Back to Journals » OncoTargets and Therapy » Volume 10
Novel role of granulocyte-macrophage colony-stimulating factor: antitumor effects through inhibition of epithelial-to-mesenchymal transition in esophageal cancer
Authors Zhang J, Liu Q, Qiao L, Hu P, Deng G, Liang N, Xie J, Luo H, Zhang J
Received 30 January 2017
Accepted for publication 17 March 2017
Published 20 April 2017 Volume 2017:10 Pages 2227—2237
DOI https://doi.org/10.2147/OTT.S133504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Jingxin Zhang,1,* Qiqi Liu,2,* Lili Qiao,3 Pingping Hu,2 Guodong Deng,2 Ning Liang,2 Jian Xie,2 Hui Luo,4 Jiandong Zhang2
1Division of Oncology, Department of Graduate, Weifang Medical College, Weifang, 2Department of Radiation Oncology, Qianfoshan Hospital Affiliated to Shandong University, Shandong University, 3Department of Oncology, The Fifth Peoples’ Hospital of Jinan, Jinan, 4Department of Radiation Oncology, Henan Cancer Hospital Affiliated to Zhengzhou University, Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
*These authors contributed equally to this work
Purpose: Recent studies demonstrate the possible antitumor effects of granulocyte-macrophage colony-stimulating factor (GM-CSF); however, the exact mechanism is still unclear. The aim of our study was to analyze the effects of GM-CSF on multiple biological functions of human esophageal cancer (EC) cell lines and to explore the potential mechanism of its antitumor effects.
Materials and methods: Eca109/9706 human EC cells were examined. Cell proliferation, apoptosis, and migration were analyzed using cell proliferation assay, flow cytometry, and transwell assay, respectively. The expression of signaling molecules were examined by reverse transcription polymerase chain reaction and Western blot.
Results: Our results provide experimental evidence that GM-CSF inhibits growth and migration, as well as induction of apoptosis in EC cells. In addition, EC cells stimulated with GM-CSF were more likely to have suppressed epithelial-to-mesenchymal transition (EMT), accompanied by increased E-cadherin and decreased vimentin expression.
Conclusion: Our data demonstrate that GM-CSF inhibits cancer cell proliferation and migration, as well as induction of apoptosis. Moreover, our findings indicate that GM-CSF may regulate EMT through JAK2-PRMT5 signaling, and thereby exhibit its antitumor effects on EC cells.
Keywords: granulocyte-macrophage colony-stimulating factor, cancer, antitumor, epithelial-to-mesenchymal transition, esophageal cancer
Introduction
Esophageal cancer (EC) is one of the most frequently diagnosed cancers and a leading cause of cancer-related death in the world.1 China’s statistics in 2015 revealed an extremely high EC-related morbidity and mortality.2 Because of the poorly defined clinical manifestations, ~70%–80% of EC patients present with locally advanced or distant metastatic disease at preliminary diagnosis, and the survival rate is extremely low.3 The efficiency of current multimodal treatments of EC, which include surgery, chemotherapy, and radiotherapy is still low. Therefore, a new effective therapy for EC patients is urgently needed.
Granulocyte-macrophage colony-stimulating factor (GM-CSF), a 22 kDa glycoprotein, was first described as an in vitro inducer of differentiation and proliferation of bone marrow progenitor cells into distinct colonies, including granulocytes and macrophages.4 GM-CSF can also act on relatively early progenitor cells and interacts with erythropoietin to stimulate eosinophil and megakaryocyte colony formation in vitro.5 Thus, recombinant GM-CSF is an important agent used to enhance bone marrow recovery after myelotoxic chemotherapy in cancer patients.6 In addition, G-CSF and GM-CSF are broadly administered to acute leukemia and other patients to accelerate bone marrow recovery, as well as induction of leukocyte proliferation and maturation in solid-organ transplantation, aplastic anemia, and neutropenia caused by acquired immune deficiency syndrome.7 In addition, recombinant GM-CSF seems likely to become an important novel therapeutic agent in several inflammatory diseases.8 Recent studies have revealed a possible antitumor effect of GM-CSF through enhancing the immune response to tumor cells.9–11 Furthermore, promising results from both experimental and clinical studies were provided for GM-CSF gene-transduced tumor cell vaccine (GVAX).12–15
Epithelial-to-mesenchymal transition (EMT) is a phenomenon whereby intercellular adhesions of epithelial cells are loosened such that the cells become isolated, mobile, and resistant to apoptosis (anoikis).16 EMT plays important roles not only in embryogenesis and tissue development but also in tumor progression and metastasis. EMT could contribute to an enhanced ability for cancer cells to migrate and to acquire greater resistance to apoptosis based on the evidence from human and mouse models of breast tumorigenesis.16 Several important hallmark processes, including loss of E-cadherin and cell polarity, as well as increased N-cadherin and vimentin expression, are needed for EMT to occur. Upon initiation of EMT, loss of E-cadherin could promote tumor progression by enhancing the capacity of cells to invade.17 These findings provide evidence that EMT is closely related to the development of highly aggressive tumors. In the present study, we demonstrate the possible correlation between GM-CSF and EMT in EC cells, and thereby explore the therapeutic potential of GM-CSF for EC patients.
Materials and methods
Cell culture
Eca109/9706 human EC cells were obtained from Shandong Academy of Medical Sciences (Shandong, People’s Republic of China). Human EC cells lines Eca109 and 9706 were cultured according to Liang et al.18 All cells were cultured at 37°C in tissue culture flasks (Corning Incorporated; Corning, NY, USA) containing Roswell Park Memorial Institute 1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific) and 1% penicillin–streptomycin (HyClone; GE Healthcare UK Ltd, Little Chalfont, UK) in a humidified incubator containing 95% air and 5% CO2. The culture medium was replaced every second day, and all cultures were periodically examined for mycoplasma contamination. Eca109/GM-CSF and 9706/GM-CSF cells were obtained by adding recombinant human GM-CSF (rhGM-CSF; R&D systems, Minnesota, USA) to 6-well culture plates in which Eca109/9706 cells were incubated, and then culturing for a further 48 h. Phosphate-buffered saline (PBS) was bought from Sigma-Aldrich Co. (St. Louis, MO, USA).
Patients
We retrospectively included 99 patients diagnosed with esophageal squamous cell carcinoma in Shandong Provincial Qianfoshan Hospital from September 2010 to June 2015. This study was performed in compliance with the 1975 Declaration of Helsinki and was approved by the local ethics committee of Qianfoshan Hospital. Paraffin-embedded samples were collected from all patients involved who received curative-intent surgical resections. Among them, some patients accepted a course of chemotherapy or radiotherapy. The protocol for radiotherapy was a median total dose of 60 Gy, using 1.8–2.2 Gy fractional doses on a five times per week schedule. The chemotherapy was mostly performed with docetaxel/cisplatin. Histological types of tumors were identified based on the World Health Organization criteria. Tumor stages were confirmed according to the 7th edition of the American Joint Committee on Cancer TNM classification system.19 The pathological characteristics and patient demographic data were obtained from medical records. Patients who died of non-tumor-related factors were excluded. All patients signed the informed consent approved by our institutional Committee on Human Rights in Research.
Cell proliferation assay
Cell counting kit-8 (CCK8; Dojindo Molecular Technologies, Inc. Rockville, MD, USA) was used to assess cell proliferation. In brief, 10% FBS was used to dissociate Eca109/9706 cells to obtain a monoplast suspension. Approximately 3,000 cells per well, in 100 μL of culture medium, were incubated in 96-well culture plates. Control wells contained the same volumes of culture media. All EC cells incubated with 10 μL of rhGM-CSF (2 μg/μL) or with 10 μL culture medium were recovered and stabilized for 24 h, and then culture was continued for 24 h, 28 h, 72 h, and 96 h in 5% CO2 at 37°C. Subsequently, 10 μL of CCK-8 reagent was added to each well and culture was continued for another 2 h. The optical density (OD) values were measured immediately at 450 nm using a microplate spectrophotometer (Epoch; Biotek, Winooski, VT, USA). Cell growth curves were plotted, with time as the horizontal axis and OD as the vertical axis.
Flow cytometry
Cells cultured for 24 h with (Eca109/GM and 9706/GM) or without (Eca109 and 9706) rhGM-CSF were washed twice with PBS and then re-suspended in 1× working solution at 1×106 cells/mL. A total of 100 μL of cell suspension was incubated with 5 μL of annexin v-phycoerythrin (fluorescein isothiocyanate; eBioscience, San Diego, CA, USA) and 5 μL of 7-amino-actinomycin (propidium iodide; eBioscience) in the dark for 20 min. After staining, 400 μL of binding buffer was added and data were acquired by flow cytometry within 4 h of staining.
Transwell assay
We used transwell polycarbonate filters (8.0 μm pore size; Costar®; Sigma-Aldrich Co.) in 24-well plates to perform the migration assay according to the manufacturer’s instructions. Eca109/9706 cells stimulated with and without GM-CSF were used for the assays. The lower chamber contained 600 μL of 10% FBS complete culture medium as chemoattractant, while 6×104 cells were incubated with 100 μL of serum-free medium in the upper chamber. After incubation in 5% CO2 atmosphere at 37°C for 36 h, the cells that invaded the lower filters were fixed in methanol for 15 min, stained with 0.25% crystal violet (Beyotime; Institute of Biotechnology, Haimen, People’s Republic of China) for 20 min at room temperature, and then washed with PBS. An independent investigator captured images of the migrated cells (magnification, ×40) and counted them (magnification, ×100) under an inverted fluorescence microscope.
Reverse transcription quantitative polymerase chain reaction
Total RNA was isolated using RNA-solv reagent (Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer’s instructions. After quantification of total RNA concentration by spectrophotometry at a wavelength of 260 nm (SpectraMax 190; Molecular Devices LLC, Sunnyvale, CA, USA), cDNA was synthesized using a Rever Tra Ace® reverse transcription quantitative polymerase chain reaction kit (Toyobo, Osaka, Japan) in a 10 μL reaction volume containing 2 μL of 5× RT buffer, 0.5 μL of enzyme mix, 0.5 μL of primer mix, 2 μL of total RNA, and 5 μL of nuclease-free water, with reaction conditions of 5 min at 95°C and 15 min at 37°C. An ABI ViiA7 Dx instrument (Applied Biosystems; Thermo Fisher Scientific) was used to perform PCR reactions. Each 10 μL reaction mixture contained 5 μL of SYBR® Green (Toyobo), 2 μL of nuclease-free water, 1 μL of forward primer, 1 μL of reverse primer, and 1 μL of cDNA. The thermo-cycling conditions were as follows: 35 cycles of 15 s at 95°C, 15 s at 61°C, and 45 s at 72°C. The primer sequences were as follows: GM-CSF, forward primer 5′-GGGAGCATGTGAATGCCATC-3′ and reverse primer 5′-GGCTCCTGGAGGTCAAACAT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward primer 5′-CAGAACATCATCCCTGCCTCTAC-3′ and reverse primer 5′-TTGAAGTCAGAGGAGACCACCTG-3′.
GAPDH served as endogenous control to account for variations in PCR products due to possible differences in the efficiency of the RT and/or PCR reactions. Each sample was amplified independently in triplicate. The cycle threshold (Ct) was defined as the PCR cycle at which an increase in the fluorescence above the baseline signal was first detected. The Ct value is inversely related to the copy number of the target sequence. PCR results were quantified using the ΔCt method according to the following formula: expression ratio =2−ΔCt, where ΔCt = ΔCt target gene − ΔCt endogenous control gene.20
Western blotting
Cells were washed twice with PBS before protein extraction. Total protein was extracted using radioimmunoprecipitation assay lysis buffer (Sigma-Aldrich Co.) according to the manufacturer’s instructions. After cell dissociation on ice for 30 min, liquid supernatant was carefully collected after centrifugation. The appropriate amount of loading buffer was added according to the protein concentration as determined using a bicinchoninic acid assay kit (Solarbio, Beijing, People’s Republic of China). Total protein samples were isolated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis at 30 μg per well and then transferred to a polyvinylidene difluoride membrane (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China). Protein bands were determined using Protein Marker (Thermo Fisher Scientific). The membrane was subsequently blocked with 5% non-fat milk at 37°C for 1 h, incubated overnight with primary antibody, including vimentin (1:1,000; Abcam, Cambridge, UK), E-cadherin (1:1,000; Abcam), and GAPDH (1:5,000; Abcam) at 4°C. After washing three times with Tris-buffered saline containing Tween-20 (Beyotime Institute of Biotechnology), the membranes were incubated with sheep anti-rat immunoglobulin G conjugated peroxidase (1:500; Sigma-Aldrich Co.). Based on the manufacturer’s instructions, immunosignals were visualized using Luminol Reagent (Merck Millipore, Billerica, MA, USA), while a FluorChem E instrument was used to capture enhanced chemiluminescence images (Cell Biosciences, Santa Clara, CA, USA). The band intensity was analyzed using ImageJ software (version 1.62; National Institutes of Health, Bethesda, MD, USA).
Immunochemistry staining
The formalin-fixed and paraffin-embedded tumor samples obtained from the pathology institution were serially sectioned at 4 μm thickness. Paraffin sections were baked overnight at 58°C, deparaffinized with xylene, and hydrated through a graded ethanol series. Subsequently, GM-CSF and E-cadherin antigens were retrieved by heat using sodium citrate buffer (pH 6.0) at 100°C for 20 min. After being immersed in 3% H2O2 for 15 min at room temperature to inactivate endogenous peroxidase, sections were incubated at 4°C overnight with either rabbit monoclonal antibodies against GM-CSF (1:100 dilution; Abcam) or E-cadherin (1:50 dilution; Proteintech Inc., Chicago, IL, USA). Immunostaining was performed using horseradish peroxidase-conjugated streptavidin (Zhongshan Inc., Beijing, People’s Republic of China). Sections were subsequently stained with 3,3′-diaminobenzidine and counterstained with hematoxylin. Finally, sections were dehydrated with an ethanol gradient and mounted with rhamsan gum.
The proportions of positive tumor cells were estimated as the number of stained cells divided by the total number of cells. Two independent pathologists who were blinded to clinical data assessed the sections. All disagreements were resolved through discussion. Expression of GM-CSF and E-cadherin was scored as absent, rare, moderate, or marked as dense. Scores of absence or rare density were considered low, whereas moderate or marked density were considered to be highly expressed, and therefore used for statistical analysis.
Statistical analysis
All statistical analyses were performed using SPSS software (version 12.0; SPSS Inc., Chicago, IL, USA). Values are expressed as mean ± standard deviation. Unpaired Student’s t-tests were utilized for comparisons between values. Spearman’s rho test was used to investigate the strength of correlation between GM-CSF and E-cadherin expression. P<0.05 was considered to indicate a statistically significant difference.
Results
GM-CSF and EMT
Western blot and RT-PCR analyses showed significantly increased E-cadherin expression and decreased vimentin expression, which are considered typical hallmarks of epithelial and mesenchymal phenotypes, respectively. Thus, there was a significant inhibition of EMT phenomenon in EC cells after GM-CSF stimulation (Figure 1). P-values for Eca109 and 9706 cells were all <0.001 for gain of E-cadherin, and <0.007 (Eca109) and <0.004 (9706) for loss of vimentin.
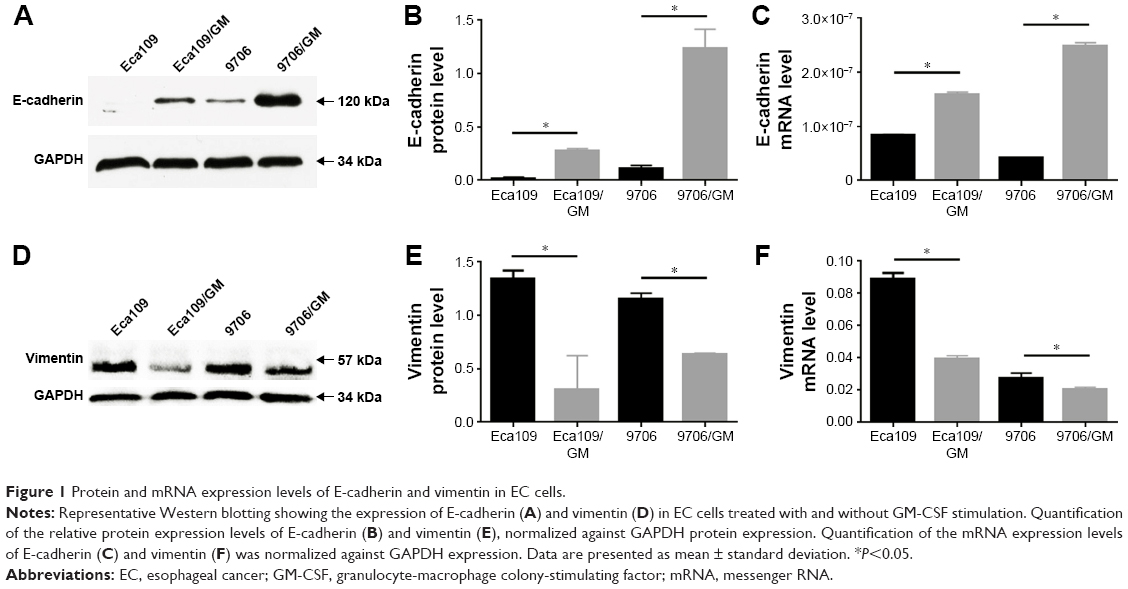 | Figure 1 Protein and mRNA expression levels of E-cadherin and vimentin in EC cells. |
GM-CSF inhibits EC-cell proliferation
The CCK-8 assay was used to assess the effects of GM-CSF on the proliferation of Eca109 and 9706 cells. The proliferation rates were significantly lower in GM-CSF-stimulated than unstimulated Eca109 cells at 72 h (P=0.011) and 96 h (P=0.003) (Figure 2). In addition, the proliferation rates for 9706 cells at 24 h (P=0.004), 72 h (P=0.040), and 96 h (P=0.003) were also significantly decreased. However, the proliferation rates of Eca109 cells incubated for 24 h and 48 h, as well as 9706 cells incubated for 48 h with or without GM-CSF, were not significantly different (P>0.05).
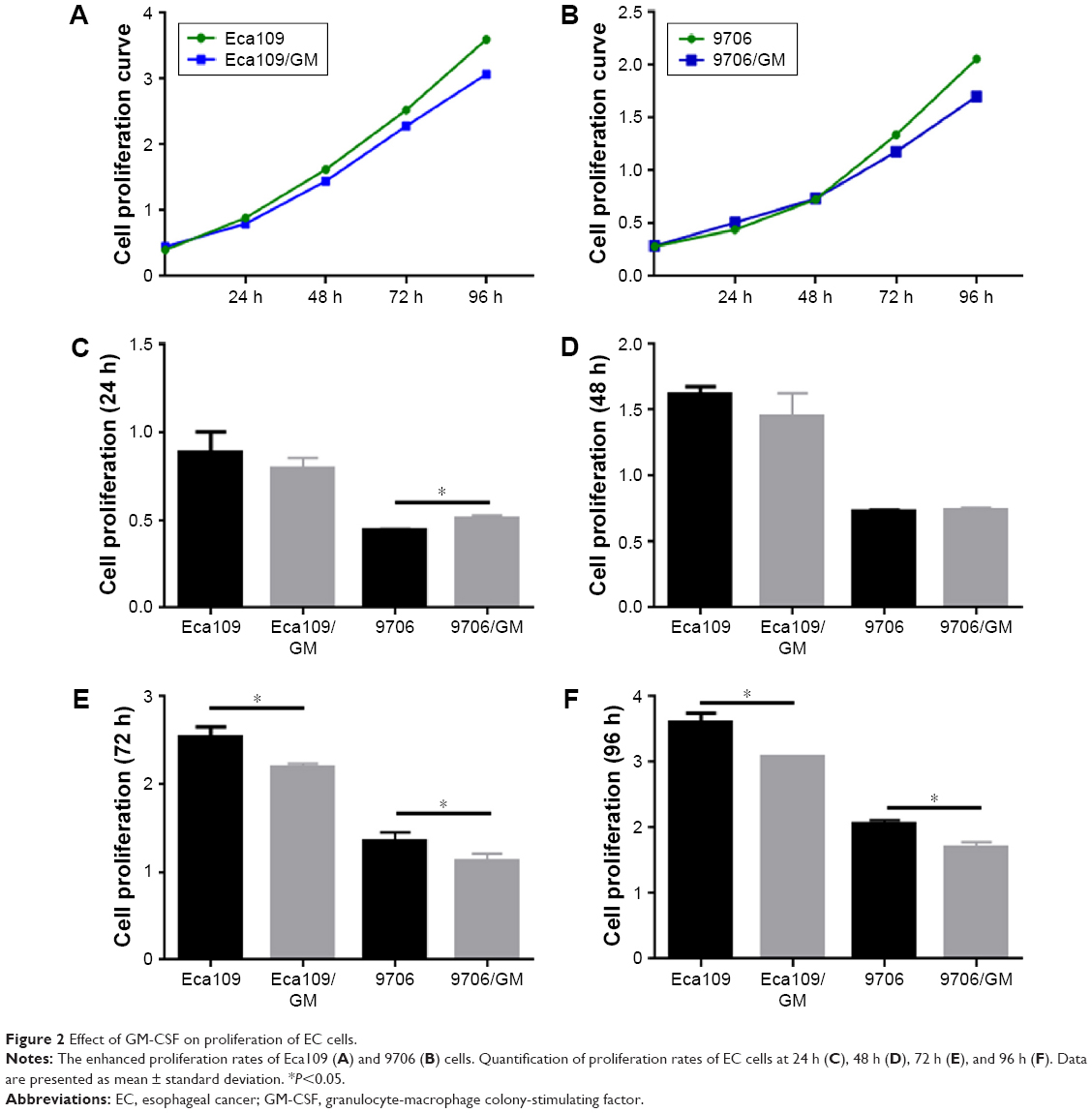 | Figure 2 Effect of GM-CSF on proliferation of EC cells. |
GM-CSF inhibits EC-cell migration
The transwell migration assay was used to evaluate the inhibitory effects of GM-CSF on EC cells. The number of invading Eca109 cells was 81.67±2.31 and 357.67±5.13 for GM-CSF stimulated and unstimulated cells, respectively. The corresponding numbers for 9706 cells were 63.67±4.16 and 369.33±2.31, respectively (Figure 3). These differences were statistically significant at P=0.023 for Eca109 and P=0.012 for 9706 cells.
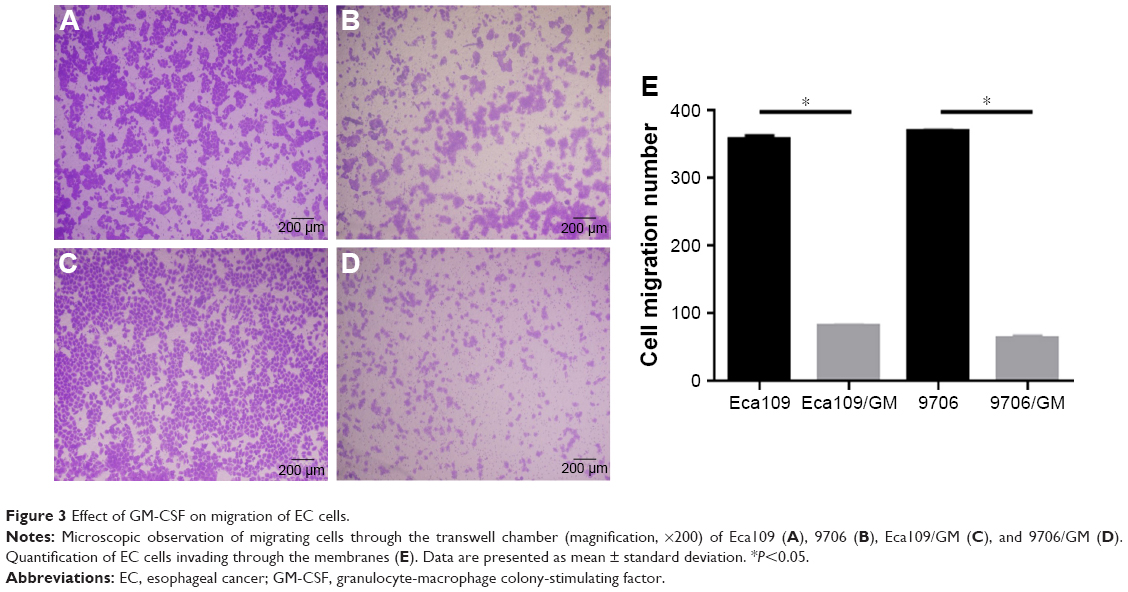 | Figure 3 Effect of GM-CSF on migration of EC cells. |
GM-CSF enhances EC-cell apoptosis
Flow cytometry was used to analyze apoptotic rates of EC cells. The apoptotic rates were higher in cells incubated for 48 h in complete culture medium than without GM-CSF. The number of apoptotic cells was 12.4±0.62 and 8.64±0.64 in the GM-CSF-stimulated and unstimulated Eca109 cells, respectively, and this difference was statistically significant at P=0.019 (Figure 4). Corresponding numbers were 7.27±0.31 and 4.13±0.21 for stimulated and unstimulated 9706 cells, respectively, which was also statistically significant at P=0.023.
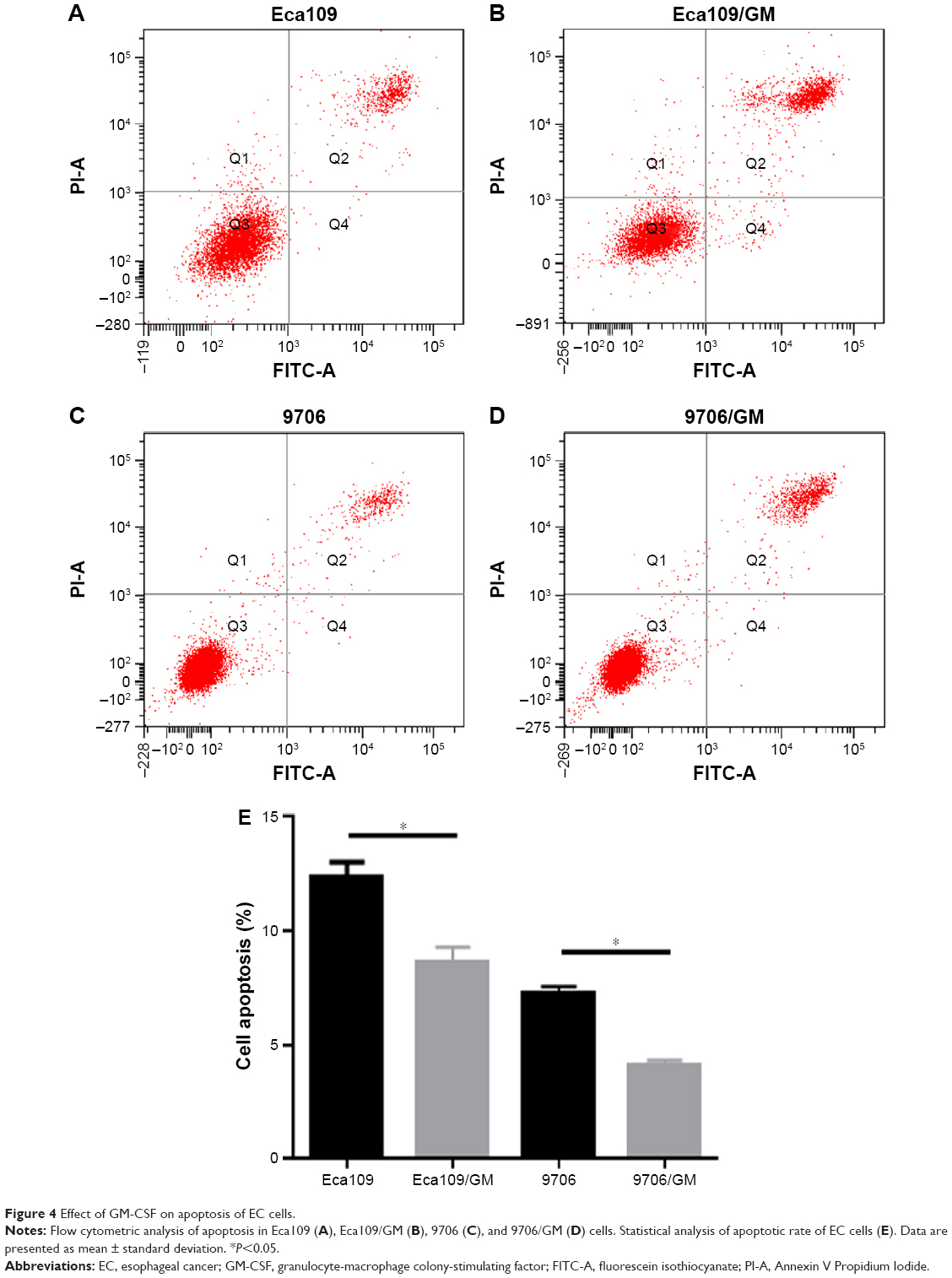 | Figure 4 Effect of GM-CSF on apoptosis of EC cells. |
Immunohistochemistry (IHC) staining
In EC tissue samples, both GM-CSF and E-cadherin expressions were increased. Based on GM-CSF staining intensities, the samples were divided into high and low groups, and the number of GM-CSF high and low groups were 47 (47.5%) and 52 (52.5%), respectively. Similarly, E-cadherin expression could be dichotomized into high (44 patients, 44.4%) and low (55 patients, 55.6%) groups. Among the GM-CSF-high group, 42 (80.8%) patients also showed high E-cadherin expression, while 34 (72.3%) patients with low E-cadherin expression identified with the GM-CSF-low group. Figure 5 shows representations of the immunostaining results. There was a significant positive correlation between GM-CSF levels and E-cadherin expression (correlation coefficient =0.473, P<0.001).
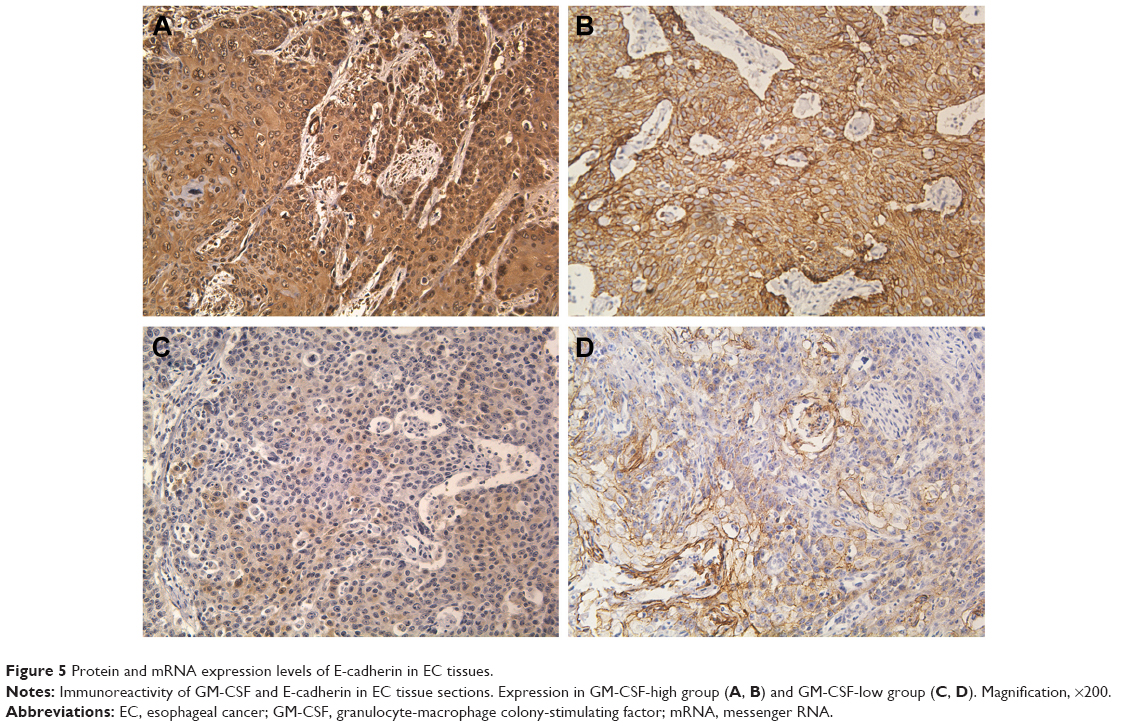 | Figure 5 Protein and mRNA expression levels of E-cadherin in EC tissues. |
Discussion
As one of the most aggressive malignant tumors, EC is generally associated with poor survival. Due to the aggressive biological behavior of EC cells, developing a novel and efficient therapy for EC patients is presently a big challenge. GM-CSF can be secreted by several cell types, including myeloid cells, dendritic cells, T cells, B cells, endothelial cells, and notably, tumor and stromal cells in the tumor microenvironment.21 Recent studies have shown that GM-CSF has a much broader range of biological activities and exerts antitumor effects on cancer patients in an immune-dependent and -independent manner. A previous publication by our group showed that increased serum GM-CSF levels during radiotherapy were associated with better survival, which indicated the potential antitumor effects of GM-CSF, although the exact mechanism was unclear.22 Many other studies of pancreatic ductal adenocarcinoma have also provided supporting evidence for the antitumor effects of GM-CSF. Urdinguio et al23 discovered that the 5-year survival rates were high in colon cancer patients overexpressing GM-CSF and their receptors. Bayne et al24 demonstrated that tumor-derived GM-CSF could act as an integral part of cancer inflammation by suppressing antigen-specific T cells, thereby blocking the growth of pancreatic ductal adenocarcinoma. Overexpression of GM-CSF had the potential to increase the susceptibility of cancer cells to anticancer drugs.25 Recent studies also reveal that the combination of GM-CSF with radiotherapy26 and/or chemotherapy27 could enhance efficacy and safety, and significantly improve the overall survival of patients with metastatic disease. These findings represent a promising strategy for antitumor therapy, and this has become an intense area of both clinical and basic research. The various research findings are consistent with an important role for GM-CSF in delaying cancer progression. However, the mechanism of tumor suppression exhibited by GM-CSF is not yet clear. Thus, the present study focused on examining the effects of GM-CSF on multiple biological functions of EC cell lines and exploring the potential mechanism of its antitumor effects.
We investigated the anti-proliferative and anti-metastatic effects of GM-CSF in EC cells by cell proliferation and transwell migration assays, respectively. The cell proliferation assay showed that GM-CSF suppressed cell proliferation in a time-dependent manner. Significantly decreased cell migration was also observed in the Eca109/GM and 9706/GM groups compared to the untreated groups. We speculated that the effect of GM-CSF might be correlated with the levels of COX-2 and iNOS. COX-2 and iNOS, which play critical roles in tumor proliferation, metastasis, prognosis, and many other biological behaviors, are highly upregulated in tumor cells.28–31 iNOS can generate a tumorigenic environment consisting of high levels of nitric oxide (NO),32 while COX-2 could delay the metabolic progression of cancer cells. The COX-2-encoding gene, PTGS2, is an early-response gene induced by carcinogens, tumor promoters, and oncogenes.33 Jiang et al34 demonstrated that the expression of GM-CSF was negatively correlated with COX-2 and iNOS levels in cervical cancer cells, and thus, tumor tissues expressing high levels of GM-CSF were likely to have lower levels of iNOS and COX-2. Thus, in cervical cancer, the mechanism of the protective role of GM-CSF is partly through the downregulation of pro-tumor factors such as COX-2 and iNOS, thereby interfering with tumor progression. However, Condamine and Gabrilovich35 discovered that iNOS expression was upregulated in peripheral blood mononuclear cells from a healthy person after being incubated with GM-CSF. Moreover, Cruz et al36 showed a synergistic function of GM-CSF and transcription factors, including IL-6 and NF-kB, in the enhancement of COX-2 expression. These contradictory results might be because GM-CSF has different effects on different cell types.
Flow cytometry was used to investigate the effect of GM-CSF on the apoptotic rates of EC cells. The apoptotic rates were higher in GM-stimulated than in unstimulated Eca109 and 9706 cells. We hypothesized that the proapoptotic effect of GM-CSF was associated with the Bcl-2 family and IL-23. The Bcl-2 family, known as anti-apoptotic proteins, plays important roles in inhibiting apoptosis and impeding the antitumor efficiency of cancer therapies.37 However, this effect could be suppressed by IL-23.38 The proapoptotic functions of GM-CSF on EC cells might correlate with IL-23 production, which sensitizes and enhances apoptosis via Bcl-2 degradation in a proteasome-dependent pathway.39 Bcl-2 downregulation enhances the activation of CASP9 and promotes proapoptotic oxidative stress. These results suggested a new pathway for the antitumor effects of GM-CSF via an IL-23-dependent signaling pathway. However, it was also reported that GM-CSF could increase the expression of Bcl-2 family members,40 probably through the activation of STAT5. The PI3K–AKT and RAS–MAPK pathways are other important signaling pathways used by GM-CSF to inhibit apoptosis in hematopoietic lineage cells.41,42 Thus, the roles of GM-CSF on cancer cells need further exploration.
Results of Western blot, RT-PCR, and IHC analysis have suggested that GM-CSF stimulation could suppress EMT in EC cells and tissues, though the exact mechanism is still under investigation. Studies suggest that GM-CSF activates JAK2, which associates with the cytoplasmic domains of a number of cytokine receptors.43 Moreover, JAK2 is an important mediator of signals triggered by several hematopoietic growth factors, including GM-CSF. Liu et al44 showed that the arginine methyltransferase, PRMT5, originally identified as JAK-binding protein 1, could be phosphorylated and attenuated by oncogenic JAK2 mutant kinase. The tumorigenic effect of PRMT5 has been demonstrated, and its overexpression correlated with poor prognosis and patient survival.45 In addition, PRMT5 has been associated with EMT, though the exact mechanism is unknown. Ibrahim et al46 demonstrated a relationship between the intracellular localization of PRMT5 and the expression of vimentin and E-cadherin. In cells expressing high levels of vimentin and low levels of E-cadherin, PRMT5 was localized in the cytoplasm; however, it became nuclear in cells expressing high E-cadherin and low vimentin. Hou et al47 discovered that PRMT5 could form a complex with AJUBA to downregulate E-cadherin in a SNAIL-dependent pathway. The depletion of PRMT5 resulted in upregulated E-cadherin expression, which indicated that PRMT5–SNAIL–AJUBA complex played an important role in E-cadherin regulation. Thus, we speculate that GM-CSF may regulate EMT through JAK2–PRMT5 signaling, and thereby exhibit its antitumor effect in EC cells.
Conclusion
In conclusion, our data are consistent with the inhibition of cancer cell proliferation and migration, as well as induction of apoptosis, by GM-CSF. The effect of GM-CSF on proliferation and migration might correlate with COX-2 and iNOS levels, while the Bcl-2 family and IL-23 might play roles in the proapoptotic effects of GM-CSF. However, the effects of GM-CSF on human EC cells and other cancer cell types require further investigation. Our data may further broaden the potential application of GM-CSF and suggest GM-CSF as a promising therapeutic agent for antitumor therapy.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (grant no 81672974 and no 81602719) and the Natural Science Foundation of Shandong Province (grant no ZR2016HQ50).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Sankaranarayanan R, Swaminathan R, Jayant K, Brenner H. An overview of cancer survival in Africa, Asia, the Caribbean and Central America: the case for investment in cancer health services. IARC Sci Publ. 2011;(162):257–291. | ||
Metcalf D. Hematopoietic cytokines. Blood. 2008;111(2):485–491. | ||
McLeish KR, Knall C, Ward RA, et al. Activation of mitogen-activated protein kinase cascades during priming of human neutrophils by TNF-alpha and GM-CSF. J Leukoc Biol. 1998;64(4):537–545. | ||
Metcalf D. The colony-stimulating factors and cancer. Nat Rev Cancer. 2010;10(6):425–434. | ||
Vilalta M, Rafat M, Giaccia AJ, Graves EE. Recruitment of circulating breast cancer cells is stimulated by radiotherapy. Cell Rep. 2014;8(2):402–409. | ||
Park BK, Zhang H, Zeng Q, et al. NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med. 2007;13(1):62–69. | ||
Aliper AM, Frieden-Korovkina VP, Buzdin A, Roumiantsev SA, Zhavoronkov A. A role for G-CSF and GM-CSF in nonmyeloid cancers. Cancer Med. 2014;3(4):737–746. | ||
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. | ||
van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;119(15):3383–3393. | ||
Chen C, Hou J, Lin Z, et al. A bystander cell-based GM-CSF secreting vaccine synergized with a low dose of cyclophosphamide presents therapeutic immune responses against murine hepatocellular carcinoma. Cell Mol Immunol. 2013;10(4):349–359. | ||
Chen G, Gupta R, Petrik S, et al. A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol Res. 2014;2(10):949–961. | ||
Gupta R, Emens LA. GM-CSF-secreting vaccines for solid tumors: moving forward. Discov Med. 2010;10(50):52–60. | ||
Seledtsov VI, Goncharov AG, Seledtsova GV. Clinically feasible approaches to potentiating cancer cell-based immunotherapies. Hum Vaccin Immunother. 2015;11(4):851–869. | ||
Savagner P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol. 2010;21 Suppl 7:vii89–vii92. | ||
Chou YS, Yang MH. Epithelial-mesenchymal transition-related factors in solid tumor and hematological malignancy. J Chin Med Assoc. 2015;78(8):438–445. | ||
Liang N, Song X, Xie J, et al. Effect of galectin-3 on the behavior of Eca109 human esophageal cancer cells. Mol Med Rep. 2015;11(2):896–902. | ||
Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471–1474. | ||
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. | ||
Stosser S, Schweizerhof M, Kuner R. Hematopoietic colony-stimulating factors: new players in tumor-nerve interactions. Mol Med (Berl). 2011;89(4):321–329. | ||
Deng G, Hu P, Zhang J, et al. Elevated serum granulocyte-macrophage colony-stimulating factor levels during radiotherapy predict favorable outcomes in lung and esophageal cancer. Oncotarget. 2016;7(51):85142–85150. | ||
Urdinguio RG, Fernandez AF, Moncada-Pazos A, et al. Immune-dependent and independent antitumor activity of GM-CSF aberrantly expressed by mouse and human colorectal tumors. Cancer Res. 2013;73(1):395–405. | ||
Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. | ||
Chaubey N, Ghosh SS. Overexpression of granulocyte macrophage colony stimulating factor in breast cancer cells leads towards drug sensitization. Appl Biochem Biotech. 2015;175(4):1948–1959. | ||
Golden EB, Chhabra A, Chachoua A, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 2015;16(7):795–803. | ||
Lawson DH, Lee S, Zhao F, et al. Randomized, placebo-controlled, phase III trial of yeast-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) versus peptide vaccination versus GM-CSF plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV melanoma: A trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). Clin Oncol. 2015;33(34):4066–4076. | ||
Chen HH, Su WC, Chou CY, et al. Increased expression of nitric oxide synthase and cyclooxygenase-2 is associated with poor survival in cervical cancer treated with radiotherapy. Int J Radiat Oncol Biol Phys. 2005;63(4):1093–1100. | ||
Licinio J, Prolo P, McCann SM, Wong ML. Brain iNOS: current understanding and clinical implications. Mol Med Today. 1999;5(5):225–232. | ||
Sanghi S, MacLaughlin EJ, Jewell CW, et al. Cyclooxygenase-2 inhibitors: a painful lesson. Cardiovasc Hematol Disord Drug Targets. 2006;6(2):85–100. | ||
Sarian LO, Derchain SF, Yoshida A, Vassallo J, Pignataro F, De Angelo Andrade LA. Expression of cyclooxygenase-2 (COX-2) and Ki67 as related to disease severity and HPV detection in squamous lesions of the cervix. Gynecol Oncol. 2006;102(3):537–541. | ||
Watanabe K, Kawamori T, Nakatsugi S, Wakabayashi K. COX-2 and iNOS, good targets for chemoprevention of colon cancer. Biofactors. 2000;12(1–4):129–133. | ||
Saldivar JS, Lopez D, Feldman RA, et al. COX-2 overexpression as a biomarker of early cervical carcinogenesis: a pilot study. Gynecol Oncol. 2007;107(1 Suppl 1):S155–S162. | ||
Jiang N, Tian Z, Tang J, Ou R, Xu Y. Granulocyte-macrophage colony-stimulating factor (GM-CSF) downregulates the expression of protumor factors cyclooxygenase-2 and inducible nitric oxide synthase in a GM-CSF receptor-independent manner in cervical cancer cells. Mediators Inflamm. 2015;2015:601604. | ||
Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25. | ||
Cruz MT, Duarte CB, Goncalo M, Figueiredo A, Carvalho AP, Lopes MC. Granulocyte-macrophage colony-stimulating factor activates the transcription of nuclear factor kappa B and induces the expression of nitric oxide synthase in a skin dendritic cell line. Immunol Cell Biol. 2001;79(6):590–596. | ||
Burlacu A. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 2003;7(3):249–257. | ||
Cocco C, Canale S, Frasson C, et al. Interleukin-23 acts as antitumor agent on childhood B-acute lymphoblastic leukemia cells. Blood. 2010;116(19):3887–3898. | ||
Subramanian M, Thorp E, Tabas I. Identification of a non-growth factor role for GM-CSF in advanced atherosclerosis: promotion of macrophage apoptosis and plaque necrosis through IL-23 signaling. Circ Res. 2015;116(2):e13–e24. | ||
Huang X, Choi JK, Park SR, et al. GM-CSF inhibits apoptosis of neural cells via regulating the expression of apoptosis-related proteins. Neurosci Res. 2007;58(1):50–57. | ||
Klein JB, Rane MJ, Scherzer JA, et al. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol. 2000;164(8):4286–4291. | ||
Kotone-Miyahara Y, Yamashita K, Lee KK, et al. Short-term delay of Fas-stimulated apoptosis by GM-CSF as a result of temporary suppression of FADD recruitment in neutrophils: evidence implicating phosphatidylinositol 3-kinase and MEK1-ERK1/2 pathways downstream of classical protein kinase C. J Leukoc Biol. 2004;76(5):1047–1056. | ||
Kamigaki M, Hide I, Yanase Y, et al. The Toll-like receptor 4-activated neuroprotective microglia subpopulation survives via granulocyte macrophage colony-stimulating factor and JAK2/STAT5 signaling. Neurochem Int. 2016;93:82–94. | ||
Liu F, Zhao X, Perna F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19(2):283–294. | ||
Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72(11):2041–2059. | ||
Ibrahim R, Matsubara D, Osman W, et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Hum Pathol. 2014;45(7):1397–1405. | ||
Hou Z, Peng H, Ayyanathan K, et al. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol. 2008;28(10):3198–3207. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.