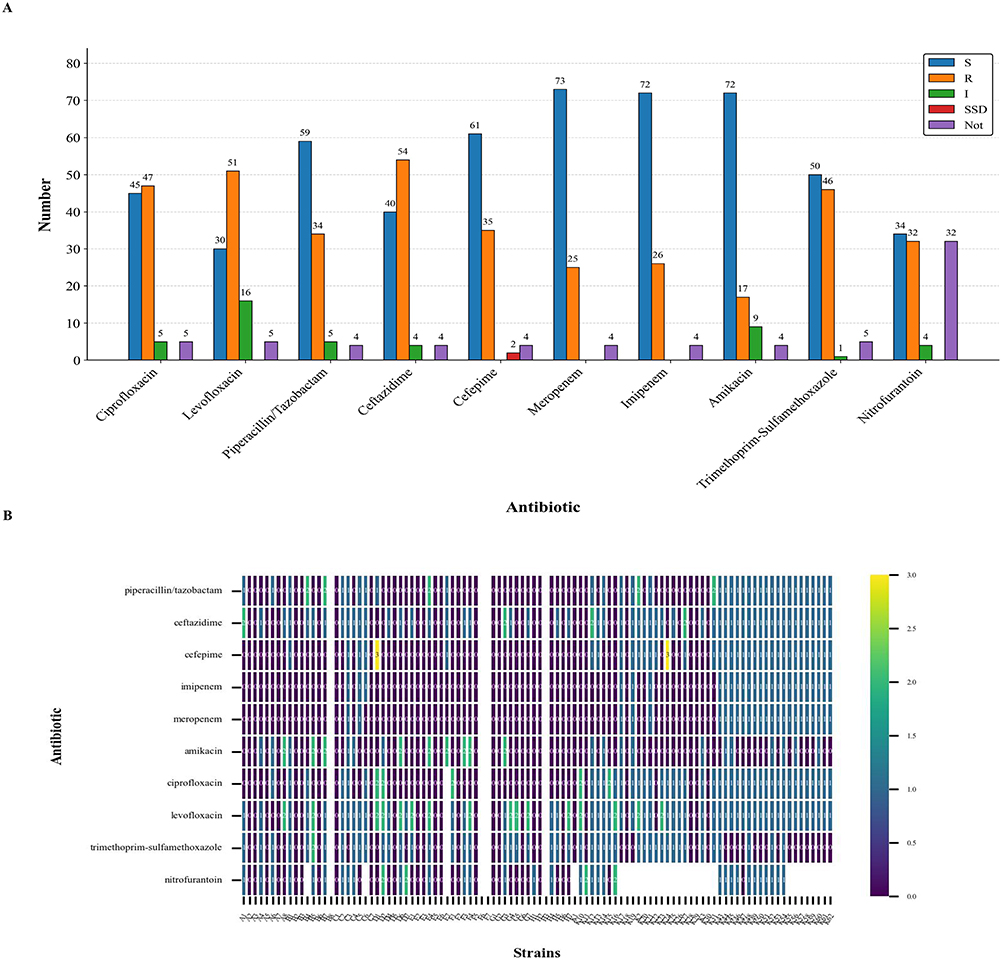 |
Figure 1 Results of Antimicrobial Susceptibility Testing. (A) Bar chart illustrating the antimicrobial susceptibility and quantity distribution of KP to different antimicrobial agents. S indicates that the bacterium is susceptible to the antimicrobial agent, R denotes that the bacterium’s susceptibility to the drug is between susceptible and resistant, I represents that the bacterium is resistant to the antimicrobial agent, and SDD means that the bacterium’s susceptibility is dose-dependent. (B) Heatmap illustrating the antimicrobial susceptibility profiles of KP to various antimicrobial agents. A value of 0 represents a susceptibility result of S, 1 denotes R, 2 indicates I, and 3 corresponds to SDD. |
Strain Typing and Phylogenetic Analysis
Whole-genome sequencing and assembly were performed for all strains. MLST typing results of the strains were obtained through Kleborate analysis. The typing results showed that a total of 41 sequence types (STs) were identified, with ST11 being significantly more prevalent than other types. Whole-genome phylogenetic analysis revealed that most ST11 strains formed a monophyletic clade with the global clone of CG258 KP, suggesting that the local ST11 strains may have originated from this globally prevalent clone (Figure 2).
 |
Figure 2 Phylogenetic tree of KP: Constructed based on the genomic sequences of 178 KP strains (local isolates and global clones provided by the Pasteur Institute). The inner circle represents different geographical regions worldwide, while the outer circle corresponds to the various clinical departments from which the hospital collected the strains. |
Virulence and Resistance Characteristics
Based on the virulence and drug resistance scores of KP using Kleborate, 33 strains were classified as hypervirulent strains (virulence score > 3). Most strains were low-drug-resistant strains, but there were still 27 strains with a drug resistance score ≥ 2, which were classified as highly drug-resistant strains (Figure 3A). Among them, 19 strains were hypervirulent and highly drug-resistant strains (Figure 3B).
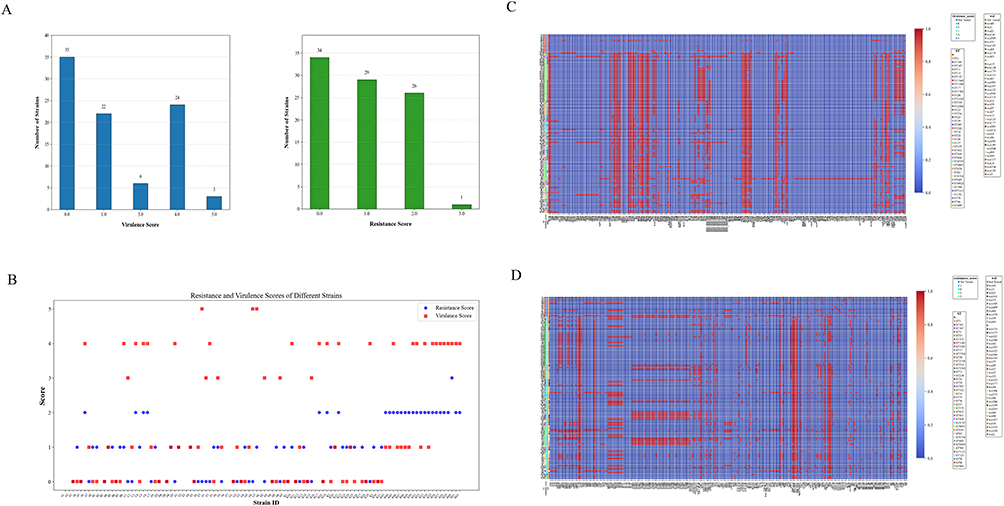 |
Figure 3 Virulence and drug resistance scores of KP. (A) Statistical count of bacterial strains with different virulence/drug resistance scores. (B) Statistical analysis of virulence/drug resistance scores for each bacterial strain. Analysis of correlations between strains and virulence/resistance genes. (C) Strains and virulence factors. (D) Strains and resistance genes. |
Correlation analysis between KP and virulence factors revealed that genes of the fim and mrk families (eg, adhesion and invasion genes such as fimA, fimD, mrkA, and mrkC) and genes of the ent family (eg, siderophore synthesis genes including entB, entC, and entD) were ubiquitous in the vast majority of strains. This suggests that these genes may constitute the core virulence modules contributing to the pathogenicity of KP.Genes of the chu family (eg, heme utilization genes such as chuA, chuS, and chuT) and genes of the csg family (eg, csgA, csgB) were prevalent in hypervirulent strains (virulence score ≥ 3), enhancing the environmental adaptability of the strains. Eighty percent of ST11 strains had a virulence score of 4, classifying them as hypervirulent strains. Strains G7, E7, and G6, which had a high virulence score of 5, not only harbored the aforementioned characteristic genes but also carried genes of the all operon (eg, allA, allB, allC) and genes of the clb operon (eg, clbA, clbB, clbC), which further enhanced the colonization and invasion capabilities of the strains (Figure 3C).
Correlation analysis between KP strains and drug resistance genes revealed that the strain samples could be roughly divided into 3 clusters. Among these, the ST11 sequence type carried the most abundant drug resistance genes, with 21 strains achieving a drug resistance score of 2. All ST11 strains harbored β-lactam resistance genes (CTX-M family, SHV family) and aminoglycoside resistance genes (ArnT). Additionally, 91% of ST11 strains carried carbapenem resistance genes (KPC-2), exhibiting an overall pattern of significantly high drug resistance (Figure 3D). Specifically, among a total of 24 ST11 isolates tested, 22 were confirmed to harbor the KPC-2 gene. Statistical analysis via Fisher’s exact test further verified that the prevalence of KPC-2 in the ST11 clone reached a statistically significant level (P = 1.646793e-15, P < 0.001), which corroborates the strikingly high carbapenem resistance rate of this sequence type.
Metabolic Trait
Biosynthetic gene clusters (BGCs) associated with the biosynthesis of redox-cofactors, azole-containing RiPPs (ribosomally synthesized and post-translationally modified peptides), terpene precursors, NRP-metallophores (non-ribosomal peptide siderophores), T1PKS (type I polyketide synthases), RiPP-like compounds and NI-siderophores (non-ribosomal peptide synthetase-independent siderophores) were identified in the bacterial genomes (Figure 4). The presence of these BGCs likely indicates the production of different types of secondary metabolites, which may enhance the environmental adaptability of the strain.
 |
Figure 4 Prediction of biosynthetic gene clusters (BGCs) in the KP genome. (a) BGCs associated with redox processes, (b and c) NRP-metallophores, (c) T1PKS, (d) azole-containing RiPPs, (e) terpene precursors, (f and h) RiPP-like, (g) NI-siderophores were annotated in the assembled genome of KP. |
Plasmid Characteristics
According to the alignment results, the plasmids with accession numbers NZ_CP186632.1 (36/102) and NZ_CP159675.1 (23/102) were the most abundant (Figure 5A). Querying the NCBI database revealed that the plasmid NZ_CP186632.1 is named pP37UNK1, while NZ_CP159675.1 remains unnamed. The plasmids of strains K23 and G6 showed the highest similarity to pP37UNK1 and NZ_CP159675.1, with identity rates of 99.31% and 99.52%, respectively. Therefore, the plasmids of these two strains (K23 and G6) were selected for subsequent analysis. The plasmid of strain K23 was designated as pKpK23_11, and that of strain G6 was designated as pKpG6_14. Plasmid pKpK23_11 has a full length of 145,397 bp, and its circular genome map indicates that the plasmid replicon carries genes such as fosA, mdtM, arcA, trpR, and osmY. Among these, fosA is a fosfomycin resistance gene. Plasmid pKpG6_14 is 127,475 bp in length; its circular genome map shows that it does not harbor any resistance genes but contains multiple functional genes, including MinC, MinD, MinE, IS150, and DprB (Figure 5B).
 |
Figure 5 Plasmid analysis results. (A) Number of different plasmids carried by bacterial strains. (B) Circular maps of plasmids pKpK23_11 and pKpG6_14. Black, green, and purple represent GC content, positive GC skew (GC skew +), and negative GC skew (GC skew -), respectively. Key functional genes are labeled in pink. RNA and tRNA are labeled in fluorescent green and blue, respectively. |
Discussion
Our KP isolates were mostly derived from the Urology ward (35/102), followed by the Intensive Care Medicine Ward (15/102) and Emergency ward (11/102), with sporadic detection in other departments. This skewed distribution is likely due to the high incidence of KP-associated urinary tract infections in the Urology ward and the frequent occurrence of mixed urinary tract and other infections in critically ill and emergency patients, who are more frequently subjected to clinical sampling for pathogen detection.
According to previous studies, ST11 is the predominant clone of KP in China and East Asia. In the present study, we found that the evolutionary characteristics of the collected clinical KP strains were consistent with those reported in previous research.17,23,24 Most strains were closely related to the CG258 clone, suggesting that adaptive transmission may have occurred, which is consistent with the previously reported global dissemination of KP clones.
Most of the strains collected in this study carried multiple virulence-associated genes, suggesting that KP possesses high pathogenic potential. Among these, iron-acquisition-related virulence factors such as ybtS, irp1, and fyuA enable the bacterium to obtain iron—an essential element for its growth—from the host environment. In-depth elucidation of the mechanisms underlying these virulence factors will provide a crucial basis for formulating targeted anti-infective strategies against KP.
Due to the inappropriate use of antibacterial agents, the problem of bacterial drug resistance has become increasingly prominent. According to the results of drug susceptibility testing, the resistance rates to ceftazidime and levofloxacin both exceed 50%. As a third-generation cephalosporin, ceftazidime is a commonly used drug for treating KP infections in community and hospital settings. Its high resistance rate suggests that such drugs should be used cautiously in clinical treatment in the future. In drug resistance gene detection, most of the collected clinical isolates of KP were found to harbor acrA, KpnE, KpnF, and KpnG, all of which belong to efflux-mediated antibiotic resistance genes.25 In addition, CTX-M-65, aadA2, aadA17, aadA25, QnrB14, QnrB15, and QnrB16 were also detected. CTX-M-65 is a β-lactamase gene, but its carriage rate is only 23%, which is inconsistent with the high resistance rate to ceftazidime in the drug susceptibility test. This may be due to the efflux of drugs by efflux pumps leading to bacterial resistance to ceftazidime. The aadA family genes are aminoglycoside resistance genes, and the enzymes encoded by them can modify aminoglycoside antibiotics, rendering them inactive and thereby mediating bacterial resistance to aminoglycoside drugs. Importantly, the aforementioned resistance genes are highly enriched in ST11 clone strains. The Qnr family genes are quinolone resistance genes, with only 15% of the strains carrying such genes. However, most strains are resistant to levofloxacin, which indicates that their resistance is not plasmid-mediated and may be caused by mutations in the quinolone resistance-determining regions (QRDR) of the gyrA and parC genes.26
The identification of BGCs in KP highlights its capacity to produce diverse secondary metabolites, and we identified redox cofactors, azole-containing RiPPs, terpene precursors, NRP-metallophores, T1PKSs, RiPP-like compounds and NI-siderophores. Redox cofactors are crucial molecules that maintain cellular redox homeostasis, facilitate intracellular catabolic and anabolic reactions, and serve as core substances for bacteria to sustain life activities.27 RiPP biosynthetic gene clusters (BGCs) typically include genes encoding precursor peptides and modification enzymes, and in some cases leader peptidases and transporters, exhibiting complex structures and antibacterial activity that enhance bacterial competitive advantages, thus creating conditions for KP to establish invasive infections.28 Terpenes produced by terpene precursor clusters display broad biological activities, including antibacterial and immunomodulatory functions.29 The immunomodulatory effects of terpenes can interfere with the secretion of host cytokines, which may not only alleviate tissue damage by inhibiting the release of excessive inflammatory factors, but also reduce the clearance efficiency of immune cells against KP under specific conditions. In addition, their antibacterial activity impairs the survival competitiveness of other bacteria by disrupting their cell membrane and inhibiting biofilm formation, there by enhancing the colonization and survival advantages of KP. Studies have shown that some terpenes can exert a synergistic effect with existing antibacterial drugs, which provides a novel research direction for alleviating antimicrobial resistance (AMR).30 Siderophore biosynthesis proceeds via two pathways: the nonribosomal peptide synthetase (NRPS) pathway and the NRPS-independent siderophore (NIS) synthase pathway. Both NRP-based and NIS-based siderophores facilitate bacterial acquisition of metal ions under metal-limited conditions, thus enabling bacteria to gain a competitive edge in specific environments. The production of siderophores may interfere with the biofilm formation process of KP and may also disrupt antibiotic activity by modulating oxidative stress mechanisms. Clinically, inhibitors targeting this biosynthetic pathway are expected to serve as promising targeted therapeutic strategies against multidrug-resistant KP infections.31,32 Type I polyketide synthases (T1PKSs) primarily function to assemble polyketides, which are pivotal for bacterial synthesis of a diverse array of important bioactive substances and can enhance bacterial viability, competitiveness, and environmental adaptability. Polyketide toxins produced by some polyketides can directly disrupt the cell membranes and organelles of host cells, trigger inflammatory responses, and thus lay the foundation for bacterial invasion and colonization. In addition, certain polyketides serve as key components of the bacterial biofilm matrix, which can reduce the permeability of antibacterial agents, thereby increasing bacterial drug resistance and the risk of persistent infections.33 The presence of these secondary metabolites indicates that KP possesses robust environmental adaptability and survival competitiveness. Its intricate metabolic characteristics affect the pathogenic mechanism to a certain extent, while also providing novel insights for clinical treatment. Further metabolomic analyses are required to clarify the specific functions of these biosynthetic gene clusters, thereby laying a scientific foundation for precise targeted therapy and the development of new antibacterial agents.34
Analysis of the plasmid map of pKpK23_11 revealed that the carried fosA gene is critical for mediating fosfomycin resistance. The protein encoded by this gene can modify the fosfomycin molecule, thereby inactivating it and limiting clinical therapeutic options. Additionally, the various functional genes carried by this plasmid provide a molecular basis for the horizontal transfer of the fosA gene, which may facilitate the spread of fosfomycin-resistant phenotypes among bacteria, thus leading to widespread fosfomycin resistance in local bacterial strains. The plasmid map of pKpG6_14 shows that although it does not carry drug resistance genes, it harbors the IS150 element. IS elements can mediate horizontal transfer between plasmids and bacterial chromosomes via their own transposases. Specifically, IS150 can insert into the mgrB gene on the bacterial chromosome, disrupting the gene’s integrity and leading to its inactivation. This inactivation relieves the negative regulatory effect on the PhoPQ signaling pathway. With the continuous activation of the PhoPQ pathway, the autophosphorylation level of PhoQ increases, which in turn upregulates the arnBCADTEF operon. The arnBCADTEF operon then catalyzes the addition of L-4-aminoarabinose (L-Ara4N) to lipid A, resulting in a reduction in the negative charge of L-Ara4N-modified lipid A. Since lipid A is not only the core component of bacterial lipopolysaccharide (LPS) but also the primary target of polymyxins, the antibacterial mechanism of polymyxins relies predominantly on the binding of their positively charged molecular moieties to the negatively charged lipid A on the bacterial outer membrane, which disrupts the outer membrane structure and increases its permeability. Therefore, the reduced negative charge of lipid A impairs the binding affinity between LPS and polymyxins, ultimately conferring polymyxin resistance on the bacterial strain.35–37 This cryptic resistance mechanism that is independent of resistance genes tends to result in false susceptibility in conventional resistance gene assays, thereby contributing to clinical treatment failure.
This study provides an effective analysis of the molecular epidemiology and pathogenic mechanisms of KP in UTIs. However, due to the small sample size and regional limitations, the results only have local representativeness. In subsequent studies, the research scale should be expanded to obtain more comprehensive epidemiological data. Additionally, it is necessary to continuously monitor changes in the drug resistance profiles of clinically isolated strains, standardize the use of antibiotics, and block the transmission chain of drug-resistant bacteria through clinical practice.
Conclusion
In this study, whole-genome sequencing and bioinformatics analyses were performed on clinically isolated KP strains, clarifying their molecular epidemiological characteristics and pathogenic patterns. The results showed that ST11 was the dominant epidemic clone in this hospital (accounting for 24%), with a high enrichment in the Intensive Care Unit and Urology Ward. This suggests that these departments are key areas for nosocomial transmission. Phylogenetic analysis indicated that most local strains are closely related to the globally prevalent CG258 clonal group, confirming the adaptive transmission trend of this clonal group in the region.
The majority of strains displayed high-level resistance to the antimicrobial agents ceftazidime and levofloxacin. The ST11 clone also exhibited a dual phenotype of hypervirulence combined with multidrug resistance. Its genome was enriched in siderophore biosynthesis genes (ybtS, irp1, irp2) and multiple antimicrobial resistance determinants (CTX-M65, fosA, aadA2). These genes directly underpin the enhanced pathogenic potential of the strains and their reduced susceptibility to antimicrobial agents. Furthermore, the biosynthetic gene clusters of secondary metabolites carried by the strains, as well as the resistance genes harbored on plasmids, further enhance the environmental adaptability of the strains and facilitate the occurrence of horizontal gene transfer events. This study provides a scientific basis for formulating targeted prevention and control strategies, emphasizing the need to strengthen the surveillance of dominant sequence types and promote the rational use of antimicrobial agents to curb the transmission of this opportunistic pathogen.
Ethics Statement
This study was approved by the Medical Ethics Committee of Chinese PLA General Hospital (No. KY2024-018). All patients or their legal guardians have signed the informed consent form.
Funding
The study was supported by Beijing Natural Science Foundation (No. M23002), the National Key Research and Development Project (No. 2024YFC2311403).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rahmat Ullah S, Jamal M, Rahman A, Andleeb S. Comprehensive insights into Klebsiella pneumoniae: unravelling clinical impact, epidemiological trends and antibiotic-resistance challenges. J Antimicrob Chemother. 2024;79(7):1484–14. doi:10.1093/jac/dkae184
2. Arato V, Raso MM, Gasperini G, et al. Prophylaxis and treatment against Klebsiella pneumoniae: current insights on this emerging anti-microbial resistant global threat[J]. Int J Mol Sci. 2021;22(8):4042. doi:10.3390/ijms22084042
3. Lee CR, Lee JH, Park KS, et al. Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front Cell Infect Microbiol. 2017;7:483. doi:10.3389/fcimb.2017.00483
4. Yang X, Sun Q, Li J, et al. Molecular epidemiology of carbapenem-resistant hypervirulent Klebsiella pneumoniae in China. Emerg Microbes Infect. 2022;11(1):841–849. doi:10.1080/22221751.2022.2049458
5. Jesudason T. WHO publishes updated list of bacterial priority pathogens. Lancet Microbe. 2024;5(9):100940. doi:10.1016/j.lanmic.2024.07.003
6. Liu Y, Dai J, Liu A, et al. Klebsiella pneumoniae pneumonia in patients with rheumatic autoimmune diseases: clinical characteristics, antimicrobial resistance and factors associated with extended-spectrum β-lactamase production. BMC Infect Dis. 2021;21(1):366. doi:10.1186/s12879-021-06055-1
7. Wang M, Earley M, Chen L, et al. Clinical outcomes and bacterial characteristics of carbapenem-resistant Klebsiella pneumoniae complex among patients from different global regions(CRACKLE-2): a prospective, multicentre, cohort study[J]. Lancet Infect Dis. 2022;22(3):401–412. doi:10.1016/S1473-3099(21)00399-6
8. Bischoff S, Walter T, Gerigk M, Ebert M, Vogelmann R. Empiric antibiotic therapy in urinary tract infection in patients with risk factors for antibiotic resistance in a German emergency department. BMC Infect Dis. 2018;18(1):56. doi:10.1186/s12879-018-2960-9
9. Stamm WE, Norrby SR. Urinary tract infections: disease panorama and challenges. J Infect Dis. 2001;183(Suppl 1):S1–4. doi:10.1086/318850
10. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13(5):269–284. doi:10.1038/nrmicro3432
11. Yang X, Dong N, Chan EW, et al. Carbapenem resistance-encoding and virulence-encoding conjugative plasmids in Klebsiella pneumoniae[J]. Trends Microbiol. 2021;29(1):65–83. doi:10.1016/j.tim.2020.04.012
12. Lee YJ, Huang CH, Ilsan NA, et al. Molecular epidemiology and characterization of carbapenem-resistant Klebsiella pneumoniae isolated from urine at a teaching hospital in Taiwan. Microorganisms. 2021;9(2):271. doi:10.3390/microorganisms9020271
13. Durdu B, Hakyemez IN, Bolukcu S, et al. Mortality markers in nosocomial Klebsiella pneumoniae bloodstream infection. Springerplus. 2016;5(1):1892. doi:10.1186/s40064-016-3580-8
14. Li D, Huang X, Rao H, et al. Klebsiella pneumoniae bacteremia mortality: a systematic review and meta-analysis. Front Cell Infect Microbiol. 2023;13:1157010. doi:10.3389/fcimb.2023.1157010
15. Shang H, Wang YS, Shen ZY. National Guide to Clinical Laboratory Procedures[M].
16. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing. CLSI M100-2022. Wayne, PA: Clinical and Laboratory Standards Institute; 2022.
17. Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. 2020;18(6):344–359. doi:10.1038/s41579-019-0315-1
18. Gardner SN, Slezak T, Hall BG. kSNP3. 0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics. 2015;31(17):2877–2878. doi:10.1093/bioinformatics/btv271
19. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296. doi:10.1093/nar/gkab301
20. Lam MMC, Wick RR, Watts SC, Cerdeira LT, Wyres KL, Holt KE. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat Commun. 2021;12(1):4188. doi:10.1038/s41467-021-24448-3
21. Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):75. doi:10.1186/1471-2164-9-75
22. Stothard P, Grant JR, Van Domselaar G. Visualizing and comparing circular genomes using the CGView family of tools. Brief Bioinform. 2019;20(4):1576–1582. doi:10.1093/bib/bbx081
23. Wang Q, Wang R, Wang S, et al. China Carbapenem-Resistant Enterobacterales (CRE) network. Expansion and transmission dynamics of high risk carbapenem-resistant Klebsiella pneumoniae subclones in China: an epidemiological, spatial, genomic analysis. Drug Resist Updat. 2024;74:101083. doi:10.1016/j.drup.2024.101083
24. Liao W, Liu Y, Zhang W. Virulence evolution, molecular mechanisms of resistance and prevalence of ST11 carbapenem-resistant Klebsiella pneumoniae in China: a review over the last 10 years. J Glob Antimicrob Resist. 2020;23:174–180. doi:10.1016/j.jgar.2020.09.004
25. Krishnan SG, Sajeev S, Sivam V, et al. Identification of serotype O3b and high-risk clone ST37 of Klebsiella pneumoniae revealed by comparative genomic analysis. Front Cell Infect Microbiol. 2025;14:1517125. doi:10.3389/fcimb.2024.1517125
26. Rezaei S, Tajbakhsh S, Naeimi B, Yousefi F. Investigation of gyrA and parC mutations and the prevalence of plasmid-mediated quinolone resistance genes in Klebsiella pneumoniae clinical isolates. BMC Microbiol. 2024;24(1):265. doi:10.1186/s12866-024-03383-5
27. Baiyun W, Xiaoyue W, Zhiwen W, Chen T, Xueming Z. Redox cofactor metabolic engineering with Escherichia coli. Prog Chem. 2014;26(09):1609–1618.
28. Ayikpoe RS, Shi C, Battiste AJ, et al. A scalable platform to discover antimicrobials of ribosomal origin. Nat Commun. 2022;13(1):6135. doi:10.1038/s41467-022-33890-w
29. Wei X, Ning W, McCadden CA, et al. Exploring and expanding the natural chemical space of bacterial diterpenes. Nat Commun. 2025;16(1):3721. doi:10.1038/s41467-025-57145-6
30. Mahizan NA, Yang SK, Moo CL, et al. Terpene derivatives as a potential agent against Antimicrobial Resistance (AMR) pathogens. Molecules. 2019;24(14):2631. doi:10.3390/molecules24142631
31. Gulick AM, Mydy LS, Patel KD. Kinetic analysis of the three-substrate reaction mechanism of an NRPS-independent siderophore (NIS) synthetase. Methods Enzymol. 2024;702:1–19. doi:10.1016/bs.mie.2024.06.012
32. Carroll CS, Moore MM. Ironing out siderophore biosynthesis: a review of non-ribosomal peptide synthetase (NRPS)-independent siderophore synthetases. Crit Rev Biochem Mol Biol. 2018;53(4):356–381. doi:10.1080/10409238.2018.1476449
33. Zhang R, Jin W, Chen Y. Bacterial inter-PKS hybrids and the biosynthetic algorithm of polyketides[J]. Synthetic Biol J. 2024;5(3):548–560.
34. Shah T, Li J, Shah Z, Lu Y, Qin W. Comprehensive genome analysis of a human-derived β-lactam-resistant Klebsiella variicola isolate from China. Infect Drug Resist. 2025;18:5223–5238. doi:10.2147/IDR.S544005
35. Lippa AM, Goulian M. Feedback inhibition in the PhoQ/PhoP signaling system by a membrane peptide. PLoS Genet. 2009;5(12):e1000788. doi:10.1371/journal.pgen.1000788
36. Liu X, Wu Y, Zhu Y, et al. Emergence of Colistin-Resistant Hypervirulent Klebsiella pneumoniae (CoR-HvKp) in China. Emerging Microbes Infect. 2022;11:648–661. doi:10.1080/22221751.2022.2036078
37. Cannatelli A, D’Andrea MM, Giani T, et al. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob Agents Chemother. 2013;57(11):5521–5526. doi:10.1128/AAC.01480-13
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The First Report of Escherichia coli and Klebsiella pneumoniae Strains That Produce Both NDM-5 and OXA-181 in Jiangsu Province, China
Tao G, Tan H, Chen Q
Infection and Drug Resistance 2023, 16:3245-3255
Published Date: 24 May 2023