Back to Journals » Journal of Inflammation Research » Volume 14
OLFM4 Regulates Lung Epithelial Cell Function in Sepsis-Associated ARDS/ALI via LDHA-Mediated NF-κB Signaling
Authors Gong F ![]() , Li R, Zheng X, Chen W, Zheng Y, Yang Z
, Li R, Zheng X, Chen W, Zheng Y, Yang Z ![]() , Chen Y, Qu H, Mao E, Chen E
, Chen Y, Qu H, Mao E, Chen E
Received 3 September 2021
Accepted for publication 3 December 2021
Published 18 December 2021 Volume 2021:14 Pages 7035—7051
DOI https://doi.org/10.2147/JIR.S335915
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Fangchen Gong,1,* Ranran Li,2,* Xiangtao Zheng,1,* Weiwei Chen,1 Yanjun Zheng,1 Zhitao Yang,1 Ying Chen,1 Hongping Qu,2 Enqiang Mao,1 Erzhen Chen1
1Department of Emergency, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, People’s Republic of China; 2Department of Critical Care Medicine, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Erzhen Chen; Ying Chen
Department of Emergency, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Ruijin 2# Road No. 197, Shanghai, 200025, People’s Republic of China
Tel +862164370045
Fax +862164333548
Email [email protected]; [email protected]
Background: Acute respiratory distress syndrome (ARDS) is one of the leading causes of death in patients with sepsis. As such, early and accurate identification of sepsis-related ARDS is critical.
Methods: Bioinformatic analysis was used to explore the GEO datasets. ELISA method was used to detect the plasma or cellular supernatant of relevant proteins. Quantitative real-time PCR was used for mRNA measurements and Western blot was applied for protein measurements. Immunohistochemistry staining and Immunofluorescence staining were used to identify the localization of OLFM4. Cecal ligation and puncture (CLP) model was used to establish sepsis model.
Results: The bioinformatic analysis results identified ten genes (CAMP, LTF, RETN, LCN2, ELANE, PGLYRP1, BPI, DEFA4, MPO, and OLFM4) as critical in sepsis and sepsis-related ARDS. OLFM4, LCN2, and BPI were further demonstrated to have diagnostic values in sepsis-related ARDS. Plasma expression of OLFM4 and LCN2 was also upregulated in sepsis-related ARDS patients compared to septic patients alone. OLFM4 expression was significantly increased in the lung tissues of septic mice and was co-localized with Ly6G+ neutrophils, F4/80+ macrophages and pro-surfactant C+ lung epithelial cells. In vitro data showed that OLFM4 expression in lung epithelial cells was downregulated upon LPS stimulation, whereas neutrophil media induced OLFM4 expression in lung epithelial cells. Overexpression of OLFM4 and treatment with recombinant OLFM4 effectively suppressed LPS-induced pro-inflammatory responses in lung epithelial cells. Furthermore, the increased levels of LDHA phosphorylation and the downstream NF-κB activation induced by LPS in epithelial cells were effectively diminished by OLFM4 overexpression and recombinant OLFM4 treatment via a reduction in ROS production and HIF1α expression.
Conclusion: OLFM4 may regulate the pro-inflammatory response of lung epithelial cells in sepsis-related ARDS by modulating metabolic disorders; this result could provide new insights into the treatment of sepsis-induced ARDS.
Keywords: sepsis, ALI/ARDS, lung epithelial cells, inflammation, OLFM4, LDHA
Introduction
Sepsis is the leading cause of death worldwide, and due to the unresolved pathophysiology of sepsis, the mortality rate among patients with sepsis-associated multiple organ dysfunction remains high. Acute respiratory distress syndrome (ARDS) is a common complication of sepsis that contributes to a prolonged recovery time and a higher risk of death among septic patients.1 Despite early recognition and advanced supportive care, the incidence of morbidity and mortality in sepsis-related ARDS remains high.2 It is well acknowledged that sepsis-induced dysregulation of inflammatory responses cause the damage of the microvascular endothelial and alveolar epithelial barriers, leading to increased pulmonary vascular permeability and tissue damage during the exudative phase in sepsis-induced ARDS.3
The lung is an immune organ. Pulmonary residential epithelial cells, alveolar macrophages, and ILCs, along with other pulmonary immune cells, play important roles in maintaining the immune function of lungs. Among others, lung epithelial cells express a broad spectrum of pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs), NOD-like receptors (NLRs), nucleic acid sensors, and C-type lectins, thus playing pivotal roles in regulating the innate and adaptive immunity in the lung.4 However, prolonged activation of lung epithelial cells may lead to increased barrier dysfunction and tissue damage, thus aggravating lung injury.5 In addition, during sepsis, the cellular metabolism is switched from oxidative phosphorylation to glycolysis or β-oxidation of fatty acids,6,7 leading to the damage of the lung tissue.8 Therefore, the abovementioned complicated pathogenesis makes it important to explore novel and potential therapeutic strategies for the treatment of sepsis-associated ARDS.
Olfactomedin (OLFM)4, also known as GW112 or hGC-1, is a granular protein that belonging to the olfactomedin family.9 Dysregulation of OLFM4 expression has been observed in various inflammatory diseases, such as inflammatory bowel disease, Helicobacter pylori infection, and gastrointestinal malignancies.10–12 An increasing number of studies have shown that OLFM4 was a marker for a subset of neutrophils.13,14 During H. pylori infection, OLFM4 is expressed by neutrophils, macrophages, and gastric epithelial cells, suggesting a possible crosstalk between these cell types. The relationship between OLFM4 and sepsis as well as ARDS which are both characterized by a dysregulated inflammatory response is a research “hotspot”. Our previous study on bioinformatics analysis integrating three GEO datasets showed that OLFM4 is a critical gene in sepsis.15 In addition, septic patients with increased expression of OLFM4 mRNA show an increased incidence of ARDS.16 In patients with hemorrhagic shock, upregulation of OLFM4 expression is associated with a longer duration of artificial ventilation.17 However, the regulatory mechanism of OLFM4 in the pro-inflammatory responses of lung epithelial cells during sepsis-related ARDS is unknown yet.
In this study, we identified the critical genes and signaling pathways associated with sepsis-related ARDS. A review of GEO datasets from whole blood RNA revealed that OLFM4 is a critical gene in sepsis and sepsis-induced ARDS. Analysis of the GSE66890 dataset showed that OLFM4 expression could be used to distinguish sepsis-induced ARDS from sepsis. In addition, the increased plasma level of OLFM4 verified its upregulation in sepsis-related ARDS patients compared with septic patients without ARDS. Further in vivo and in vitro experiments in this study demonstrated that OLFM4 plays a regulatory role in the immune response of lung epithelial cells in sepsis-induced ARDS by modulating metabolic disorders.
Materials and Methods
Data Sources
Previously, we analyzed the differentially expressed genes (DEGs) between septic patients and healthy controls from datasets downloaded from the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/). GSE95233, GSE57065, and GSE28750 datasets were used as representative datasets of patients with sepsis. The three datasets included 156 whole blood RNA samples from 89 patients with sepsis and 67 healthy controls. GSE32707 is a dataset of 32 control cases and 18 patients with sepsis-related ARDS. Patients from the GSE66890 dataset were divided into sepsis (n=28) and sepsis-related (n=29) ARDS groups. Data from day-1 of whole blood sample collection were used to analyze gene expression.
DEG Identification
Correction of background expression and normalization of raw data were performed using Affy 1.64.0 in R (R Institute for Statistical Computing, Vienna, Austria). The Linear Models for Microarray Analysis (limma) package (version 3.42.2) in R were used to analyze the differential expression. DEGs were defined as those with a Student’s t-test P <0.05 and log fold-change (FC) >2. DEGs overlapping between datasets were obtained using an online tool for analyses of Venn diagrams (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Enrichment Analyses
The functional roles of gene sets were explored using the Gene Ontology (GO) database. Enrichment of the signaling pathways of the involved genes was investigated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. For comprehensive functional annotation, analyses of the identified DEGs using the GO and KEGG databases were conducted using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tool ((https://david.ncifcrf.gov/). A false discovery rate of <0.05, using GO and KEGG databases, was set as the threshold for significant enrichment.
Construction of a Protein–Protein Interaction (PPI) Network and Hub-Gene Analysis
DEGs were uploaded to the Search Tool for the Retrieval of Interacting Genes (STRING; https://string-db.org/) to analyze the interactions among the proteins encoded by the identified DEGs. Results with a minimum interaction score of 0.4 were visualized using Cytoscape (https://cytoscape.org/). The PPI network for hub genes was computed using the maximal clique centrality (MCC) method and cytoHubba.
Patient Enrollment
Patients diagnosed with sepsis in the emergency department of Shanghai Ruijin Hospital from May 31, 2021, to October 31, 2021, were enrolled. This study was approved by the Ethics Committee of Ruijin Hospital (No.20210101). The investigation was based on the institution’s guidelines for human studies and conformed to the ethics guidelines of the Declaration of Helsinki. Informed consent was obtained from each participant. The enrollment criteria were as follows: (1) age: 18–90 years old; (2) adherence to the sepsis 3.0 sepsis diagnostic criteria; (3) hospital stay > 24h. The corresponding exclusion criteria were as follows: (1) discharge or death within 24 h after admission; (2) participation in other clinical research; (3) emergency surgery after admission; and (4) malignant tumor; (5) pregnant or lactating patients; (6) lack of necessary clinical data. Finally, a total of 20 healthy volunteers, 19 septic patients, and 19 sepsis-related ARDS patients were enrolled.
Patients and Data Records
The following data were collected for each patient. Baseline data: age, sex, BMI index, sepsis type, SOFA score, and Acute Physiology and Chronic Health Evaluation II score. Clinical data: source of infection, white blood cell (WBC), neutrophil, C-reactive protein (CRP), procalcitonin (PCT), lactic acid, and whether mechanical ventilation was used during treatment. Primary endpoint: whether ARDS developed; secondary endpoint: in-hospital mortality, ICU stay, hospital stay, and whether mechanical ventilation was performed.
ELISA method was used to detect the plasma levels of OLFM4, LCN2 and BPI in enrolled patients on day 1 of admission.
Animals
Male C57BL/6 mice (6–10 weeks; 22–28 g) were purchased from Slac Lab Animals (Shanghai, China). Mice were allowed to acclimatize to their surroundings and were maintained in a temperature-controlled (25°C±0.5°C) and air-conditioned (humidity 50–60%) environment with a 12-h light–dark cycle. The mice were provided food and water ad libitum. The protocols for animal experiments were approved by the Animal Ethics Committee of Ruijin Hospital Affiliated to Shanghai Jiaotong University School of Medicine (No. 092) and were in line with the International Guidelines for Care and Use of Laboratory Animals (National Academy of Sciences Health Publication No. 85–23, revised in 1996).
Cecal Ligation and Puncture (CLP) Model
Mice were anesthetized using 3% isoflurane in an anesthetic chamber and maintained with 2% isoflurane via a face mask. An electric razor was used to shave abdominal fur, and 70% ethanol was used to clean the skin. A 0.5 cm midline incision was made on the abdomen to expose the cecum, which was ligated fully below the ileocecal valve. An 18-G needle was used to puncture two holes at the top and bottom of the cecum, and a similar amount of feces was squeezed out. The cecum was returned to the abdomen in its anatomical position, and 50 mL/kg of physiologic (0.9%) saline administered (s.c.) after the surgical procedure. Animals were sacrificed at the indicated times. Tissues and serum were isolated, flash-frozen, and stored at −80°C. Bronchoalveolar lavage fluid (BALF) was collected three times using a sterile tracheal cannula. The total volume of phosphate-buffered saline (PBS) administered was 1.5 mL. The BALF was centrifuged at 1500 rpm for 5 min at 4 °C.
Cell Culture
The murine lung epithelial cell line (MLE-12) and human bronchial epithelial cells (BEAS-2B) were procured from the Institute of Stem Cell Research within the Chinese Academy of Sciences (Shanghai, China). MLE-12 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)-f12 (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal calf serum (Gibco), 1% penicillin, and streptomycin (catalog number: TMS-AB2-C; Millipore, Waltham, MA, USA) in a humidified atmosphere of 5% CO2 at 37 °C. BEAS-2B cells were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum, 1% penicillin, and streptomycin and grown under the same conditions. For murine bone marrow-derived neutrophil (BMDN) purification, bone marrow cells were resuspended in 1mL of ice-cold PBS and layered on top of a pre-layered solution of Histopaque 1119 (Sigma, Missouri, USA) on the bottom and Histopaque 1077 (Sigma) on top. Cells were centrifuged at 500 xg for 30 min at room temperature, without deceleration. BMDNs were collected at the interface of the Histopaque layers, and purity was assessed by flow cytometry. Freshly isolated murine neutrophils were plated in serum-free DMEM with or without LPS stimulation for 4h. Conditioned media were collected, centrifuged at 500 xg for 5 min, and then cultured with MLE-12 cells. Serum-free DMEM was used in the negative control group.
Plasmid Transfection
The full-length coding sequence of OLFM4 was cloned into the pcDNA3.1 vector (Genepharma, Shanghai, China), and Lipofectamine™ 3000 (Invitrogen, Carlsbad, CA, USA) reagent was used to transfect plasmids into MLE-12 cells according to the manufacturer’s protocol. Transfection confirmed using Western blotting.
Quantitative Real-Time PCR
Total RNA was extracted from cells using TRIzol® Reagent (Vazyme, Nanjing, China). RNA concentration and purity were measured using a Gen5 microplate spectrophotometer (BioTek, Winooski, VT, USA). Reverse transcription was performed using HiScript III RT SuperMix for qPCR (+gDNA wiper) (Vazyme, Nanjing, China). qPCR was performed using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China). PCR amplification consisted of 10 min of an initial denaturation step at 95°C, followed by 40 cycles of PCR at 95°C for 15 s and 60°C for 1 min. Relative mRNA expression was determined using the 2−ΔCT method relative to the housekeeping gene GAPDH. Data are presented as fold changes relative to the control groups. The primer pairs used in the experiments are listed as supplementary material (Supplementary Table S1).
Western Blot
Samples of total protein from lung tissue or cells were subjected to ultrasound grinding with RIPA lysis buffer, protease inhibitors, and a phosphatase inhibitor cocktail. Proteins (20 μg) were denatured with sample buffer (sodium dodecyl sulfate), separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (using 10% gels), and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, MA, USA). After blocking in 5% bovine serum albumin for 1 h at room temperature, PVDF membranes were incubated overnight with primary antibodies at 4°C. Subsequently, membranes were washed and incubated with horseradish peroxidase-labeled secondary antibodies (1:10,000 dilution) for 1 h at 37°C. Then, the PVDF membranes were washed thrice for 10-min each. Finally, signals were detected using an electrochemiluminescence substrate (Tanon, Shanghai, China), according to the manufacturer’s instructions. Immunoreactive protein bands were visualized using a specialized imaging system (Bio-Rad Laboratories, Carlsbad, CA, USA). The primary antibodies used were purchased from Cell Signaling Technology (Danvers, MA, USA): OLFM4 (1:1000; mouse-specific; 39141), OLFM4 (1:1000; 14369), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:5000; 5174), hypoxia-inducible factor (HIF)-1α (1:1000; 36169), phosphorylated (p)-LDHA (1:1000; 8176), and phosphorylated nuclear factor-kappa B (p-NF-κB; 1:1000; 3033).
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentrations of interleukin (IL)-6, C-X-C motif chemokine ligand (CXCL)-1, and chemokine ligand (CCL)-2 in mouse sera, BALF, and cell supernatants were measured using an ELISA kit (MultiSciences Biotechnology, Hangzhou, China), according to the manufacturer’s instructions. The concentrations of OLFM4 in mouse plasma, BALF and cell supernatant were measured using an ELISA kit (Cloud-clone, Wuhan, China). The lipocalin (LCN)2 concentration was measured using ELISA (ab119601; Abcam, Cambridge, UK). The concentrations of OLFM4 and LCN2 in human plasma were measured using ELISA kits (OLFM4, ab267805; LCN2, ab215541; Abcam, Cambridge, UK). The human BPI concentrations was measured by ELISA kit (HK314-01, HyCult, Uden, The Netherlands).
Immunohistochemistry Staining
For immunohistochemical detection of OLFM4, fixed lung tissues were dehydrated in an ascending series of alcohol concentrations, embedded in paraffin, and sectioned at a thickness of 5 μm. The sections were treated with endogenous peroxidase and blocked with nonspecific proteins. Sections were then incubated with primary antibody against OLFM4 (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C. PBS was used as a negative control. Sections were then incubated with a biotinylated secondary antibody for 1 h at room temperature. Finally, sections were stained with diaminobenzidine for microscopic examination (DS-U3; Nikon, Tokyo, Japan).
Immunofluorescence Staining
Deparaffinized and dehydrated lung sections were boiled at 98°C for 10 min for antigen retrieval, followed by blocking with 10% normal goat serum for 1 h at room temperature. Sections were incubated with anti-F4/80 (Servicebio, Wuhan, China), anti-Ly6G (Servicebio), and anti-prosurfactant protein C (pro-SPC) (ab211326, Abcam) at 4°C overnight. Sections were then incubated with Alexa Fluor-conjugated isotype-specific secondary antibodies (Servicebio) for 1 h at room temperature. Nuclei were stained with 4,6-diamidino-2-phenylindole (Servicebio) for 5 min at room temperature, and samples were mounted with mounting media (Sigma, Missouri, USA). Finally, sections were stained with diaminobenzidine for microscopic examination (DS-U3; Nikon, Tokyo, Japan).
Measurement of ROS Levels
Intracellular ROS production was detected using the Reactive Oxygen Species Assay Kit from the Beyotime Institute of Biotechnology (Shanghai, China). Cells were seeded into six-well plates at a density of approximately 50–70% and treated with lipopolysaccharide (LPS; 5 μg/mL) for 24 h. Upon removal of the medium, 5 μM of dichloro-dihydro-fluorescein diacetate was added to each well and incubated for 30 min at 37°C. MLE-12 cells were washed twice with phosphate-buffered saline (PBS). Intracellular ROS was quantified using a Synergy Neo2 Multi-Mode Reader with 488 nm excitation and 525 emissions (BioTek, Winooski, Vermont, USA).
Statistical Analysis
All data were analyzed using SPSS (version 23.0; IBM, Armonk, NY, USA). Kolmogorov–Smirnov was used for sample normality testing. Continuous variables were expressed as mean ± standard deviation or median and interquartile range. Categorical variables were expressed as numbers and percentages. For continuous variables, data that met the normal distribution were compared using the t-test for comparison between the two groups and ANOVA among three or more groups. For categorical variables, methods such as Pearson’s chi-square test, continuity correction, Fisher’s exact, or likelihood ratio were used. Statistical significance was set at P < 0.05. Analyses of ROC curves were performed to determine the specificity, sensitivity, likelihood ratio, positive predictive value, and negative predictive value of all possible thresholds. The diagnostic value of genes and proteins were predicted based on the ROC curve analyses.
Results
Identification of DEGs in Septic Patients and Patients with Sepsis-Related ARDS
Previously, we analyzed DEGs between healthy controls and septic patients from three GEO datasets (GSE95233, GSE57065, and GSE28750). GSE32707 is a dataset comprising healthy controls and patients with sepsis-related ARDS. The GSE32707 dataset was initially analyzed to identify DEGs unique to sepsis-related ARDS. In total, 28 DEGs were found to overlap between the three datasets we evaluated previously and GSE32707, which suggested their potential role in both sepsis and sepsis-related ARDS (Figure 1A).
 |
Figure 1 OLFM4 is increased in septic and sepsis-induced ARDS patients. (A) Venn diagram of DEGs from our previous datasets and the GSE32707 dataset. Each colored circle represents the DEGs of corresponding datasets. The overlapped area indicates 28 DEGs shared between septic patients and patients with sepsis-induced ARDS. Significant DEGs were defined as those having p < 0.05 and [log FC] > 1.5 as cutoff criteria. (B) Establishment of a PPI network and identification of hub genes. Twenty-two DEGs were filtered into a DEG–PPI-network complex. The line between proteins denotes an interaction score ≥0.4. The more interactions with other proteins, the more important that protein is. (C) Ten genes were chosen as critical genes according to the MCC method using the CytoHubba plugin. |
Enrichment analyses were performed using the GO and KEGG databases on the 28 common DEGs (Table 1). The molecular functions enriched in this dataset are primarily associated with the inflammatory response. A PPI network was established to better understand which shared DEGs were most likely to be most essential for the development from sepsis to sepsis-related ARDS (Figure 1B). The top 10 genes according to the MCC method were selected using the CytoHubba plugin and are sequentially listed as follows: CAMP, LTF, RETN, LCN2, ELANE, PGLYRP1, BPI, DEFA4, MPO, and OLFM4 (Figure 1C).
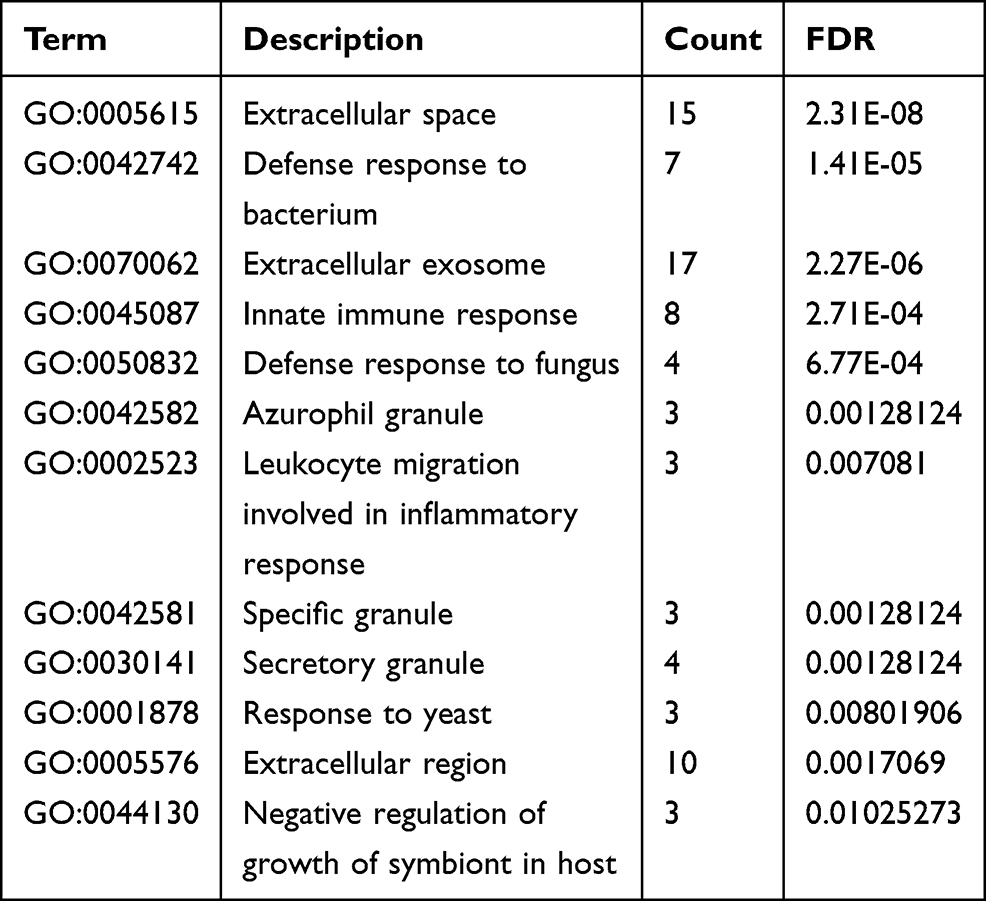 |
Table 1 Functional Enrichment of 28 DEGs |
We further explored whether the selected ten genes participate in sepsis-related ARDS and could be used to distinguish sepsis-related ARDS from sepsis. The GSE66890 dataset, which comprised patients with sepsis and sepsis-related ARDS, was used for further analyses. Heatmaps revealed the expression profiles of ten genes in patients with sepsis and sepsis-related ARDS (Figure 2A). Statistical analyses of these genes are shown in Figure 2B. Five of these ten genes showed significant differences between the sepsis group and sepsis-related ARDS group: OLFM4, LCN2, BPI, DEFA4, and LTF. ROC curves were generated, and three genes showed an AUC (area under the ROC curve) of > 0.7,0.716 for OLFM4, 0.719 for LCN2, and 0.735 for BPI) (Figure 2C).
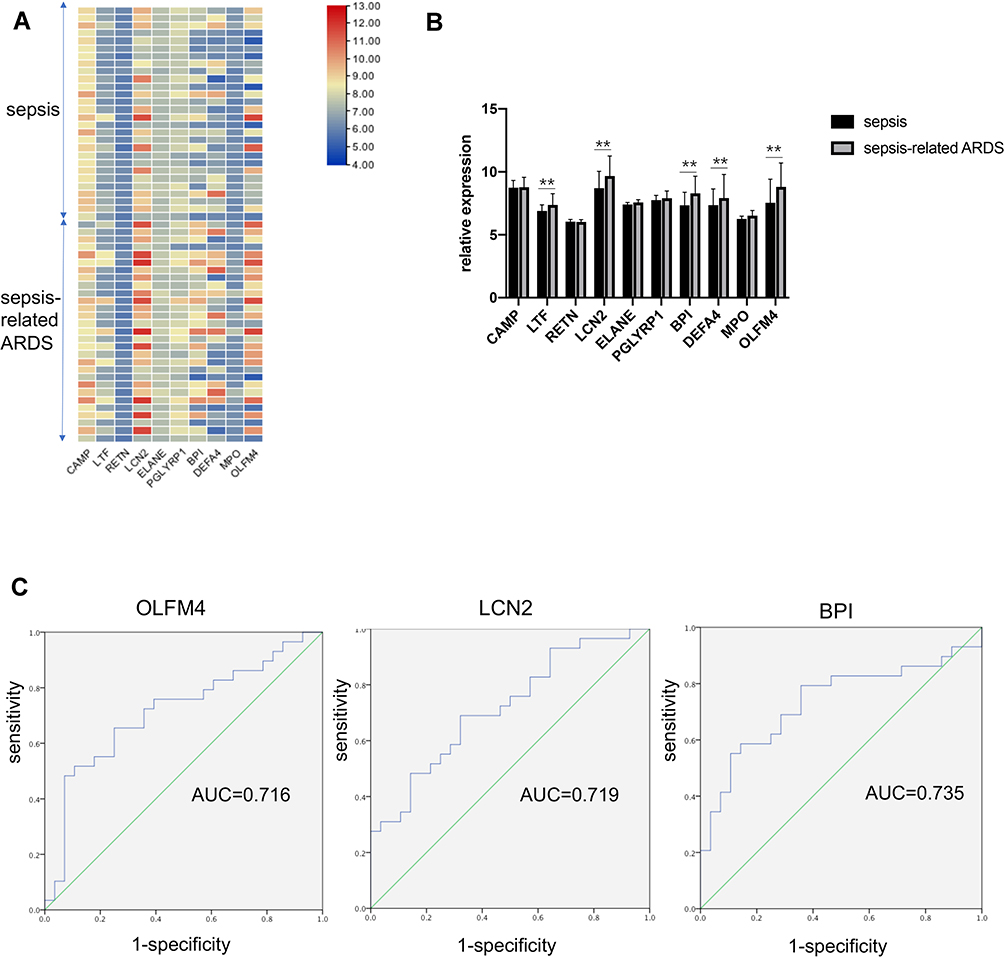 |
Figure 2 OLFM4 expression was different between septic and sepsis-induced ARDS patients. (A and B) Heatmap and statistical analyses of expression of the ten hub genes in the GSE66890 dataset. Red = upregulated. Blue = downregulated. **p < 0.01. (C) Analyses of ROC curves of critical DEGs in the GSE66890 dataset. ROC curves were generated and the area under the ROC was used to compare the ten genes in the sepsis group and sepsis-related ARDS group. Three DEGs showed AUC >0.7: 0.716 for OLFM4, 0.719 for LCN2, and 0.735 for BPI. |
Critical Genes Were Validated at Transcriptional and Protein Levels
The expression of ten key genes in the whole blood RNA samples from healthy controls and septic patients with or without ARDS (n = 5) was measured using quantitative real-time PCR (Figure 3A). The results showed that the expression of most of the genes was consistent with the trend observed in the microarray analysis, while CAMP and DEFA4 showed no significant difference.
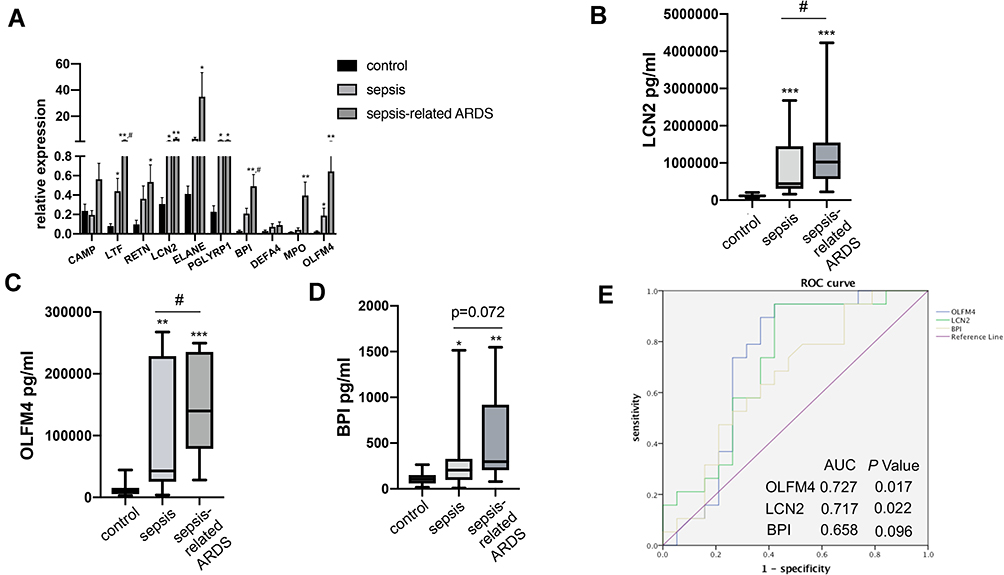 |
Figure 3 Validation of OLFM4 expression at the transcriptional and protein level. (A) Expression of ten critical genes was compared between healthy controls, septic patients, and sepsis-related ARDS patients by quantitative real-time PCR. Differences between two groups were analyzed. *p < 0.05, **p < 0.01 versus CT group, #p < 0.05 versus sepsis group. (B, C and D) Plasma OLFM4, LCN2 and BPI expressions were compared between healthy controls, septic patients, and sepsis-related ARDS patients using ELISA kit. **p < 0.01 versus control group, ***p < 0.001 versus control group, #p < 0.05 versus sepsis group. (E) Analyses of ROC curves of plasma OLFM4, LCN2 and BPI expression. ROC curves were generated and the area under the ROC was used to compare plasma OLFM4, LCN2 and BPI expressions in the sepsis group and sepsis-related ARDS group. |
Since OLFM4, LCN2, and BPI are secreted proteins, the plasma levels of OLFM4, LCN2, and BPI were measured by ELISA to evaluate their diagnostic value for sepsis-related ARDS. Table 2 showed characteristics of patients at the time of blood collection. Compared to patients with sepsis alone, sepsis-related ARDS patients had lower BMI and higher SOFA scores without other significant differences. As shown in Figure 3B–D, plasma levels of OLFM4, LCN2 and BPI in sepsis as well as sepsis-related ARDS group were significantly higher than the control group. In addition, plasma levels of OLFM4 and LCN2 exhibited significant elevation in sepsis-related ARDS group compared with that in sepsis group. ROC curves were generated to evaluate the diagnostic value of OLFM4, LCN2, and BPI in sepsis-related ARDS, and the area under the curves was 0.727 for OLFM4 and 0.717 for LCN2 (Figure 3E).
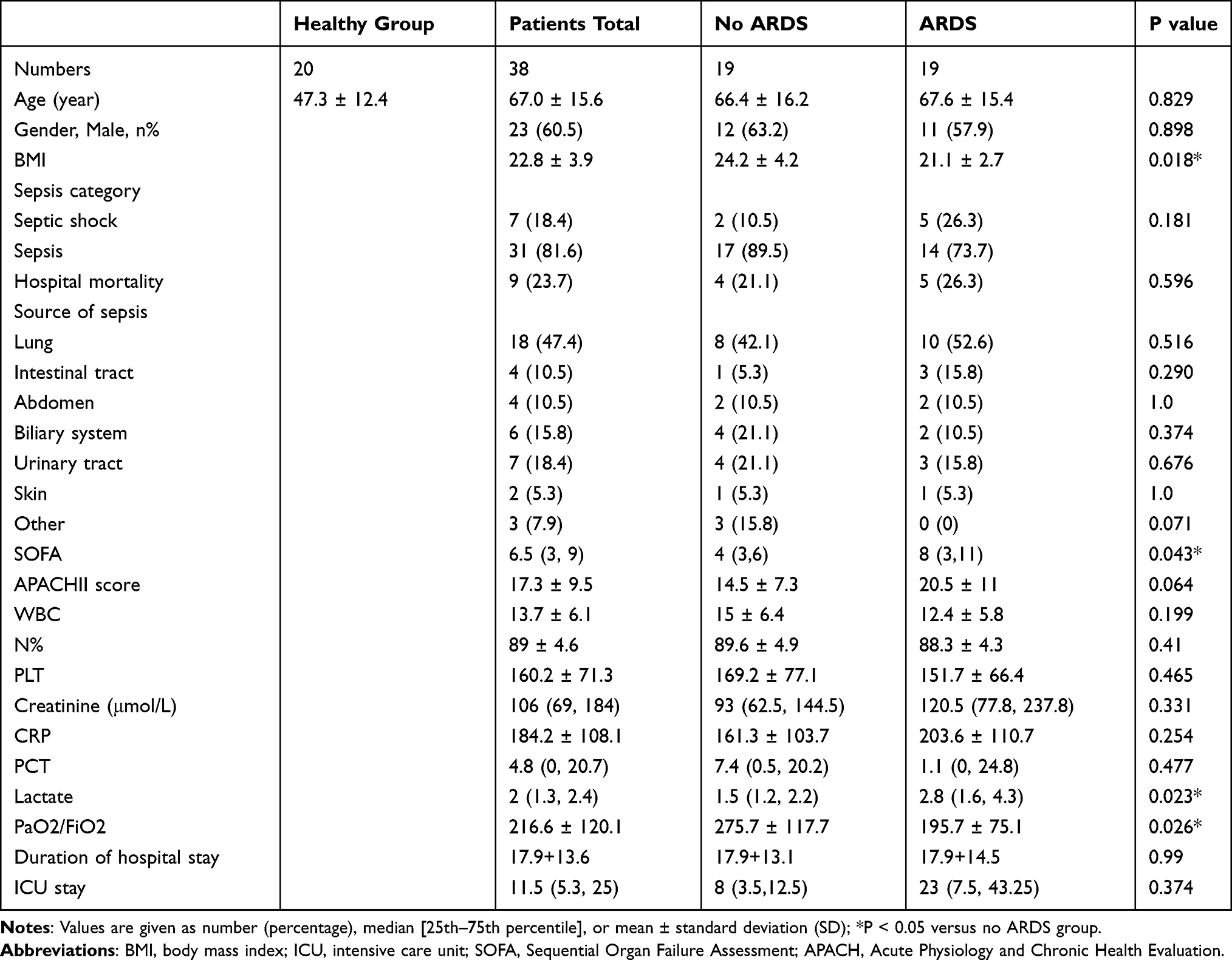 |
Table 2 Characteristics of Septic Patients with or Without ARDS |
OLFM4 Was Associated with Lung Injury in Septic Mice
Among the three markers (OLFM4, LCN2, BPI) obtained from the bioinformatic analysis, OLFM4 was focused on in our further experiments since the role of OLFM4 in sepsis as well as sepsis-related ARDS remains elusive. In CLP-induced mouse model of sepsis, sepsis-induced lung injury was confirmed by H&E staining as well as increased IL-6 levels both in sera and BALF (Figure S1A and B). The protein level of OLFM4 was significantly increased both in the lung tissue as well as in BALF compared to the sham control (Figure 4A and B). IHC staining showed the increased expression of OLFM4 in the lung tissue of septic mice (Figure 4C). Moreover, as shown in Figure 5, OLFM4 was primarily colocalized with neutrophils marked by Ly6G, whereas the co-localization of OLFM4 with F4/80+ macrophages and pro-SPC+ lung epithelial cells was also observed, although to a less extent.
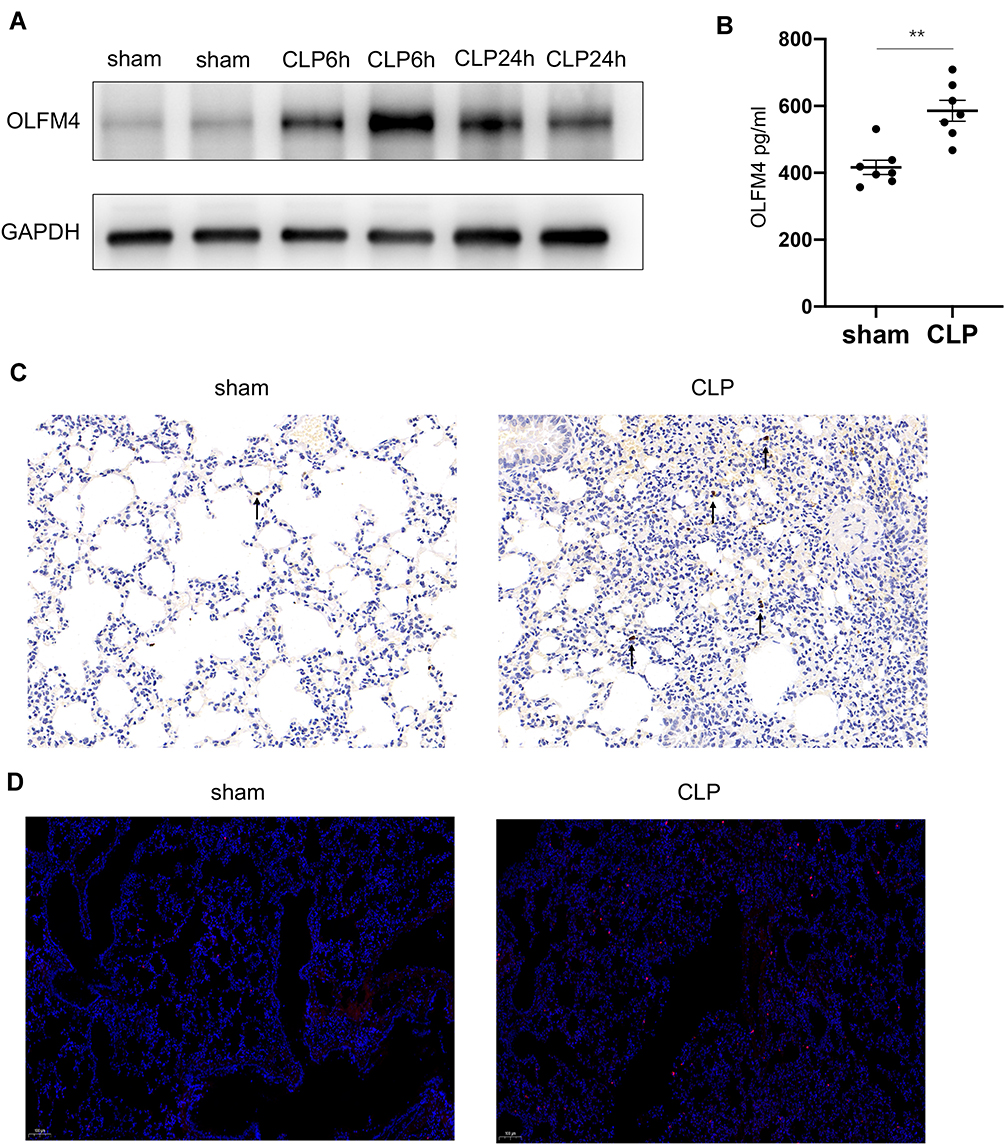 |
Figure 4 OLFM4 expression was increased in CLP-induced lung injury. (A). OLFM4 protein expression in CLP-induced lung injury at 6 h and 24 h was detected by Western blot. (B). OLFM4 expression in the BALF of sham mice and CLP mice was measured by ELISA. Data are presented as mean ± SEM (n = 7). **p < 0.01. (C and D). OLFM4 expression in the lungs of sham mice and CLP mice was detected by immunohistochemical staining and immunofluorescence staining. OLFM4 was stained red, and nuclei was stained with DAPI in IF images. |
 |
Figure 5 OLFM4 expression was localized in neutrophils, macrophages, and lung epithelial cells. (A–C) Lung tissues of sham mice and CLP mice were double stained to determine the detailed localization of OLFM4. OLFM4 was stained red. Neutrophils, macrophages and lung epithelial cells were marked with Ly6G, F4/80 and pro-SPC respectively in green, and nuclei were stained with DAPI. |
To verify OLFM4 expression in epithelial cells, MLE-12 were treated with different doses of LPS or incubated with LPS for different time periods. Interestingly, we found that the protein level of OLFM4 was downregulated in a time-dependent and LPS dose-dependent manner (Figure 6A and B), while OLFM4 level in the supernatant was not altered by detection of ELISA kit. These results were further confirmed by the decreased OLFM4 expression in BEAS-2B, representing bronchial epithelial cells, upon treatment of LPS at different timepoints (Figure 6C).
 |
Figure 6 OLFM4 expression in lung epithelial cells was induced by neutrophil media. (A) MLE-12 cells were incubated with LPS (5 µg/mL) for 6, 12, 24, and 48 h, and the expression level of OLFM4 was detected by Western blot. (B) MLE-12 cells were incubated with different doses of LPS (500 ng/mL, 1 µg/mL, 5 µg/mL, 10 µg/mL) for 24 h, and OLFM4 expression was measured by Western blot. (C) BEAS-2B cells were incubated with LPS (5 µg/mL) for 6, 12, 24, or 48 h, and OLFM4 expression was measured by Western blot. (D) Murine BMDNs were purified and the purity was assessed by flow cytometry. (E) BMDNs were stimulated with or without LPS for 4h, and BMDN media was collected. MLE-12 cells were treated with BMDN media or DMEM and OLFM4 expression was measured by Western blot. |
To investigate whether neutrophil-released OLFM4 can be taken up by lung epithelial cells, bone marrow-derived neutrophils (BMDN) were treated with or without LPS, and the conditioned neutrophil media was added to MLE-12. Intriguingly, the expression level of OLFM4 was increased in MLE-12 cells upon incubation with the conditioned supernatants from neutrophils with and without LPS compared to the negative control group (Figure 6E). The results suggested that the uptake of exogenous OLFM4 by lung epithelial cells may play a role in regulating epithelial function.
OLFM4 Inhibited the Pro-Inflammatory Responses of Lung Epithelial Cells Induced by LPS
To further investigate the effect of OLFM4 on the pro-inflammatory responses of lung epithelial cells, OLFM4 was overexpressed in MLE-12 before LPS stimuli and pro-inflammatory cytokines as well as LCN2 were evaluated accordingly. Notably, LPS treatment increased the mRNA expression of IL-6, CXCL-1, and LCN2 in MLE-12 cells, while OLFM4 overexpression effectively downregulated their expression (Figure 7A). In addition, LPS-induced increase of NF-κB/p65 phosphorylation, a key transcription factor involved in sepsis-induced inflammation, was inhibited by OLFM4 overexpression (Figure 7B). Furthermore, BEAS-2B cells were pre-treated with human recombinant OLFM4 at different doses before LPS stimuli. The results showed that recombinant OLFM4 inhibited LPS-induced phosphorylation of p65 as well as the expression of pro-inflammatory cytokines (Figure 7C and D). These data indicated that OLFM4 has anti-inflammatory effects on LPS-induced lung epithelial cells.
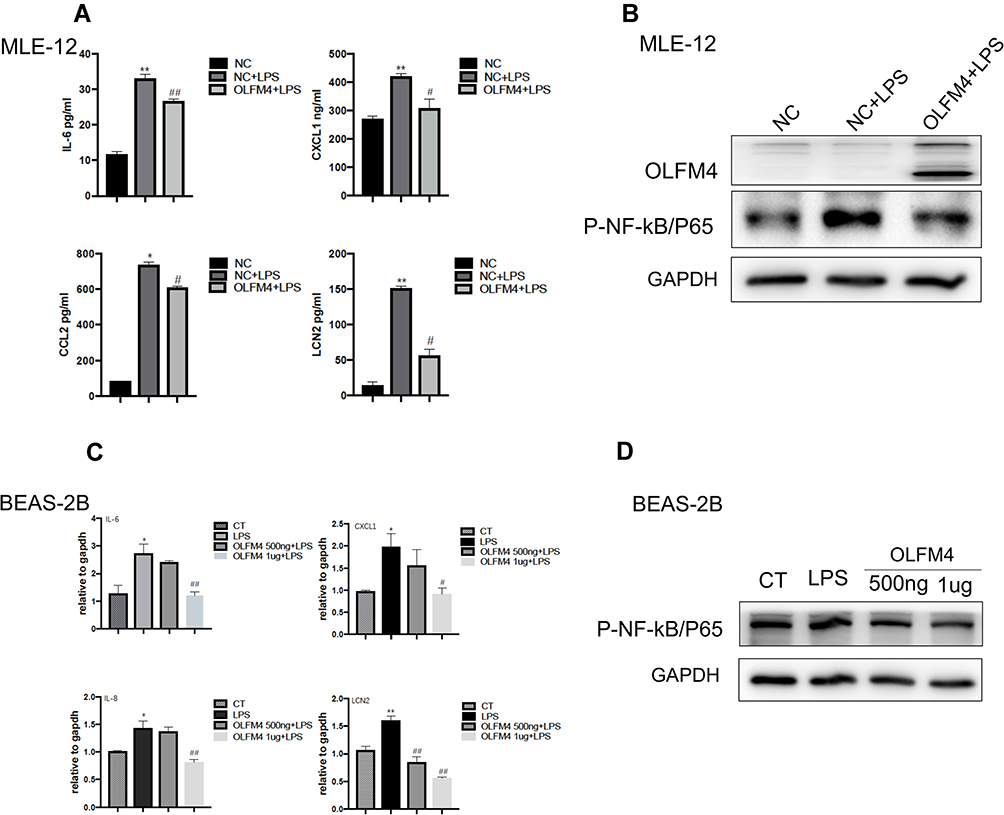 |
Figure 7 OLFM4 inhibited the pro-inflammatory responses of lung epithelial cells by blocking NF-κB activation. (A) MLE-12 cells were transfected with OLFM4 plasmids or negative control (NC) for 24 h before LPS stimulation. Expression of IL-6, CCL2, CXCL1, and LCN2 in the supernatants of MLE-12 cells was measured using ELISA. *p < 0.05 versus NC group, **p < 0.01 versus NC group, #p < 0.05 versus NC+LPS group, ##p < 0.01 versus NC+LPS group. (B) OLFM4 expression and the level of phosphorylated NF-κB/P65 in MLE-12 cells were examined by Western blot. (C) BEAS-2B cells were pretreated with human recombinant OLFM4 (500 ng/mL or 1 µg/mL) for 30 min before stimulated with LPS. The mRNA expression of IL-6, CXCL1, IL-8, and LCN2 was measured by real-time RT-qPCR. *p < 0.05 versus CT group, **p < 0.01 versus CT group, #p < 0.05 versus LPS group, ##p < 0.01 versus LPS group. (D) OLFM4 expression and the level of phosphorylated NF-κB/P65 in BEAS-2B cells were examined by Western blot. |
OLFM4 Alleviated LPS-Induced Lung Epithelial Inflammation by Inhibiting LDHA/NF-κB Signaling
Our previous study has reported that mitochondrial dysfunction and glycolysis play indispensable roles in sepsis-related epithelial injury.18 MLE-12 cells treated with LPS showed increased ROS production, and OLFM4 overexpressed MLE-12 cells exhibited less ROS production upon exposure to LPS (Figure 8A). Considering the association of ROS production with HIF-1α, we further examined the expression of HIF-1α. As shown in Figure 8B, OLFM4 overexpression effectively inhibited LPS-induced upregulation of HIF-1α. In addition, BEAS-2B cells were treated with different doses of recombinant OLFM4 prior to LPS stimulation. Consistently, the results showed that LPS-induced HIF-1α expression in BEAS-2B cells was greatly decreased by OLFM4 treatment in a dose-dependent manner (Figure 8C and D).
 |
Figure 8 OLFM4 alleviated mitochondrial dysfunction in lung epithelial cells. (A) MLE-12 cells were transfected with OLFM4 plasmids or NC for 24 h before LPS stimulation. ROS production in MLE-12 cells was evaluated using a microplate reader. (B) The expression of HIF-1α and phosphorylated LDHA (p-LDHA) in MLE-12 cells was detected by Western blot. (C) BEAS-2B cells were pre-treated with human recombinant OLFM4 for 30 min before stimulated with LPS. ROS production in BEAS-2B cells was evaluated using a microplate reader. (D) The expression of HIF-1α and phosphorylated LDHA (p-LDHA) in BEAS-2B cells was detected by Western blot. **p < 0.01 versus CT group, ##p < 0.01 versus LPS group. |
HIF-1α plays an important role in the activation of myeloid cells and the regulation of the glycolytic capacity during inflammation. Eman et al found that HIF-1α induced downstream LDHA expression and led to neutrophil mobilization.19 Therefore, we examined the influence of OLFM4 overexpression on LDHA in MLE-12 cells. LDHA expression in MLE-12 was not significant altered upon LPS stimuli, whereas the phosphorylation level of LDHA was greatly increased which was highly inhibited by OLFM4 overexpression (Figure 8B). To further verify whether LDHA phosphorylation plays a role in mediating the pro-inflammatory response in lung epithelial cells, MLE-12 was treated with the LDHA-specific inhibitor FX-11 before LPS stimulation. We found that FX-11 dramatically diminished the LPS-induced phosphorylation of LDHA and p65 as well as the pro-inflammatory responses in MLE-12 cells (Figure 9A and B). These results were verified in BEAS-2B cells as well (Figure 9C and D). These data indicated that OLFM4 exhibits its anti-inflammatory effects via modulating metabolic disorders in LPS-induced lung epithelial cells.
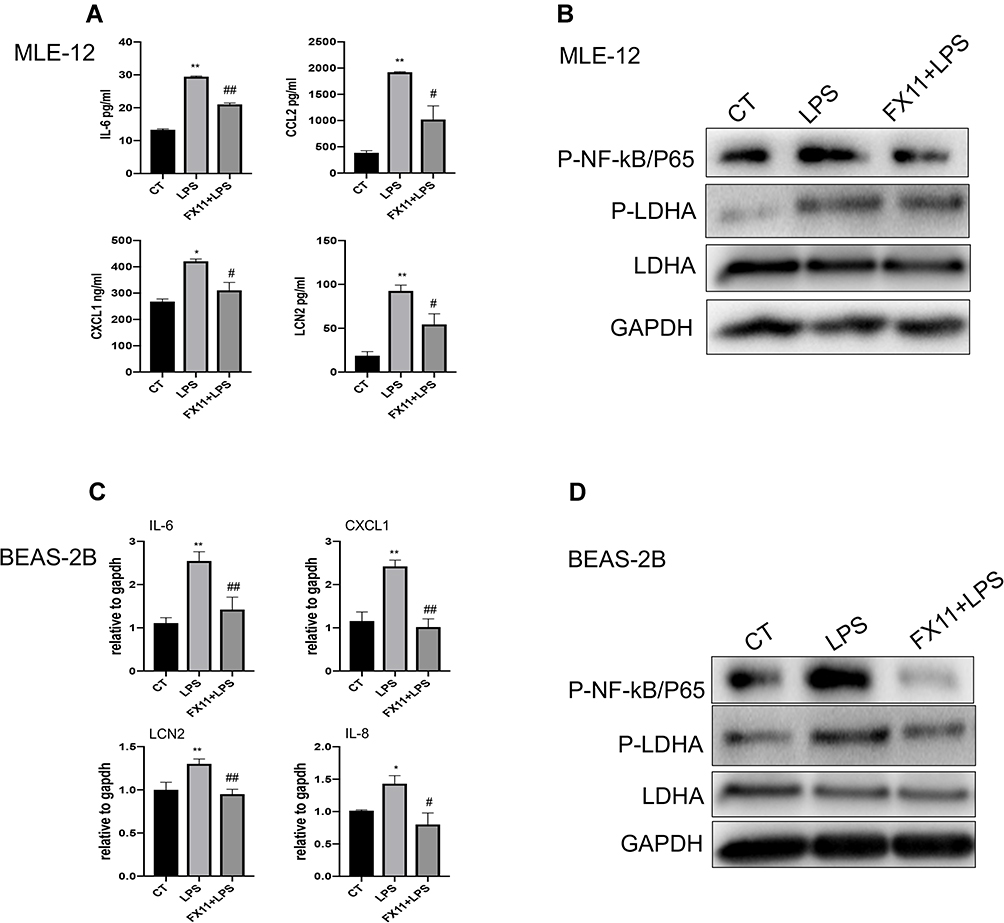 |
Figure 9 LDHA inhibition effectively alleviated LPS-induced inflammation in lung epithelial cells. (A) MLE-12 cells were pre-treated with FX-11 (LDHA inhibitor) for 30 min before LPS stimulation. The concentrations of IL-6, CCL2, CXCL1, and LCN2 in supernatants were measured using ELISA. N=3. (B) MLE-12 cells were pre-treated with FX-11 (LDHA inhibitor) for 30 min before LPS stimulation. The expression of phosphorylated NF-κB/P65, phosphorylated LDHA (p-LDHA) and LDHA in MLE-12 cells was detected by Western blot. (C) BEAS-2B cells were pre-treated with FX-11 for 30 min before stimulated with LPS. The mRNA levels of IL-6, CXCL1, IL-8, LCN2 were determined by real-time RT-qPCR. (D) BEAS-2B cells were pre-treated with FX-11 for 30 min before stimulated with LPS. The expression of phosphorylated NF-κB/P65, phosphorylated LDHA (p-LDHA) and LDHA in BEAS-2B cells was detected by Western blot. *p < 0.05 versus CT group, **p < 0.01 versus CT group, #p < 0.05 versus LPS group, ##p < 0.01 versus LPS group. |
Discussion
Sepsis-related ARDS is the leading cause of death in critically ill patients. In this study, we compared the differences of gene expression profiles between septic patients and patients with sepsis-induced ARDS to identify the key genes involved in the progression of sepsis-related ARDS. We found that OLFM4, LCN2, and BPI were the critical genes that were significantly different between the two groups. Plasma levels of OLFM4 and LCN2 was also upregulated in sepsis-related ARDS patients compared to septic patients without ARDS. Among them, although the role of OLFM4 in immune cells has been extensively studied. The regulatory effects as well as the underlying mechanisms of OLFM4 on the inflammatory activation in lung epithelial cells have not been reported yet. In this study, we demonstrated that OLFM4 was strongly increased in the lung tissue of CLP-induced septic mice, and it was primarily observed in Ly6G+ neutrophils whereas in macrophages and lung epithelial cells less OLFM4 was observed. In vitro, we found that OLFM4 expression in lung epithelial cells was downregulated upon LPS stimulation, while the treatment of lung epithelial cells with conditioned neutrophil media increased the expression of OLFM4. OLFM4 overexpression significantly inhibited the pro-inflammatory responses in lung epithelial cells. Furthermore, we found that the increased level of LDHA phosphorylation and the downstream NF-κB activation upon LPS stimuli were effectively diminished by OLFM4 overexpression and recombinant OLFM4 treatment via reducing ROS production and HIF-1α expression.
The selected critical genes in our present study were mainly associated with the innate immune responses and tissue damage. ELANE, encoded as neutrophil elastase, has been reported to cause tissue damage and alter the remodeling process in sepsis and acute lung injury.19 Myeloperoxidase (MPO) is a major component of active neutrophils and is widely used to evaluate neutrophil infiltration during lung injury.20 It is intriguing that, most of the identified genes, including ELANE, LCN2, MPO, OLFM4, and BPI, are associated with neutrophil activity, implying the importance of neutrophil-related mechanisms in the development of sepsis-associated ARDS.21–23 Neutrophils are the first line of defense against invading microorganisms, whereas excessive neutrophil activation may lead to tissue damage and increase vascular permeability.24 Thus, the identification of several neutrophil-related genes in this study has biological plausibility and may provide insights into the understanding of the pathogenesis during sepsis-related ARDS. The analysis of GEO datasets could also give us an avenue to carry out a predictive model for sepsis-related ARDS. The increase of plasma OLFM4 and LCN2 further confirmed their diagnostic values for sepsis-related ARDS. The relatively low AUCs could be due to small size of patients enrolled. Considering plasma sample is more convenient to be obtained, more patients will be enrolled to verify the diagnostic potential of these two molecules.
Emerging studies have focused on the role of OLFM4 in sepsis as well as sepsis-related organ dysfunction. We have previously revealed that OLFM4 expression was increased in patients suffering from sepsis.15 Upregulation of OLFM4 expression at the transcriptional level has also been reported to be associated with the occurrence of septic shock-associated acute kidney injury.25 Pediatric septic shock patients with a high ratio of OLFM4+ neutrophils had a higher risk of organ failure and mortality.26 Here we showed for the first time the association of OLFM4 with sepsis-induced ARDS. In CLP mice, OLFM4 expression was upregulated in the injured lung tissue. Consistent with a previous study showing that OLFM4 was expressed by a subset of neutrophils,14 the majority of upregulated OLFM4 was localized in the neutrophils during sepsis-related acute lung injury. The increase of OLFM4 expression in lung epithelial cells stimulated with conditioned neutrophil media further suggests a possibly novel finding of neutrophil-epithelial talk.
The contradictory effects of OLFM4 on inflammation have been reported in several studies. On the one hand, OLFM4 knockout mice showed higher expression of IL-1β, TNFα, IL-5, IL-12α, MIP-1α, and MCP-1 than that in wild-type mice 2 weeks after H. pylori infection.10 In addition, in mice challenged with Staphylococcus aureus in chronic granulomatous disease, the activity of cathepsin C, along with its downstream proteases and serum level of proinflammatory cytokines, was higher in OLFM4-deficient mice compared with that in wild-type mice.27 Conversely, in an intestinal ischemia-reperfusion model in mice, OLFM4-deficient mice survived longer and had less intestinal barrier dysfunction than wild-type mice. However, the infusion of OLFM4-null mice with OLFM4-positive neutrophils exacerbated intestinal injury and reduced the survival rate of mice. It has also been found that neutrophils secret OLFM4 into the environment and stimulated a downstream inflammatory response by inducing macrophages to produce inducible nitric oxide synthase (iNOS).28 Moreover, different origins of OLFM4 (eg, epithelium, neutrophils, stem cells) might exert different effects during inflammation. Kuno et al showed that OLFM4 secretion led to antimicrobial activity, while cytoplasmic OLFM4 played an anti-apoptotic role in human intestinal epithelial cells.11 Therefore, the OLFM4 producing cell type needs to be clarified in order to determine the role of OLFM4 in sepsis-related ARDS. Subsequently, cell type-specific OLFM4 knockout model can be constructed to explore the detailed mechanism of OLFM4 in sepsis-related ARDS.
NF-κB is a key transcription factor involved in pro-inflammatory activation in sepsis and ARDS. In our study, OLFM4 overexpression and recombinant OLFM4 treatment decreased the phosphorylation level of NF-κB/p65 in lung epithelial cells induced by LPS. In accordance with our study, Liu et al demonstrated that OLFM4 negatively regulated the activation of NF-κB signaling.10 However, the regulatory mechanisms of NF-κB by OLFM4 is not well known yet. It has been shown that OLFM4 inhibited NF-κB activation as well as subsequent production of cytokines and chemokines by directly associating with nucleotide-binding oligomerization domain (NOD)1 and NOD2.10 Whereas in our study, NOD signaling was not affected by OLFM4, suggesting the involvement of other mechanisms. HIF-1α is well known to play diverse roles in regulating inflammation and metabolism.29 We found that OLFM4 overexpression in lung epithelial cells downregulated the production of ROS as well as the expression of HIF-1α upon LPS stimuli, suggesting the potential effect of OLFM4 on metabolic disorders. Furthermore, HIF-1α was demonstrated to regulate LDHA, the key enzyme for the conversion of pyruvate to lactate during glycolysis.30 LDHA plays a pivotal role in regulating host immune responses. Under tumor conditions, LDHA can affect macrophage polarization, differentiation of T-helper cells, and tumor immune-surveillance. LDHA inactivation can heighten production of type-1 interferon to protect mice from viral infection.31 Natural killer cell-specific deletion of LDHA leads to defective anti-viral and anti-tumor activity of natural killer cells.32 Moreover, during acute inflammation, inhibition of LDHA by specific inhibitors markedly reduced the number of neutrophils or ROShigh neutrophils in blood.30 Our results showed that the inhibition of LDHA effectively inhibited NF-κB activation in lung epithelial cells induced by LPS, indicating the potential role of LDHA in sepsis-associated ARDS. Posttranslational modification such as phosphorylation increases the enzymatic activity of LDHA.33 In our research, LPS stimulation highly increased the phosphorylation level of LDHA in MLE-12 cells and BEAS-2B cells, which was effectively reduced by OLFM4 overexpression and recombinant OLFM4 treatment. Our finding for the first time suggested LDHA-mediated regulation of pro-inflammatory responses in lung epithelial cells by OLFM4.
Our study has several limitations. First, the datasets used to identify the genes associated with sepsis and sepsis-related ARDS were not consistent in diagnosis criteria, which may be due to their different study implantation times and study backgrounds. Samples of new enrolled patients have been analyzed to evaluate the applicability of the results from bioinformatic analysis. Second, the direct role of neutrophil-derived OLFM4 in regulating lung epithelial cell function was not explored by cell co-culture partially due to the complicated cytokine profiles released by neutrophils. In addition, the short life period after neutrophil isolation makes it difficult to employ OLFM4 knock down or knock out strategy in neutrophils.
Conclusions
In this study, we identified critical genes involved in sepsis and sepsis-related ARDS, including CAMP, LTF, RETN, LCN2, ELANE, PGLYRP1, BPI, DEFA4, MPO, and OLFM4. Among these, OLFM4, LCN2, and BPI showed diagnostic value in distinguishing sepsis-related ARDS from sepsis. Increased expression of OLFM4 was observed in the lung tissues of CLP mice. In lung epithelial cells, we found that OLFM4 expression effectively ameliorated LPS-induced pro-inflammatory responses. Furthermore, we found that OLFM4 inhibited ROS production and HIF-1α, thereby blocking NF-κB activation by reducing the phosphorylation-dependent activation of LDHA. Our present study revealed that OLFM4 plays a role in linking the immune responses to metabolic changes in sepsis-induced ARDS and indicates that OLFM4 may have therapeutic potential in the treatment of sepsis-related ARDS.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China (81772107, 81772040), Program for Outstanding Medical Academic, Shanghai Shenkang Hospital Development Center of China (grant numbers SHDC2020CR1028B), Shanghai Municipal Committee of Science and Technology (18411950900), Project of Shanghai Municipal Commission of Health and Family Planning ZY(2018-4012020)-FWTX-1108 and Shanghai Jiao Tong University School of Medicine (DLY201803) and Shanghai Sailing Program (21YF1440600).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43(3):304–377. doi:10.1007/s00134-017-4683-6
2. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562–572. doi:10.1056/NEJMra1608077
3. Reiss LK, Schuppert A, Uhlig S. Inflammatory processes during acute respiratory distress syndrome: a complex system. Curr Opin Crit Care. 2018;24(1):1–9. doi:10.1097/MCC.0000000000000472
4. Leiva-Juárez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018;11(1):21–34. doi:10.1038/mi.2017.71
5. Croasdell Lucchini A, Gachanja NN, Rossi AG, Dorward DA, Lucas CD. Epithelial cells and inflammation in pulmonary wound repair. Cells. 2021;10(2):339. doi:10.3390/cells10020339
6. Escoll P, Buchrieser C. Metabolic reprogramming of host cells upon bacterial infection: why shift to a Warburg-like metabolism? FEBS J. 2018;285(12):2146–2160. doi:10.1111/febs.14446
7. Galván-Peña S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi:10.3389/fimmu.2014.00420
8. Gong Y, Lan H, Yu Z, et al. Blockage of glycolysis by targeting PFKFB3 alleviates sepsis-related acute lung injury via suppressing inflammation and apoptosis of alveolar epithelial cells. Biochem Biophys Res Commun. 2017;491(2):522–529. doi:10.1016/j.bbrc.2017.05.173
9. Zhang J, Liu WL, Tang DC, et al. Identification and characterization of a novel member of olfactomedin-related protein family, hGC-1, expressed during myeloid lineage development. Gene. 2002;283(1–2):83–93. doi:10.1016/S0378-1119(01)00763-6
10. Liu W, Yan M, Liu Y, et al. Olfactomedin 4 down-regulates innate immunity against Helicobacter pylori infection. Proc Natl Acad Sci U S A. 2010;107(24):11056–11061. doi:10.1073/pnas.1001269107
11. Kuno R, Ito G, Kawamoto A, et al. Notch and TNF-α signaling promote cytoplasmic accumulation of OLFM4 in intestinal epithelium cells and exhibit a cell protective role in the inflamed mucosa of IBD patients. Biochem Biophys Rep. 2021;25:100906. doi:10.1016/j.bbrep.2020.100906
12. Wang XY, Chen SH, Zhang YN, Xu CF. Olfactomedin-4 in digestive diseases: a mini-review. World J Gastroenterol. 2018;24(17):1881–1887. doi:10.3748/wjg.v24.i17.1881
13. Alder MN, Mallela J, Opoka AM, Lahni P, Hildeman DA, Wong HR. Olfactomedin 4 marks a subset of neutrophils in mice. Innate Immun. 2019;25(1):22–33. doi:10.1177/1753425918817611
14. Mortaz E, Alipoor SD, Adcock IM, Mumby S, Koenderman L. Update on neutrophil function in severe inflammation. Front Immunol. 2018;9:2171. doi:10.3389/fimmu.2018.02171
15. Gong FC, Ji R, Wang YM, et al. Identification of potential biomarkers and immune features of sepsis using bioinformatics analysis. Mediators Inflamm. 2020;2020:3432587. doi:10.1155/2020/3432587
16. Kangelaris KN, Prakash A, Liu KD, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol. 2015;308(11):L1102–L1113. doi:10.1152/ajplung.00380.2014
17. Kassam AF, Levinsky NC, Mallela JP, et al. Olfactomedin 4-positive neutrophils are upregulated after hemorrhagic shock. Am J Respir Cell Mol Biol. 2021;64(2):216–223. doi:10.1165/rcmb.2020-0276OC
18. Ji R, Chen W, Wang Y, et al. The Warburg effect promotes mitochondrial injury regulated by uncoupling protein-2 in septic acute kidney injury. Shock. 2020. doi:10.1097/SHK.0000000000001351
19. Polverino E, Rosales-Mayor E, Dale GE, Dembowsky K, Torres A. The role of neutrophil elastase inhibitors in lung diseases. Chest. 2017;152(2):249–262. doi:10.1016/j.chest.2017.03.056
20. Aratani Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47–52. doi:10.1016/j.abb.2018.01.004
21. Young RE, Thompson RD, Larbi KY, et al. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. J Immunol. 2004;172(7):4493–4502. doi:10.4049/jimmunol.172.7.4493
22. Holweg A, Schnare M, Gessner A. The bactericidal/permeability-increasing protein (BPI) in the innate defence of the lower airways. Biochem Soc Trans. 2011;39(4):1045–1050. doi:10.1042/BST0391045
23. Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H. Lipocalin-2: a master mediator of intestinal and metabolic inflammation. Trends Endocrinol Metab. 2017;28(5):388–397. doi:10.1016/j.tem.2017.01.003
24. Ginsburg I, Korem M, Koren E, Varani J. Pro-inflammatory agents released by pathogens, dying host cells, and neutrophils act synergistically to destroy host tissues: a working hypothesis. J Inflamm Res. 2019;12:35–47. doi:10.2147/JIR.S190007
25. Wong HR, Cvijanovich NZ, Anas N, et al. A multibiomarker-based model for estimating the risk of septic acute kidney injury. Crit Care Med. 2015;43(8):1646–1653. doi:10.1097/CCM.0000000000001079
26. Alder MN, Opoka AM, Lahni P, Hildeman DA, Wong HR. Olfactomedin-4 is a candidate marker for a pathogenic neutrophil subset in septic shock. Crit Care Med. 2017;45(4):e426–e432. doi:10.1097/CCM.0000000000002102
27. Liu W, Yan M, Liu Y, McLeish KR, Coleman WG, Rodgers GP. Olfactomedin 4 inhibits cathepsin C-mediated protease activities, thereby modulating neutrophil killing of Staphylococcus aureus and Escherichia coli in mice. J Immunol. 2012;189(5):2460–2467. doi:10.4049/jimmunol.1103179
28. Levinsky NC, Mallela J, Opoka AM, et al. The olfactomedin-4 positive neutrophil has a role in murine intestinal ischemia/reperfusion injury. FASEB J. 2019;33(12):13660–13668. doi:10.1096/fj.201901231R
29. Fitzpatrick SF. Immunometabolism and sepsis: a role for HIF? Front Mol Biosci. 2019;6:85. doi:10.3389/fmolb.2019.00085
30. Khatib-Massalha E, Bhattacharya S, Massalha H, et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat Commun. 2020;11(1):3547. doi:10.1038/s41467-020-17402-2
31. Zhang W, Wang G, Xu ZG, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178(1):176–189.e115. doi:10.1016/j.cell.2019.05.003
32. Sheppard S, Santosa EK, Lau CM, et al. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 2021;35(9):109210. doi:10.1016/j.celrep.2021.109210
33. Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate dehydrogenase A: a key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018;7(12):6124–6136. doi:10.1002/cam4.1820
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.