Back to Journals » Journal of Inflammation Research » Volume 14
Overview on the Discovery and Development of Anti-Inflammatory Drugs: Should the Focus Be on Synthesis or Degradation of PGE2?
Authors Mahesh G, Anil Kumar K, Reddanna P ![]()
Received 27 August 2020
Accepted for publication 12 November 2020
Published 3 February 2021 Volume 2021:14 Pages 253—263
DOI https://doi.org/10.2147/JIR.S278514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Video abstract presented by Pallu Reddanna.
Views: 975
Gopa Mahesh, Kotha Anil Kumar, Pallu Reddanna
Department of Animal Biology, School of Life Sciences, University of Hyderabad, Hyderabad, 500046, India
Correspondence: Pallu Reddanna Tel +91-40-23134542
Email [email protected]
Abstract: Inflammation is a protective response that develops against tissue injury and infection. Chronic inflammation, on the other hand, is the key player in the pathogenesis of many inflammatory disorders including cancer. The cytokine storm, an inflammatory response flaring out of control, is mostly responsible for the mortality in COVID-19 patients. Anti-inflammatory drugs inhibit cyclooxygenases (COX), which are involved in the biosynthesis of prostaglandins that promote inflammation. The conventional non-steroidal anti-inflammatory drugs (NSAIDs) are associated with gastric and renal side-effects, as they inhibit both the constitutive COX-1 and the inducible COX-2. The majority of selective COX-2 inhibitors (COXIBs) are without gastric side-effects but are associated with cardiac side-effects on long-term use. The search for anti-inflammatory drugs without side-effects, therefore, has become a dream and ongoing effort of the Pharma companies. As PGE2 is the key mediator of inflammatory disorders, coming up with a strategy to reduce the levels of PGE2 alone without affecting other metabolites may form a better choice for the development of next generation anti-inflammatory drugs. In this direction the options being explored are on synthesis of PGE2-mPGES-1; PGE2 degradation through a specific PG dehydrogenase, 15-PGDH, and by blocking its activity mediated through a specific PGE receptor, EP4. As leukotrienes formed via the 5-lipoxygenase (5-LOX) pathway also play an important role in the mediation of inflammation, efforts are also being made to target both COX and LOX pathways. This review focuses on addressing the following three points: 1) How NSAIDs and COXIBs are associated with gastric, renal and cardiac side-effects; 2) Should the focus be on the targets upstream or downstream of PGE2; and 3) the status of alternative targets being explored for the discovery and development of anti-inflammatory drugs without side-effects.
Keywords: inflammation, cyclooxygenase, COX, NSAIDs, COXIBs, microsomal PGE synthase-1, mPGES-1, 15-hydroxy prostaglandin dehydrogenase, 15-PGDH, 5-lipoxygenase, 5-LOX
Introduction
Inflammation is the body’s general response to protect itself against injury or infection. An acute inflammation process is a short-term reaction, which occurs in response to injury or infection. During this phase the immune system releases an army of white blood cells to surround and defend the area, leading to a visible redness and swelling. In view of this, inflammation is crucial; without it, simple infections could be dangerous. A prolonged or chronic inflammation, on the other hand, is the root cause of many inflammatory disorders, including cancer.
The prostaglandin (PG) biosynthesis or cyclooxygenase (COX) pathway is an essential component of inflammatory responses, generating a class of physiologically active lipid mediators called prostanoids. The COX pathway is activated by lipopolysaccharides (LPS),1 cytokines,2 xenobiotics, including metal compounds3,4 and fluoride.5,6 Membrane bound unsaturated 20-carbon containing fatty acid, arachidonic acid (AA), is the most predominant precursor for PG biosynthesis. A variety of stimuli, both physiological as well as pathological, lead to the release of AA by the action of various phospholipases, gets oxygenated via COX, lipoxygenase (LOX), and cytochrome P450 dependent monooxygenase (MOX) enzymes7 to generate prostaglandins (PGs), leukotrienes (LTs) and epoxy eicosatrienoic acids (EETs), collectively termed as eicosanoids. In the case of PGs formed via the COX pathway, AA is initially converted to an unstable prostaglandin G2 (PGG2), which is further reduced by the peroxidase activity of the same enzyme to a stable prostaglandin H2 (PGH2). PGH2 is further converted to biologically active PGs such as prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), prostacyclin (PGI2), and thromboxane A2 (TxA2) by the action of various cell-specific terminal synthases.8
Among these PGs, PGE2 is the major inflammatory mediator and its biological actions are mediated by signaling via various G-protein coupled receptors, which include EP1 (405 aa), EP2 (362 aa), EP3, and EP4 (513 aa). EP3 has multiple splicing isoforms such as EP3α (366 aa), EP3β (362 aa), and EP3γ (365 aa). The distribution and relative abundance of these receptors vary, for example, in mouse EP2 is the least abundant receptor, while EP3 and EP4 are the most dominant (widely distributed) receptors, and EP1 mRNA is restricted to organs such as the kidneys, stomach, and lungs.9,10 Their signaling mechanism involves G-protein mediated activation of downstream targets such as kinases via second messengers such as cAMP, Ca2+, and inositol phosphates. EP1 induces Ca2+ elevation with activation by agonist via unidentified G protein, whereas EP2 and EP4 receptors couple Gs for their biological action by a raise in cAMP levels. EP3, which exists in multiple splicing isoforms,9 inhibits adenylyl cyclase by coupling with Gi, leading to a decrease in cAMP levels. In humans, PGs are important physiological mediators, hence the dysregulation of its synthesis has links with inflammation, pain, cancer, cardiovascular disease, asthma, osteoporosis, male sexual dysfunction, etc.11 As a result, the COX, catalyzing the first step in the biosynthesis of PGs, has become the target for developing anti-inflammatory drugs.
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) and Their Targets
Many of the available anti-inflammatory drugs are non-steroidal in nature (NSAIDs) and target COX isoforms as part of their therapeutic action. The Greek physician Hippocrates used an extract of willow bark and leaves in the treatment of fever and inflammation for the first time about 3,500 years ago. Later on, the active compound of the extract was identified as salicylic acid in the 17th century. Acetylsalicylic acid, the acetyl derivative of salicylic acid (aspirin), was introduced in 1869 by Bayer,12 but the mode of action was unknown when the drug was out in the market. After almost a century, the molecular mechanistic role of aspirin was studied by Vane13 and colleagues, and the findings that all these NSAIDs inhibit the COX enzyme have now become a platform for developing novel NSAIDs. The most common side-effects associated with these NSAIDs, the extent of which varies depending on the type of NSAID, are gastrointestinal symptoms such as stomach pain, constipation, diarrhea, stomach ulcers, etc. In addition, kidney and liver problems have also been reported.14 Though these classical NSAIDs such as aspirin, indomethacin, ibuprofen, etc. have been in use for more than a century, it is not clear how these therapeutic and side-effects are being mediated by NSAIDs simultaneously.
With the discovery of an inducible isoform of COX, COX-2,15,16 it became very clear that there are two isoforms of COX, the constitutive isoform, COX-1, which is cytoprotective and mediates various physiological functions while the inducible isoform; and COX-2, which is the mediator of inflammation. The classical NSAIDs inhibit both the isoforms of COX – COX-1 and COX-2. This discovery of COX-2 has led to the development of selective COX-2 inhibitors (COXIBs); celecoxib, co-developed by G. D. Searle and Pfizer in 1999, followed by rofecoxib by Merck, and many others subsequently, which became the block buster drugs.
Current Targets and Their Limitations
As pointed out above the NSAIDs in general have gastric and renal side-effects. Aspirin was shown to cause damage in the gastric system in a rat model by decreasing the plasma membrane protein of tight junctions, occludin expression, which was reversed by pretreatment with another drug mosapride.17 High-dose of ibuprofen caused jejunal perforations even in short-courses.18 Another anti-inflammatory drug, naproxen, causes gastric antral ulcerations, and an increase of lipid peroxide levels, but the effect was reversed with curcumin.19 Similarly perforated jejunal ulcers20 and small bowel ulcerations21 have been reported with the usage of NSAIDs. The simultaneous use of proton pump inhibitors and non-selective short-course NSAIDs increases the risk of bowel injury.22 Similarly a combination of ibuprofen and acetaminophen causes kidney and liver problems even at a therapeutic dosage.23,24 Another drug, rofecoxib, is associated with increased renal and arrhythmia risks.25 Diclofenac, an NSAID, has also been found to increase the risk of heart attacks and strokes by 50% within days of ingestion. This study, performed in around 6.3 million cases over a period of 20 years from 1996–2016,26 also reported that the drug causes a lot of acidity and gastric intolerance in Indian patients. The adverse reactions with NSAIDs are high in patients who are under long-term medication and these adverse effects were attributed due to the targeting COX-1 enzyme, which is cytoprotective.
The COX-2 selective drugs, COXIBs, became the most preferred anti-inflammatory drugs as they have good anti-inflammatory activity, and some exhibited reduced gastrointestinal toxicity.27,28 However, very soon it became clear that COXIBs are associated with cardiac side-effects on long-term use in arthritic patients.29,30 As a result, some of the COXIBs such as rofecoxib were withdrawn from the market while others like celecoxib are still on the market with a warning label for use in patients with cardiovascular problems.31 Though COX-2 was originally identified in inflammatory tissues, it is now evident that it is also expressed constitutively in tissues like kidney, brain, and testis.32,33
The side-effects associated with COXIBs were attributed to the simultaneous reduction of prostacyclin, also a product of COX-2, which is a natural inhibitor of platelet aggregation and also involved in renal hemodynamics, and the control of blood pressure.30 Also the inhibition of COX was shown to shunt the arachidonic acid metabolism towards the 5-LOX pathway, leading to increased levels of leukotrienes, which were also a contributing factor for the side-effects of COX inhibitors.34–36 With these developments the search has begun for anti-inflammatory drugs without gastric and cardiac side-effects. The new era of anti-inflammatory agents that have emerged include NO-NSAIDs, Biologicals, cytokine inhibitors, etc, which are out of the scope of the present review. This review will focus particularly on the targets of eicosanoid pathways, which are at different stages of development.37
COX-2/5-LOX Dual Inhibitors Spare Gastrointestinal and Cardiac Side-Effects?
In addition to the key role of biologically active prostaglandins formed through the cyclooxygenase pathway in inflammation, there is a growing body of information on the involvement of other arachidonic acid metabolites such as leukotrienes formed via the 5-lipoxygenase pathway.38 Though several molecules have been developed with the objective of COX/LOX dual inhibitors, none of them reached the market in view of their unfavorable toxicity.34,39
In this direction the German pharmaceutical company, Merckle GmbH, together with Euro Alliance partners Alfa Wassermann and Lacer, developed licofelone (ML3000), which is a COX/LOX dual inhibitor, the first member of this new class of analgesic and anti-inflammatory drugs.40 It was shown to decrease the production of proinflammatory PGs and LTs with the potential to combine good analgesic and anti-inflammatory effects with excellent gastrointestinal tolerability.41 It is currently under evaluation for the treatment for osteoarthritis (OA), the most common form of arthritis. Although Phase III trials have been successfully completed in OA patients no dates for regulatory submission have been given (https://www.clinicaltrialsarena.com/projects/licofelone/).42 Later studies indicate that licofelone suppresses PGE2 formation by inhibiting mPGES-1, in addition to inhibition of COX-1 and COX-2.43 Our studies in this direction have resulted in the isolation of a natural product, chebulagic acid isolated from Terminalia chebula and 6-hydroxy salvinolone isolated from the roots of Premna integrifolia with COX-2/5-LOX dual inhibition and with potent anti-inflammatory and anticancer effects.44–46 However, further well-designed pre-clinical and clinical trials are required for evaluation of their efficacy and safety and further development.
Thus, the evolution of anti-inflammatory drugs, targeting COX and/or LOX enzymes, ie, from NSAIDs to COXIBs to CLOXIBs, have not resulted in the development of anti-inflammatory drugs without side-effects.47 Hence there is a need to explore novel targets for the development of anti-inflammatory drugs without side-effects.
Prostaglandins and Inflammation: Search for Novel Anti-Inflammatory Drug Targets
In view of several complications reported with anti-inflammatory drugs targeting COX and LOX enzymes, there is a need for alternate target(s) for treatment of inflammatory disorders. The COX enzyme is responsible for the formation of a stable intermediate, PGH2, which forms the precursor for the formation of prostaglandins (PGE2, PGF2a, PGD2, TXB2, PGI2, etc.) by tissue-specific enzymes.8 By targeting COX and/or LOX pathways, as seen in NSAIDs, COXIBs, and CLOXIBs, they not only block the formation of PGE2-, the major mediator of inflammation, but also block the formation of other downstream metabolites, which are essential for homeostatic functions.48,49 Also targeting these PLA2 or COX pathways is known to affect the production of histamine.50 Hence, the selection of a target and its validation in terms of other linked functions is very essential to come up with safe and effective drugs.
Targeting PGE2: The Possible Effects and Limitations
It is recognized that the key lipid mediator that is involved in chronic inflammation is PGE2, which has been implicated in tumor development.51 Also, PGE2 is a key PG that mediates several biological functions, such as blood pressure, fertility, immune responses, etc. Its action is mediated through the G-protein-coupled receptors, EP1-4.52 Dysregulation of PGE2 production is also linked with several complications.53 As a result, PGE2 metabolism and signaling formed as one of the areas being investigated actively so as to identify the most ideal therapeutic target (Figure 1). The significant elevation in microsomal PGE Synthase (mPGES) level has been observed in patients with myositis54 and gastric ulcers.55 Based on these studies, it is very clear that PGE2 is the major mediator of inflammation and hence the enzymes involved in PGE synthesis are being explored for development of anti-inflammatory drugs.56
 |
Figure 1 Cyclooxygenase pathway showing the formation of PGE2 and other metabolites. Inhibition of both COX-1 and COX-2 non-selectively by NSAIDs (green triangles) and selective inhibition of COX-2 by COXIBs (red triangles) is also shown. The potential targets in the COX pathway at various levels – Synthesis (1), Catabolism (2) and Signaling (3) are also indicated. Abbreviations: COX, cyclooxygenase; PGG2, prostaglandin G2; PGD2, prostaglandin D2; PGE2, prostaglandin E2; PGI2, prostacyclin; PGF2, prostaglandin F2α; TXA2, thromboxane A2. |
The synthesis of PGE2 from endoperoxide PGH2 is mediated by prostaglandin E synthases (PGES). There are three different PGE synthases such as cytosolic PGE synthase (cPGES) and two membrane-bound PGE synthases, mPGES-1 and mPGES-2. Of these, mPGES-2 and cPGES are constitutive enzymes, whereas mPGES-1 is an inducible enzyme. While mPGES-2 and cPGES are responsible for maintenance of constitutive levels of PGE2, mPGES-1 is mainly responsible for the synthesis of enhanced levels of PGE2 during inflammation. While COX-1 and cPGES function in coordination to maintain constitutive levels of PGE2, COX-2 and mPGES-1 are responsible for elevated levels of PGE2 under inflammatory conditions.57 Unlike NSAIDs, inhibitors of mPGES-1, which reduces the PGE2 production without affecting the synthesis of other PGs, may form better anti-inflammatory drug candidates without side-effects. Especially, mPGES-1 is the only inducible enzyme and in many pathological conditions it is co-induced with COX-2.58 Also, mPGES-1 is having an association in other inflammatory diseases of the brain such as ischemic stroke, Alzheimer’s disease, Parkinson’s disease, epilepsy, brain cancer, etc.58
Overexpression of mPGES-1 was reported in several types of cancers such as colorectal, gastric adenoma, gastric cancer, breast cancer, pancreatic cancer, papillary thyroid, ovarian cancer, head and neck cancer, lung cancer, etc.59 The in vitro/in vivo models with mPGES-1 knockout, RNA interference with siRNA or genetic deletion have a negative effect on tumor growth, which collectively indicates a role for mPGES-1 in cancer.59 The rest of the PGE synthases are constitutively expressing enzymes and may not form good targets for anti-inflammatory drug development. Bergqvist et al60 reported recently how selective inhibitors of mPGES-1 are beneficial. They have shown that mPGES-1 inhibitor, CIII, decreased PGE2 production and increased PGF2α and TXB2 whereas NS-398 (COX-2 inhibitor) blocked the entire PGs production in A549 lung cancer cells. Hence, the selective inhibitors of mPGES-1, reducing the production of only PGE2 with no effects on other PGs, may form a better target for the development of anti-inflammatory drugs without side-effects.
mPGES-1: A Better Target?
The mPGES-1, the terminal enzyme involved in the production of inducible prostaglandin E2, has become an attractive target for development of anti-inflammatory drugs. It is a homo trimer with three subunits of 16 kDa each with 152 amino acids. Microsomal prostaglandin E synthase-1, a membrane associated protein, is a member of the Membrane Associated Proteins in Eicosanoid and Glutathione metabolism (MAPEG) super family.59 MAPEG proteins are small proteins of 14–18 kDa and have a similar 3D structure. mPGES-1 is the terminal enzyme in the biosynthesis of PGE2. The first report on selective inhibitors of mPGES-1 came from the group of Riendeau et al61 in 2005, where they have shown a reduction in levels of PGE2 in IL-1β induced A549 cells.61 In contrast to COX-2 inhibition, mPGES-1 inhibition was associated with lower risk of cardiovascular side-effects, as it results in increasing PGI2 levels.62 Recently, Eli Lilly developed a compound inhibiting mPGES-1, LY3031207, but the study was terminated after in Phase I trial in view of drug induced liver injury.63 Glenmark Pharmaceuticals molecule, GRC 27864, a potent, selective, and orally bioavailable inhibitor of mPGES-1, is currently in Phase II clinical trials for evaluation of efficacy and safety in patients with moderate osteoarthritic pain (https://www.prnewswire.com/in/news-releases/glenmark-initiates-phase-iib-dose-range-finding-study-for-novel-molecule-grc-27864-669501853.html).64 These details and the progress in the development of mPGES-1 inhibitors over time have been reviewed recently.65
As of today there are no approved inhibitors of mPGES-1 in clinical practice due to different issues with the compounds themselves.66 In another study, Ding et al67 have developed a molecule based on a structure-based design of mPGES-1 inhibitors, which was found to be orally bioavailable and effective against both human and mouse mPGESs. Also, DREAM-in-CDM (Drug Repurposing Effort Applying Integrated Modeling-in vitro/in vivo-Clinical Data Mining), is another novel approach for structure-based screening of FDA-approved drugs against mPGES-1 drug repurposing.68 For example, lapatinib, a known anti-cancer drug, was identified as a potent inhibitor of mPGES-1, using this novel approach, and it can be used in treatment of inflammation, pain, and inflammation-related diseases.68 Design and development of a specific inhibitor of mPGES-1 is a challenging task as there are interspecies differences in the structure of mPGES-1 in humans and rodents. Also, the structural similarity of the mPGES-1 with others like Huntingtin interacting protein 12, microsomal glutathione-S-transferase (GST)-1-like 1 and protein tyrosine kinase 2 beta (PTK2B),59 makes the task much more challenging.
mPGES-1/5-LOX Dual Inhibitors
As discussed, the NSAIDs, COXIBs, and COX/5-LOX dual inhibitors (CLOXIBs), being used in the treatment of many inflammatory diseases, are associated with gastrointestinal, renal, and cardiovascular problems, as they are involved in blocking not only the PGE2 formation but also are inhibiting the formation of cytoprotective prostacyclin and other eicosanoids. In light of the above, attempts are being made to target microsomal prostaglandin E synthase-1 (mPGES-1) and 5-LOX, so as to spare COXs and thus the cytoprotective PGs. Several mPGES-1 inhibitors are being developed as potential candidates for the treatment of inflammatory disorders, including cancer.59,69,70 Licofelone, originally identified as a dual inhibitor blocking both COX and 5-LOX pathways, has also been shown to inhibit mPGES-1 activity with an IC50 value of 6 μM.43 Compared to licofelone, which inhibits both COXs and 5-LOX, mPGES-1/5-LOX dual inhibitors spare COX1/2 and thus may form better candidates for the development of next generation anti-inflammatory drugs. Further in-depth studies, however, are required to evaluate their efficacy in treating various inflammatory disorders and their safety.
Limiting the PGE2 Levels: Role of 15-Hydroxyprostaglandin Dehydrogenase (15-PGDH)
As pointed above, PGE2 has been identified as the key mediator of inflammation and as a result the enzymes limiting the formation of PGE2 have been exploited as the targets for the discovery and development of anti-inflammatory drugs: at PLA2 level, steroidal; COX-1 and/or -2 -non-steroidal/COX-2 specific; mPGES level. However, the effective level of PGE2 in the tissues ultimately depends on its rate of formation or anabolism and its rate of degradation or catabolism. The key enzyme involved in the catabolism of PGE2 has been identified as 15-PGDH (15-Hydroxyprostaglandin dehydrogenase). In fact, recent reports indicate the importance of PGE2 catabolism, on various inflammatory disorders, including cancer.71 15-hydroxy prostaglandin dehydrogenase (15-PGDH), a dimer with a molecular weight of 25–29 kDa, catalyzes the reversible oxidation of the 15-hydroxyl group of prostaglandins to produce a 15-keto metabolite with reduced biological activity.72 It exists in two isoforms, PGDH-1 and 15-PGDH-2. PGDH-1, NAD+ dependent enzyme, catalyzes the conversion of PGE2 to 15-keto-PGE2. 15-PGDH-2, on the other hand, is NADP+ dependent and has a much broader range of substrates and can also use NAD+ as a cofactor.72 Contrary to COX-2 which is being overexpressed in lung and many other tumors, 15-PGDH was found to be under-expressed. In inflammation associated human colon cancer upregulation of PGE2 is considered as a key event.73 15-PGDH a PGE2 catabolizing enzyme is downregulated in colon cancer.74 15d-PGJ2, a peroxisome proliferator activated receptor-γ ligand, which has anticarcinogenic activities, upregulates the expression of 15-PGDH in the HCT119 human colon cancer cell line.75 15-PGDH and miR-21 are inversely correlated in colorectal cancer (CRC) patients. It has been reported that dysregulated expression of miR-21 contributes to the loss of PGDH expression and promotes CRC progression via accumulation of PGE2.76 miR-21 also induces loss of 15-PGDH in early gastric tubular adenocarcinoma.77 It was shown that inhibition of 15-PGDH enhances KRAS-driven tumor progression via all -trans retinoic acid (ATRA) depletion in the pancreas.78 Thus, downregulation of 15-PGDH expression in lung and other tumors suggest that this enzyme is a tumor suppressor. Docosahexaenoic acid (DHA), a polyunsaturated fatty acid of omega 3 class, significantly reduces PGE2 levels in line with upregulation of 15-PGDH in endothelial cells.79 It was revealed that NAD+ triggered reactive oxygen species (ROS) mediate degradation of 15-PGDH and drove cells to undergo EMT transition.80 It was shown that 15-PGDH-derived 15-keto-PGE2 from hepatocytes is able to activate PPAR-γ and inhibit inflammatory cytokine production in Kupffer cells and that this paracrine mechanism negatively regulates the LPS-induced necro-inflammatory response in the liver.81
In addition to its pro-inflammatory and pathogenic role, PGE2 is an important mediator for bone formation, gastric ulcer healing, and dermal wound healing. Elevation of PGE2 using a 15-PGDH inhibitor was proved to be valuable for the management of diseases that require elevated PGE2.82 Inhibitors of 15-PGDH are anticipated to elevate the in vivo levels of PGE2 and promote healing and tissue regeneration.83 PGDH inhibition attenuated acute inflammation and weight loss and decreased mortality in murine pulmonary fibrosis.84 7,3ʹ,4ʹ-Trimethoxyflavone (TMF), a potent inhibitor of 15-PGDH, facilitated in vitro wound healing in a HaCaT cell scratch model, which was completely inhibited by adding both 15-PGDH and NAD+ as a cofactor, confirming the involvement of PGE2 in its wound healing effect.85 Recently it was shown that inhibition of 15-PGDH could alleviate LPS-induced acute kidney injury by regulating the apoptosis, autophagy, and oxidative stress rather than inflammation in mice.86
The foregoing studies clearly indicate that 15-PGDH is weakly expressed in many types of inflammatory disorders and cancers, where COX-2 and mPGES-1 are overexpressed. Such a situation eventually leads to accumulation of PGE2 levels and disease manifestation. As a result, the pharmacological inhibition of 15-PGDH, which elevates PGE2 levels, may not be a right choice for treating inflammatory disorders. On the contrary agents which induce 15-PGDH could be explored for the treatment of inflammatory disorders. This in fact appears to be the mechanism by which many of the disease modifying anti-rheumatic drugs (DMARDs) like hydroxychloroquine bring in therapeutic efficacy in osteo arthritis and rheumatoid arthritis, where 15-Hydroxyprostaglandin dehydrogenase is upregulated.87 Similarly, the beneficial effects of hydroxychloroquine in the case of COVID-19 patients may also be mediated through similar induction of 15-PGDH and decreased PGE2 levels. However, the impact of long-term use of such DMARDs on normal physiological functions requiring high levels of PGE2 such as bone formation, gastric ulcer healing, and dermal wound healing needs to be assessed. In such a scenario the alternative target which will not affect other normal functions could be the specific receptors of PGE2, which mediate inflammatory responses.
PGE2 (EP) Receptors as Potential Targets
The targets discussed so far, starting from PLA2, COX, and/or LOX, mPGES-1, 15-PGDH, mediate not only inflammatory pathogenesis but also are involved in key homeostatic functions. As a result, it is quite expected to have therapeutic efficacy on one side and side-effects on the other. Hence there is a need to identify a target that is exclusively involved in mediating inflammatory pathologies. In this direction the PGE2 receptor(s) mediating the inflammatory responses may form the right choice.
Prostaglandins mediate their effects through a class of G-protein-coupled receptors (GPCRs). PGE2 also mediates its effects through GPCRs, EP 1–4, which are expressed tissue specifically.52 Among these four receptors, the EP3 and EP4 have high affinity towards PGE2, while the EP1 and EP2 receptors are activated only by the high levels of PGE2. In view of their pathogenic link with inflammation and cancer, the antagonists of EP receptors have become potential drug targets and constitute an intense area of research in anti-inflammatory drug discovery.88
The developments on the therapeutic roles of EP receptor ligands have recently been reviewed10 in diseases such as Ulcerative colitis (EP4 agonists: rivenprost, KAG-308, ONO-AE1-329), Glaucoma and intraocular hypertension (EP2 agonist: taprenepag isopropyl), Solid tumors (EP4 antagonists: grapiprant, ONO-AE3-208, GW627368X, AH23848), Colorectal cancer (EP1 antagonist: ONO-8711 and EP4 antagonist: ONO-AE-227), Cardiovascular diseases (EP3 agonists: GR 63799X, MB-28767, ONO-AE-248, TEI-3356, and EP4 agonists: EP4RAG), etc. From these studies it is observed that EP4 receptor antagonists show greater potential in treating pain, inflammation, and solid tumors.10 Hence it is important to solve the EP receptors (even ligand-bound EP receptors) structures in high resolution for rational drug design. It is also important to develop antagonists specific to EP4, which is the key receptor mediating inflammatory disorders, without affecting other receptors of PGE2, which mediate homeostatic functions of PGE2. Targeting EP receptors thus appear promising, but safety profiles of these targets have not been studied.89 Further in-depth studies on the efficacy and safety of targeting EP receptors may form rewarding exercise for the Pharma companies.
Conclusions and Future Directions
In conclusion, the discovery and development of anti-inflammatory drugs is mostly based on targeting prostaglandin biosynthesis, to limit the formation of Prostaglandin E2 via the cyclooxygenases. This approach though resulted in putting forth a number of drugs into the market, it has major limitations due to the undesirable side-effects. Hence there is now a shift in identifying the targets downstream of COXs, which limit the levels of PGE2 or block its specific receptors. Also, efforts are being made for simultaneous targeting of the 5-LOX pathway which is involved in the formation of inflammatory leukotrienes. The efforts made so far in this direction include:
a. Inhibitors of Phospholipases, which are involved in the release of arachidonic acid from membrane phospholipids.
b. Inhibitors of the COX enzymes – (NSAIDs).
c. Specific COX-2 (COXIBs) and COX-2 and 5-LOX dual inhibitors (CLOXIBs).
The drugs developed in this direction, though approved and in the market, have undesirable side-effects, as they affect the formation of other key metabolites mediating physiological functions. The focus, therefore, is now on downstream targets such as:
- PGE synthases, specifically the inducible isoform – mPGES-1.
- Enzymes involved in the metabolism of PGE2, specifically 15-PGDH.
- PGE receptors, specifically EP4 which mediates inflammation.
Figure 2 summarizes the past, present, and future drug targets for the discovery and development of anti-inflammatory drugs. Hopefully, some of these novel approaches might lead to next generation anti-inflammatory drugs with enhanced efficacy and safety in the near future.
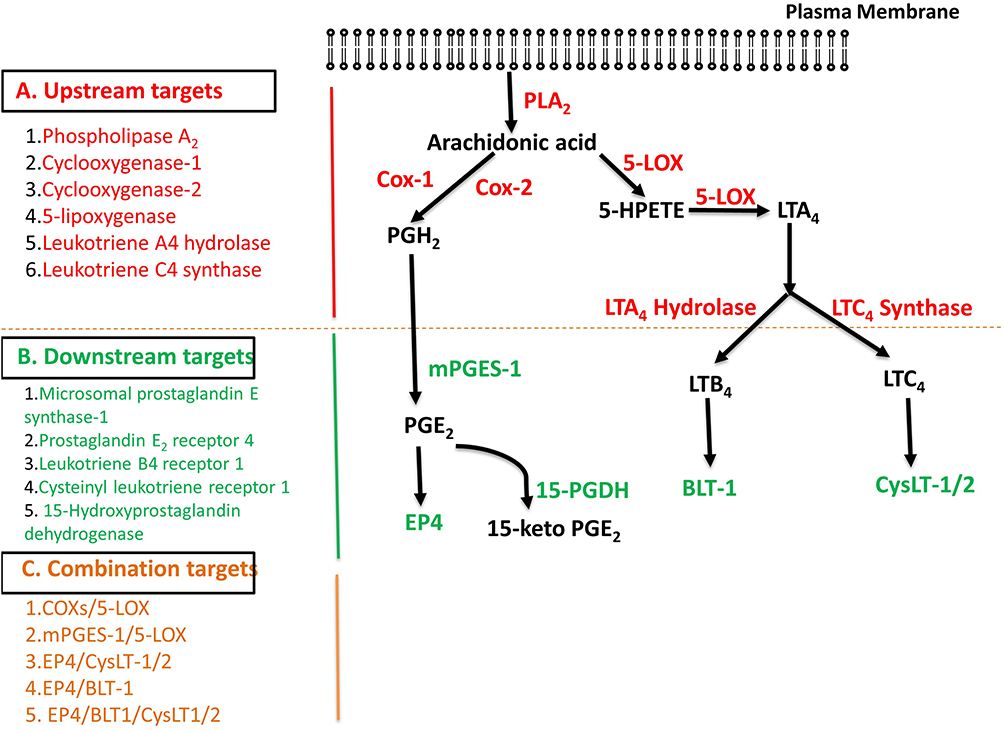 |
Figure 2 The metabolism of arachidonic acid via the cyclooxygenase and lipoxygenase pathways showing the possible up-stream (A), down-stream (B), and combination targets (C) for the discovery and development of anti-inflammatory drugs. Abbreviations: COX1, cyclooxygenase 1; COX2, cyclooxygenase 2; NSAIDs, non-steroidal anti-inflammatory drugs; COXIBs, COX-2 inhibitors; mPGES, microsomal prostaglandin E synthase; cPGES, cytosolic prostaglandin E2 synthase; 15-PGDH, 15-hydroxyprostaglandin dehydrogenase; EP4, prostaglandin E2 (PGE2) receptor-4. |
Abbreviations
AA, arachidonic acid; COX, cyclooxygenase; COX-1, cyclooxygenase-1; COX-2, cyclooxygenase-2; mPGES-1, microsomal prostaglandin E synthase-1; mPGES-2, microsomal prostaglandin E synthase-2; cPGES, cytosolic prostaglandin E2 synthase; PGs, prostaglandins; PGE2, prostaglandin E2; 15-PGDH, 15-hydroxyprostaglandin dehydrogenase; LOX, lipoxygenase; NSAIDs, non-steroidal anti-inflammatory drugs; COXIBs, cyclooxygenase-2 specific inhibitors; EP1, EP2, EP3 & EP4, E prostanoid receptor subtypes 1,2,3 and 4; PGG2, prostaglandin G2; PGH2, prostaglandin H2; PGD2, prostaglandin D2; PGF2α, prostaglandin F2α; PGI2, prostacyclin; TxA2, thromboxane.
Acknowledgments
The authors gratefully acknowledge the financial support received from the Department of Science and Technology (# VI, D&P/560/2016-17/TDT) and BSR Faculty Fellowship from the Ministry of Human Resources Development to PR (Award Letter No. F. 18-1/2011 (BSR) dated June 26, 2018). Dr. K. Anil Kumar acknowledges Senior Research Associate Fellowship from Council of Scientific and Industrial Research (CSIR-SRA Pool No (13(8956-A)/2017Pool).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPARgamma. J Biol Chem. 2000;275(36):28028–28032. doi:10.1074/jbc.M001387200
2. Newton R, Kuitert LM, Bergmann M, Adcock IM, Barnes PJ. Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem Biophys Res Commun. 1997;237(1):28–32. doi:10.1006/bbrc.1997.7064
3. Korbecki J, Baranowska-Bosiacka I, Gutowska I, Piotrowska K, Chlubek D. Cyclooxygenase-1 as the main source of proinflammatory factors after sodium orthovanadate treatment. Biol Trace Elem Res. 2015;163(1–2):103–111. doi:10.1007/s12011-014-0176-4
4. Olszowski T, Gutowska I, Baranowska-Bosiacka I, et al. The effect of Cadmium on COX-1 and COX-2 gene, protein expression, and enzymatic activity in THP-1 macrophages. Biol Trace Elem Res. 2015;165(2):135–144. doi:10.1007/s12011-015-0234-6
5. Chen R, Zhao LD, Liu H, et al. Fluoride induces neuroinflammation and alters Wnt signaling pathway in BV2 microglial cells. Inflammation. 2017;40(4):1123–1130. doi:10.1007/s10753-017-0556-y
6. Gutowska I, Baranowska-Bosiacka I, Safranow K, et al. Fluoride in low concentration modifies expression and activity of 15 lipoxygenase in human PBMC differentiated monocyte/macrophage. Toxicology. 2012;295(1–3):23–30. doi:10.1016/j.tox.2012.02.014
7. Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi:10.1146/annurev.biochem.69.1.145
8. Hara S. Prostaglandin terminal synthases as novel therapeutic targets. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(9):703–723. doi:10.2183/pjab.93.044
9. Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282(16):11613–11617. doi:10.1074/jbc.R600038200
10. Markovic T, Jakopin Z, Dolenc MS, Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug Discov Today. 2017;22(1):57–71. doi:10.1016/j.drudis.2016.08.003
11. M.Metters MK. Prostanoid receptors. In: Annual Reports in Medicinal Chemistry. Vol. 33. Elsevier; 1998.
12. Vane JR. The fight against rheumatism: from willow bark to COX-1 sparing drugs. J Physiol Pharmacol. 2000;51(4 Pt 1):573–586.
13. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231(25):232–235. doi:10.1038/newbio231232a0
14. Horl WH. Nonsteroidal anti-inflammatory drugs and the kidney. Pharmaceuticals (Basel). 2010;3(7):2291–2321. doi:10.3390/ph3072291
15. Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A. 1991;88(7):2692–2696. doi:10.1073/pnas.88.7.2692
16. Lee SH, Soyoola E, Chanmugam P, et al. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem. 1992;267(36):25934–25938. doi:10.1016/S0021-9258(18)35698-9
17. Liu C, Duan Z, Guan Y, et al. Increased expression of tight junction protein occludin is associated with the protective effect of mosapride against aspirin-induced gastric injury. Exp Ther Med. 2018;15(2):1626–1632. doi:10.3892/etm.2017.5550
18. Yehiyan A, Barman S, Varia H, Pettit S. Short-course high-dose ibuprofen causing both early and delayed jejunal perforations in a non-smoking man. BMJ Case Rep. 2017:
19. Kim JH, Jin S, Kwon HJ, Kim BW. Curcumin blocks naproxen-induced gastric antral ulcerations through inhibition of lipid peroxidation and activation of enzymatic scavengers in rats. J Microbiol Biotechnol. 2016;26(8):1392–1397. doi:10.4014/jmb.1602.02028
20. Risty GM, Najarian MM, Shapiro SB. Multiple indomethacin-induced jejunal ulcerations with perforation: a case report with histology. Am Surg. 2007;73(4):344–346. doi:10.1177/000313480707300406
21. Kessler WF, Shires GT, Fahey TJ. Surgical complications of nonsteroidal antiinflammatory drug-induced small bowel ulceration. J Am Coll Surg. 1997;185(3):250–254. doi:10.1016/s1072-7515(97)00067-7
22. Washio E, Esaki M, Maehata Y, et al. Proton pump inhibitors increase incidence of nonsteroidal anti-inflammatory drug-induced small bowel injury: a randomized, placebo-controlled trial. Clin Gastroenterol Hepatol. 2016;14(6):809–815. doi:10.1016/j.cgh.2015.10.022
23. Zaffanello M, Brugnara M, Angeli S, Cuzzolin L. Acute non-oliguric kidney failure and cholestatic hepatitis induced by ibuprofen and acetaminophen: a case report. Acta Paediatr. 2009;98(5):903–905. doi:10.1111/j.1651-2227.2008.01209.x
24. Yue Z, Jiang P, Sun H, Wu J. Association between an excess risk of acute kidney injury and concomitant use of ibuprofen and acetaminophen in children, retrospective analysis of a spontaneous reporting system. Eur J Clin Pharmacol. 2014;70(4):479–482. doi:10.1007/s00228-014-1643-8
25. Zhang J, Ding EL, Song Y. Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials. JAMA. 2006;296(13):1619–1632. doi:10.1001/jama.296.13.jrv60015
26. Schmidt M, Sorensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018;362:k3426. doi:10.1136/bmj.k3426
27. Zarghi A, Arfaei S. Selective COX-2 inhibitors: a review of their structure-activity relationships. Iran J Pharm Res. 2011;10(4):655–683.
28. Hegazy R, Alashhab M, Amin M. Cardiorenal effects of newer NSAIDs (Celecoxib) versus classic NSAIDs (Ibuprofen) in patients with arthritis. J Toxicol. 2011;2011:862153. doi:10.1155/2011/862153
29. Mendes RT, Stanczyk CP, Sordi R, Otuki MF, Dos Santos FA, Fernandes D. Selective inhibition of cyclooxygenase-2: risks and benefits. Rev Bras Reumatol. 2012;52(5):767–782.
30. Patrono C. Cardiovascular effects of cyclooxygenase-2 inhibitors: a mechanistic and clinical perspective. Br J Clin Pharmacol. 2016;82(4):957–964. doi:10.1111/bcp.13048
31. Bello AE, Holt RJ. Cardiovascular risk with non-steroidal anti-inflammatory drugs: clinical implications. Drug Saf. 2014;37(11):897–902. doi:10.1007/s40264-014-0207-2
32. Neeraja S, Sreenath AS, Reddy PR, Reddanna P. Expression of cyclooxygenase-2 in rat testis. Reprod Biomed Online. 2003;6(3):302–309. doi:10.1016/S1472-6483(10)61849-4
33. Kirkby NS, Chan MV, Zaiss AK, et al. Systematic study of constitutive cyclooxygenase-2 expression: role of NF-kappaB and NFAT transcriptional pathways. Proc Natl Acad Sci U S A. 2016;113(2):434–439. doi:10.1073/pnas.1517642113
34. Martel-Pelletier J, Lajeunesse D, Reboul P, Pelletier JP. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann Rheum Dis. 2003;62(6):501–509. doi:10.1136/ard.62.6.501
35. Brune K. Safety of anti-inflammatory treatment—new ways of thinking. Rheumatology. 2004;43(suppl_1):i16–i20. doi:10.1093/rheumatology/keh104
36. Ye YN, Wu WK, Shin VY, Bruce IC, Wong BC, Cho CH. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis. 2005;26(4):827–834. doi:10.1093/carcin/bgi012
37. Agarwal S, Reddy GV, Reddanna P. Eicosanoids in inflammation and cancer: the role of COX-2. Expert Rev Clin Immunol. 2009;5(2):145–165. doi:10.1586/1744666X.5.2.145
38. Sala A, Zarini S, Bolla M. Leukotrienes: lipid bioeffectors of inflammatory reactions. Biochemistry (Mosc). 1998;63(1):84–92.
39. Manju SL, Ethiraj KR, Elias G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: a structure-based approach. Eur J Pharm Sci. 2018;121:356–381. doi:10.1016/j.ejps.2018.06.003
40. Kulkarni SK, Singh VP. Licofelone–a novel analgesic and anti-inflammatory agent. Curr Top Med Chem. 2007;7(3):251–263. doi:10.2174/156802607779941305
41. Cicero AF, Laghi L. Activity and potential role of licofelone in the management of osteoarthritis. Clin Interv Aging. 2007;2(1):73–79. doi:10.2147/ciia.2007.2.1.73
42. Gaur K, Kori ML, Tyagi LK, et al. Licofelone- Novel Analgesic and Anti-Inflammatory Agent for Osteoarthritis: An Overview. Journal of Young Pharmacists. 2009;1(1):67–71. doi:10.4103/0975-1483.51884.
43. Koeberle A, Siemoneit U, Buhring U, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326(3):975–982. doi:10.1124/jpet.108.139444
44. Reddy DB, Reddanna P. Chebulagic acid (CA) attenuates LPS-induced inflammation by suppressing NF-kappaB and MAPK activation in RAW 264.7 macrophages. Biochem Biophys Res Commun. 2009;381(1):112–117. doi:10.1016/j.bbrc.2009.02.022
45. Reddy DB, Reddy TC, Jyotsna G, et al. Chebulagic acid, a COX-LOX dual inhibitor isolated from the fruits of Terminalia chebula Retz., induces apoptosis in COLO-205 cell line. J Ethnopharmacol. 2009;124(3):506–512. doi:10.1016/j.jep.2009.05.022
46. Azad R, Babu NK, Gupta AD, Reddanna P. Evaluation of anti-inflammatory and immunomodulatory effects of Premna integrifolia extracts and assay-guided isolation of a COX-2/5-LOX dual inhibitor. Fitoterapia. 2018;131:189–199. doi:10.1016/j.fitote.2018.10.016
47. Mahipal V. Suraneni GVR, and Reddanna Pallu. NSAIDs, COXIBs, CLOXIBs and Cancer Pain. Horizons Cancer Res. 2011;47.
48. Hunter LJ, Wood DM, Dargan PI. The patterns of toxicity and management of acute nonsteroidal anti-inflammatory drug (NSAID) overdose. Open Access Emerg Med. 2011;3:39–48. doi:10.2147/OAEM.S22795
49. Yakovleva OO, Zhamba AO, Doroshkevych IO, Vitruk TK. Cardiac toxicity of coxibs: mechanisms of development and their prevention. Pain Med. 2018;3(3):27–32. doi:10.31636/pmjua.v3i3.3
50. Morita Y, Aida N, Miyamoto T. Role of phospholipase A2 activation in histamine release from human basophils. Allergy. 1983;38(6):413–418. doi:10.1111/j.1398-9995.1983.tb05084.x
51. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181–193. doi:10.1038/nrc2809
52. Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol. 2009;49:123–150. doi:10.1146/annurev.pharmtox.011008.145616
53. Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int J Biochem Cell Biol. 2010;42(2):198–201. doi:10.1016/j.biocel.2009.09.015
54. Korotkova M, Helmers SB, Loell I, et al. Effects of immunosuppressive treatment on microsomal prostaglandin E synthase 1 and cyclooxygenases expression in muscle tissue of patients with polymyositis or dermatomyositis. Ann Rheum Dis. 2008;67(11):1596–1602. doi:10.1136/ard.2007.079525
55. Gudis K, Tatsuguchi A, Wada K, et al. Microsomal prostaglandin E synthase (mPGES)-1, mPGES-2 and cytosolic PGES expression in human gastritis and gastric ulcer tissue. Lab Invest. 2005;85(2):225–236. doi:10.1038/labinvest.3700200
56. Koeberle A, Werz O. Perspective of microsomal prostaglandin E2 synthase-1 as drug target in inflammation-related disorders. Biochem Pharmacol. 2015;98(1):1–15. doi:10.1016/j.bcp.2015.06.022
57. Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59(3):207–224. doi:10.1124/pr.59.3.1
58. Ikeda-Matsuo Y. The role of mPGES-1 in inflammatory brain diseases. Biol Pharm Bull. 2017;40(5):557–563. doi:10.1248/bpb.b16-01026
59. Chang HH, Meuillet EJ. Identification and development of mPGES-1 inhibitors: where we are at? Future Med Chem. 2011;3(15):1909–1934. doi:10.4155/fmc.11.136
60. Bergqvist F, Ossipova E, Idborg H, et al. Inhibition of mPGES-1 or COX-2 results in different proteomic and lipidomic profiles in A549 lung cancer cells. Front Pharmacol. 2019;10:636. doi:10.3389/fphar.2019.00636
61. Riendeau D, Aspiotis R, Ethier D, et al. Inhibitors of the inducible microsomal prostaglandin E-2 synthase (mPGES-1) derived from MK-886. Bioorg Med Chem Lett. 2005;15:3352–3355. doi:10.1016/j.bmcl.2005.05.027
62. Ozen G, Gomez I, Daci A, et al. Inhibition of microsomal PGE synthase-1 reduces human vascular tone by increasing PGI2: a safer alternative to COX-2 inhibition. Br J Pharmacol. 2017;174(22):4087–4098. doi:10.1111/bph.13939
63. Jin Y, Regev A, Kam J, et al. Dose-dependent acute liver injury with hypersensitivity features in humans due to a novel microsomal prostaglandin E synthase 1 inhibitor. Br J Clin Pharmacol. 2018;84(1):179–188. doi:10.1111/bcp.13423
64. Glenmark initiates phase IIb dose range finding study for novel molecule GRC 27864 [press release]. PRNewswire; 2018.
65. Bergqvist F, Morgenstern R, Jakobsson PJ. A review on mPGES-1 inhibitors: from preclinical studies to clinical applications. Prostaglandins Other Lipid Mediat. 2020;147:106383. doi:10.1016/j.prostaglandins.2019.106383
66. Zhou Z, Yuan Y, Zhou S, Ding K, Zheng F, Zhan CG. Selective inhibitors of human mPGES-1 from structure-based computational screening. Bioorg Med Chem Lett. 2017;27(16):3739–3743. doi:10.1016/j.bmcl.2017.06.075
67. Ding K, Zhou Z, Hou S, et al. Structure-based discovery of mPGES-1 inhibitors suitable for preclinical testing in wild-type mice as a new generation of anti-inflammatory drugs. Sci Rep. 2018;8(1):5205. doi:10.1038/s41598-018-23482-4
68. Zhou S, Zhou Z, Ding K, et al. DREAM-in-CDM approach and identification of a new generation of anti-inflammatory drugs targeting mPGES-1. Sci Rep. 2020;10(1):10187. doi:10.1038/s41598-020-67283-0
69. Nakanishi M, Gokhale V, Meuillet EJ, Rosenberg DW. mPGES-1 as a target for cancer suppression: A comprehensive invited review “Phospholipase A2 and lipid mediators”. Biochimie. 2010;92(6):660–664. doi:10.1016/j.biochi.2010.02.006
70. Kock A, Larsson K, Bergqvist F, et al. Inhibition of microsomal prostaglandin E synthase-1 in cancer-associated fibroblasts suppresses Neuroblastoma tumor growth. EBioMedicine. 2018;32:84–92. doi:10.1016/j.ebiom.2018.05.008
71. Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013;35(2):123–137. doi:10.1007/s00281-012-0342-8
72. Charles MarkEnsor -H-H. 15-Hydroxyprostaglandin dehydrogenase. J Lipid Mediat Cell Signal. 1995;12(2–3):313–319. doi:10.1016/0929-7855(95)00040-W
73. Greenhough A, Smartt HJ, Moore AE, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30(3):377–386. doi:10.1093/carcin/bgp014
74. Myung SJ, Rerko RM, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(32):12098–12102. doi:10.1073/pnas.0603235103
75. Park JM, Na HK. Erratum: 15-deoxy-delta(12,14)-prostaglandin J2 upregulates the expression of 15-hydroxyprostaglandin dehydrogenase by inducing AP-1 activation and heme oxygenase-1 expression in human colon cancer cells. J Cancer Prev. 2019;24(4):245. doi:10.15430/JCP.2019.24.4.245
76. Monteleone NJ, Moore AE, Iacona JR, Lutz CS, Dixon DA. miR-21-mediated regulation of 15-hydroxyprostaglandin dehydrogenase in colon cancer. Sci Rep. 2019;9(1):5405. doi:10.1038/s41598-019-41862-2
77. Park YS, Lee JH, Jung DB, et al. MicroRNA-21 induces loss of 15-hydroxyprostaglandin dehydrogenase in early gastric tubular adenocarcinoma. Sci Rep. 2018;8(1):17717. doi:10.1038/s41598-018-36139-z
78. Arima K, Ohmuraya M, Miyake K, et al. Inhibition of 15-PGDH causes Kras-driven tumor expansion through prostaglandin E2-ALDH1 signaling in the pancreas. Oncogene. 2019;38(8):1211–1224. doi:10.1038/s41388-018-0510-y
79. Yin J, Xia W, Zhang Y, et al. Role of dihydroartemisinin in regulating prostaglandin E2 synthesis cascade and inflammation in endothelial cells. Heart Vessels. 2018;33(11):1411–1422. doi:10.1007/s00380-018-1190-9
80. Wang W, Hu Y, Wang X, Wang Q, Deng H. ROS-mediated 15-hydroxyprostaglandin dehydrogenase degradation via cysteine oxidation promotes NAD(+)-mediated epithelial-mesenchymal transition. Cell Chem Biol. 2018;25(3):255–261 e254. doi:10.1016/j.chembiol.2017.12.008
81. Yao L, Chen W, Song K, et al. 15-hydroxyprostaglandin dehydrogenase (15-PGDH) prevents lipopolysaccharide (LPS)-induced acute liver injury. PLoS One. 2017;12(4):e0176106. doi:10.1371/journal.pone.0176106
82. Karna S. In-vitro wound healing effect of 15-hydroxyprostaglandin dehydrogenase inhibitor from plant. Pharmacogn Mag. 2017;13(Suppl 1):S122–S126. doi:10.4103/0973-1296.203971
83. Antczak MI, Zhang Y, Wang C, et al. Inhibitors of 15-prostaglandin dehydrogenase to potentiate tissue repair. J Med Chem. 2017;60(9):3979–4001. doi:10.1021/acs.jmedchem.7b00271
84. Smith JNP, Witkin MD, Jogasuria AP, et al. Therapeutic targeting of 15-PGDH in murine pulmonary fibrosis. Sci Rep. 2020;10(1):11657.
85. Sun KH, Karna S, Moon YS, Cho H, Choi CH. The wound-healing effect of 7,3ʹ,4ʹ-trimethoxyflavone through increased levels of prostaglandin E2 by 15-hydroxyprostaglandin dehydrogenase inhibition. Biotechnol Lett. 2017;39(10):1575–1582. doi:10.1007/s10529-017-2386-2
86. Miao S, Lv C, Liu Y, et al. Pharmacologic blockade of 15-PGDH protects against acute renal injury induced by LPS in mice. Front Physiol. 2020;11:138. doi:10.3389/fphys.2020.00138
87. Kim HJ, Lee S, Lee HY, Won H, Chang SH, Nah SS. 15-hydroxyprostaglandin dehydrogenase is upregulated by hydroxychloroquine in rheumatoid arthritis fibroblast-like synoviocytes. Mol Med Rep. 2015;12(3):4141–4148. doi:10.3892/mmr.2015.3931
88. O’Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br J Pharmacol. 2015;172(22):5239–5250. doi:10.1111/bph.13331
89. Chen L. Current trends in PGE2 targeting for anti-inflammatory therapy. Pharm Bioprocess. 2016;4(3).
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.