Back to Journals » Cancer Management and Research » Volume 11
OSM-induced CD44 contributes to breast cancer metastatic potential through cell detachment but not epithelial-mesenchymal transition
Authors Covert H, Mellor LF, Wolf CL, Ankenbrandt N, Emathinger JM ![]() , Tawara K, Oxford JT
, Tawara K, Oxford JT ![]() , Jorcyk CL
, Jorcyk CL
Received 14 March 2019
Accepted for publication 4 July 2019
Published 15 August 2019 Volume 2019:11 Pages 7721—7737
DOI https://doi.org/10.2147/CMAR.S208721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Hunter Covert,1 Liliana F Mellor,2,3 Cody L Wolf,1 Nicole Ankenbrandt,1 Jacqueline M Emathinger,2 Ken Tawara,1 Julie Thom Oxford,1,2 Cheryl L Jorcyk1,2
1Boise State University, Biomolecular Sciences Program, Boise, ID 83725, USA; 2Boise State University, Department of Biological Sciences, Boise, ID 83725, USA; 3Oncología Molecular, Centro Nacional de Investigaciones Oncologicas (CNIO), Madrid 28029, Spain
Background: Hormone receptor status in human breast cancer cells is a strong indicator of the aggressiveness of a tumor. Triple negative breast cancers (TNBC) are aggressive, difficult to treat, and contribute to high incidences of metastasis by possessing characteristics such as increased tumor cell migration and a large presence of the transmembrane protein, cluster of differentiation 44 (CD44) on the cell membrane. Estrogen receptor-positive (ER+) cells are less aggressive and do not migrate until undergoing an epithelial-mesenchymal transition (EMT).
Methods: The relationship between EMT and CD44 during metastatic events is assessed by observing changes in EMT markers, tumor cell detachment, and migration following cytokine treatment on both parental and CD44 knockdown human breast tumor cells.
Results: ER+ T47D and MCF-7 human breast cancer cells treated with OSM demonstrate increased CD44 expression and CD44 cleavage. Conversely, ER- MDA-MB-231 human breast cancer cells do not show a change in CD44 expression nor undergo EMT in the presence of OSM. In ER+ cells, knockdown expression of CD44 by shRNA did not prevent EMT but did change metastatic processes such as cellular detachment and migration. OSM-induced migration was decreased in both ER+ and ER- cells with shCD44 cells compared to control cells, while the promotion of tumor cell detachment by OSM was decreased in ER+ MCF7-shCD44 cells, as compared to control cells. Interestingly, OSM-induced detachment in ER- MDA-MB-231-shCD44 cells that normally don’t detach at significant rates.
Conclusion: OSM promotes both EMT and tumor cell detachment in ER+ breast cancer cells. Yet, CD44 knockdown did not affect OSM-induced EMT in these cells, while independently decreasing OSM-induced cell detachment. These results suggest that regulation of CD44 by OSM is important for at least part of the metastatic cascade in ER+ breast cancer.
Keywords: epithelial to mesenchymal transition, Oncostatin M, cluster of differentiation 44, breast tumor metastasis
Background
The primary cause of mortality in patients with breast cancer is not from the primary tumor itself but its metastasis.1–4 Continued studies addressing breast cancer metastasis are important for improved patient survival and to gain a better understanding of the multitude of events that take place during metastatic disease. In order for tumor cells to metastasize they must undergo a phenotypic change known as an epithelial-mesenchymal transition (EMT), which allows for the intravasation of the tumor cell into a nearby blood vessel or lymphatic channel.5–7 Once the primary breast tumor cells are within the network of vessels they undergo a homing process to specific organs within the body,8,9 such as the bones, lung, liver, and brain.10–18 The tumor microenvironment (TME) plays a major role in the metastatic cascade,19 and signaling from cytokines present in the TME lead to tumor cell phenotypic changes necessary for metastasis to occur.20 Inflammatory cytokines of the interleukin-6 (IL-6) family such as oncostatin M (OSM) have been shown to be important in driving tumor invasion and metastasis.21
OSM is a pleiotropic, IL-6-family cytokine, which is important in inflammation exhibited during breast tumor development.22–24 Tumor cells, as well as monocytes, macrophages, and neutrophils, secrete OSM.25 OSM activates the JAK/STAT, MAPK, and PI3K/AKT pathways via binding its receptors, OSM receptor beta (OSMRβ) or leukemia inhibitory factor receptor beta (LIFRβ), each dimerized with a common gp130 subunit.21,26,27 Our previous data and data from the literature have shown that OSM inhibits proliferation of breast cancer cells, while increasing metastatic capacity.28,29 OSM causes EMT, cell detachment, migration, and an overall increase in tumor cell aggressiveness. Proteins and events involved in breast tumor metastasis are directly affected by OSM signaling, which creates a more aggressive phenotype through the upregulation of proteins such as CD44.
Cluster of differentiation 44 (CD44) is a transmembrane protein involved in cell adhesion and migration after it undergoes cleavage from proteolytic activity.30 Standard CD44 (CD44s) is roughly 80–85 kDa depending on post-translational modifications.31–33 CD44 contains a carboxy-terminal portion known as the cytoplasmic tail and an amino-terminal portion outside the membrane, folded together by post-translational glycosylation.34,35 CD44 is a major receptor for binding hyaluronic acid (HA) in the extracellular matrix,36,37 and it binds HA by its stem region that is directly above the surface of the membrane and includes domains derived from alternatively spliced exons.38,39 Alternatively spliced CD44, such as CD44v6 has been found in multiple cancers types including gastric and head and neck carcinoma.40,41 Depending on the structure of CD44 with its multiple combinations of exons, signaling differs based on how it is recognized by an upstream ligand.42,43 The varying roles of CD44 raises the complexity while studying the protein. The protein itself is not a potential therapeutic target from a global knockdown scale because signaling through CD44 is crucial to the health and function of many different cells including T cells,44 while others have shown a potential role for CD44 during metastatic processes such as EMT.
CD44 consists of 20 exons; 1–5 and 16–20 comprise CD44s, whereas 6–15 provide the exons which are capable of undergoing alternative splicing.45–47 Variant exons 6–15 are termed v1, v2, v3, etc. through v10, and many CD44 variant (CD44v) isoforms exist.47,48 CD44v6 contains only variant exon 6 plus the CD44 standard and is associated with highly aggressive tumors.49 CD44v3-10 contains the standard exons plus variant exons v3-v10 and is less common than CD44v6.50 ER+ cells possessing an epithelial phenotype display CD44v8-10 known as CD44E (CD44 epithelial).51 Regardless of the combination of exons comprising CD44v, exons from the CD44s remain present despite alternative splicing.52
As the metastatic process begins, CD44 expression is upregulated and CD44 protein is cleaved, which correlates within the same time frame that EMT takes place.53 EMT and the upregulation of CD44 have been shown to be involved together in breast tumor metastasis.54–56 The correlation between EMT and CD44 during metastasis has been shown as the standard form of CD44 becomes the dominant form of the CD44 protein while EMT takes place.57 The direct association and timing of EMT and cellular detachment has yet to be linked to CD44 expression. CD44s and CD44v have been shown to be strong biomarkers commonly found in higher percentages of more aggressive tumor populations,58 that are assumed to have previously undergone EMT. Although both CD44 and EMT are implicated in breast tumor progression and metastasis, establishing a direct connection between the two has proved to be challenging. A connection between CD44 and tumor progression exists as high CD44 expression is considered to be hallmark for breast cancer stem cells (BCSCs).
Cancer stem cells pose a challenge during treatment as they are capable of controlled cell proliferation.59 CD44 expression is an important marker for a BCSC identification within a diverse population of tumor cells.60 The most common BCSC population studied in human breast cancer is CD44+/CD24low/-. Metastasis occurs though EMT, cellular detachment, migration and potentially BCSCs evading common therapeutic treatment.
In this study, we demonstrate a connection between OSM-induced metastatic events and CD44 during tumor cellular detachment and migration. Both EMT and CD44 have been linked to metastasis, but there has been a question as to whether they are directly or indirectly associated. Here, we provided data showing that OSM induces EMT and upregulates CD44 expression, but that OSM-induced EMT is independent of CD44. Conversely, we show that CD44 plays a very specific role in OSM-induced detachment and migration, both important events in the metastatic cascade that follows EMT. Furthermore, CD44 knockdown in aggressive ER- TNBC cells contributed to the cells behaving as a less aggressive (ER+-like) cell during detachment. These findings delineate another important mechanism by how OSM induces breast cancer metastatic potential.
Materials and methods
Cell culture and cytokine stimulation
MCF7 and T47D ER+ luminal A human breast cancer cells were obtained directly from the American Type Culture Collection (Rockville, MD), maintained in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) and 100 U/mL each of penicillin and streptomycin. Cells were maintained at 37 °C, 5% carbon dioxide, and 95% humidity. All media and supplements were obtained from Hyclone (Logan, UT). Human oncostatin M (OSM) (Peprotech, Rocky Hill, NJ) was added at 25 ng/mL.61,62
Immunofluorescence staining and microscopy
2.5×105 cells were plated onto glass coverslips (Thermo Fisher Scientific). After 24 hrs, cells were treated with or without OSM for 3 days. After experimental treatment, cells were rinsed once with PBS and then fixed in ice-cold methanol for 20 mins. Cells were rinsed 3 times with PBS for 10 mins. Cells were blocked in PBSAT (1X PBS/1% BSA/0.5%Triton X-100) for 30 mins and then incubated in PBSAT containing the primary antibodies human E-cadherin (1:200, same as immunoblotting), α-catenin (1:200; Abcam, Cambridge, UK), Phalloidin for actin (1:200, Thermo Fisher Scientific, Waltham, MA) overnight. Cells were washed 3 times in PBS and incubated with PBSAT containing goat anti-mouse Alexa Flour 488 (1:1000) and donkey anti-rabbit Alexa Flour 546 (1:500; Molecular Probes, Invitrogen, Carlsbad, CA) secondary antibodies for 2 hrs. Cells were washed 3 times in PBS and mounted using Prolong Gold DAPI (Molecular Probes). Stains were visualized and confocal microscopy images were taken using a Zeiss LSM 510 Meta system combined with the Zeiss Axiovert Observer Z1 inverted microscope and ZEN 2009 imaging software (Carl Zeiss, Inc., Thornwood, NY).
Real time qPCR
Total RNA was collected using RNA STAT-60 and isolated per the manufacture’s protocol (Tel-Test; Friendswood, TX). cDNA was synthesized according to the manufacture’s protocol (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems, In/Life Technologies). A LightCycler 96 (Roche Diagnostic Corporation, Indianapolis, IN) with program settings of preincubation 1 cycle, 2 step amplification 45 cycles, and high resolution melting 1 cycle; steps were set at 95 degrees C for 120 seconds. Primers used are shown in Table 1. ΔΔCq method was used for analyzation of the data as described by Livak and Schmittgen.63 Statistical analysis was performed using GraphPad Prism software for 1 way ANOVA tests.
 |
Table 1 The list of primers used for qPCR |
Western blot analysis
For whole cell lysates, 5×104 cells were seeded into 24-well plates (Thermo Fischer Scientific, Rockford, IL), unless otherwise stated. At the end of the experiment, plates were rinsed with ice-cold PBS, then placed on ice with 125 μL of RIPA buffer (Thermo Fischer Scientific) supplemented with 1:100 dilution of Sigma Protease Inhibitor Cocktail and 1 µL/mL phenylmethylsulfonyl fluoride (PMSF) (Sigma, St. Louis, MO) for 15 mins, mixing every 5 mins. Plates were scraped and the contents transferred to an Eppendorf tube. Tubes were then centrifuged at 14,000 rpm for 20 mins.
Lysates were run on a 7.5–10% SDS-PAGE gel and transferred to a nitrocellulose membrane. Immunoblot analysis was performed using primary antibodies for human E-cadherin (1:2000), β-actin (1:5,000), α-catenin (1:2,500) (Cell Signaling Secondary, Danvers, MA), CD44 (1:5000) (kindly provided by Dr. Liliana Mellor and Dr. Knudson). Antibodies were HRP-conjugated donkey anti-rabbit (1:5,000).
Flow cytometry
A FACSCaliburTM flow cytometer (BD Biosciences, San Jose, CA) was used to observe populations of BCSCs present within a homogenous population. MCF-7 and T47D cells were treated with OSM (25 ng/mL) for 72 hrs before performing flow cytometry with antibodies (BD Biosciences, San Jose, CA) to CD44 (1:5 FACS buffer), CD24 (1:5 FACS buffer), FITC (1:50 FACS buffer), and PE (1:50 FACS buffer) after blocking cells for 10 mins with 1% BSA. Antibodies bound to their appropriate targets for 30 mins on ice in the dark. Following incubation cells were washed 3x with FACS buffer (10% FBS, 0.1% sodium azide in PBS). Software analysis was performed using BD FACSCalibur software: CellQuest Pro, version 6.0 by BD Biosciences. Statistical analysis was performed using GraphPad Prism software for 1 way ANOVA tests.
shRNA transduction
Gene knockdown of CD44 and a non-targeting control was conducted using shERWOOD shRNA lentiviral constructs (Transomic Technologies, Huntsville, AL). Viral transduction of MDA-MB-231 luc and MCF-7 luc cells was performed according to manufacturer’s protocol. Puromycin was used for initial selection followed by fluorescence microscopy to isolate transduced cell population. CD44 expression was confirmed by Western blot analysis.
Detachment assay
A Detachment assay was used to compare relative tumor cell detachment in the presence or absence of recombinant OSM (25 ng/mL). Cells were plated at 10,000 cells per well in a 24-well plate and allowed to attach overnight. Once plated, cells were placed in 1% FBS RPMI media for 4 hrs without treatment. After a 4 hr incubation, cells were then treated with or without OSM in 1% media for the allotted time (1, 3, or 5 days). To assess detached cells, media was collected and centrifuged. The pellets were resuspended in equal parts of RPMI media and Trypan Blue. Detached cells were then counted. Cells stained with Trypan blue were excluded from the counts. Proliferation was also measured, by trypsinizing adherent cells, pelleting cells, resuspending pellets in equal parts of RPMI media and Trypan blue, and counting. Detachment was measured by comparing the number of proliferating cells and detached cells in the presence or absence of OSM, and fold detachment was calculated. Statistical analysis was performed using GraphPad Prism software for 1 way ANOVA tests.
Wound healing assay
A wound healing assay was used to measure mobility of breast cancer cell with or without OSM treatment. MCF-7 luc and MDA-MB-231 luc shCD44 and shNTC control cells were plated at 200,000 cells per well in a 6-well plate with 10% FBS RPMI media. Once cells were 80–85% confluent, a scratch was made with a sterile p1000 micropipette tip. Wells were washed with PBS and then 10% media was placed on each well with or without OSM (25 ng/mL). Photomicrographs were taken at 72 hrs. The area of each scratch was measured using ImageJ and fold-migration was assessed. Statistical analysis was performed using GraphPad Prism software for 1 way ANOVA tests.
Chemical inhibitor
A MMP inhibitor (GM6001; Santa Cruz Biotechnology), was added to the cell growth media (10 nM) 2 days before cytokine treatment in serum-free media; followed by the addition of complete media and OSM for 2 more days.
Results
OSM induces EMT and CD44+/CD24−/low BCSC population
The EMT process is commonly studied in vitro using ER+ human breast cells because, compared to ER- cells, they are less aggressive, phenotypically epithelial, and respond to metastatic inducers such as TGF-β and OSM.64,65 Our studies showed that ER+ MCF7 human breast cancer cells treated with OSM (25 ng/mL) underwent an EMT over a 3-day period, as displayed by a loss of E-cadherin expression as measured by immunofluorescence, as well as a complete dissociation of individual cells breaking away from the initial colony and subsequently take on a more spindle-like mesenchymal appearance (Figures 1A and S1). On the other hand, ER- MDA-MB-231 cells grew with a mesenchymal phenotype and were not capable of undergoing an OSM-induced EMT. Previous studies have shown that developmental transcription factors such as Slug and Snail are responsible for repressing epithelial marker proteins, which creates a seesaw effect as cells progress through EMT.66 ER+ MCF-7 cells treated with OSM (25 ng/mL) for one hour upregulated both transcription factors Slug and Snail by qRT-PCR (Figure 1B). OSM treatment (25 ng/mL) of MCF-7 cells also resulted in a slight decrease in expression of the cell-cell adhesion proteins E-cadherin and α-catenin over time, as measured by Western blot analysis (Figure 1C). These results demonstrated that OSM was able to initiate an EMT in ER+ breast cancer cells by upregulating transcription factors, down regulating cell-cell adhesion proteins, and promoting a change in cell morphology.
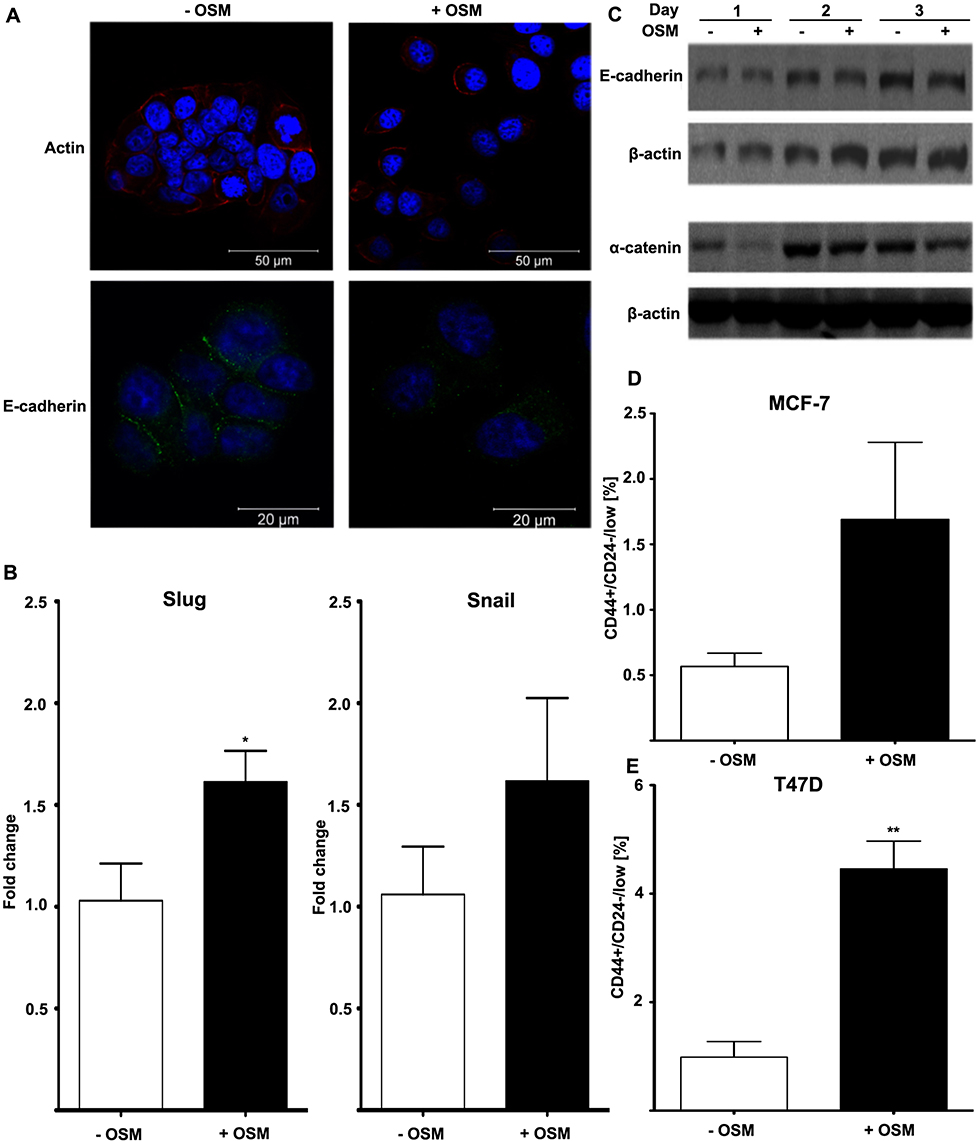 |
Figure 1 OSM induces EMT and a BCSC phenotype in human breast cancer cells (A) MCF-7 luc human breast cancer cells were treated with OSM (25 ng/mL) for 72 hrs. OSM induces an increased mesenchymal phenotype and a decrease in E-cadherin expression, as measured by immunofluorescence using antibodies for E-cadherin and actin (63× magnification). (B) MCF-7 cells were treated with OSM (25 ng/mL) for 1 hr, and qPCR was performed with primers for the EMT transcription factors Slug and Snail (*P<0.05, unpaired t-test). (C) MCF-7 cells were treated with OSM (25 ng/mL) for 24, 48 and 72 hrs, and cell lysates were assessed by Western blot analysis for E-cadherin and α-catenin (D and E). ER+ MCF-7 and T47D cells were treated with OSM (25 ng/mL) for 72 hrs before flow cytometry was performed. In T47D cells, OSM induces the CD44+/CD24− BCSC population 3-fold, while a small non-significant increase was seen in MCF-7 cells. **P<0.01, *P<0.05, unpaired t-test. |
Another common feature associated with tumor progression and overall metastatic burden is the appearance of a breast cancer stem cell (BCSC) phenotype, observed as cells that are CD44+/CD24low within a heterogeneous tumor cell population. CD44+/CD24low cells have been shown to possess features of developmental stem cells with controlled cell division and presence at a defined location.67 Treatment with OSM (25 ng/mL) for 72 hrs increased the CD44+/CD24low BCSC population in ER+ MCF-7 cells from 1% to over 1.5% (Figure 1D), and in T47D cells from 1% to over 4% (Figure 1E). The CD44+/CD24low BCSC cell population in MCF-7 was not significant because of the high basal expression of CD24 observed during FACS analysis. Taken together, OSM induces both an EMT and the CD44+/CD24low population of BCSCs in ER+ breast cancer cells, confirming previous results that OSM is a driver of metastatic potential.
OSM induces both CD44 expression and cleavage
CD44 expression fluctuates during tumor progression and metastasis. Our studies showed that OSM induced CD44 expression at the protein level over time in ER+ but not ER- cells (Figure 2). Treatment of ER+ MCF7 cells with OSM (25 ng/mL) for 24, 48, and 72 hrs resulted in an increase in high molecular weight CD44 alternatively spliced (top panels) isoforms and low molecular weight cleavage products (lower panels) at each time point (Figure 2A and B), as measured by Western blot analysis of whole cell lysates using an antibody specific to the cytoplasmic fragment of CD44. For ER+ T47D cells, OSM-induced CD44 high molecular weight CD44 isoforms were most prevalent at 48 and 72 hrs (Figure 2C and D). CD44 expression in the more aggressive ER- MDA-MB-231 cells showed a high basal level, which did not change in the presence of OSM during the same time period (Figure 2E and F). In these experiments, we observed an OSM-induced increase of high molecular weight bands larger than the CD44s (70–85 kDa) band.
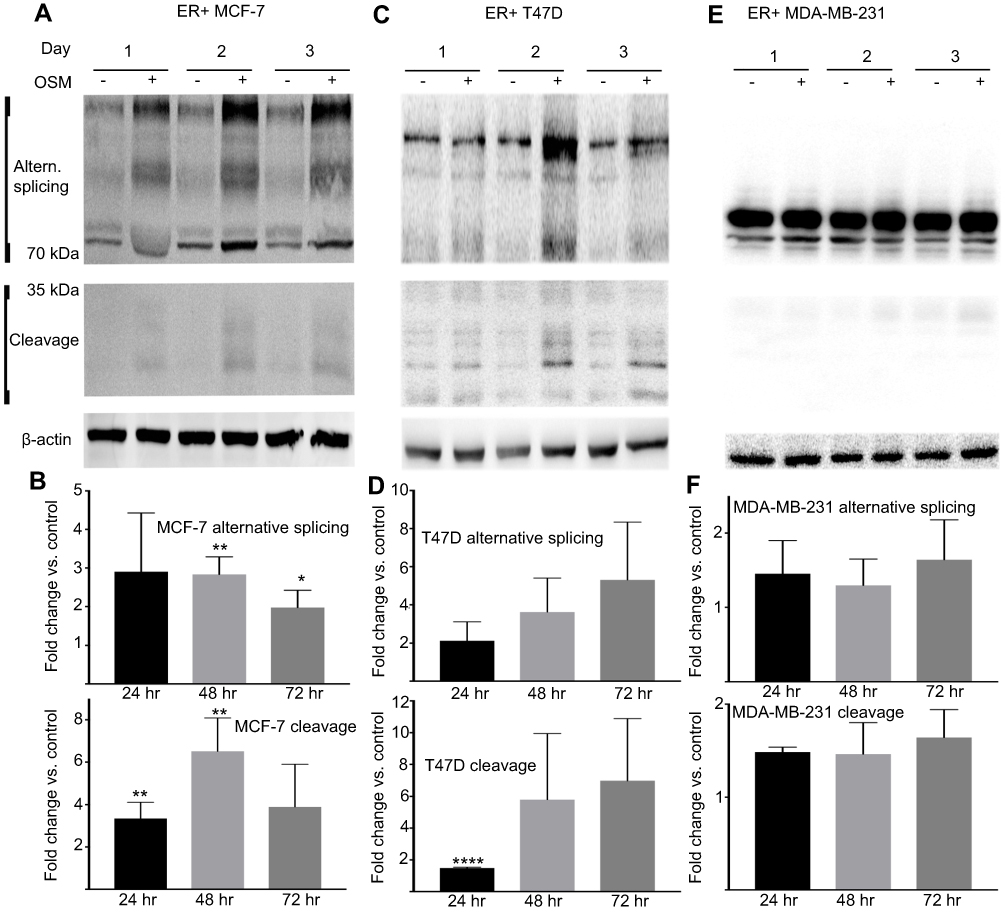 |
Figure 2 OSM induces CD44s and CD44 isoforms (above 85 kDa) and cleaved products (below 50 kDa) in ER+ cells, but not ER- cells (A) tumor cells were treated with OSM for 24, 48, and 72 hrs and whole cell lysates were collected followed by immunoblotting for CD44 using a CD44 antibody kindly provided by Dr. Mellor CD44 expression in MCF-7 (ER+), and (B) quantification of (A). (C) CD44 expression in T47D (ER+), and (D) quantification of (C). (E) CD44 expression in MDA-MB-231 (ER-), and (F). quantification of (E). All OSM treatment time points were compared to the corresponding non-treatment time points after normalizing to ß-actin. Unpaired t-tests were performed for each time point. **P<0.01, *P<0.05. |
To support our finding that OSM induced CD44s and variant CD44 protein expression in ER+ breast cancer cells, we investigated OSM-induced CD44 mRNA levels by qRT-PCR. While a non-significant increase in OSM-induced CD44s expression was observed at 8 hrs in MCF-7 cells (Figure S2A), OSM did not significantly induce the mRNA levels of individual CD44 variants (v2 through v10) at the 8 hr time point, it was observed that OSM slightly increased CD44 variants (Figure S2B). Together, these studies suggest that while OSM does not induce CD44 mRNA expression, it does result in increased levels of CD44s and variant proteins. These results showed that in ER+ breast cancer cells, which have a lower basal level of CD44 expression, OSM increased the expression of high molecular weight CD44 isoforms, while the more aggressive TNBC cells that express high basal levels of CD44, do not respond to OSM.
Proteolytic cleavage of CD44 by matrix metalloproteinases (MMPs) and γ-secretase occurs in close proximity to the cell membrane and produces low molecular CD44 fragments. Specifically, CD44 is first cleaved by MMP-9 and subsequently undergoes a second cleavage by γ-secretase producing a smaller fragment known as the intracellular domain (ICD) CD44.68 Our lab has previously shown that OSM induces MMP activity in breast tumor cells.21 T47D cells treated with the global MMP inhibitor GM6001 for 48 hrs followed by OSM (25 ng/mL) treatment for 48 hrs decreased the amount of OSM-induced MMP cleavage of CD44 (Figure S3A and B). Proteolytic activity of MMP cleavage produces molecular weight bands of 30 kDa and smaller. Inhibiting OSM-induced MMP activity decreased the amount of cleaved CD44. To summarize, OSM promotes EMT in ER+ breast cancer cells, increases CD44 expression, and induces proteolytic cleavage of CD44, presumably through OSM-induced MMP activity.
OSM-induced EMT is independent of CD44
The relationship between OSM-driven EMT and OSM-upregulation of CD44 expression was explored by establishing stable ER+ MCF-7 luc and ER- MDA-MB-231 luc cells with CD44 expression knockdown using shCD44 lentiviral transduction. MCF-7 luc-shCD44 cells showed a 70% reduction in CD44s expression by Western blot analysis compared to control shNTC (non-targeting control) cells, while MDA-MB-231 luc-shCD44 cells showed over an 80% reduction in CD44s (Figure 3A and B).
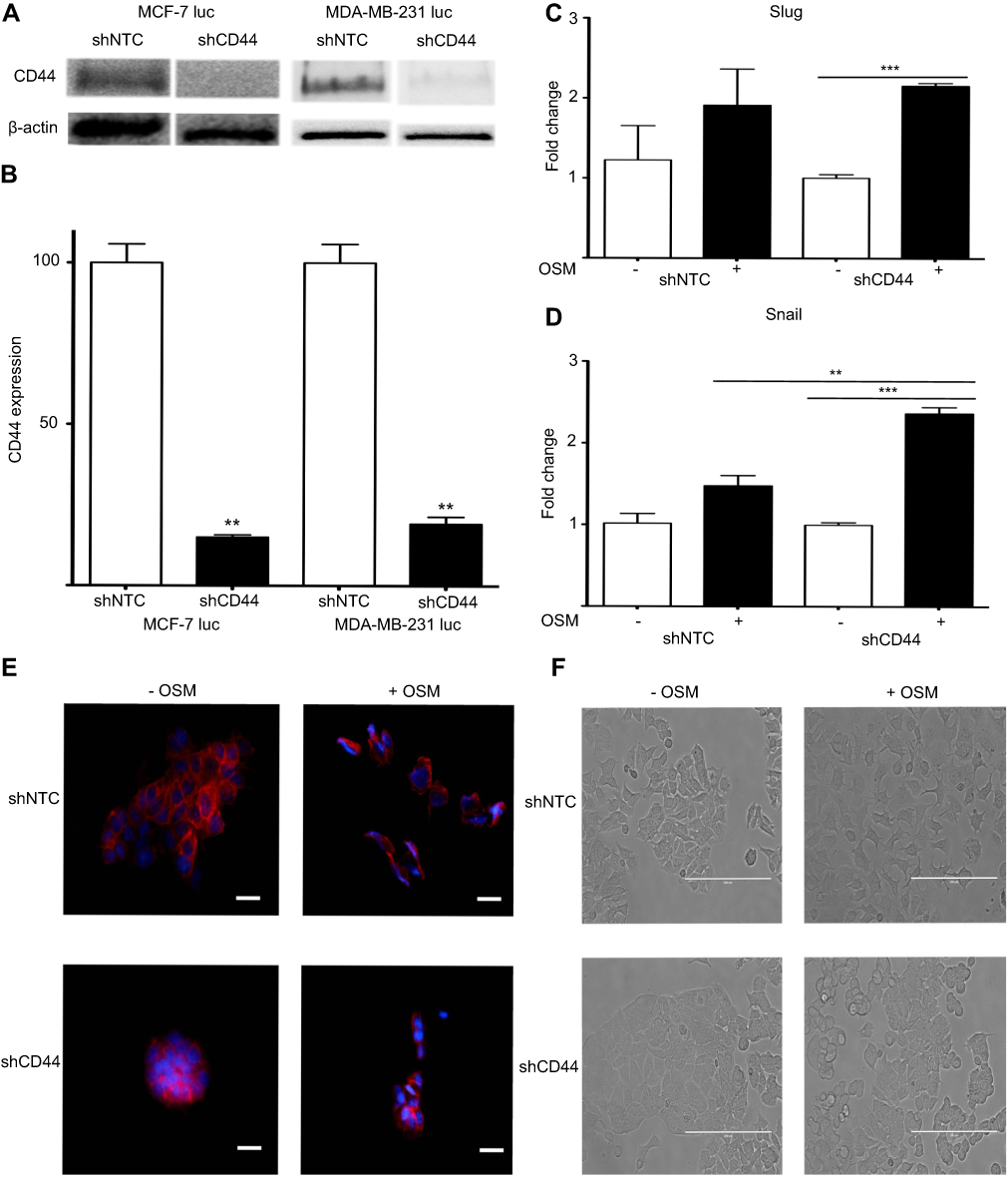 |
Figure 3 Reduced CD44 expression does not abrogate OSM-induced EMT in ER+ cells (A) Both MCF-7 luc-shCD44 and MDA-MB-231 luc-shCD44 cells show knockdown of CD44 protein expression compared to non-targeting control (shNTC) cells by Western blot analysis, and quantification in (B). MCF7 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) for one hour, and qPCR analysis for Slug and Snail was performed. Knockdown of CD44 expression did not decrease OSM-induced Slug (C), or Snail (D) in these cells (n.s. = no significance, *p<0.05, **P<0.005, ***P<0.001, One way Anova). (E) MCF7 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) for 72 hrs, and E-cadherin was observed via immunofluorescence (20x magnification, scale bar-20 nm). (F) MCF7 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) for 72 hrs to observe EMT morphology by phase contrast images (20x magnification, scale bar-100 µm). |
Next, we investigated two transcription factors, Slug and Snail, which have been shown to drive EMT by repressing E-cadherin expression that results in loss of cell-cell adhesion. MCF-7 luc-shCD44 and control shNTC cells were treated with OSM (25 ng/mL) for 1 hr and qRT-PCR was performed. As expected, OSM increased both Slug and Snail over 1.5-fold compared to no treatments in control MCF-7 luc-shNTC cells (Figure 3C and D). However, OSM treatment of MCF-7 luc-shCD44 cells resulted in no significant change in Slug expression compared to control shNTC cells, suggesting that CD44 is not involved in the upregulation of Slug by OSM (Figure 3C). Interestingly, CD44 knockdown resulted in a significant increase in OSM-induced Snail expression compared to control shNTC to cells, suggesting that the absence of CD44 increased OSM-induced Snail (Figure 3D).
OSM-repressed E-cadherin expression during EMT was observed by confocal microscopy immunostaining in MCF-7 luc-shCD44 and shNTC cells (Figure 3E). In the presence of OSM (25 ng/mL) for 72 hrs, shNTC and shCD44 cells both showed a reduction in the cell-cell adhesion protein, E-cadherin (Figure 3E). Photomicrographs of MCF-7 luc-shCD44 and shNTC cells treated with OSM for 72 hrs were also taken to observe the phenotypic EMT process (Figure 3F). Although both cell lines underwent characteristic EMT processes, the MCF-7 shCD44 cells appeared to form much tighter colonies prior to OSM treatment. Together, these results suggest that OSM-induced EMT takes place independently of CD44 in ER+ MCF-7 luc cells.
OSM-induced tumor cell detachment is dependent on CD44
As part of the metastatic cascade, tumor cells must detach from their established three-dimensional location. We have previously shown OSM to be a strong inducer of tumor cell detachment, especially in ER+ breast cancer cells.21 MCF-7 luc-shCD44 and MDA-MB-231 luc-shCD44 cells were treated with OSM (25 ng/mL), and fold-detachment was measured over a 5-day period (Figure 4). OSM induced a 1.8- to 3-fold increase in cell detachment using the less aggressive ER+ MCF-7 luc-shNTC cells compared to untreated cells (Figure 4A; left). When MCF-7 luc-shCD44 cells were treated with OSM, no significant increase in tumor cell detachment was measured at days 1 and 3 (Figure 4A; right), suggesting that knockdown of CD44 eliminated or delayed the ability of OSM to induce ER+ MCF-7 breast tumor cell detachment. The more aggressive ER- MDA-MB-231 luc-shNTC control cells did not detach in the presence or absence of OSM (Figure 4B; left). Interestingly, MDA-MB-231 luc-shCD44 cells treated with OSM for 5 days showed a 6-fold increase in tumor cell detachment compared to control cells, thus mimicking MCF-7 luc shNTC cells (Figure 4B; right). Therefore, it appears that MDA-MB-231 luc-shCD44 cells reverted from aggressive and basically OSM-unresponsive cells to less aggressive cells that responded to OSM when CD44 expression was reduced. Together, these results suggest that OSM-induced ER+ MCF-7 tumor cell detachment is dependent on CD44 expression.
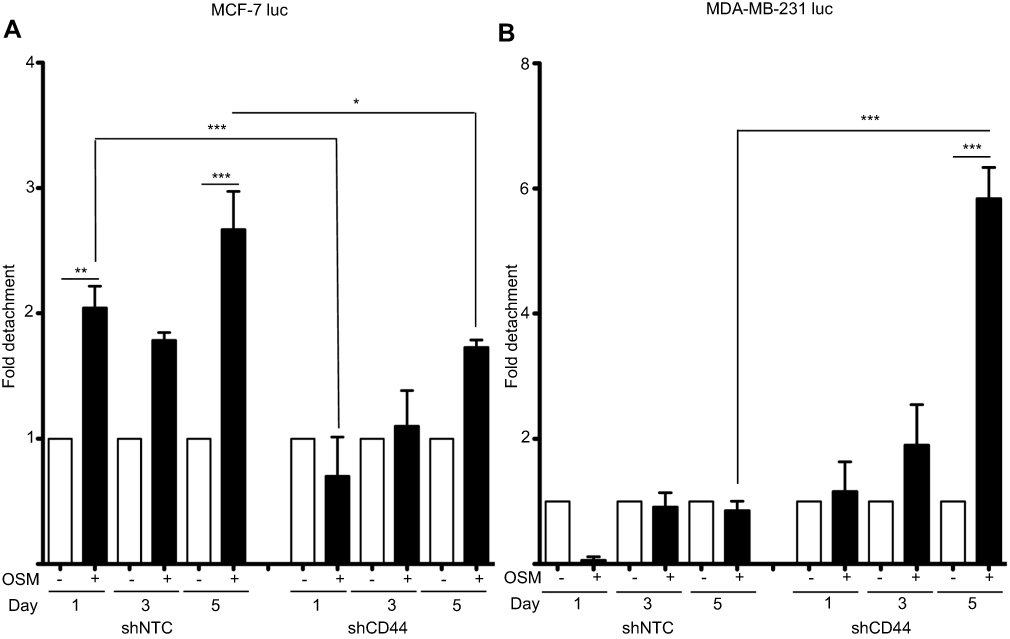 |
Figure 4 Reduced CD44 expression decreases OSM-induced tumor cell detachment in ER+ cells (A) ER+ MCF7 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) over a 5-day period, detached cells were counted on days 1, 3, and 5, and data is represented as fold-change in graphical representation (*P<0.05, **P<0.005, ***P<0.001, one-way Anova). (B) ER- MDA-MB-231 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) over a 5-day period, detached cells were counted on days 1, 3, and 5, and data is represented as fold change calculated by comparing non-OSM treated cells to OSM treated cells and differences between OSM treated shNTC and shCD44 as shown in graphical representation. ***P<0.001, one-way Anova. |
OSM-induced tumor cell migration is dependent on CD44
Following EMT, a tumor cell must utilize two-dimensional movement in order to initiate invasion and metastasis. Wound healing assays were performed using ER+ MCF-7 luc-shCD44 and MDA-MB-231 luc-shCD44 cells to quantitate tumor cell migration. MCF-7 luc shNTC cells treated with OSM (25 ng/mL) for 3 days demonstrated a slight 1.5-fold increase in migration compared to untreated cells (Figure 5A), Importantly, MCF-7 luc-shCD44 cells migrated to a significantly less extent when compared to shNTC control cells in the presence of OSM (Figure 5A, black bars). When treated with OSM for 3 days, the ER- MDA-MB-231 luc-shCD44 cells showed a nearly 2.5-fold reduction in the number of migrating cells compared to MDA-MB-231 luc-shNTC cells (Figure 5B, black bars). This striking difference reiterates the importance of CD44 in tumor cell migration for aggressive ER- breast cancer cells, while demonstrating a role for OSM-induced CD44 in ER+ MCF-7 cell migration. In conclusion, OSM-induced CD44 appears to be important in tumor cell detachment and migration, both necessary for tumor cells undergoing metastasis.
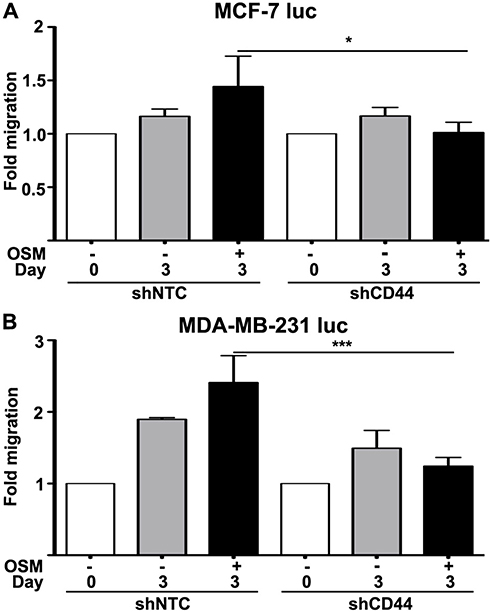 |
Figure 5 Reduced CD44 expression decreases OSM-induced tumor cell migration in ER- cells greater than ER+ cells (A) ER+ MCF-7 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) for 3 days, wound healing was analyzed using ImageJ software, and data was interpreted as fold-change. (B) ER- MDA-MB-231 luc-shNTC and shCD44 cells were treated with OSM (25 ng/mL) for 3 days, wound healing was analyzed using Image J software, and data was interpreted as fold-change. ***P<0.001, one-way Anova. |
Discussion
Breast cancer metastasis requires that individual tumor cells undergo multiple changes and survive in diverse environments before reaching a secondary location and expanding. It is speculated that the metastatic cascade consists of a number of specifically ordered and timed events that must take place, starting with EMT and ending in mesenchymal-epithelial transition (MET) and clonal expansion of each metastasis. CD44 has been previously shown to be an important driver of invasion and the metastatic process. In this research, we show that OSM induces an EMT that is not dependent on CD44. Rather, OSM-induced CD44 is necessary for breast tumor cell detachment and migration regulated by OSM.
Our results presented here compare to previously published data on OSM-induced EMT by showing a decrease in epithelial markers and a phenotypic change to cells in culture.69 We also demonstrated the ability of OSM to increase a BCSC phenotype, which has been previously published.70 OSM-induced breast tumor cell detachment has also been published.71 In this study, we show for the first time that OSM induces alternatively spliced and cleaved CD44 protein expression.
Other cytokines have been shown to increase metastatic characteristics in breast cancer, and here we showed that OSM increased CD44 expression, but did not affect its alternative splicing. Previous studies have shown cytokines such as TGF-β are strong inducers of CD44.72,73 Our results show OSM induces CD44 expression in a similar manner to that of TGF-β by increasing CD44 expression and subsequent CD44 cleavage. Although ER- cells did not respond to OSM in the same manner of ER+, PR+, HER2- luminal A cells, it was shown CD44 is actually expressed at such a high basal level that an increase was not observed and therefore TNBC MDA-MB-231 luc cells were not used to study induction of CD44 via OSM, as other publications have shown the same using this ER- cell line.74 Furthermore, OSM-induced MMP-9 CD44 cleavage was reduced by using the non-specific MMP inhibitor GM6001 as shown by the reduction of low molecular weight bands. Our lab and others have previously shown that OSM induces the expression of a family of MMPs.75,76 Our lab was not able to show that OSM was capable of inducing secretase, suggesting that OSM’s ability to cleave CD44 is limited to MMP activity.
The connection between OSM-induced EMT and CD44 expression was examined using MCF-7 luc and MDA-MB-231 luc shCD44 cell lines with knockdown expression of CD44. Non-targeting control (shNTC) cell lines responded similarly to parental MCF-7 cells in terms of OSM-induced EMT, transcriptional activity, and EMT-related proteins. In CD44 knockdown cell lines, EMT occurred following OSM treatment, but the knockdown of CD44 in MCF-7 luc cells displayed less detachment compared to control cell lines. MDA-MB-231 luc shCD44 cells detached at a much larger number compared to control. Migration was reduced in both MCF-7 luc and MDA-MB-231 luc shCD44 cells compared to control shNTC cells suggesting CD44 contributes to migration during metastasis. Furthermore, with reduced CD44 expression, MCF7 luc cells grew and responded much differently compared to the control cells in terms of morphological changes as they appeared to contain an increased number of rounded up cell populations and they also took much longer to adhere to cell culture flasks. These findings suggest the potential role for CD44 as an adhesion molecule and cell-cell attachment protein. MCF-7 luc-shCD44 cells also grew in much tighter colonies, with small groups broken away from the primary colony following 72 hrs of OSM treatment. Cells that broke away from the primary colony following OSM treatment did not undergo an EMT as they appeared to be display tighter cell-cell adhesion. All of this indicates that CD44 was not directly responsible for an early event in metastasis, EMT. Although previous studies have displayed a correlation between an increase in CD44 expression and EMT,77 here we show they are ordered events as CD44 participates in later metastatic events, but not in the initial EMT.
Metastatic events such as cellular detachment and migration have often thought to be steps necessary in the early to middle stages of metastasis. In the presence of OSM, control cells migrated more than shCD44 cells and a similar trend was observed for detachment. Aggressive cells like the MDA-MB-231 luc cells do not detach in the presence of OSM, but the knockdown of CD44 promoted OSM-induced detachment. The opposite was shown in the less aggressive MCF-7 luc cells where OSM-induced detachment was reduced in the shCD44 group suggesting CD44 is used by less aggressive cells to become more metastatic. Others have shown cytokines such as TGF-β increase detachment and migrationbut our data shows both events can occur through OSM signaling and the expression of CD44.
Together our studies show the binding of OSM to its receptor leads to the promotion of EMT, migration, and detachment in less aggressive ER+ breast tumor cells (Figure 6). OSM signaling also leads to an increase in the upregulation of CD44 expression and CD44 cleavage in these cells. OSM-induced CD44 cleavage via MMP proteolytic action promotes migration and detachment in less aggressive cells thus making them more aggressive. CD44 knockdown expression in highly aggressive MDA-MB-231 luc cells allows for the cells to become receptive to OSM and adapt to ER+ cell-like sequence of metastatic events.
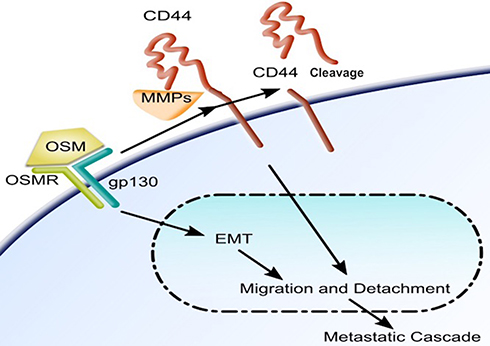 |
Figure 6 OSM signaling induces an EMT and upregulates CD44 and cleavage of CD44 in breast tumor cells: Once bound to its receptor, OSM induces cells to undergo EMT and subsequent detachment and migration. OSM signaling increases expression of the transmembrane protein CD44 and also cleavage via MMPs. OSM-induced CD44 is involved in migration and detachment as well as docking, which is the step immediately before extravasation during the metastatic cascade. |
Conclusion
The results presented in this manuscript demonstrate that the inflammatory cytokine OSM promotes CD44 expression in ER+ breast cancer cells. Furthermore, this CD44 seems to play a central role in OSM-induced tumor cell detachment but not in EMT. Together, our results suggest that the regulation of CD44 by OSM is important for at least part of the metastatic cascade in ER+ breast cancer. These findings should provide useful in future studies of human metastatic breast cancer with the goal of developing potential therapeutics to reduce mortality rates.
Abbreviations list
EMT, epithelial to mesenchymal transition; CD44, cluster of differentiation 44; OSM, oncostatin M; BCSC, breast cancer stem cell; shRNA, short hairpin RNA.
Consent for publication
All authors have consented to publication.
Data sharing statement
Original data available on request.
Acknowledgments
The following people have contributed to this work in more ways than one. Raquel Brown provided great insight and technical experience for immunofluorescent imaging. Hannah Scott provided data analysis and contributed to the growth and propagation of cells used for this study. This study was partially funded by the following grants: NIH/NCI R15CA137510, NIH/NCRR P20RR016454, NIH/NIGMS P20GM103408, NIH/NIGMS P20GM109095, Susan G. Komen Foundation KG100513, and American Cancer Society RSG-09-276-01-CSM.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Scully OJ, Bay B-H, Yip G, Yu Y. Breast cancer metastasis. Cancer Genomics Proteomics [Internet]. 2012;9:311–320. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22990110.
2. Weigelt B, Peterse JL, van’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. doi:10.1038/nrc1670
3. Lu X, Kang Y. Organotropism of breast cancer metastasis. J Mammary Gland Biol Neoplasia. 2007;12:153–162. doi:10.1007/s10911-007-9047-3
4. Jung SY, Rosenzweig M, Sereika SM, Linkov F, Brufsky A, Weissfeld JL. Factors associated with mortality after breast cancer metastasis. Cancer Causes Control. 2012;23:103–112. doi:10.1007/s10552-011-9859-8
5. Shapiro IM, Cheng AW, Flytzanis NC, et al. An emt-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7. doi:10.1371/journal.pgen.1002138
6. Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:253–260. doi:10.1007/s10911-010-9173-1
7. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi:10.1007/s10555-008-9169-0
8. Müller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi:10.1038/35065016
9. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (80-). 2011;331:1559–1564. doi:10.1126/science.1203543
10. Ma R, Feng Y, Lin S, et al. Mechanisms involved in breast cancer liver metastasis. J Transl Med. 2015;13:64. doi:10.1186/s12967-015-0541-x
11. Tabariès S, Dong Z, Annis MG, et al. Claudin-2 is selectively enriched in and promotes the formation of breast cancer liver metastases through engagement of integrin complexes. Oncogene. 2011;30:1318–1328. doi:10.1038/onc.2010.518
12. Tabariès S, Siegel PM. Breast cancer liver metastasis. Cancer Metastasis - Biol Treat [Internet]. 2011:273–303. Available from: http://www.embase.com/search/results?subaction=viewrecord&from=export&id=L366194192%5Cndoi:10.1007/978-94-007-0292-9_10.
13. Bos PD, Zhang XH-F, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19421193&retmode=ref&cmd=prlinks%5Cnpapers3://publication/doi/10.1038/nature08021.
14. Cheng X, Hung M-C. Breast cancer brain metastases. Cancer Metastasis Rev. 2007;26:635–643. doi:10.1007/s10555-007-9083-x
15. Zhang L, Ridgway LD, Wetzel MDA, et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5:180ra48. doi:10.1126/scitranslmed.3005109
16. Kang Y, Siegel PM, Shu W, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi:10.1016/S1535-6108(03)00132-6
17. Chen Y-C, Sosnoski DM, Mastro AM. Breast cancer metastasis to the bone: mechanisms of bone loss. Breast Cancer Res. 2010;12:215. doi:10.1186/bcr2722
18. Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived Jagged1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19:192–205. doi:10.1016/j.ccr.2010.12.022
19. Dvorak HF, Weaver VM, Tlsty TD, Bergers G. Tumor microenvironment and progression. J Surg Oncol. 2011;103:468–474. doi:10.1002/jso.v103.6
20. Sánchez-Tilló E, Liu Y, de Barrios O, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci [Internet]. 2012 [cited October 18, 2013];69:3429–3456. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22945800.
21. Jorcyk CL, Holzer RG, Ryan RE. Oncostatin M induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine. 2006;33:323–336. doi:10.1016/j.cyto.2006.03.004
22. Ryan RE, Martin B, Mellor L, et al. Oncostatin M binds to extracellular matrix in a bioactive conformation: implications for inflammation and metastasis. Cytokine. 2015;72:71–85. doi:10.1016/j.cyto.2014.11.007
23. Mozaffarian A, Brewer AW, Trueblood ES, et al. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J Immunol. 2008;181:7243–7253. doi:10.4049/jimmunol.181.5.3177
24. Langdon C, Kerr C, Tong L, Richards CD. Oncostatin M regulates eotaxin expression in fibroblasts and eosinophilic inflammation in C57BL/6 mice. J Immunol. 2003;170:548–555. doi:10.4049/jimmunol.170.1.548
25. Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res. 2005;65:8896–8904. doi:10.1158/0008-5472.CAN-04-4557
26. Pastuschek J, Poetzsch J, Morales-Prieto DM, Schleußner E, Markert UR, Georgiev G. Stimulation of the JAK/STAT pathway by LIF and OSM in the human granulosa cell line COV434. J Reprod Immunol. 2015;108:48–55. doi:10.1016/j.jri.2015.03.002
27. Mosley B, De IC, Friend D, et al. Dual Oncostatin M (OSM) receptors. J Biol Chem. 1996;271:32635–32643. doi:10.1074/jbc.271.51.32635
28. Hutt JA, DeWille JW. Oncostatin M induces growth arrest of mammary epithelium via a CCAAT/enhancer-binding protein delta-dependent pathway. Mol Cancer Ther [Internet]. 2002;1:601–610. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12479220.
29. Grant SL, Douglas AM, Goss GA, Begley CG. Oncostatin M and leukemia inhibitory factor regulate the growth of normal human breast epithelial cells. Growth Factors. 2001;19:153–162. doi:10.3109/08977190109001083
30. Marrero-Diaz R, Bravo-Cordero JJ, Megìas D, et al. Polarized MT1-MIVIP-CD44 interaction and CD44 cleavage during cell retraction reveal an essential role for MT1-MMP in CD44-mediated invasion. Cell Motil Cytoskeleton. 2009;66:48–61. doi:10.1002/cm.20378
31. Stevens JW, Kurriger GL, Carter AS, Maynard JA. CD44 expression in the developing and growing rat intervertebral disc. Dev Dyn. 2000;219:381–390. doi:10.1002/(ISSN)1097-0177
32. Goebeler M, Kaufmann D, Bröcker EB, Klein CE. Migration of highly aggressive melanoma cells on hyaluronic acid is associated with functional changes, increased turnover and shedding of CD44 receptors. J Cell Sci [Internet]. 1996;109(Pt 7):1957–1964. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8832418.
33. Fitzgerald KA, L A O. Characterization of CD44 induction by IL-1: a critical role for Egr-1. J Immunol [Internet]. 1999;162:4920–4927. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10202038.
34. Dasgupta A, Takahashi K, Cutler M, Tanabe KK. O-linked glycosylation modifies CD44 adhesion to hyaluronate in colon carcinoma cells. Biochem Biophys Res Commun. 1996;227:110–117. doi:10.1006/bbrc.1996.1475
35. Lesley J, Hyman R. CD44 structure and function. Front Biosci. 1998:d616–30. doi:10.2741/A306
36. Skelton TP, Zeng C, Nocks A, Stamenkovic I. Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. J Cell Biol. 1998;140:431–446. doi:10.1083/jcb.140.2.431
37. Wu RL, Sedlmeier G, Kyjacova L, et al. Hyaluronic acid-CD44 interactions promote BMP4/7-dependent Id1/3 expression in melanoma cells. Sci Rep. 2018;8:1–12.
38. Zoller M, Herrlich P, Giinthert U, Herrlich P. CD44 splice variants confer metastatic behavior in rats: homologous sequences are expressed in human tumor cell lines. Cancer Res. 1991;51:5292–5297.
39. Rodriguez C, Monges G, Rouanet P, Dutrillaux B, Lefrançois D, Theillet C. CD44 expression patterns in breast and colon tumors: a PCR-based study of splice variants. Int J Cancer. 1995;64:347–354. doi:10.1002/(ISSN)1097-0215
40. Xie J-W, Chen P-C, Zheng C-H, et al. Evaluation of the prognostic value and functional roles of CD44v6 in gastric cancer. J Cancer Res Clin Oncol. 2015;141:1809–1817. doi:10.1007/s00432-015-1964-8
41. Van Hal NLW, Van Dongen GAMS, Ten Brink CBM, et al. Evaluation of soluble CD44v6 as a potential serum marker for head and neck squamous cell carcinoma. Clin Cancer Res. 1999;5:3534–3541.
42. Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood). 2013;238:324–338. doi:10.1177/1535370213480714
43. Okamoto I, Kawano Y, Murakami D, et al. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. 2001;155:755–762. doi:10.1083/jcb.200108159
44. Mckallip RJ, Do Y, Fisher MT, Robertson JL, Nagarkatti PS, Nagarkatti M. Role of CD44 in activation-induced cell death: CD44-deficient mice exhibit enhanced T cell response to conventional and superantigens. Int Immunol. 2002;14:1015–1026. doi:10.1093/intimm/dxf068
45. Iida J, Clancy R, Dorchak J, et al. DNA aptamers against exon v10 of CD44 inhibit breast cancer cell migration. PLoS One. 2014;9. doi:10.1371/journal.pone.0088712
46. Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319.
47. Yu Q, Toole BP. A new alternatively spliced exon between v9 and v10 provides a molecular basis for synthesis of soluble CD44. J Biol Chem. 1996;271:20603–20607. doi:10.1074/jbc.271.34.20603
48. Reedeer JA, Gotley DC, Walsh MD, Fawcett J, Antalis TM. Expression of antisense CD44 variant 6 inhibits colorectal tumor metastasis and tumor growth in a wound environment. Cancer Res. 1998;58:3719–3726.
49. Todaro M, Gaggianesi M, Catalano V, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14:342–356. doi:10.1016/j.stem.2014.01.009
50. Hao J, Chen H, Madigan MC, et al. Co-expression of CD147 (EMMPRIN), CD44v3-10, MDR1 and monocarboxylate transporters is associated with prostate cancer drug resistance and progression. Br J Cancer. 2010;103:1008–1018. doi:10.1038/sj.bjc.6605822
51. Hiraga T, Nakamura H. Comparable roles of CD44V8-10 and CD44S in the development of bone metastases in a mouse model. Oncol Lett. 2016;12:2962–2969. doi:10.3892/ol.2016.4985
52. Iida N, Bourguignon LY. New CD44 splice variants associated with human breast cancers. J Cell Physiol. 1995;162:127–133. doi:10.1002/jcp.1041620115
53. Mashita N, Yamada S, Nakayama G, et al. Epithelial to mesenchymal transition might be induced via CD44 isoform switching in colorectal cancer. J Surg Oncol. 2014;110:745–751. doi:10.1002/jso.23705
54. Shang Z, Cai Q, Zhang M, et al. A switch from CD44+ cell to EMT cell drives the metastasis of prostate cancer. Oncotarget. 2015;6:1202–1216. doi:10.18632/oncotarget.2841
55. Klingbeil P, Isacke CM. The “alternative” EMT switch. Breast Cancer Res. 2011;13:313. doi:10.1186/bcr3052
56. Cho SH, Park YS, Kim HJ, et al. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int J Oncol. 2012;41:211–218.
57. Brown RL, Reinke LM, Damerow MS, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064–1074. doi:10.1172/JCI57873
58. Orian-Rousseau V. CD44, a therapeutic target for metastasising tumours. Eur J Cancer. 2010;46:1271–1277. doi:10.1016/j.ejca.2010.02.024
59. Valent P, Bonnet D, De Maria R, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767–775. doi:10.1038/nrc3368
60. Ricardo S, Vieira AF, Gerhard R, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64:937–946. doi:10.1136/jcp.2011.090456
61. Holzer RG, Ryan RE, Tommack M, Schlekeway E, Jorcyk CL. Oncostatin M stimulates the detachment of a reservoir of invasive mammary carcinoma cells: role of cyclooxygenase-2. Clin Exp Metastasis. 2004;21:167–176. doi:10.1023/B:CLIN.0000024760.02667.db
62. Bolin C, Tawara K, Sutherland C, et al. Oncostatin M promotes mammary tumor metastasis to bone and osteolytic bone degradation. Genes Cancer. 2012;3:117–130. doi:10.1177/1947601912458284
63. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi:10.1006/meth.2001.1262
64. David CJ, Huang YH, Chen M, et al. TGF-?? Tumor Suppression through a Lethal EMT. Cell. 2016;164:1015–1030. doi:10.1016/j.cell.2016.01.009
65. Pollack V, Sarközi R, Banki Z, et al. Oncostatin M-induced effects on EMT in human proximal tubular cells: differential role of ERK signaling. Am J Physiol Renal Physiol. 2007;293:F1714–26. doi:10.1152/ajprenal.00130.2007
66. Elloul S, Elstrand MB, Nesland JM, et al. Snail, slug, and smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103:1631–1643. doi:10.1002/cncr.21069
67. Giatromanolaki A, Sivridis E, Fiska A, Koukourakis MI. The CD44+/CD24- phenotype relates to “triple-negative” state and unfavorable prognosis in breast cancer patients. Med Oncol. 2011;28:745–752. doi:10.1007/s12032-010-9530-3
68. Mellor L, Knudson CB, Hida D, Askew EB, Knudson W. Intracellular domain fragment of CD44 alters CD44 function in chondrocytes. J Biol Chem. 2013;288:25838–25850. doi:10.1074/jbc.M113.494872
69. Junk DJ, Bryson BL, Jackson MW. HiJAK’d signaling; the STAT3 paradox in senescence and cancer progression. Cancers (Basel). 2014;6:741–755. doi:10.3390/cancers6020741
70. Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-B and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176.
71. Ouhtit A, Madani S, Gupta I, et al. TGF-B2: A novel target of CD44-promoted breast cancer invasion. J Cancer. 2013;4:566–572. doi:10.7150/jca.6638
72. Wang L, Zhang D, Zhang C, et al. A microRNA expression signature characterizing the properties of tumor-initiating cells for breast cancer. Oncol Lett. 2012;3:119–124. doi:10.3892/ol.2011.444
73. Nagata T, Kai H, Shibata R, Koga M, Yoshimura A, Imaizumi T. Oncostatin M, an interleukin-6 family cytokine, upregulates matrix metalloproteinase-9 through the mitogen-activated protein kinase kinase-extracellular signal-regulated kinase pathway in cultured smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:588–593. doi:10.1161/01.ATV.0000071350.78872.C4
74. Lilja A, Nordborg C, Brun A, Salford LG, Aman P. Expression of the IL-6 family cytokines in human brain tumors. Int J Oncol. 2001;19:495–499.
75. Xu H, Tian Y, Yuan X, et al. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. 2015;8:3783–3792.
76. Li J, Zhou BP. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer. 2011;11:49. doi:10.1186/1471-2407-11-49
77. Gao J, Zhu Y, Nilsson M, Sundfeldt K. TGF-β isoforms induce EMT independent migration of ovarian cancer cells. Cancer Cell Int. 2014;14:72. doi:10.1186/1475-2867-14-67
Supplementary materials
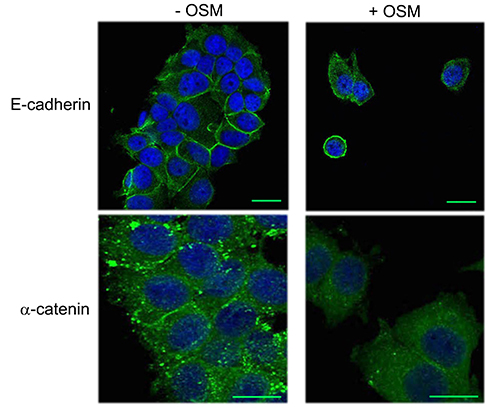 |
Figure S1 OSM induces an EMT in MCF-7 cells. MCF-7 breast tumor cells were treated with OSM (25 ng/mL) for 72 hrs. OSM induces an increased mesenchymal phenotype, a redistribution in E-cadherin expression (green, upper panels scale bar-20 nm), and a slight decrease in α-catenin expression (green, lower panels scale bar-20 nm), as measured by immunofluorescence (DAPI = blue). |
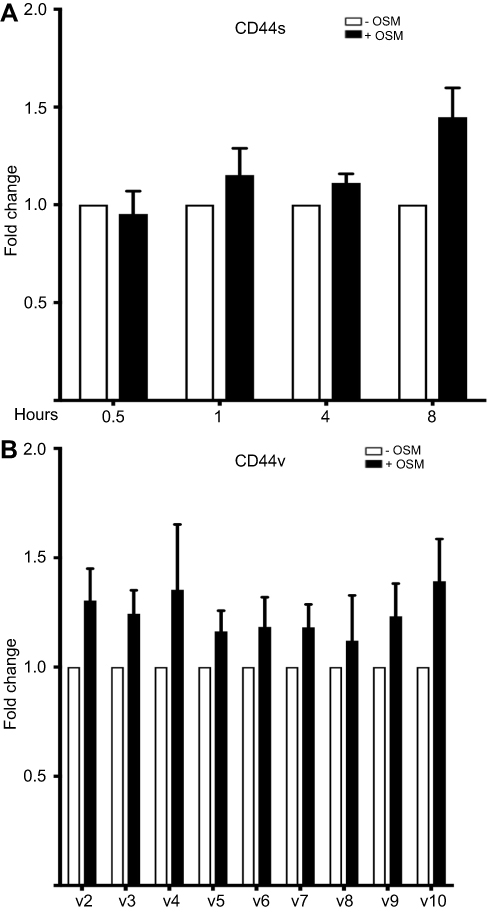 |
Figure S2 OSM has no effect on different CD44 variant mRNA levels in breast cancer cells (A) MCF-7 cells were treated with OSM (25 ng/mL) for 0.5, 1, 4, and 8 hrs followed by RNA collection and qPCR analysis for CD44s (n=3). (B) MCF-7 cells were treated with OSM (25 ng/mL) for 8 hrs followed by RNA collection and qPCR analysis for different CD44 splice variants. (n=3). |
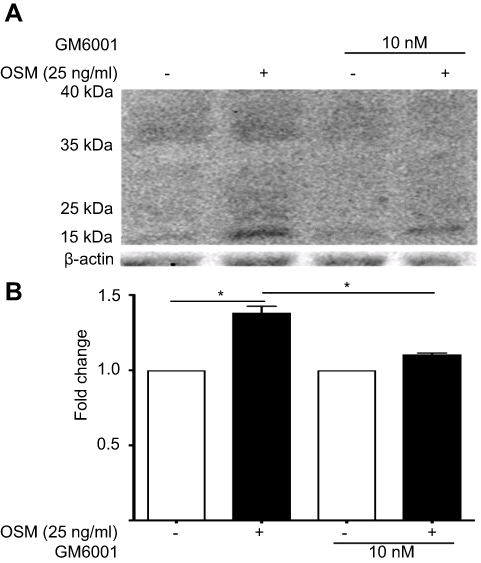 |
Figure S3 OSM-induced CD44 cleavage is reduced by a general MMP inhibitor in ER+ breast tumor cells (A) T47D cells were treated with 10 nM of GM6001 (global MMP inhibitor) for 48 hrs prior to the addition of OSM (25 ng/mL) for an additional 48 hrs. Whole cell lysates were collected and CD44 Western blot analysis was performed to observe cleaved products. OSM induces CD44 in T47D cells after 48 hrs; inhibition of MMP activity reduces OSM-induced CD44 cleavage. (B) Quantitation of the 15–35 kDa bands by ImageJ demonstrate that GM6001 significantly reduces OSM-induced CD44 cleaved products (n=3) (*P<0.05, unpaired t-test). Fold change was calculated using non-OSM treatment as a baseline control compared to OSM treatment. GM6001 with OSM fold change was calculated by comparing it to OSM treatment in the absence of GM6001. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.