Back to Journals » Nature and Science of Sleep » Volume 7
Obstructive sleep apnea as a risk factor for type 2 diabetes mellitus
Authors Rajan P, Greenberg H
Received 20 June 2015
Accepted for publication 10 August 2015
Published 5 October 2015 Volume 2015:7 Pages 113—125
DOI https://doi.org/10.2147/NSS.S90835
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Steven Shea
Preethi Rajan, Harly Greenberg
Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, Hofstra-North Shore LIJ School of Medicine, New Hyde Park, NY, USA
Abstract: Obstructive sleep apnea (OSA) is independently associated with cardiovascular and cardiometabolic risk in several large epidemiologic studies. OSA leads to several physiologic disturbances such as intermittent hypoxia, sleep fragmentation, and increase in autonomic tone. These disturbances have been associated with insulin resistance and type 2 diabetes mellitus (T2DM) in animal and human studies. Studies also suggest a bidirectional relationship between OSA and T2DM whereby T2DM itself might contribute to the features of OSA. Moreover, successful treatment of OSA may reduce these risks, although this is controversial. The purpose of this article is to review 1) the links and bidirectional associations between OSA and T2DM; 2) the pathogenic mechanisms that might link these two disease states; 3) the role of continuous positive airway pressure therapy in improving glucose tolerance, sensitivity, and resistance; and 4) the implications for clinical practice.
Keywords: Insulin resistance, metabolic syndrome, sleep disordered breathing, intermittent hypoxia
Introduction
Obstructive sleep apnea (OSA) is a growing medical problem, with moderate-to-severe cases affecting 10%–17% of men and 3%–9% of women between the ages of 30 years and 70 years.1 OSA is characterized by repeated upper airway occlusions during sleep that result in specific physiologic perturbations, including sleep fragmentation and chronic intermittent hypoxia (CIH). These disturbances can lead to a cascade of events related to the activation of the sympathoadrenal system, oxidative stress, systemic inflammation, and changes in adipokines – all of which can be important in increasing the risk of cardiovascular disease, hypertension, metabolic syndrome, and diabetes.2–4 The present review will focus on the associations and pathophysiologic mechanisms that link OSA with the development of type 2 diabetes mellitus (T2DM).
Epidemiological links between OSA and T2DM
Prevalence of T2DM in patients with OSA
Several large-scale, cross-sectional, and epidemiological studies have suggested that OSA is an independent risk factor for the development of T2DM, and that as many as 15%–30% of patients with OSA have this comorbidity.5 Moreover, as the severity of OSA increases, so does the likelihood T2DM incidence and of worse glycemic control in patients with T2DM.6
The Sleep Health Heart Study (SHHS) was a large, prospective cohort study designed to investigate the role of OSA as a risk factor for the development of cardiovascular and other chronic diseases.7 This study provided evidence of an independent association between OSA severity and both impaired glucose tolerance and insulin resistance. OSA severity was measured by the apnea–hypopnea index (AHI), which reflects the number of apneas/hypopneas per hour of sleep and whereby AHI <5/h is considered normal, AHI 5–15/h is mild, AHI 15–29/h is moderate, and AHI >30/h is severe. Impaired glucose tolerance was defined as a fasting plasma glucose level of 110–125 mg/dL and a 2-hour oral glucose tolerance test (OGTT) between 140 mg/dL and 200 mg/dL. After adjustment for multiple potential confounders, including age, body mass index (BMI), waist circumference, and self-reported sleep duration, subjects with mild- or moderate-to-severe OSA had odds ratios (ORs) of 1.27 (95% confidence interval [CI] 0.98–1.64) and 1.46 (95% CI 1.09–1.97), respectively, for impaired glucose tolerance compared with subjects with normal AHIs. The severity of nocturnal hypoxemia was also independently associated with glucose intolerance. In addition, OSA severity was associated with increased insulin resistance, which was measured by the homeostasis model assessment insulin resistance (HOMA-IR) index.7
The Wisconsin Sleep Cohort was another large, prospective study that found a significant correlation between the severity of OSA and the prevalence of diabetes: 2.8% of subjects with an AHI of <5/h had the diagnosis of diabetes, compared to 14.7% of subjects with an AHI of ≥15/h. The OR for the diagnosis of diabetes was 2.30 (95% CI 1.28–4.11; P=0.005) in subjects with an AHI of ≥15/h, compared with subjects with an AHI of <5/h, after adjustment for age, sex, and body habitus. On the other hand, a 4-year longitudinal analysis of 978 participants without diabetes at entry did not demonstrate a statistically significant association of diabetes incidence with severity of OSA at baseline, after adjustment for potential confounders at the end of the 4-year period.8
The findings of the Busselton Health Study also noted an association between OSA and higher T2DM prevalence over a 4-year follow-up period. After adjustment for age, sex, BMI, waist circumference, HDL cholesterol, and mean arterial pressure, moderate-to-severe OSA was found to be an independent risk factor for 4-year incident diabetes (OR 13.45, 95% CI 1.59–114.11) and a univariate risk factor for prevalent diabetes (OR 4.37, 95% CI 1.12–17.12).9
Prevalence of OSA in subjects with T2DM
The studies described previously demonstrated independent associations of OSA with insulin resistance and T2DM. Conversely, studies assessing the prevalence of OSA in patients with existing T2DM have found remarkably high rates of OSA.5,10,11 These findings have raised the possibility of T2DM as a risk factor for the development of OSA in a bidirectional fashion. The prevalence of OSA in obese subjects with T2DM was assessed with ambulatory nocturnal respiratory monitoring in The Sleep Action for Health in Diabetes (AHEAD) study, a four-site ancillary study of the Look AHEAD Trial. This is a 16-center trial investigating the long-term health impact of lifestyle intervention designed to achieve and maintain weight loss in over 5,000 obese adults with type 2 diabetes. Sleep testing was performed in 306 participants in the Sleep AHEAD study; surprisingly, 86.6% of obese subjects with type 2 diabetes in this study had an AHI indicative of sleep apnea (AHI ≥5/h). The mean AHI in this cohort was in the moderate range at 20.5±16.8/h. Of these patients, 30.5% had moderate OSA (AHI 15–29/h) and 22.6% had severe OSA (AHI ≥30/h). Severe OSA was most likely as BMI increased (OR 1.1; 95% CI 1.0–1.2; P=0.03).10 These findings could be secondary to the common risk factor for obesity but also raise the possibility that T2DM might contribute to OSA, as will be discussed in future sections.
Association of OSA with metabolic syndrome
OSA is closely associated with metabolic syndrome because of shared risk factors. The term “Syndrome Z” has been developed to describe the links between obesity, insulin resistance, hypertension, and dyslipidemia with OSA. The OR for the presence of metabolic syndrome in patients with OSA ranges from fivefold to as high as ninefold, when compared to subjects without OSA, independent of age and BMI.12–14 In a Chinese population-based study of 255 subjects, severity of OSA correlated with an increasing prevalence of the metabolic syndrome.14 A Japanese case-control study analyzed lean men of normal BMI with and without OSA and demonstrated an association of OSA with the following three components of the metabolic syndrome: insulin resistance, hypertension, and dyslipidemia.15 Although it is difficult to exclude obesity as major contributing factor for the association of OSA with metabolic syndrome, these studies provide evidence that factors other than obesity may mediate this relationship. Some of these potential mediators are discussed in the following sections.
Associations of OSA with nonalcoholic fatty liver disease and insulin resistance
Another clinical condition that is closely linked with the metabolic syndrome is nonalcoholic fatty liver disease (NAFLD), a common liver disease characterized by excessive fatty deposits in the liver. NAFLD is closely related to insulin resistance and has also been recently associated with OSA. The CIH induced by OSA can result in structural damage to the liver with subsequent hepatic fibrosis and inflammation.16–19 These changes appear to be independent of obesity since they are seen among both obese and nonobese patients with OSA.18 Moreover, there is an independent association between the severity of the nocturnal hypoxemia and steatosis that is exacerbated by preexisting obesity. These results were subsequently confirmed in pediatric patients with OSA.20
Potential pathogenic mechanisms linking OSA to insulin resistance and T2DM
OSA can lead to insulin resistance and pancreatic β-cell dysfunction through many intermediary pathways. The upper airway occlusion during sleep that is characteristic of OSA can be partial, resulting in hypopneas, or complete, resulting in apneas. These disordered breathing events result in several pathophysiological perturbations, including sleep fragmentation, activation of the autonomic system, and CIH21,22 (see Figure 1).
 | Figure 1 Links between obstructive sleep apnea and the development of glucose intolerance and T2DM. |
Sleep fragmentation
Sleep fragmentation in the face of normal sleep duration is a common consequence of OSA and results from frequent arousals that often occur at the termination of sleep-disordered breathing events. These are detected on the cortical electroencephalogram and can contribute to elevated sympathetic activity and the symptom of daytime somnolence, which is a characteristic clinical feature of OSA.23 Sleep deprivation and fragmentation are likely risk factors for obesity via effects on metabolism and inflammation. Sleep fragmentation associated with OSA most likely also plays an important role in the development of insulin resistance in many of these patients. Exposing human subjects to acute sleep fragmentation was shown to decrease insulin sensitivity.24,25 A recent animal study also found decreases in visceral and adipose cell insulin sensitivity in mice that were exposed to sleep disruption during their natural sleep period. Importantly, this study also provided a potential mechanistic link between sleep fragmentation and insulin resistance. It posited that sleep fragmentation reduced insulin sensitivity through observed increases in macrophage number and infiltration in visceral fat along with increases in Nox2 (nicotinamide adenine dinucleotide phosphate oxidase) activity, both of which are markers of increased oxidative stress.26
Autonomic nervous system activation
Activation of the autonomic nervous system occurs in association with obstructive apneas and hypopneas; parasympathetic activity tends to predominate during apneas; and sympathetic tone increases at the termination of apneic events. Sympathetic neural drive may be increased at the termination of disordered breathing events as a result of hypoxia-related activation of peripheral chemoreceptors as well as from the effects of sudden arousal from sleep. In addition, elevated levels of circulating and urinary catecholamines have been observed in OSA. Interestingly, elevated sympathetic tone not only is evident during sleep but also seems to persist during the day, even when breathing is normal in patients with OSA.27,28
Intermittent hypoxia
Another very important pathophysiological feature of OSA relates to episodes of intermittent hypoxia and reoxygenation, which are associated with disordered breathing events. These periods of oxyhemoglobin desaturation and resaturation can lead to intermittent tissue hypoxia followed by reoxygenation, which has physiologic consequences that differ from those of chronic hypoxia. The repetitive decreases and increases in oxygen saturation contribute to the formation of reactive oxygen and nitrogen species that increase oxidative stress and can activate redox-sensitive cellular signaling pathways important in inflammation.29–33
To assess the impact of chronic exposure of intermittent hypoxia on various physiological parameters, many small animal models have been developed. Most of these protocols entailed placing rodents in chambers during their sleep periods that are flushed with room air (fraction of inspired oxygen [FIO2] 0.21) followed by different gas mixtures with FIO2 typically at 5% or less with varying cycle times. The resultant hypoxemia may be analogous to that which occurs in severe human OSA. However, this model has been criticized because the extreme reductions in FIO2 might lead to hypoxemia that is more severe than that seen in mild-to-moderate OSA in humans.34 Further, the model is also typically associated with hypocapnia, which might not be apparent in OSA in a clinical situation. Nevertheless, this model has provided important insights, regarding the impact of one of the main pathophysiological features of OSA, namely CIH. CIH in the animal model has been shown to activate the proinflammatory transcription factor nuclear factor-κβ (NF-κβ) in cardiovascular tissues.35 Activation of NF-κβ has also been demonstrated in circulating leukocytes in patients with OSA and is reversible with the treatment of OSA.31
In summary, sleep fragmentation, changes in autonomic tone, and CIH all have the combined effects of increasing sympathetic activation, causing alterations in the HPA axis, increasing oxidative stress, and activating inflammatory pathways. These in turn can result in insulin resistance and pancreatic β-cell dysfunction.21 The impact of CIH on overall insulin sensitivity, sympathetic neural activation, and organ-specific tissues will be discussed in the subsequent sections.
CIH decreases overall insulin sensitivity
Many studies have suggested a link between impaired insulin sensitivity and intermittent hypoxemia, which is an important component of OSA. Both animal and human data indicate that CIH can impair glucose tolerance and clearance and can also increase the HOMA index, a marker of insulin resistance.36
In order to mimic the CIH associated with severe OSA, an animal study used a model of CIH in mice with exposure to CIH during their sleep period, with a return to room air conditions for the remainder of the day. Exposure protocols ranged from hours to several months. The hyperinsulinemic–euglycemic clamp technique was then used to assess insulin sensitivity during exposure to CIH. The CIH group had a 21% reduction in the amount of exogenous glucose necessary to maintain euglycemia during the hyperinsulinemic–euglycemic clamp, indicating that CIH induced insulin resistance in these mice. They also exhibited elevated fasting glucose, providing strong evidence for a causal relationship between exposure to CIH and insulin resistance, independent of obesity. Similar findings were also observed in mice with diet-induced obesity and genetic obesity.16,36
Human data have subsequently confirmed these findings. Exposing healthy human volunteers to a hypoxic alternating with normoxic gas mixture (to mimic the CIH often seen with patients with moderate sleep apnea) resulted in decreased insulin sensitivity without a commensurate increase in insulin secretion, indicative of insulin resistance. This study also noted a decrease in “glucose effectiveness”: the ability of glucose to stimulate its uptake by peripheral tissues and to suppress hepatic glucose production independent of an insulin response.37
CIH causes sympathetic activation
A potential mechanism linking CIH with peripheral insulin resistance is the increased sympathetic neural activity with elevation in catecholamines that occurs with exposure to CIH.28 These catecholamines both decrease peripheral insulin-mediated glucose uptake and increase insulin resistance.38 In addition, activation of the hypothalamic–pituitary–adrenal axis impairs insulin sensitivity and also increases mobilization of glucose. Animal studies have also assessed the impact of CIH-induced sympathetic nervous system activation on insulin resistance using a ganglionic blocker (hexamethonium) to prevent autonomic activation. However, blockade of autonomic activity had no impact on the development of insulin resistance in response to CIH. Therefore, mechanisms other than sympathetic neural activation seem to be responsible for the development of insulin resistance, at least in animal models of CIH. Nevertheless, in a clinical setting, overactivation of the sympathetic nervous system, as well as the hypothalamic–pituitary–adrenal axis, occurring as a result of sleep apnea and its associated sleep fragmentation, might contribute to insulin resistance in OSA along with other factors.
The effects of CIH on organ-specific tissue
CIH and the pancreas
Insulin resistance is an important factor in the pathophysiology and evolution of diabetes. However, clinical diabetes develops when pancreatic β-cells are unable to compensate for increasing insulin resistance. β-cell dysfunction may lead to impairment of the compensatory increases in insulin secretion that are required to maintain normal levels of blood glucose in the setting of progressive insulin resistance. Recent data have suggested various mechanisms for pancreatic β-cell dysfunction. Lean mice exposed to CIH during their sleep period had elevated plasma fasting insulin levels without a change in glucose, suggesting the presence of insulin resistance. Despite this, there was no compensatory pancreatic β-cell proliferation or hypertrophy in these animals. Rather, insulin content was decreased in the pancreatic islets due to downregulation of the enzyme prohormone convertase 1 that converts proinsulin to insulin. Furthermore, the animals exposed to CIH had impaired insulin secretion with impairment of insulin synthesis and processing in the pancreatic β-cells.39 CIH can also result in β-cell apoptosis, through the interaction between apoptosis-related proteins (Bcl-2 and Bax). CIH-mediated oxidative stress results in a downregulation of Bcl-2 and upregulation of Bax, and this imbalance promotes apoptosis.40
Additional animal studies have also suggested a possible role for mitochondrial-derived ROS in CIH-induced pancreatic β-cell injury and dysfunction.39 Finally, cellular studies in vitro have demonstrated CIH-induced downregulation of CD38 gene expression, which is an important gene involved in insulin secretion through the mobilization of Ca2+.41
Impact of CIH on the liver
In both animal and human studies, CIH has been shown to induce structural liver damage and increase liver enzyme levels such as serum alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase.18,19 In animal studies, several weeks of CIH exposure resulted in liver steatosis, necrosis, and inflammation with resultant neutrophil accumulation and collagen deposits. The mechanisms may involve increased synthesis of lipid biosynthesis enzymes, proinflammatory cytokines, and oxidative processes, resulting in DNA damage and apoptosis.17–19
A study showed that after 5 weeks of CIH, both hypoxia-inducible factor 1α (HIF-1α) and NF-κβ transcription factors were upregulated in the liver.42 CIH exposure results in increased lipid biosynthesis enzymes in the liver, such as sterol regulatory element–binding protein-1 (SREBP-1), sterol-coenzyme A desaturase-1 (SCD-1), and high-density lipoprotein (HDL) receptor,43 and thus plays an important role in the development of NAFLD and the metabolic syndrome. CIH also increases the expression of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and macrophage inflammatory protein 2 in obese mice exposed to 4 weeks of CIH. Interestingly, there was no observed increase in these cytokines among lean mice.16 Another study observed increased liver proinflammatory cytokines such as interleukin (IL) 1β, IL-6, and macrophage inflammatory protein 2 in lean mice exposed to longer periods of CIH.18 CIH also results in upregulation of nitric oxide metabolites and reduced activity of liver antioxidant enzymes, which can contribute to DNA damage and apoptosis.42 In addition, CIH can not only increase glucose output from hepatocytes but also upregulate gene expression and protein levels of several gluconeogenic enzymes in the liver,44 which can contribute to fasting hyperglycemia and development of T2DM.
CIH and skeletal muscle
Skeletal muscle is responsible for the majority of insulin-induced glucose uptake. Despite this, few studies have examined the effect of CIH on skeletal muscle metabolism and glucose uptake. A study using a mouse model of CIH observed not only decreases in whole-body insulin sensitivity but also reduces glucose utilization and insulin sensitivity in the soleus muscle, suggesting a clear decrease in glucose metabolism and uptake in this muscle. The impact of CIH was most pronounced in oxidative muscle fibers, while glycolytic muscle fibers were relatively unaffected. Thus, glucose uptake in oxidative muscle tissue is impaired by CIH and this effect appears independent of obesity.45
CIH and adipose tissue
There have been many recent advances in our understanding of the role of white adipose tissue (WAT) in normal physiological function and in response to endogenous stressors.46–48 WAT not only serves as a reservoir of stored energy in the form of triacylglycerols but also serves as an important endocrine organ involved in a variety of metabolic activities. It is especially important for its role in insulin resistance through the release of free fatty acids (FFAs) during lipolysis.49 These induce insulin resistance through their effects on liver, muscle, and adipose tissue itself. CIH can cause dyslipidemia through an increased FFA release; interestingly, this effect is normalized by oxygen supplementation in humans.50 CIH can cause decreased lipoprotein clearance through HIF-1 and angiopeoitin-like 4 inhibition of lipoprotein lipase.51 CIH also downregulates the potent insulin-sensitizing hormone adiponectin in T3-L1 adipocytes. This increases adipose tissue production of resistin, which contributes to further insulin resistance through inflammatory pathways involving cytokines such as TNF-α and IL-6.52
There is also increasing evidence that regions with enlarged areas of visceral adipose tissue experience hypoxia53,54 as a result of reduced adipose tissue blood flow. This occurs as adipocytes hypertrophy and their size exceeds the diffusion capacity of oxygen, thus reducing regional perfusion of adipose tissue.49 This hypoxic stress also activates inflammatory signaling pathways, including HIF-1α and NF-κβ.53 OSA in this population can exacerbate tissue hypoxia and further contribute to adipose tissue inflammation, providing an additional pathway leading to insulin resistance and cardiometabolic morbidity. Adipose tissue hypoxia also leads to the expression and release of various “adipokines”, which are hormones and cytokines that have important functions in health and disease. Proinflammatory adipokines (including cytokines, chemokines, and acute-phase proteins such as haptoglobin and plasminogen activator inhibitor 1) play particularly important roles in the development of obesity-related insulin resistance. These substances are associated with obesity-induced proinflammatory states and they are elevated in the circulation of obese subjects with insulin resistance, whereas the antiinflammatory adipokine, adiponectin, is diminished in the circulation of these subjects. As adipocytes increase in size, macrophages are attracted to and retained within adipose tissue through the actions of chemokines, MCP-1, and MIF, respectively. Consequently, infiltration of type M1-macrophages occurs, and these in turn secrete the proinflammatory cytokines IL-6 and TNF-α. Hence, the M1-macrophage arrival in adipose tissues increases the degree of inflammation in already inflamed tissues. Such inflammatory processes play a significant role in the development of insulin resistance through inhibition of adipocyte storage of lipids, secretions of adipokines, enhanced lipolysis, and reduced reesterification of FFAs resulting in elevation in FFAs in the circulation.
Potential pathogenic mechanisms linking T2DM to OSA
As mentioned previously, there is a higher prevalence of OSA in patients with T2DM than in nondiabetic patients. This has led to the question of whether a reverse causality exists, whereby diabetes itself might lead to some of the features of OSA. Potential mechanisms for the association of OSA among diabetic populations include altered ventilatory control and increased oxidative stress.55,56
Some studies have shown that insulin resistance is associated with a reduced hypercapnic and hypoxic ventilatory response that is reversed with insulin treatment.57 However, it is unclear whether this reduced ventilatory response can exacerbate apneas and hypopneas. Yet other studies have found an association between diabetes and increased risk of central sleep apnea, possibly mediated by autonomic dysfunction that can in turn cause increased central chemoreceptor responsiveness to hypercapnia, thereby predisposing patients to periodic breathing and central sleep apnea.11,58–60 Indeed, diabetic patients with autonomic neuropathy have a higher prevalence of OSA, more severe OSA, longer duration of sleep disordered breathing events, and more severe oxygen desaturations when compared with diabetic patients without autonomic neuropathy.59
Chronic hyperglycemia can also contribute to the development of OSA by increasing oxidative stress. This in turn can result in structural nerve damage and dysfunction with worsening autonomic dysfunction.59
In summary, various possible pathogenic mechanisms might contribute to the association of OSA among diabetic populations, suggesting that there is a bidirectional relationship between OSA and T2DM.
Continuous positive airway pressure therapy in OSA improves glucose control
Continuous positive airway pressure (CPAP) is the gold-standard treatment of OSA and is highly effective in relieving the symptoms of OSA. However, its impact on comorbidities, such as diabetes, is less clear with studies demonstrating variable effects on markers of insulin resistance and insulin sensitivity. A recent meta-analysis of randomized controlled studies examining the effects of CPAP on measures of glycemic control suggested that while CPAP does not decrease hemoglobin A1c (HbA1c) level or BMI in patients who have OSA and T2DM, it may improve insulin sensitivity.61 The variable results may be related to differences in methods of assessment of insulin sensitivity, variation in study population characteristics, and inconsistent adherence to and duration of CPAP therapy. Nevertheless, there are data to suggest that use of CPAP can increase insulin sensitivity and decrease insulin resistance.
CPAP in OSA might improve insulin sensitivity
Several studies have suggested that CPAP improves insulin sensitivity. An observational study demonstrated significant improvement in sensitivity (measured by the hyperinsulinemic–euglycemic clamp technique) after 2 days and 3 months of CPAP therapy in patients with moderate-to-severe OSA. Interestingly, this improvement was most pronounced in the subgroup of subjects with a BMI <30 kg/m2.62 A meta-analysis of 12 prospective observational studies of nondiabetic adults who were newly diagnosed with moderate-to-severe OSA demonstrated that CPAP treatment for 3–24 weeks resulted in a significant decrease in insulin resistance as assessed by HOMA-IR.63
Several other randomized controlled trials have also shown significant improvements in insulin sensitivity in patients with OSA who are treated with CPAP, compared with sham-CPAP. In these studies, insulin sensitivity was assessed by the Gutt index, the quantitative insulin sensitivity check index, the short insulin tolerance test, and the hyperinsulinemic–euglycemic clamp technique.64–66 One such study showed a trend toward improvement in insulin sensitivity after CPAP therapy using the hyperinsulinemic–euglycemic clamp; however, the degree of improvement did not reach statistical significance.67 It is possible that the failure to obtain a statistically significant result was because mean nightly usage of CPAP in that study was only 3.6 hours. In another study, nightly CPAP therapy for OSA resulted in a significant increase in Kitt (glucose disappearance rate) in as little as 1 week of nightly use.65 A review and meta-analysis by Feng et al obtained information from prospective studies examining the effects of CPAP on markers of diabetes in patients with OSA and T2DM. Their findings indicate that CPAP does not improve BMI or glycemic control (measured by HbA1c level) but confirm that it may improve insulin sensitivity in patients with OSA and T2DM, as assessed by the hyperinsulinemic–euglycemic clamp method.61
The differing methodologies utilized to assess insulin sensitivity, patient characteristics, and CPAP adherence rates in prior studies may explain some of the variability in their results. For example, the quantitative insulin sensitivity check index test has a substantially better linear correlation with hyperinsulinemic–euglycemic clamp than HOMA-IR and performs better in patients with insulin resistance. Likewise, HOMA-IR is a good surrogate for the effect of insulin on hepatic gluconeogenesis but may not accurately measure other facets of insulin response and may be less accurate, particularly in the setting of severely impaired pancreatic β-cell function.68 Furthermore, OSA, and CPAP treatment itself, may alter and interact with various aspects of insulin and glucose metabolism such as skeletal muscle insulin sensitivity and pancreatic β-cell function that may not be adequately assessed by the metrics used in these studies.65
Importantly, a common limitation in most of these trials was the limited hours of CPAP use. On average, CPAP use ranged from 3.3 hours/night to 6.2 hours/night. In order to evaluate the role of increasing hours of usage of CPAP, another randomized placebo-controlled study demonstrated incremental improvement in the insulin sensitivity index with each additional hour of nightly CPAP use. However, in this study, significant improvements in insulin sensitivity were observed only in patients with severe OSA (AHI >30/h).66 In order to further assess the impact of increased hours of usage, a recent control study randomized patients either to receive CPAP 8 hours nightly or to receive an oral placebo. Adherence to CPAP was monitored with continuous supervision in a sleep laboratory. Glucose metabolism was measured with the 2-hour OGTT. In the 8-hour/night CPAP group, glucose levels were reduced and insulin sensitivity was increased when compared to placebo. In addition, circulating norepinephrine levels and 24-hour blood pressure were also reduced in the CPAP group as compared to placebo.69 This study highlighted the importance of adherence to CPAP therapy when assessing its impact on these parameters.
CPAP in OSA improves HbA1c
The percentage of HbA1c, a marker of long-term glucose control in diabetic individuals, has been shown to be positively correlated with the severity of OSA in patients with T2DM. HbA1c increases with the severity of OSA after adjusting for age, sex, BMI, race, number of antidiabetes medications, exercise, duration of diabetes, and total sleep time compared to patients without OSA.70 A number of studies have shown an improvement in HbA1c after 3 months of CPAP therapy.71–73 A recent meta-analysis by Gallegos et al demonstrated significant improvement in HbA1c levels and/or increase in insulin sensitivity with >3 months of CPAP therapy among patients who had OSA and either prediabetes or T2DM as determined by laboratory evaluation.74
Predictably, the magnitude of improvement in HbA1c was strongly correlated with the number of hours of nightly usage of CPAP.71 In one study, patients who utilized CPAP for >4 hours/night (mean 6.6 hours/night) achieved the greatest improvement in HbA1c.71 In contrast, another investigation where the mean duration of nightly CPAP use was only 3.6±2.8 hours/night noted no significant improvements in HbA1c or BMI. However, there was significant improvement in insulin sensitivity.67
These findings indicate that long-term CPAP therapy for OSA may produce significant improvements in glucose metabolism and control in T2DM and even prediabetes, but adequate nightly adherence to CPAP is essential to achieve this outcome.
Impact of therapy for OSA on metabolic syndrome
As discussed in a previous section, OSA might be an independent risk factor for the development of metabolic syndrome.75 Elevated systemic arterial hypertension, hyperglycemia, hypertriglyceridemia, hypercholesterolemia, abdominal and visceral obesity, and insulin resistance are all components of metabolic syndrome and have the potential to significantly increase the risk of diabetes, cardiovascular, and cerebrovascular disease.75
Several studies have explored the effect of CPAP therapy for OSA on metabolic syndrome and its specific components.64,76,77 The greatest impact of CPAP therapy appears to be on systemic arterial pressure. Several randomized placebo controlled studies have demonstrated significant reductions in arterial blood pressure with CPAP therapy for OSA.65,76,78 One such study even showed that systemic arterial pressure significantly increased upon CPAP withdrawal among patients with OSA who were previously treated with CPAP, providing further evidence for the impact of CPAP therapy on hypertension in OSA.78
Hyperlipidemia is another component of metabolic syndrome associated with OSA32 and can potentially improve with CPAP therapy. Animal models have shown increases in serum triglyceride and low-density lipoprotein-cholesterol levels with exposure to CIH, possibly through the increased activity of SREBP-1 and SCD-1. This increased activity enhances conversion of saturated to monounsaturated fatty acids, increases serum triglycerides, and promotes lipoprotein secretion.79 Another study has shown that lipid profiles in patients with OSA can improve with CPAP therapy with improvement seen in serum triglycerides, low-density lipoprotein, nonhigh-density lipoprotein, total cholesterol, and the high-density lipoprotein to total cholesterol ratio.65
Abdominal and visceral obesity is another important feature of metabolic syndrome linked with increased cardiovascular risk in patients with OSA. This has been posited to improve with CPAP therapy. However, randomized controlled studies of nondiabetic patients with OSA failed to demonstrate a significant impact of CPAP therapy on visceral, subcutaneous, or hepatic fat distribution.63,64,80
Despite the contradictory and conflicting nature of some of the data, there is mounting evidence that CPAP therapy for moderate-to-severe OSA may improve components of metabolic syndrome, and this may ultimately reduce the cardiovascular and cerebrovascular risk associated with OSA.
Treatments other than CPAP are available for OSA. Mandibular advancement oral appliance therapy is an effective treatment of OSA and has also been shown to improve some outcomes related to the metabolic syndrome. Oral appliance therapy for OSA has been shown to improve markers of lipid peroxidation that are linked to endothelial dysfunction.81 Markers of endothelial dysfunction, an important event that precedes the development of atherosclerosis and might predict future cardiovascular events, may also improve with oral appliance therapy for OSA.82 Some studies have also shown improvement in hypertension with this therapeutic modality in OSA.83 A parallel group study showed similar reduction in morning diastolic blood pressure between CPAP and dental appliance therapy after 10 weeks of use.84
The role of weight loss in improving features of the metabolic syndrome such as insulin resistance has been assessed among patients undergoing bariatric surgery. Weight loss following bariatric surgery has been associated with improvement in insulin resistance, as well as partial or complete remission of T2DM in a subset of patients.85 However, an independent impact of improved OSA versus weight loss on improvement in manifestations of T2DM has not been established.
Clinical implications
Screening for OSA in patients with T2DM
With the increasing prevalence of T2DM and obesity in the aging population, there is a growing need to identify those patients with T2DM who might also be at risk of OSA, since both are independent risk factors for cardiovascular and cerebrovascular disease and are often comorbid chronic conditions.75 The identification of OSA in patients with T2DM is therefore of paramount importance, and this task often falls to the primary care physician.
Assessing risk of OSA might be challenging in primary care settings because many patients with OSA do not report, in routine office visits, “typical” OSA-related symptoms, such as heavy snoring, witnessed pauses of breathing during sleep, and excessive daytime somnolence. Other risk factors might be considered, including anatomic features such as obesity (BMI >30 kg/m2), large neck circumference (>16 in for females; >17 in for males), a crowded oropharynx with a low lying soft palate, large base of tongue and tonsillar hypertrophy, as well as craniofacial abnormalities including retrognathia.86 Evaluation of OSA symptoms should ideally be part of routine patient history and physical examinations. However, this might not always be practical or feasible, especially in primary care settings. Several self-administered screening tools and questionnaires have been developed to facilitate identification of patients who may require referral or testing for OSA.
The Epworth Sleepiness Scale is one such tool that measures subjectively reported tendency to fall asleep during a variety of situations. However, it lacks sensitivity for detection of moderate-to-severe OSA. The STOP-Bang questionnaire assesses various risk factors for OSA and has reasonable sensitivity and specificity when a cutoff score of 5–8 is used.87,88 However, this tool was specifically developed for use in presurgical testing populations. As a result, its validation may not apply to primary care or other settings. The Berlin questionnaire is another short, ten-question survey comprising three different categories. A high risk of OSA is identified by positive answers in two or more of the categories, which yields a 78.6% sensitivity with a 50.5% specificity, for detection of moderate-to-severe OSA.87 The Sleep Apnea Clinical Score is a longer questionnaire (36 items) that has been validated for calculation of likelihood ratios for the presence of OSA. A score of ≥15 yields a likelihood ratio of 4.45 of moderate–to-severe sleep apnea.89 The sensitivities and specificities of these tools are listed in Table 1.90 The STOP-Bang questionnaire and the Berlin questionnaire can each be completed in <5 minutes, allowing them to be used as effective OSA screening tools in a busy clinical setting. Although the sensitivity of the Epworth Sleepiness Scale for OSA is relatively low, it can also provide useful data regarding the degree of daytime somnolence and its improvement with treatment.
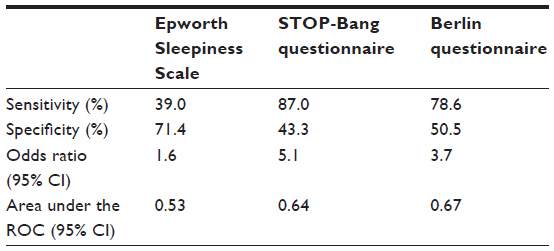 | Table 1 Predictive value of screening questionnaires for moderate-to-severe OSA |
Utilization of these tools in primary care offices might aid in raising the suspicion for OSA in diabetic populations in which the risk is already higher than in the average population. This can ultimately facilitate referral to sleep practices where further evaluation for OSA can occur.
Diagnosis of OSA
Once a patient is referred for further sleep evaluation, the diagnosis of OSA and its severity can be assessed by recording physiologic parameters during sleep. The gold standard is attended polysomnography (PSG) performed in a sleep laboratory. This comprehensively assesses sleep and breathing with recordings of the electroencephalogram, electromyogram, electrooculogram, electrocardiogram, nasal/oral airflow surrogate, thoracic and abdominal respiratory effort, oxygen saturation, and an audio recording of snoring throughout the night.91 Home sleep testing (HST), with more limited respiratory assessment, has recently gained popularity because of its ease of use and reduced cost when compared to standard PSG. This may be useful in cases where the pretest probability of moderate or severe OSA is high.92,93 However, it must be noted that HST has many important limitations that reduce its utility in certain settings. It usually only monitors a surrogate for airflow, respiratory effort, and oxygen saturation and provides no objective measure of total sleep duration or sleep quality. This may lead to underestimation of the severity and impact of sleep disordered breathing, particularly in those patients with milder forms of OSA. In addition, automated scoring of HST may further underestimate the severity of OSA compared with manual scoring of these studies.94
Patient selection for HST is also important as false-negative results may occur in patients with coexisting insomnia who might spend a large portion of the test night awake, since the AHI is calculated in reference to recording time rather that total sleep time, which is usually not measured. HSTs are also inadequate for accurate assessment of more complex sleep disorders, especially in those patients with significant comorbid cardiopulmonary disease. Therefore, patients suspected of having OSA on a clinical basis who then have negative HST results might need referral for confirmatory in-laboratory PSG.93
Treatment of OSA
Various treatment modalities are available for OSA. In order to maximize the chances of achieving successful therapeutic outcomes, strong consideration must also be given to patient-specific needs, expectations, and comorbidities such as cardiac, pulmonary, and cerebrovascular disease, as well as to coexisting sleep disorders such as insomnia.
CPAP therapy remains the gold standard for the treatment of OSA, with randomized, placebo-controlled trials clearly demonstrating significant improvement in quality of life, daytime somnolence, and neurobehavioral performance. These improvements have been observed for all degrees of OSA, including the milder forms.95,96 Several alternatives to CPAP therapy can also be considered and include oral appliance therapy for mandibular advancement,97,98 surgeries of the upper airway; maxillofacial surgery for jaw advancement,99 and bariatric surgery for weight loss100 in appropriately selected patients. As OSA is a chronic condition, long-term disease management with monitoring of compliance and treatment efficacy is essential to achieving optimal functional outcomes, as well as for long-term cardiovascular risk reduction.101
Conclusion
In summary, there is a high prevalence of insulin resistance and T2DM in patients with OSA. An even higher prevalence of OSA has been documented in those patients with T2DM who are obese. The multiple physiologic disturbances in OSA, including sleep fragmentation, activation of the sympathetic nervous system, and CIH secondary to recurrent apneas, may contribute to abnormal glucose and insulin metabolism. CIH, with its associated systemic inflammation and oxidative stress, has been demonstrated in animal models to contribute to hepatic and peripheral insulin resistance as well as to pancreatic β-cell dysfunction, independent of obesity.
Given the links between T2DM and OSA, screening for OSA in this population is important as effective treatment of OSA with CPAP may not only improve sleep apnea-related symptoms and quality of life but also improve components of the metabolic syndrome that contribute to long-term cardiovascular and cerebrovascular risk.
Disclosure
The authors report no conflicts of interest in this work.
References
Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–1014. | |
Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–1384. | |
Punjabi NM, Caffo BS, Goodwin JL, et al. Sleep-disordered breathing and mortality: a prospective cohort study. PLoS Med. 2009; 6(8):e1000132. | |
Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep. 2008;31(8):1071–1078. | |
Pamidi S, Tasali E. Obstructive sleep apnea and type 2 diabetes: is there a link? Front Neurol. 2012;3:126. | |
Kent BD, Grote L, Ryan S, et al; ESADA collaborators. Diabetes mellitus prevalence and control in sleep-disordered breathing: the European Sleep Apnea Cohort (ESADA) study. Chest. 2014;146(4):982–990. | |
Punjabi NM, Shahar E, Redline S, et al; Sleep Heart Health Study Investigators. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol. 2004;160(6):521–530. | |
Reichmuth KJ, Austin D, Skatrud JB, Young T. Association of sleep apnea and type II diabetes: a population-based study. Am J Respir Crit Care Med. 2005;172(12):1590–1595. | |
Marshall NS, Wong KK, Phillips CL, Liu PY, Knuiman MW, Grunstein RR. Is sleep apnea an independent risk factor for prevalent and incident diabetes in the Busselton Health Study? J Clin Sleep Med. 2009;5(1):15–20. | |
Foster GD, Sanders MH, Millman R, et al; Sleep AHEAD Research Group. Obstructive sleep apnea among obese patients with type 2 diabetes. Diabetes Care. 2009;32(6):1017–1019. | |
Resnick HE, Redline S, Shahar E, et al; Sleep Heart Health Study. Diabetes and sleep disturbances: findings from the Sleep Heart Health Study. Diabetes Care. 2003;26(3):702–709. | |
Coughlin SR, Mawdsley L, Mugarza JA, Calverley PM, Wilding JP. Obstructive sleep apnoea is independently associated with an increased prevalence of metabolic syndrome. Eur Heart J. 2004;25(9):735–741. | |
Gruber A, Horwood F, Sithole J, Ali NJ, Idris I. Obstructive sleep apnoea is independently associated with the metabolic syndrome but not insulin resistance state. Cardiovasc Diabetol. 2006;5:22. | |
Lam JC, Lam B, Lam CL, et al. Obstructive sleep apnea and the metabolic syndrome in community-based Chinese adults in Hong Kong. Respir Med. 2006;100(6):980–987. | |
Kono M, Tatsumi K, Saibara T, et al. Obstructive sleep apnea syndrome is associated with some components of metabolic syndrome. Chest. 2007;131(5):1387–1392. | |
Drager LF, Li J, Reinke C, Bevans-Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity (Silver Spring). 2011;19(11):2167–2174. | |
Rosa DP, Martinez D, Picada JN, Semedo JG, Marroni NP. Hepatic oxidative stress in an animal model of sleep apnoea: effects of different duration of exposure. Comp Hepatol. 2011;10(1):1. | |
Savransky V, Nanayakkara A, Vivero A, et al. Chronic intermittent hypoxia predisposes to liver injury. Hepatology. 2007;45(4):1007–1013. | |
Savransky V, Bevans S, Nanayakkara A, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293(4):G871–G877. | |
Sundaram SS, Sokol RJ, Capocelli KE, et al. Obstructive sleep apnea and hypoxemia are associated with advanced liver histology in pediatric nonalcoholic fatty liver disease. J Pediatr. 2014;164(4):699–706. e1. | |
Briançon-Marjollet A, Weiszenstein M, Henri M, Thomas A, Godin-Ribuot D, Polak J. The impact of sleep disorders on glucose metabolism: endocrine and molecular mechanisms. Diabetol Metab Syndr. 2015;7:25. | |
Morgenstern M, Wang J, Beatty N, Batemarco T, Sica AL, Greenberg H. Obstructive sleep apnea: an unexpected cause of insulin resistance and diabetes. Endocrinol Metab Clin North Am. 2014;43(1):187–204. | |
Koren D, O’Sullivan KL, Mokhlesi B. Metabolic and glycemic sequelae of sleep disturbances in children and adults. Curr Diab Rep. 2015;15(1):562. | |
Broussard JL, Ehrmann DA, Van Cauter E, Tasali E, Brady MJ. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann Intern Med. 2012;157(8):549–557. | |
Stamatakis KA, Punjabi NM. Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest. 2010;137(1):95–101. | |
Khalyfa A, Wang Y, Zhang SX, Qiao Z, Abdelkarim A, Gozal D. Sleep fragmentation in mice induces nicotinamide adenine dinucleotide phosphate oxidase 2-dependent mobilization, proliferation, and differentiation of adipocyte progenitors in visceral white adipose tissue. Sleep. 2014;37(5):999–1009. | |
Chandra S, Sica AL, Wang J, Lakticova V, Greenberg HE. Respiratory effort-related arousals contribute to sympathetic modulation of heart rate variability. Sleep Breath. 2013;17(4):1193–1200. | |
Narkiewicz K, Somers VK. Sympathetic nerve activity in obstructive sleep apnoea. Acta Physiol Scand. 2003;177(3):385–390. | |
Arnaud C, Poulain L, Lévy P, Dematteis M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis. 2011;219(2):425–431. | |
Drager LF, Yao Q, Hernandez KL, et al. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am J Respir Crit Care Med. 2013;188(2):240–248. | |
Htoo AK, Greenberg H, Tongia S, et al. Activation of nuclear factor kappaB in obstructive sleep apnea: a pathway leading to systemic inflammation. Sleep Breath. 2006;10(1):43–50. | |
Lavie L. Intermittent hypoxia: the culprit of oxidative stress, vascular inflammation and dyslipidemia in obstructive sleep apnea. Expert Rev Respir Med. 2008;2(1):75–84. | |
Savransky V, Nanayakkara A, Li J, et al. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175(12):1290–1297. | |
Morgan BJ. Intermittent hypoxia: keeping it real. J Appl Physiol (1985). 2009;107(1):1–3. | |
Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T, Liu SF. Chronic intermittent hypoxia activates nuclear factor-kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun. 2006;343(2):591–596. | |
O’Donnell CP. Metabolic consequences of intermittent hypoxia. Adv Exp Med Biol. 2007;618:41–49. | |
Louis M, Punjabi NM. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol (1985). 2009;106(5):1538–1544. | |
Deibert DC, DeFronzo RA. Epinephrine-induced insulin resistance in man. J Clin Invest. 1980;65(3):717–721. | |
Wang N, Khan SA, Prabhakar NR, Nanduri J. Impairment of pancreatic β-cell function by chronic intermittent hypoxia. Exp Physiol. 2013;98(9):1376–1385. | |
Fang Y, Zhang Q, Tan J, Li L, An X, Lei P. Intermittent hypoxia-induced rat pancreatic β-cell apoptosis and protective effects of antioxidant intervention. Nutr Diabetes. 2014;4:e131. | |
Ota H, Tamaki S, Itaya-Hironaka A, et al. Attenuation of glucose-induced insulin secretion by intermittent hypoxia via down-regulation of CD38. Life Sci. 2012;90(5–6):206–211. | |
da Rosa DP, Forgiarini LF, Baronio D, Feijó CA, Martinez D, Marroni NP. Simulating sleep apnea by exposure to intermittent hypoxia induces inflammation in the lung and liver. Mediators Inflamm. 2012;2012:879419. | |
Li J, Thorne LN, Punjabi NM, et al. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res. 2005;97(7):698–706. | |
Polak J, Shimoda LA, Drager LF, et al. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. Sleep. 2013;36(10):1430–1490; 1490A–1490B. | |
Iiyori N, Alonso LC, Li J, et al. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med. 2007;175(8):851–857. | |
Hajer GR, van Haeften TW, Visseren FL. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J. 2008;29(24):2959–2971. | |
Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89(6):2548–2556. | |
Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne). 2013;4:71. | |
Ye J. Adipose tissue vascularization: its role in chronic inflammation. Curr Diab Rep. 2011;11(3):203–210. | |
Jun JC, Drager LF, Najjar SS, et al. Effects of sleep apnea on nocturnal free fatty acids in subjects with heart failure. Sleep. 2011;34(9):1207–1213. | |
Yao Q, Shin MK, Jun JC, et al. Effect of chronic intermittent hypoxia on triglyceride uptake in different tissues. J Lipid Res. 2013;54(4):1058–1065. | |
Borst SE, Conover CF, Bagby GJ. Association of resistin with visceral fat and muscle insulin resistance. Cytokine. 2005;32(1):39–44. | |
Trayhurn P, Wang B, Wood IS. Hypoxia and the endocrine and signalling role of white adipose tissue. Arch Physiol Biochem. 2008;114(4):267–276. | |
Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand. 2005;184(4):285–293. | |
Aurora RN, Punjabi NM. Obstructive sleep apnoea and type 2 diabetes mellitus: a bidirectional association. Lancet Respir Med. 2013;1(4):329–338. | |
Martínez Cerón E, Casitas Mateos R, García-Río F. Sleep apnea-hypopnea syndrome and type 2 diabetes. A reciprocal relationship? Arch Bronconeumol. 2015;51(3):128–139. | |
Hein MS, Schlenker EH, Patel KP. Altered control of ventilation in streptozotocin-induced diabetic rats. Proc Soc Exp Biol Med. 1994;207(2):213–219. | |
Bottini P, Scionti L, Santeusanio F, Casucci G, Tantucci C. Impairment of the respiratory system in diabetic autonomic neuropathy. Diabetes Nutr Metab. 2000;13(3):165–172. | |
Bottini P, Dottorini ML, Cristina Cordoni M, Casucci G, Tantucci C. Sleep-disordered breathing in nonobese diabetic subjects with autonomic neuropathy. Eur Respir J. 2003;22(4):654–660. | |
Tantucci C, Scionti L, Bottini P, et al. Influence of autonomic neuropathy of different severities on the hypercapnic drive to breathing in diabetic patients. Chest. 1997;112(1):145–153. | |
Feng Y, Zhang Z, Dong ZZ. Effects of continuous positive airway pressure therapy on glycaemic control, insulin sensitivity and body mass index in patients with obstructive sleep apnoea and type 2 diabetes: a systematic review and meta-analysis. NPJ Prim Care Respir Med. 2015;25:15005. | |
Harsch IA, Schahin SP, Radespiel-Tröger M, et al. Continuous positive airway pressure treatment rapidly improves insulin sensitivity in patients with obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 2004;169(2):156–162. | |
Yang D, Liu Z, Yang H, Luo Q. Effects of continuous positive airway pressure on glycemic control and insulin resistance in patients with obstructive sleep apnea: a meta-analysis. Sleep Breath. 2013;17(1):33–38. | |
Hoyos CM, Killick R, Yee BJ, Phillips CL, Grunstein RR, Liu PY. Cardiometabolic changes after continuous positive airway pressure for obstructive sleep apnoea: a randomised sham-controlled study. Thorax. 2012;67(12):1081–1089. | |
Lam JC, Lam B, Yao TJ, et al. A randomised controlled trial of nasal continuous positive airway pressure on insulin sensitivity in obstructive sleep apnoea. Eur Respir J. 2010;35(1):138–145. | |
Weinstock TG, Wang X, Rueschman M, et al. A controlled trial of CPAP therapy on metabolic control in individuals with impaired glucose tolerance and sleep apnea. Sleep. 2012;35(5):617B–625B. | |
West SD, Nicoll DJ, Wallace TM, Matthews DR, Stradling JR. Effect of CPAP on insulin resistance and HbA1c in men with obstructive sleep apnoea and type 2 diabetes. Thorax. 2007;62(11):969–974. | |
Muniyappa R, Lee S, Chen H, Quon MJ. Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Endocrinol Metab. 2008;294(1):E15–E26. | |
Pamidi S, Wroblewski K, Stepien M, et al. Eight hours of nightly CPAP treatment of obstructive sleep apnea improves glucose metabolism in prediabetes: a randomized controlled trial. Am J Respir Crit Care Med. 2015;192(1):96–105. | |
Aronsohn RS, Whitmore H, Van Cauter E, Tasali E. Impact of untreated obstructive sleep apnea on glucose control in type 2 diabetes. Am J Respir Crit Care Med. 2010;181(5):507–513. | |
Babu AR, Herdegen J, Fogelfeld L, Shott S, Mazzone T. Type 2 diabetes, glycemic control, and continuous positive airway pressure in obstructive sleep apnea. Arch Intern Med. 2005;165(4):447–452. | |
Hassaballa HA, Tulaimat A, Herdegen JJ, Mokhlesi B. The effect of continuous positive airway pressure on glucose control in diabetic patients with severe obstructive sleep apnea. Sleep Breath. 2005;9(4):176–180. | |
Shpirer I, Rapoport MJ, Stav D, Elizur A. Normal and elevated HbA1c levels correlate with severity of hypoxemia in patients with obstructive sleep apnea and decrease following CPAP treatment. Sleep Breath. 2012;16(2):461–466. | |
Gallegos L, Dharia T, Gadegbeku AB. Effect of continuous positive airway pressure on type 2 diabetes mellitus and glucose metabolism. Hosp Pract (1995). 2014;42(2):31–37. | |
Tasali E, Ip MS. Obstructive sleep apnea and metabolic syndrome: alterations in glucose metabolism and inflammation. Proc Am Thorac Soc. 2008;5(2):207–217. | |
Coughlin SR, Mawdsley L, Mugarza JA, Wilding JP, Calverley PM. Cardiovascular and metabolic effects of CPAP in obese males with OSA. Eur Respir J. 2007;29(4):720–727. | |
Kritikou I, Basta M, Vgontzas AN, et al. Sleep apnoea, sleepiness, inflammation and insulin resistance in middle-aged males and females. Eur Respir J. 2014;43(1):145–155. | |
Kohler M, Stoewhas AC, Ayers L, et al. Effects of continuous positive airway pressure therapy withdrawal in patients with obstructive sleep apnea: a randomized controlled trial. Am J Respir Crit Care Med. 2011;184(10):1192–1199. | |
Savransky V, Jun J, Li J, et al. Dyslipidemia and atherosclerosis induced by chronic intermittent hypoxia are attenuated by deficiency of stearoyl coenzyme A desaturase. Circ Res. 2008;103(10):1173–1180. | |
Sivam S, Phillips CL, Trenell MI, et al. Effects of 8 weeks of continuous positive airway pressure on abdominal adiposity in obstructive sleep apnoea. Eur Respir J. 2012;40(4):913–918. | |
Itzhaki S, Dorchin H, Clark G, Lavie L, Lavie P, Pillar G. The effects of 1-year treatment with a herbst mandibular advancement splint on obstructive sleep apnea, oxidative stress, and endothelial function. Chest. 2007;131(3):740–749. | |
Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104(2):191–196. | |
Gotsopoulos H, Kelly JJ, Cistulli PA. Oral appliance therapy reduces blood pressure in obstructive sleep apnea: a randomized, controlled trial. Sleep. 2004;27(5):934–941. | |
Lam B, Sam K, Mok WY, et al. Randomised study of three non-surgical treatments in mild to moderate obstructive sleep apnoea. Thorax. 2007;62(4):354–359. | |
Courcoulas AP, Belle SH, Neiberg RH, et al. Three-Year outcomes of bariatric surgery vs lifestyle intervention for type 2 diabetes mellitus treatment: a randomized clinical trial. JAMA Surg. 2015. | |
Gami AS, Caples SM, Somers VK. Obesity and obstructive sleep apnea. Endocrinol Metab Clin North Am. 2003;32(4):869–894. | |
Chung F, Subramanyam R, Liao P, Sasaki E, Shapiro C, Sun Y. High STOP-Bang score indicates a high probability of obstructive sleep apnoea. Br J Anaesth. 2012;108(5):768–775. | |
Silva GE, Vana KD, Goodwin JL, Sherrill DL, Quan SF. Identification of patients with sleep disordered breathing: comparing the four-variable screening tool, STOP, STOP-Bang, and Epworth Sleepiness Scales. J Clin Sleep Med. 2011;7(5):467–472. | |
Flemons WW, Whitelaw WA, Brant R, Remmers JE. Likelihood ratios for a sleep apnea clinical prediction rule. Am J Respir Crit Care Med. 1994;150(5 pt 1):1279–1285. | |
Mulgrew AT, Fox N, Ayas NT, Ryan CF. Diagnosis and initial management of obstructive sleep apnea without polysomnography: a randomized validation study. Ann Intern Med. 2007;146(3):157–166. | |
Kirsch DB. In-home testing for obstructive sleep apnea. Continuum (Minneap Minn). 2013;19(1 Sleep Disorders):223–228. | |
Collop NA, Anderson WM, Boehlecke B, et al; Portable Monitoring Task Force of the American Academy of Sleep Medicine. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2007;3(7):737–747. | |
Gay PC, Selecky PA. Are sleep studies appropriately done in the home? Respir Care. 2010;55(1):66–75. | |
Aurora RN, Swartz R, Punjabi NM. Misclassification of OSA severity with automated scoring of home sleep recordings. Chest. 2015;147(3):719–727. | |
Gay P, Weaver T, Loube D, et al; Positive Airway Pressure Task Force; Standards of Practice Committee; American Academy of Sleep Medicine. Evaluation of positive airway pressure treatment for sleep related breathing disorders in adults. Sleep. 2006;29(3):381–401. | |
Weaver TE, Mancini C, Maislin G, et al. Continuous positive airway pressure treatment of sleepy patients with milder obstructive sleep apnea: results of the CPAP Apnea Trial North American Program (CATNAP) randomized clinical trial. Am J Respir Crit Care Med. 2012;186(7):677–683. | |
Jayesh SR, Bhat WM. Mandibular advancement device for obstructive sleep apnea: an overview. J Pharm Bioallied Sci. 2015; 7(Suppl 1):S223–S225. | |
Randerath WJ. Mandibular advancement therapy for obstructive sleep apnea: answers and (more) questions. JAMA Intern Med. 2015;175(8):1285–1287. | |
Smith DF, Cohen AP, Ishman SL. Surgical management of OSA in adults. Chest. 2015;147(6):1681–1690. | |
Ashrafian H, Toma T, Rowland SP, et al. Bariatric surgery or non-surgical weight loss for obstructive sleep apnoea? a systematic review and comparison of meta-analyses. Obes Surg. 2015;25(7):1239–1250. | |
Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med. 2009;5(3):263–276. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.