Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Parkinson’s disease managing reversible neurodegeneration
Authors Hinz M, Stein A, Cole T, McDougall B, Westaway M
Received 15 October 2015
Accepted for publication 8 December 2015
Published 5 April 2016 Volume 2016:12 Pages 763—775
DOI https://doi.org/10.2147/NDT.S98367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
This paper has been retracted.
Marty Hinz, 1 Alvin Stein, 2 Ted Cole, 3 Beth McDougall, 4 Mark Westaway 5
1Clinical Research, NeuroResearch Clinics, Inc., Cape Coral, FL, 2Stein Orthopedic Associates, Plantation, FL, 3Cole Center for Healing, Cincinnati, OH, 4CLEARCenter of Health, Mill Valley, CA, USA; 5Four Pillars Health, Brendale, QLD, Australia
Abstract: Traditionally, the Parkinson’s disease (PD) symptom course has been classified as an irreversible progressive neurodegenerative disease. This paper documents 29 PD and treatment-induced systemic depletion etiologies which cause and/or exacerbate the seven novel primary relative nutritional deficiencies associated with PD. These reversible relative nutritional deficiencies (RNDs) may facilitate and accelerate irreversible progressive neurodegeneration, while other reversible RNDs may induce previously undocumented reversible pseudo-neurodegeneration that is hiding in plain sight since the symptoms are identical to the symptoms being experienced by the PD patient. Documented herein is a novel nutritional approach for reversible processes management which may slow or halt irreversible progressive neurodegenerative disease and correct reversible RNDs whose symptoms are identical to the patient’s PD symptoms.
Keywords: Parkinson’s disease, L-dopa, carbidopa, B6, neurodegeneration
Corrigendum for this paper has been published
Expression of Concern for this paper has been published
Introduction
This study does not document a new Parkinson’s disease (PD) treatment, but discusses effective and novel side effect management associated with the most effective PD treatment known: L-dopa (L-3,4-dihydroxyphenylalanine). The following approach definitively addresses PD, L-dopa, and carbidopa-associated side effects and adverse reactions which interfere with achieving optimal L-dopa results.
PD is classified as a “progressive neurodegeneration” (PN) disease.1,2 With PD, irreversible brain damage involving the post-synaptic substantia nigra dopamine neurons3 induces fine motor control dysfunction4 (herein referred to as “electrical damage”).
A relative nutritional deficiency (RND) occurs when an optimal diet does not meet nutritional requirements.5–11 This paper demonstrates how the reversible PD symptoms induced by newly identified RNDs have been allowed to accumulate because of the disease process or traditional treatment. These symptoms have traditionally been exclusively attributed to irreversible PN. This novel approach breaks PN down into three subcategories: irreversible PN, reversible facilitated PN (FPN), and reversible pseudo-neurodegeneration (RPN).
PD electrical damage dysfunction is classically limited to the post-synaptic dopamine neurons.12,13 Electrical flow, regulating fine motor control, flows from the pre-synaptic neurons, across the synapse, then through the post-synaptic neurons. It is the novel primary hypothesis that if PD symptoms from post-synaptic neuron damage develop, then compromise at any point in the electrical event chain, to include outside the post-synaptic dopamine neuron focus, may exacerbate and mimic PD symptoms and/or disease progression.
In PD patients, reversible RNDs may induce PD symptoms which may be inappropriately managed when treated as PN.9–11 Figure 1 illustrates 29 primary system depletions associated with PD and its treatment. Each depletion induces and represents a significant underlying RND which contributes to PD symptom exacerbation and/or overall irreversible disease collapse. The novel hypothesis is that if a dysfunction resulting from nutrient depletion exists, then one or more underlying RNDs is always present.
 | Figure 1 The 29 primary RND-inducing depletions resulting from PD, L-dopa, and carbidopa. |
With regard to Figure 1, the 29 PD and treatment associated depletions are listed immediately below. The RNDs associated with each are discussed in the following sections prior to the “Materials and methods” section.
- PD-associated dopamine depletion.12,13
- PD-associated norepinephrine depletion.14,15
- PD-associated epinephrine depletion.15
- PD-associated glutathione depletion prior to symptom onset.16–18
- PD-associated S-adenosylmethionine depletion.19–21
- PD-associated L-cysteine depletion.22
- PD is associated with glutathione depletion.16–18 Glutathione depletion may induce L-methionine depletion.23
- PD-associated vitamin B6 deficiency.24
- PD-associated serotonin depletion.25,26
- PD associated with 5-hydroxytryptophan depletion.27
- PD-associated tyrosine depletion.28
- PD-associated L-dopa depletion.29–39
- PD associated with L-tryptophan depletion.31,32
- L-dopa-associated L-tyrosine depletion.33
- L-dopa-associated L-tryptophan depletion.33
- L-dopa-associated S-adenosylmethionine depletion.34–36
- L-dopa-associated L-methionine depletion.36
- L-dopa-associated L-cysteine depletion.37
- L-dopa-associated glutathione depletion.37
- L-dopa-associated serotonin depletion.33,34,38–40
- Carbidopa-associated vitamin B6 depletion.10,11,41,42
- Carbidopa associated serotonin depletion.43,44
- Carbidopa-associated dopamine depletion.43
- Carbidopa-associated norepinephrine depletion.41
- Carbidopa-associated epinephrine depletion.41
- Carbidopa-associated vitamin B6 depletion.10,11,41,42 Vitamin B6 depletion may deplete glutathione.45,46
- Carbidopa-associated vitamin B6 depletion.10,11,41,42 Vitamin B6 depletion may deplete glutathione.45,46 Glutathione depletion may deplete S-adenosylmethionine.47
- Carbidopa-associated vitamin B6 depletion.10,11,41,42 Vitamin B6 depletion may deplete glutathione.45,46 Glutathione depletion may deplete L-methinonine.48
- Carbidopa-associated vitamin B6 depletion.10,11,41,42 Vitamin B6 depletion may deplete glutathione.45,46 Glutathione depletion may deplete L-cysteine.48
As illustrated in Figure 1, patients with PD suffer from one or more RNDs involving L-cysteine and methionine, vitamin B6, serotonin precursors (L-tryptophan, 5-HTP), and dopamine precursors (L-tyrosine, L-dopa).
Drug-nutrient perspective
The following definitions are required to understand this approach. L-dopa and 5-hydroxytryptophan (5-HTP) are defined as dopamine and serotonin nutrient amino acid precursors, respectively.
The following documented drug-nutrient reference point is required for optimal RND management:
A nutrient is any substance that facilitates normal system function. A drug is any substance that induces abnormal system function. A nutrient may become a drug. A drug may not become a nutrient. When the nutrient 5-HTP is administered as a single agent, dopamine depletion may occur. If dopamine depletion is induced, 5-HTP is no longer functioning as a nutrient; it is a drug. When L-dopa is administered as a single agent, it may deplete serotonin, and would then be considered a drug, not a nutrient.11
The following novel hypothesis is required to address the 29 PD RNDs which cause reversible FPN and RPN. If when administered properly drugs have side effects and nutrients have no side effects, then nutrients that display side effects are functioning as drugs.
Under the previous definitions, drugs are substances not normally found in the body which induce abnormal effects. If these abnormal effects are desirable, then the drug is administered. Undesirable effects are labeled side effects. When undesirable effects outweigh desirable ones, drugs may be discontinued. Normal body functions require nutrients. Properly administered L-dopa and other nutrients facilitate normal function without side effects. If L-dopa or 5-HTP administration induces side effects, then it is nutrient to drug conversion evidence.
Drugs cannot treat RNDs. In the USA L-dopa is concomitantly packaged with carbidopa as a drug and administered with side effect expectations.47 In the USA there is no single ingredient prescription L-dopa form without carbidopa available. When L-dopa is indicated while intolerable carbidopa side effects develop the non-prescription nutritionally sourced L-dopa found in Mucuna pruriens (synonym for Mucuna Cochinchinensis) is the only option.10,11 Ayurvedic medicine has prescribed M. pruriens for 3,500 years.48
Competitive inhibition
Competitive inhibition is the interaction between serotonin and dopamine along with their precursors in synthesis, transport, and metabolism. In the endogenous state, where no or insufficient serotonin or dopamine precursors are ingested, competitive inhibition does not exist.5–11,49–56
Synthesis
Aromatic-L-amino acid decarboxylase (AADC) metabolizes L-dopa to dopamine, 5-HTP to serotonin, histidine to histamine, and phenylalanine to phenylethylamine. Competitive inhibition may exist between the four precursors for metabolism by AADC. Administering L-dopa without balanced serotonin precursor concentrations may decrease AADC serotonin synthesis. This causes a L-dopa-induced serotonin precursor RND, Figure 2.5–11,49–56
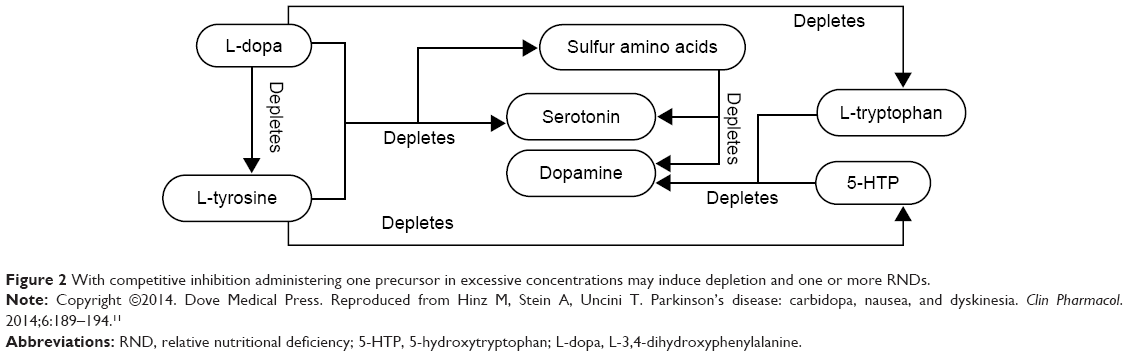 | Figure 2 With competitive inhibition administering one precursor in excessive concentrations may induce depletion and one or more RNDs. |
Transport
Organic Cation Transporters (OCT) transport the centrally acting monoamines (serotonin, dopamine, norepinephrine, and epinephrine) and their precursors bidirectionally across cell membranes. OCT transports precursors into the cellular structures where AADC metabolism occurs. Newly synthesized centrally acting monoamines are transported extracellularly by OCT to effect functional regulation. There is a direct correlation between L-dopa administration and dopamine concentrations. Increasing only L-dopa and dopamine concentrations induces competitive inhibition at the transporter, an event that may exclude serotonin precursor transport leading to synthesis inhibition. Subsequent serotonin depletion represents a serotonin precursor RND.5–11,49–56
Metabolism
The monoamine oxidase-A catalyzes centrally acting monoamine metabolism. Increasing dopamine concentrations with L-dopa enhances monoamine oxidase-A activity which may deplete serotonin, inducing a serotonin precursor RND.5–11,49–56
Reversible PD phenomenon
The PD clinical course is typically described as irreversible PN. The following is based on 18 years of data collection from several hundred medical clinics. This data was used to define reversible RNDs whose symptoms are identical to PD symptoms and/or facilitate irreversible PN.
The problems
This approach rests on the hypothesis that dysfunction resulting from inadequate nutrient concentrations, even when an optimal diet is consumed, results in a nutritional precursor RND. The RNDs associated with PD and its precursors include:
- “Total glutathione loss” which is known to occur before PD symptoms develop.16,57 This represents a novel glutathione precursor RND involving L-cysteine and/or L-methionine,57 the normal dietary glutathione precursor source.
- PD-induced serotonin, dopamine, norepinephrine, and epinephrine depletion represents novel serotonin and dopamine precursor RNDs.5
- L-dopa-induced competitive inhibition depletes serotonin with an associated novel serotonin precursor RND.5
- Melanin steal, which has been documented and discussed in the “Other impacts” section, induces dopamine fluctuations that represent an L-dopa and L-tyrosine RND.51
- L-dopa induces a glutathione precursor RND.36
The most potentially devastating RND is caused by carbidopa which irreversibly binds to and then deactivates vitamin B6 (B6) and all B6-dependent enzymes causing a drug-induced B6 RND that may compromise function involving over 300 enzymes and proteins. When the B6-dependent enzyme AADC collapses, this may compromise metabolism of L-dopa to dopamine, 5-HTP to serotonin, histidine to histamine, and phenylalanine to phenylethylamine. Carbidopa-induced B6 depletion may compromise the two B6-dependent enzymes (histadine decarboxylase and AADC) which metabolize histadine to histamine inducing a profound antihistamine effect. Carbidopa-induced B6 depletion can deplete glutathione. The B6-dependent enzymes cystathione-beta-synthetase and cystathione-gamma-lyase metabolize homocysteine to L-cysteine. L-cysteine is the rate limiting step in glutathione synthesis.10,11,43,58
The primary PD RND
The primary PD RND involves the inability to obtain adequate dopamine precursors from the diet. Under normal conditions dietary L-tyrosine and L-dopa metabolism meet the synaptic dopamine requirements for optimal electrical transfer across the synapse which facilitates optimal fine motor control regulation. When PD symptoms are present, an optimal diet cannot facilitate dopamine synthesis at the levels required for adequate increase in post-synaptic electrical flow.5
Increased L-dopa intake may facilitate synaptic dopamine levels which enhance post-synaptic electricity flow. The subsequent decrease in PD symptoms is due to the dopamine-induced improvement in fine motor control regulation. Increasing synaptic dopamine levels is analogous to increasing the post-synaptic voltage which regulates fine motor control.10
An exception to the assumed “more is better concept” was documented in 2014. In the competitive inhibition state, administering too much or too little L-dopa can display exactly the same PD symptoms with identical intensity. The previously documented pill stop technique is required to determine optimal dosing.9
AADC freely metabolizes L-dopa to dopamine without biochemical feedback regulation. With adequate AADC, ingesting unlimited L-dopa will yield unlimited dopamine. Under the traditional PD treatment medical care standard, the L-dopa daily dosing value limiting factor is usually L-dopa-induced side effects and/or tachyphylaxis. Typically, less effective drugs, such as dopamine agonists, are prescribed first to delay dealing with inevitable and escalating L-dopa side effects. This is a practice that ignores the primary PD RND, inadequate dopamine precursor intake, which is a reversible dietary deficiency. As discussed in the following section, this RND has RPN symptoms that are identical to irreversible PN.5,49
Facilitated irreversible progressive neurodegeneration
It is postulated that the primary PD etiology is lipophilic neurotoxin-induced post-synaptic dopamine neuron damage.56,59–66 The body’s most powerful and abundant protection against lipophilic neurotoxins is glutathione.67,68 Total glutathione loss prior to PD symptom onset is documented.16,57,68 The hypothesis is, if glutathione depletion facilitates and potentiates lipophilic neurotoxin damage, then the first step in halting or slowing the irreversible PN is establishing glutathione at optimal levels. The medical care standard does not address the glutathione precursor RND. To the contrary, as discussed in the following section, many medical actions and inactions facilitate glutathione collapse which in turn may potentiate and accelerate FPN. FPN occurs when a reversible process, if left unchecked, enhances and/or accelerates irreversible PN.
Glutathione depletion
With regard to Figure 3, L-cysteine is the rate-limiting step in glutathione synthesis.70 Under normal conditions the glutathione precursor L-cysteine and/or its precursor L-methionine are obtained from the diet in adequate amounts to facilitate optimal glutathione synthesis.
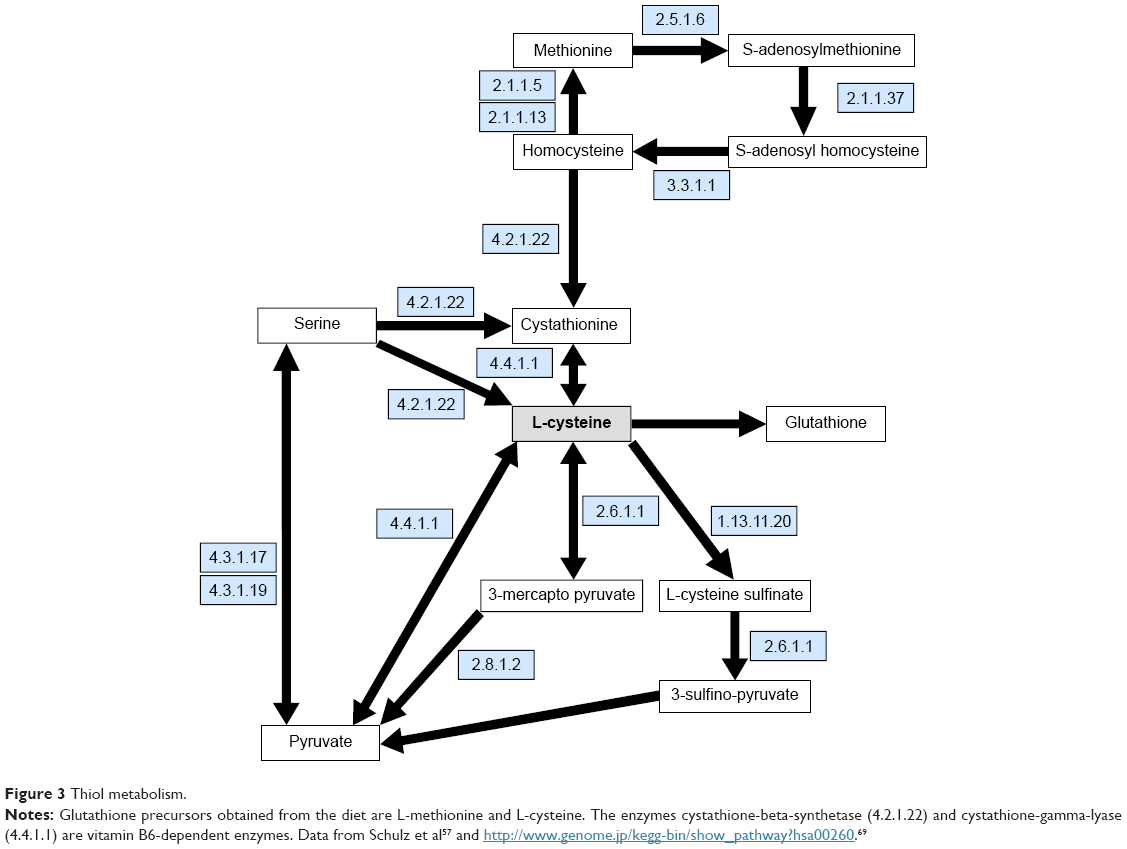 | Figure 3 Thiol metabolism. |
Inadequate glutathione levels can be caused by one or two conditions; The first is an L-cysteine RND, Figure 3. The second is vitamin B6 RND. The B6-dependent enzymes cystathionine-beta-synthase (4.2.1.22) and cystathionine-gamma-lyase (4.4.1.1) metabolize homocysteine to L-cysteine, Figure 3. A B6 RND may profoundly inhibit glutathione synthesis.45,46
To properly manage glutathione depletion from carbidopa or other sources, glutathione precursor status and/or B6 status must be addressed. Under the current PD medical care standard, nothing is done. The hypothesis is, if common PD treatment approaches deplete glutathione, then each facilitates the neurotoxin-induced electrical damage which leads to irreversible PN. The novel hypothesis is if glutathione depletion facilitates irreversible PN, then administering carbidopa may facilitate irreversible PD symptom progression and collapse.
With regard to PD patients, 89% take carbidopa.71,72 Carbidopa’s only indication is L-dopa-induced nausea management.46 It has no PD efficacy. Carbidopa has one mechanism: it causes a drug-induced B6 RND through irreversible binding to both B6 and the B6 active site found on all B6-dependent enzymes. This includes the B6-dependent enzyme AADC that catalyzes the L-dopa to dopamine reaction and the 5-HTP to serotonin reaction. The two B6 enzymes illustrated in Figure 3 metabolize homocysteine to L-cysteine.10,11,41,42 If carbidopa is not inducing a B6 RND state its clinical effects will not be observed. Supplementing B6 may hinder, reverse or cancel carbidopa’s one benefit, its L-dopa anti-nausea effect.47
Referring to Figure 2, glutathione depletion may be induced by L-dopa administration which can deplete the glutathione sulfur amino acid (thiol) precursor S-adenosyl-methionine, Figure 3.34,35,73
With regard to the 29 depletion etiologies and seven primary RNDs illustrated in Figure 1, the medical care standard only addresses the dopamine precursor RND by incorporating L-dopa in a manner that accelerates other depletions while inducing RNDs and converts it to a drug with side effects, Figure 2.10,11
To prevent facilitated irreversible PN, carbidopa needs to be stopped while B6 and glutathione precursors need to be properly administered.
L-dopa may also deplete L-tyrosine, L-tryptophan, all thiols, and serotonin.33–40 Significant depletion induces an RND.5
Reversible pseudo-neurodegeneration
In the endogenous state there is an inverse relationship between central dopamine concentrations and PD symptom severity.47 Dopamine synthesis requires L-dopa and AADC.5,49 When B6 activates AADC it becomes the enzyme’s active site.41,42 The novel hypothesis is, if the post-synaptic electrical damage or compromise is no longer progressing, then dopamine precursor RND and/or B6 RND exacerbations will be responsible for the worsening PD symptoms due to inadequate dopamine concentrations. This novel RPN induced by L-dopa RND and B6 RND symptoms has been hiding in plain sight for years with symptoms identical to PD symptoms. In 1941, Baker documented that B6 administration yielded significant PD symptom relief in some patients.24 Dopamine collapse secondary to dopamine precursor RND and/or the B6 RND are RPN examples with symptoms that are identical to PD symptoms induced by irreversible PN.
The hypothesis is, if RPN is induced by carbidopa and carbidopa is discontinued without adequate B6 administration, then RND components may be misdiagnosed as irreversible PN.
B6 crosses the blood–brain barrier.74 At equilibrium, peripheral and central B6 represent one pool. Carbidopa-induced central B6 depletion has been known since 1978. With carbidopa administration the resulting B6 depletion may lead to collapse of central and peripheral B6-dependent enzymes including AADC which metabolizes L-dopa to dopamine. When L-dopa appears to stop working while carbidopa is administered, B6 driven AADC collapse needs to be considered as the etiology, not L-dopa tachyphylaxis. This is a reversible event which may wrongly be interpreted as irreversible PN.41
Other impacts
Carbidopa’s irreversible binding to B6 induces a double assault on B6-dependent enzyme integrity. It irreversibly binds to and then depletes the free B6 required to activate the enzyme. It irreversibly binds to the active site then irreversibly deactivates all B6-dependent enzymes.10,11
Carbidopa’s molecular weight is 244.3.47 Pyridoxal-5′-phosphate molecular weight is 247.140.74 One carbidopa molecule binds with one pyridoxal-5′-phosphate molecule. Administering carbidopa 100 mg has the potential to irreversibly deactivate B6 101.16 mg in the free and B6-dependent enzyme bound forms. A patient taking carbidopa 100 mg per day while ingesting the 2 mg B6 per day United States Recommended Dietary Allowance (US-RDA) for 5 years will have a potential 181,925 mg B6 deficit.
L-dopa was introduced in 1959. From its introduction until 1976 there was a year-to-year decrease in the PD mortality rate. B6 depletion is documented to increase death rates from heart failure, coronary artery disease, colorectal cancer, stroke, peripheral vascular disease, and atherosclerosis. Literature from 2014 linked the increasing PD death rate to carbidopa-induced B6-depletion. Carbidopa, whose mechanism of action is B6 depletion, was introduced in 1975. PD mortality increased over 328% between 1976 and 2011.11
Prior to 1976, an era with no carbidopa administration, irreversible dyskinesias were not reported. In 2014, the authors documented that irreversible dyskinesias are caused by carbidopa, not L-dopa. The mechanism of action is a carbidopa-induced B6 RND which compromises the two B6-dependent enzymes, histidine decarboxylase and AADC, which metabolize histidine to histamine. B6 depletion may induce profound carbidopa-induced antihistamine dyskinesias which have been wrongly described as L-dopa-induced dyskinesias in the past. Managing these dyskinesias requires stopping carbidopa and administering adequate B6. If adequate B6 is not administered the dyskinesias may be perceived as permanent and irreversible.11
L-dopa enhances melanin steal. Melanin precursor metabolism is catalyzed by tyrosinase which preferentially metabolizes L-tyrosine and L-dopa to dopaquinone at dopamine synthesis expense. In the competitive inhibition state, unstable melanin synthesis is induced by L-dopa which causes an L-tyrosine RND. It was documented that dopamine fluctuations have a direct correlation with PD clinical symptom fluctuations and on/off effect.51
5-HTP versus carbidopa
Literature documents that 5-HTP administration for L-dopa-induced nausea management is “superior” to carbidopa in several ways. L-dopa-induced nausea etiology is due to peripheral dopamine dominating serotonin and its precursors in competitive inhibition synthesis (AADC), metabolism, and transport.10,11
5-HTP controls L-dopa-induced nausea by the same mechanism as carbidopa: AADC inhibition. Carbidopa inhibition is irreversible. AADC inhibition by 5-HTP is reversible. 5-HTP and L-dopa freely cross the blood–brain barrier and are metabolized by AADC to serotonin and dopamine respectively without biochemical feedback inhibition. With adequate AADC availability, administering 5-HTP will yield unlimited serotonin.5,8,10,11,49,51
L-dopa taken alone, without concomitant 5-HTP administration, is prone to function as a drug with side effects and induces serotonin depletion. 5-HTP is highly effective in controlling L-dopa-induced nausea which removes the need for carbidopa.11
5-HTP may facilitate L-dopa functioning as a nutrient without side effects as opposed to concomitant carbidopa administration where L-dopa is a drug. 5-HTP does not deplete B6, induce PD symptoms due to RPN or facilitate irreversible PN as carbidopa has the potential for.10,11
Normally, L-tyrosine and L-tryptophan dietary intake provides optimal competitive inhibition balance between dopamine and serotonin precursors with no side effects. However, for proper balance, like force must balance like force. L-tyrosine must be paired with L-tryptophan and L-dopa must be paired with 5-HTP. Generally, L-dopa and 5-HTP from the diet are available in small amounts. Pairing increased L-dopa administration with dietary L-tryptophan is a competitive inhibition mismatch which converts L-dopa from a nutrient to a drug with its accompanying side effects. To compensate for this imbalance and the ensuing nausea, standard medical care administers another drug, carbidopa, with all its inherent undesirable consequences.5,8,10,11,49,51
To recap, with 5-HTP, the PD patient is not needlessly exposed to carbidopa-induced problems solely for L-dopa-induced nausea control. Unlike carbidopa, 5-HTP does not interfere with glutathione synthesis which facilitates irreversible PN. It does not irreversibly deplete B6 which compromises dopamine, norepinephrine, epinephrine, or histamine synthesis. 5-HTP does not interfere with serotonin precursor transport, reduce serotonin synthesis, nor does it have the potential to deactivate all free B6 and all B6-dependent enzymes. Furthermore, it has not been linked to the PD mortality rate. It does not induce irreversible dyskinesias, nor interfere with over 300 enzyme and protein functions.
Materials and methods
These nutrient studies were carried out in the USA. All nutrients studied were classified by the USA Food and Drug Administration as “Generally Recognized as Safe” and sold over-the-counter without a prescription. Institutional review board oversight required of drug studies was not required. This PD research using L-dopa without carbidopa started in 2002. In order to exactly replicate these results, the following disclosures are made.
Since there is no single ingredient prescription L-dopa without carbidopa available in the USA, M. Pruriens- sourced L-dopa was used.
The following products were obtained from CHK Nutrition, Inc., Duluth, MN, USA. Two L-dopa sources were employed: D5 Mucuna 40% standardized L-dopa 300 mg veggie caps and D5 Mucuna 40% standardized L-dopa powder. The following 5-HTP sources were employed: 1) 99%+ pure 5-HTP, 100 mg 5-HTP per capsule; 2) NeuroReplete, one capsule contains 37.5 mg 5-HTP, 375 mg L-tyrosine, 9.375 mg pyridoxal phosphate, 125 mg ascorbic acid, 62.5 mg lysine, 27.5 mg calcium carbonate, 50 mcg folic acid; and 3) RepleteExtra, one capsule contains 75 mg 5-HTP, 250 mg L-tyrosine. The thiol precursors were obtained from CysReplete, one capsule contains 750 mg L-cysteine, 67 mcg selenium, 66.5 mcg folic acid. Supplemental L-tyrosine was obtained from TyrosineReplete, one capsule contains 500 mg L-tyrosine. CHK Nutrition sourced vitamin B6, one capsule contains 100 mg pyridoxal phosphate, was also employed.
Urine samples were collected after the patient had taken a static daily dosing value of amino acids for a minimum of 5 days. Urine samples were collected 6 hours prior to sleep cycle, and 4 pm was the most frequent time selected for this purpose. To create an environment with a pH less than 3.0 in order to stabilize serotonin and dopamine 6 N HCl was employed. Samples were processed by DBS Laboratories (Duluth, MN, USA) directed by a board certified hospital-based laboratory medicine pathologist. Urinary dopamine and serotonin were assayed utilizing commercially produced RIA kits (3-CAT RIA IB88501 and IB89527; Immuno Bio Lab, Inc., Minneapolis, MN, USA).
If there was no carbidopa ingestion history, the PD patient was started on vitamin B6 100 mg three times a day. If the patient had a carbidopa ingestion history, B6 200 mg three times a day for thirty days was administered then decreased to 300 mg per day. If the patient was taking a carbidopa/L-dopa preparation and presented with head-bobbing dyskinesias or choreiform movements, the carbidopa preparation was discontinued and B6 400 mg three times a day was started until dyskinesias ceased, at which point B6 was lowered to 100 mg three times a day.
Patients who were not taking carbidopa/L-dopa preparations at the treatment onset were initiated as follows. They were started on D5 Mucuna 40% two pills three times a day, NeuroReplete one pill twice a day, CysReplete two pills three times a day and vitamin B6 300 mg per day. The D5 Mucuna 40% pills were increased weekly in six pill increments until 24 pills per day (six pills four times a day) was established, depending on patient symptom relief. A change from D5 Mucuna 40% pills to D5 Mucuna 40% powder generally occurred when patients were taking 24 pills per day or sooner if the patient experienced issues with pill swallowing. If the daily dosing level reached 24 pills, eight pill per day incremental increases were implemented as needed to achieve symptom relief. Pill stops, under the protocol documented in 2014, were utilized at the caregiver’s discretion and generally were introduced when the daily dosing value was between 24 and 42 pills.9
If the patient was taking any carbidopa/L-dopa preparation, those medications were stopped upon treatment initiation. Patients were then started on NeuroReplete one pill twice a day with CysReplete two pills three times a day along with B6 as previously discussed. The total daily L-dopa dose from the carbidopa/L-dopa preparation was calculated. The resulting value, multiplied by the factor 12.5, determined the 40% mucuna daily dosing value (in milligrams) when the carbidopa/L-dopa was discontinued. Pill stops were initiated the following week.
Patients were instructed to report any nausea incidence or intestinal disturbance, no matter how slight. L-dopa-induced nausea onset usually occurred by the time the patients were taking 24–300 mg D5 Mucuna pills per day. Nausea was controlled by first adjusting the NeuroReplete daily dosing value. If the nausea was severe or vomiting was reported, the D5 Mucuna dose was lowered until the patient reported comfort. The NeuroReplete was adjusted to achieve optimal balance between the 5-HTP and L-dopa as evidenced by complete nausea resolution. Until nausea has been brought under control, no further upward D5 Mucuna 40% adjustments should be attempted.
If nausea was reported on the one pill twice a day NeuroReplete daily starting dose, the dose was decreased to one pill in the morning. If nausea persisted after 4 days, the NeuroReplete was increased to one pill three times a day. If after 4 days nausea was still present, the NeuroReplete was increased to two pills three times a day. Persistent nausea unresolved in 4 additional days required the NeuroReplete to be increased to four pills twice a day. If after 4 additional days nausea remained, the patient was placed on four NeuroReplete in the morning and noon with four RepleteExtra at 4 pm. Increases in D5 Mucuna 40% with pill stops were resumed once nausea was under control.
Results
Since 2002, through July 31, 2015, this research project has databased information on 1,207 PD patients treated with L-dopa without carbidopa. This section reports PD data N=388 obtained between August 1, 2014 and July 31, 2015.
Dyskinesias
Since 2002, and consistent with pre-1976 literature, there have been no new onset L-dopa-induced irreversible dyskinesias reported among the 1,207 PD patients taking L-dopa without carbidopa.11
Between August 1, 2014 and July 31, 2015, 17 patients with carbidopa-induced head bobbing dyskinesias presented for treatment. Their dyskinesias involved the upper shoulder and neck muscles and all patients were taking a carbidopa/L-dopa preparation. Sinemet® (Merck & Co. Whitehouse Station, NJ, USA) was the most frequently prescribed formula. The carbidopa/L-dopa preparation was stopped and the protocol described in the materials and methods section was started with 1,200 mg B6 per day until dyskinesias resolved, at which time the B6 was decreased to 300 mg per day. The time range reported to symptom resolution was 14 to 163 days with a 26-day mean and a 18.2-day standard deviation. In no cases were the dyskinesias refractory. One patient experienced dyskinesia abatement after 29 days of ingesting 1,200 mg B6 per day, then 6 days after stopping B6 300 mg per day experienced dyskinesia relapse. Permanent dyskinesia lysis was established in this patient with 1,200 mg B6 per day for 9 additional days.11
L-dopa tachyphylaxis
Under competitive inhibition between serotonin and dopamine, too much or too little L-dopa may present as a PD relapse with identical symptoms. L-dopa tachyphylaxis has been documented to occur secondary to a B6 RND, dopamine precursor RND and/or a serotonin precursor RND. With symptom relapse pill stops were required to determine if the daily L-dopa dosing value was under dosed or overdosed.9
Under this approach, in all cases pill stops revealed that the waning symptom control resulted from overdose and less L-dopa was required. It was postulated that correcting the B6 deficiency facilitated dopamine synthesis and the decreased L-dopa daily dosing requirement. No true L-dopa tachyphylaxis has been observed with this nutritional approach.
Melanin steal-related RND
As previously documented, urinary dopamine greater than 40,000 μg/gcr were interpreted as retrograde phase 1 dopamine fluctuations. This was attributed to melanin steal which was managed with L-tyrosine in 7,500 mg incremental additions. The daily L-tyrosine dosing requirement ranges up to 75,750 mg per day, Table 1. Generally, L-tyrosine was well tolerated only when adjusted under lab guidance. All on/off effect occurrences responded to L-tyrosine dosing optimization. No refractory on/off cases were reported.51
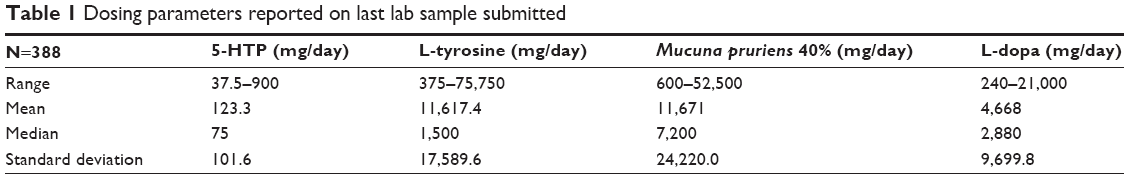 | Table 1 Dosing parameters reported on last lab sample submitted |
L-dopa-induced nausea
5-HTP was substituted for carbidopa. If nausea was experienced, no further increases in the D5 Mucuna 40% were performed until the NeuroReplete was established at the daily dosing value required for complete nausea relief. 5-HTP dosing parameters are found in Tables 1 and 2. With the 5-HTP administration there were no refractory L-dopa-induced nausea cases reported.
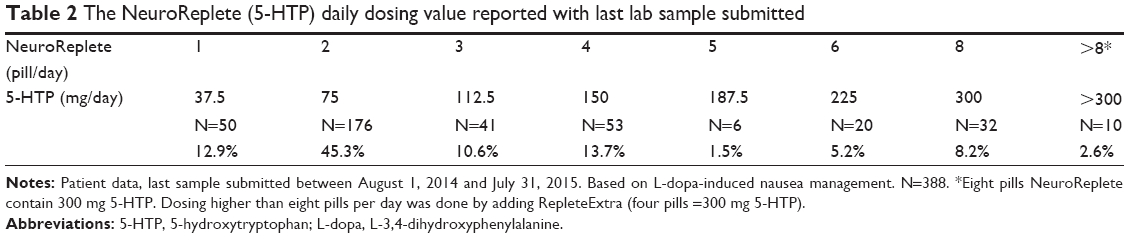 | Table 2 The NeuroReplete (5-HTP) daily dosing value reported with last lab sample submitted |
Discussion
The novel assertion is when nutrients display side effects they are functioning as drugs. These side effects are not just acute events experienced by the patient. They may represent slow and insidious systemic dysfunction and/or collapse which evolves over long time periods.
Under the usual and customary medical approach, there is no consideration given to RNDs. When L-dopa is turned into a drug, side effects and waning efficacy define the maximum improvement point. If RNDs relating to L-dopa precursors, serotonin precursors, thiols, and B6 depletion are induced and/or ignored by standard medical care, then the current standard treatment methodology is facilitating irreversible PN and inducing reversible pseudo-neurodegeneration, which causes patient deterioration under the belief that a single etiology model, irreversible PN, is the cause of all clinical deterioration.10,11
This novel amino acid approach avoids turning nutrients into drugs with side effects. This amino acid approach does not utilize one size fits all dosing. For optimal results, L-dopa, 5-HTP, L-tyrosine, L-cysteine, and B6 must be administered simultaneously at dosing values uniquely defined for each patient.5,8,10,11,49,51
The relentless neurotoxin-induced PN is facilitated by glutathione collapse, a process that can be slowed or halted. The novel assertion is if glutathione collapse occurs long before the first clinical PD symptom appears, then the neurotoxin-induced damage prior to symptoms is facilitated.
Other new assertions are, if carbidopa’s B6 depletion may compromise the two B6-dependent enzymes which metabolize homocysteine to L-cysteine, the rate limiting step in glutathione synthesis, then carbidopa administration may deplete glutathione and facilitate irreversible PN. If L-dopa administration depletes the glutathione precursor S-adenosyl-methionine,34–36 then it is another novel assertion that L-dopa administration may deplete glutathione and facilitate irreversible PN.
If the PD symptom breadth and severity is the reference point, then it is a novel assertion that carbidopa-induced B6 depletion is the most damaging component of standard PD treatment. It is not possible to address B6 depletion until carbidopa is completely discontinued. Reviewing carbidopa pharmacokinetics reveals that if enough B6 is administered to compensate for carbidopa depletion, then L-dopa-induced nausea control will no longer be observed.16,57,68
With regard to PD, carbidopa-induced B6 depletion may collapse AADC function. This has the same novel effect on dopamine as decreasing L-dopa intake. Dopamine synthesis is decreased while PD symptoms increase.10,11
Carbidopa irreversibly binds to and deactivates B6 and all B6-dependent enzymes.41,42 Literature documents that 5-HTP is a superior alternative to carbidopa without the deleterious side effects.11 The novel hypothesis is, if when administered properly 5-HTP has no side effects, then there is no justifiable reason to not substitute 5-HTP for carbidopa. The current medical care standard not only fails to recognize the vast array of carbidopa-induced B6 deficiency issues, it is actively facilitating permanent electrical damage progression and inducing reversible PD symptom deterioration every day carbidopa is prescribed and taken. Carbidopa administration is the glutathione and nutritional optimization antithesis.10,11 It deactivates and prevents activating enzymes that are critical in metabolizing homocysteine to L-cysteine, the rate limiting step in glutathione synthesis.45,46 Carbidopa-induced B6 depletion may collapse AADC L-dopa and 5-HTP metabolism to dopamine and serotonin, respectively.5,10,11,49 Typically, the medical care standard treats PD with carbidopa/L-dopa then simply documents the progressive neurodegenerative course with no attention paid to the reversible components.
L-dopa is the most effective PD treatment; however, its typical long-term monotherapy administration depletes all thiols including glutathione.34–38 This depletion exacerbates and facilitates irreversible lipophilic neurotoxin-induced electrical damage. Glutathione depletion is reversible.
L-dopa metabolism to dopamine, through competitive inhibition, induces serotonin depletion that leads to a serotonin precursor RND. As previously documented, under this balanced competitive inhibition based approach, in order for L-dopa to not be converted to a drug it must be titrated with pill stops in the 600 to 52,500 mg per day D5 Mucuna 40% daily dosing range in combination with proper 5-HTP, L-tyrosine, a thiol and B6 levels.10,11
Controlling L-dopa-induced nausea with 5-HTP is higher priority than achieving optimal symptom control. Under this approach, if at any point nausea is encountered, symptom optimization needs to be abandoned until nausea has fully resolved secondary to establishing the individualized optimal 5-HTP daily dosing value.9 This research notes the novel finding that as treatment progresses, if other issues related to serotonin and dopamine imbalance such as headache, anxiety, insomnia, etc, occur, 5-HTP may be secondarily titrated within 37.5 to 900 mg per day range.
Serotonin depletion is associated with PD,25,26 carbidopa administration which leads to B6-induced AADC collapse and from competitive inhibition induced by dopamine precursors.5,10,11,49 Reversible serotonin depletion symptoms have been wrongly attributed to irreversible PN. Previously documented were the observations that severe serotonin depletion compromises dopamine’s clinical effects.49
L-cysteine, the immediate glutathione precursor and rate-limiting step in glutathione synthesis, is a one dose fits all approach.10,11,43,58 As previously documented, the optimal adult L-cysteine daily dosing value is 4,500 mg per day in divided doses.9,51,52,55 Due to toxicity of methyl mercury to central nervous system, selenium 400 μg per day is added.74–76 If the leading PD post-synaptic damage cause is neurotoxins and glutathione is the most powerful protective mechanism, then administrating adequate glutathione precursors at proper levels will positively impact the clinical course. If glutathione is administered intravenously (IV), on a molecular equivalent basis, 11,414.25 mg per day needs to be administered. YouTube and other anecdotal sources document the extraordinary response some PD patients achieve with IV glutathione administration.77,78 These results are short lived generally lasting 36 hours or less. The hypothesis is, if this IV glutathione response is observed, then it demonstrates severe glutathione precursor RND. Severe glutathione precursor RNDs caused by PD, L-dopa, and carbidopa only progress to the level noted on these YouTube videos when severe nutritional neglect has occurred. It is the novel assertion that glutathione precursor RND collapse on this scale should never happen. Ideally, proper nutritional glutatione precursor intervention should be started long before the first PD symptoms are observed.
Dopamine fluctuations that induce on/off effect are documented as being caused by melanin steal. Resolving existing or developing L-tyrosine driven RNDs can effectively address this issue. However, this should not be undertaken without proper laboratory assay guidance. Failure to do so may convert L-tyrosine into a drug with side effects.51
The observations discussed in this manuscript are based on 18 years first hand clinical patient experience. Optimal results with this nutritional approach cannot be achieved by self-treatment or while cared for by one who lacks a medical license and in-depth training in general PD care and this methodology. Even though this is a nutritionally based approach, achieving optimal outcomes is far more complex than any previous drug approach. It is not just following protocols but rather knowing, identifying, and understanding when the actions prescribed may create long-term nutritional harm to the patient. Negative consequences on other systems and side effects due to nutritional imbalance can convert the nutrient to a drug, and unwittingly accelerate PD symptom deterioration. It is incumbent upon the caregiver to recognize and properly manage these nutritional deficiencies in order to gain the optimal outcomes for their PD patients and not contribute to reversible and irreversible PN.
Conclusion
This paper does not discuss a new treatment approach. It discusses side effect management for the most effective known PD treatment, L-dopa. When nutrients to include L-dopa and 5-HTP are displaying side effects they have been converted to a drug.
Prior to this research project’s documented results, the PD clinical course was referred to exclusively as irreversible PN. This paper outlines two reversible states: reversible FPN and RPN. Optimal results in PD treatment cannot be achieved until the reversible RND etiologies and collapses are properly addressed.
Removing carbidopa from treatment has the greatest positive outcome impact. Carbidopa’s ability to irreversibly bind with B6 and all B6-dependent enzymes can induce glutathione depletion, dopamine collapse, serotonin collapse, antihistamine-induced dyskinesias, and RNDs contributing to these collapses. In turn, B6 depletion induces secondary function collapse involving over 300 enzyme and protein systems. Carbidopa has been linked to the 328% increase in PD mortality which started the year after its introduction.
While all RNDs need to be managed simultaneously, the highest priority must be given to managing B6 deficiency, something that cannot be done effectively while the patient is taking carbidopa. As demonstrated by AB Baker in 1941, for some PD patients all that may be needed for adequate treatment may be ingesting proper B6 levels.24 L-dopa and AADC are both critical to establishing dopamine levels required for maximum PD improvement. L-dopa RNDs and AADC dysfunction induced by B6 depletion are equally powerful in compromising dopamine synthesis. While carbidopa does not cross the blood–brain barrier, B6 does. It has been known since 1978 that B6 depletion by peripheral carbidopa depletes central dopamine by compromising the B6 enzyme AADC,41 an event that induces PD reversible symptom deterioration and facilitates irreversible PN.
The central PD event is permanent substantia nigra post-synaptic dopamine neuron damage.3 When neurotoxins are the cause, the standard of care in medicine has traditionally ignored the associated glutathione collapse which may facilitate further neurotoxic damage. Medicine compounds the problem by continuing to prescribe treatment that facilitates neurotoxin-induced collapse. Administering thiols (glutathione) without serotonin and dopamine precursors induces a dopamine and serotonin collapse which represents a precursor RND.5,11 Documentation exists that ultra-early in the disease course, prior to symptoms, glutathione precursors are in an RND state.16,57,68 This glaring potential thiol intervention has never been embraced.
As a strategy to deal with the side effects associated with improperly administered L-dopa, standard medical care delays starting L-dopa until other less effective drugs fail. This ignores the dopamine precursor RNDs which only facilitates and accelerates irreversible PN. On/off effect has puzzled medicine for years; proper management with L-tyrosine under the melanin-steal model has been documented as a simple and safe laboratory guided method to reverse this effect.51
Traditionally, PD exacerbations were exclusively attributed to substantia nigra post-synaptic neurons undergoing irreversible PN. This served as a single etiology to explain the source, magnitude, and clinical course. While it is known in the endogenous state that increasing synaptic dopamine concentrations has an inverse correlation with PD symptoms, carbidopa’s B6 inactivation ability compromises L-dopa metabolism to dopamine by AADC thereby exacerbating PD symptoms. Thiol depletion and collapse induced by L-dopa facilitates post-synaptic damage progression which fuels a vicious cycle that increases the L-dopa dosing needs leading to increased thiol collapse.
The key to optimal long-term PD treatment results is to completely avoid carbidopa while administering thiols, L-tyrosine, L-dopa, 5-HTP and B6 in a manner where they continue to function as nutrients without inducing short-term or long-term problems or side effects. This stated goal may appear deceptively simple on the surface. Until this novel approach, there was no prior documentation claiming that L-dopa needed to be conceptualized as a nutrient which may be converted to a drug if it generates side effects, depletion, or dysfunction. When nutrient-associated side effects develop it is proof that one or more nutrients have been converted to a drug.
This paper documents the superiority of the individual patient tailored treatment approach which comprehensively addresses the multisystem nutritional collapses that contribute to irreversible PN and RPN. The paper focuses on the shortcomings of the current medical care standard.
Disclosure
MH discloses his relationship with DBS Laboratory services and NeuroResearch Clinics, Inc. The other authors report no conflicts of interest in this work.
References
Neurodegenerative diseases. Mayo Clinic 2015. Available from: http://www.mayo.edu/research/labs/neurodegenerative-diseases/overview. Accessed October 8, 2015. | ||
Suremeier J, Guzman JN, Sanchez-Padilla J, Schumacker PT. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience. 2011;198:221–231. | ||
Phani S, Loike JD, Przedborski S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S207–S209. | ||
Yu N-Y, Chang S-H. Comparisons of fine motor control functions in subjects with Parkinson’s disease and essential tremor. Int J Med Health Biomed Bioeng Pharm Eng. 2013;7(10):343–346. | ||
Hinz M, Stein A, Uncini T. Relative nutritional deficiencies. Int J Gen Med. 2012;5:413–430. | ||
Hinz M, Stein A, Uncini T. Monoamine depletion by reuptake inhibitors. Drug Healthc Patient Saf. 2011;3:69–77. | ||
Hinz M, Stein A, Uncini T. The discrediting of the monoamine hypothesis. Int J Gen Med. 2012;5:135–142. | ||
Hinz M, Stein A, Uncini T. 5-HTP efficacy and contraindications. Neuropsychiatr Dis Treat. 2012;8:323–328. | ||
Hinz M, Stein A, Uncini T. Management of L-dopa overdose in the competitive inhibition state. Drug Healthc Patient Saf. 2014;6:93–99. | ||
Hinz M, Stein A, Uncini T. The Parkinson’s disease death rate: carbidopa and vitamin B6. Clin Pharmacol. 2014;6:161–169. | ||
Hinz M, Stein A, Uncini T. Parkinson’s disease: carbidopa, nausea, and dyskinesia. Clin Pharmacol. 2014;6:189–194. | ||
Gröger A, Kolb R, Schäfer R, Klose U. Dopamine reduction of the substantia nigra of Parkinson’s disease patients confirmed in vivo magnetic resonance spectroscopic imaging. PLoS One. 2014;9(1):1–6. | ||
Venkateshappa C, Harish G, Mythri RB, Mahadevan A, Bharath MM, Shankar SK. Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson’s Disease. Neurochem Res. 2012;37:358–369. | ||
Charlton CG, Crowell B Jr. Striatal dopamine depletion, tremors, and hypokenesia following the intracranial, injection of S-adenosylmethionine. Mol Chem Neuropathol. 1995;26:269–284. | ||
Eldrup E, Mogensen P, Jacobsen J, Pakkenberg H, Christensen NJ. CSF and plasma concentrations of free norepinephrine, dopamine, 3,4-dihydroxyphenylacetatic acid (DOPAC), 3,4-dihydroxyphenylalanine (DOPA) and epinephrine in Parkinson’s disease. Acta Neurol Scand. 1995;92(2):116–121. | ||
Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Exp Neurol. 2007;203:512–520. | ||
Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress, and neurodegeneration. Eur J Biochem. 2000;267:4909–4911. | ||
Martin HL, Teismann P. Glutathione – a review of its role and significance in Parkinson’s disease. FASEB J. 2009;23:3263–3272. | ||
Newman PE. Alzheimer’s disease revisited. Med Hypothesis. 2000;54(5):774–776. | ||
Corrales F, Ochoa P, Rivas C, Martin-Lomas M, Mato JM, Pajares MA. Inhibition of glutathione synthesis in the liver leads to S-adenosyl-L-methionine synthetase reduction. Hepatology. 1991;14:528–533. | ||
Cheng H, Gomes-Trolin C, Aquilonius SM, et al; Levels of L-methionine S-adenosyltranferase activity in erythrocytes and concentrations of S-adenosylmethionine and S-adenosylhomocysteine in whole blood of patients with Parkinson’s disease. Exp Neurol. 1997;145(2):580–585. | ||
Baek M, Choy JH, Choi SJ. Montmorillonite intercalated with glutathione for antioxidant delivery: synthesis, characterization, and bioavailability evaluation. Int J Pharm. 2012;425(1–2):29–34. | ||
Lertratanangkoon K, Orkiszewski RS, Scimeca JM. Methyl-donor deficiency due to chemically induced glutathione depletion. Cancer Res. 1996;56:995–1005. | ||
Baker AB. Treatment of paralysis agitans with vitamin B6 (pyridoxine hydrochloride). JAMA. 1941;116(22):2484–2487. | ||
Scatton B, Javoy-Agid F, Rouquier L, Dubois B, Agid Y. Reduction of cortical dopamine, noradrenaline, serotonin and their metabolites in Parkinson’s disease. Brain Res. 1983;275(2):321–328. | ||
Halliday GM, Blumbergs PC, Cotton RG, Blessing WW, Geffen LB. Loss of brainstem serotonin- and substance P-containing neurons in Parkinson’s disease. Brain Res. 1990;510(1):104–107. | ||
Jenner P, Sheehy M, Marsden CD. Noradrenaline and 5-hydroxytryptamine modulation of brain dopamine function: implications for the treatment of Parkinson’s disease. Br J Clin Pharmacol. 1983;15:277S–289S. | ||
Pan T, Li X, Jankovic J. The association between Parkinson’s disease and melanoma. Int J Cancer. 2011;128:2251–2260. | ||
Stansley BJ, Yamamoto BK. L-dopa and brain serotonin system dysfunction. Toxics. 2015;3:75–88. | ||
Miguelez C, Morera-Herreras T, Torrecilla M, Ruiz-Ortega JA, Ugedo L. Interaction between the 5-HT system and the basal ganglia: functional implication and therapeutic perspective in Parkinson’s disease. Front Neural Circuits. 2014;8:1–9. | ||
Widner B, Leblhuber F, Fuchs D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J Neural Transm. 2002;109(2):181–189. | ||
Nemeth H, Toldi J, Vécsei L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm. 2006;(70 Suppl):285–304. | ||
Karobath M, Díaz JL, Huttunen MO. The effect of L-dopa on concentrations of tryptophan, tyrosine, and serotonin in rat brain. Euro J Pharmacol. 1971;14:393–396. | ||
Charlton CG, Crowell B Jr. Parkinson’s disease-like effects of S-adenosyl-L-methionine: effects of L-dopa. Pharmacol Biochem Behav. 1992;43(2):423–431. | ||
Liu XX, Wilson K, Charlton CG. Effects of L-dopa treatment on methylation in mouse brain: implications for the side effects of L-dopa. Life Sci. 2000;66(23):2277–2288. | ||
Muller T, Woitalla D, Hauptmann B, Fowler B, Kuhn W. Decrease of methionine and S-adenosylmethionine and increase of homocysteine in treated patients with Parkinson’s disease. Neurosci Lett. 2001; 308(1):54–56. | ||
Spencer JP, Jenner P, Daniel SE, Lees AJ, Marsden DC, Halliwell B. Conjugates of catecholamines with cysteine and GSH in Parkinson’s disease: possible mechanisms of formation involving reactive oxygen species. J Neurochem. 1998;71:2112–2122. | ||
Borah A, Mohanakumar KP. Long-Term L-DOPA treatment causes indiscriminate increase in dopamine levels at the cost of serotonin synthesis in discrete brain regions of rats. Cell Mol Neurobiol. 2007;27:985–996. | ||
Wuerthele SM, Moore KE. Studies of the mechanism of L-dopa-induced depletion of 5-hydroxytryptamine in the mouse brain. Life Sci. 1977;20:1675–1680. | ||
Ritvo ER, Yuwiler A, Geller E, et al; Effects of L-dopa in autism. J Autism Dev Disord. 1971;1(2):190–205. | ||
Airoldi L, Watkins CJ, Wiggins JF, Wurtman RJ. Effect of pyridoxine on the depletion of tissue pyridoxal phosphate by carbidopa. Metabolism. 1978;27(7):771–779. | ||
Daidone F, Montioli R, Paiardini A, et al; Identification by virtual screening and in vitro testing of human DOPA decarboxylase inhibitors. PLoS One. 2012;7(2):1–15. | ||
Lado-Abeal J, Graña M, Rey C, Cabezas-Cerrato J. L-5-hydroxytryptophan does not stimulate LH secretion directly from the pituitary in patients with gonadotrophin releasing hormone deficiency. Clin Endocrinol (Oxf). 1998;49(2):203–207. | ||
Bartlett MG. Biochemistry of the water soluble vitamins: a lecture for first year pharmacy students. Am J Pharm Educ. 2003;67(2):Article 64. | ||
Kery V, Bukovska G, Kraus JP. Transsulfuration depends on heme in addition to pyridoxal-5′-phosphate. J Biol Chem. 1994;269(41):25283–25288. | ||
Bruinenberg PG, De Roo G, Limsowtin G. Purification and characterization of cystathionine g-lyase from lactococcus lactis subsp. cremoris SK11: possible role in flavor compound formation during cheese maturation. Appl Environ Microbiol. 1997;63(2):561–566. | ||
Sinemet prescribing information. Merck website. Available from: https://www.merck.com/product/usa/pi_circulars/s/sinemet/sinemet_pi.pdf. Accessed October 8, 2015. | ||
Katzenchlager R, Evans A, Manson A, et al; Mucuna Pruriens in Parkinson’s disease: a double blind clinical and pharmacologic study. J Neurol Neurosurg Psychiatr. 2004;75:1672–1677. | ||
Hinz M, Stein A, Uncini T. APRESS: apical regulatory super system, serotonin and dopamine interaction. Neuropsychiatr Dis Treat. 2011;7:457–463. | ||
Hinz M, Stein A, Uncini T. Validity of monoamine assay sales under the “spot baseline urinary neurotransmitter testing marketing model”. Int J Nephrol Renovasc Dis. 2011;4:101–113. | ||
Hinz M, Stein A, Cole T. Parkinson’s disease-associated melanin steal. Neuropsychiatr Dis Treat. 2014;10:2331–2337. | ||
Hinz M, Stein A, Uncini T. Amino acid management of Parkinson’s disease: a case study. Int J Gen Med. 2011;4:165–174. | ||
Hinz M, Stein A, Uncini T. Urinary neurotransmitters testing: considerations of spot baseline norepinephrine and epinephrine. Open Access J Urol. 2011;3;19–24. | ||
Hinz M, Stein A, Neff R, Weinberg R, Uncini T. Treatment of attention deficit hyperactivity disorder with monoamine amino acid precursors and organic cation transporter assay interpretation. Neuropsychiatr Dis Treat. 2011;7:31–38. | ||
Stein A, Hinz M, Uncini T. Amino acid-responsive Crohn’s disease: a case study. Clin Exp Gastroenterol. 2010;3:171–177. | ||
Hinz M, Stein A, Uncini T. The dual-gate lumen model of renal monoamine transport. Neuropsychiatr Dis Treat. 2010;6:387–392. | ||
Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. | ||
Kegg cysteine methionine metabolism-homosapiens. Available from: http://www.genome.jp/kegg-bin/show_pathway?org_name=hsa&mapno=00270&mapscale=&show_description=hide. Accessed October 9, 2015. | ||
Blum D, Torch S, Lambeng N, et al; Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Progr Neurobiol. 2001;65: 135–172. | ||
Curtin K, Fleckenstein AE, Robison RJ, Crookston MJ, Smith KR, Hanson GR. Methamphetamine/amphetamine abuse and risk of Parkinson’s disease in Utah: a population-based assessment. Drug Alcohol Depend. 2015;146:30–38. | ||
Bove J, Perier C. Neurotoxin-based models of Parkinson’s disease. Neuroscience. 2012;211:51–76. | ||
Blesa J, Phani S, Jackson-Lewis V, Przedborski S. Classic and new animal models of Parkinson’s disease. J Biomed Biotechnol. 2012;2012:1–10. Article ID 845618. | ||
Tieu K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb Perspect Med. 2011;1:a009316. | ||
Perier C, Vila M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;4:a009332. | ||
Bezard E, Yue Z, Kirik D, Spillantini MG. Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Mov Disord. 2013;28(1):61–70. | ||
Kanthasamy A, Jin H, Anantharam V, et al; Emerging neurotoxic mechanisms in environmental factors-induced neurodegeneration. Neurotoxicology. 2012;33(4):833–837. | ||
Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet. 1998;352(9137):1344–1346. | ||
Gunjima K, Tomiyama R, Takakura K, et al; 3,4-dihydroxybenzalacetone protects against Parkinson’s disease-related neurotoxin 6-OHDA through Akt/Nrf2/glutathione pathway. J Cell Biochem. 2014; 115:151–160. | ||
Kegg glycine, serine, threonine metabolism-homosapiens. Available from: http://www.genome.jp/kegg-bin/show_pathway?hsa00260. Accessed October 9, 2015. | ||
Badaloo A, Reid M, Forrester T, Heird WC, Jahoor F. Cysteine supplementation improves the erythrocyte glutathione synthesis rate in children with severe edematous malnutrition. Am J Clin Nutr. 2002; 76:646–652. | ||
National Parkinson Foundation. The National Parkinson Foundation’s Helpline Speaks: Lessons from the 2011 Sinemet Shortage. Available from: http://c.ymcdn.com/sites/www.worldpdcoalition.org/resource/resmgr/docs/npfreport_sinimetshortage201.pdf. Accessed October 10, 2015. | ||
Benson R, Crowell B, Hill B, Doonquah K, Charlton C. The effects of L-dopa on the activity of methionine adenosyltransferase: relevance to L-dopa therapy and tolerance. Neurochem Res. 1993;18(3):325–330. | ||
Giardina G, Montioli R, Gianni S, et al; Open conformation of human DOPA decarboxylase reveals the mechanism of PLP addition to Group II decarboxylases. PNAS. 2011;108(51):20514–20519. | ||
Pubchem, pyridoxal phosphate. Available from: http://pubchem.ncbi.nlm.nih.gov/compound/1051#section=Top. Accessed October 11, 2015. | ||
Goyer RA. Toxic and essential metal interactions. Annu Rev Nutr. 1997; 17:37–50. | ||
Park ST, Lim KT, Chung YT, Kim SU. Methylmercury-induced neurotoxicity in cerebral neuron culture is blocked by antioxidants and NMDA receptor antagonists. Neurotoxicology. 1996;17(1):37–45. | ||
YouTube, Dr David Perlmutter, MD FACN Glutathione Therapy for Parkinson’s Part 2. Available from: https://www.youtube.com/watch?v=YYvd5-xN-CM. Accessed October 11, 2015. | ||
YouTube, Parkinson’s- Glutathione Therapy – Dr. David Perlmutter, MD FACN. – Protandim prescribed. Available from: https://www.youtube.com/watch?v=KWuOezgVHdI. Accessed October 11, 2015. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.