Back to Journals » ImmunoTargets and Therapy » Volume 4
Novel perspectives on non-canonical inflammasome activation
Authors Diamond C, Khameneh HJ, Brough D, Mortellaro A ![]()
Received 26 February 2015
Accepted for publication 6 May 2015
Published 24 July 2015 Volume 2015:4 Pages 131—141
DOI https://doi.org/10.2147/ITT.S57976
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Video abstract presented by Catherine Diamond
Views: 2235
Catherine Emma Diamond,1,2 Hanif Javanmard Khameneh,1 David Brough,2 Alessandra Mortellaro,1
1Singapore Immunology Network (SIgN), Agency for Science, Technology and Research (A*STAR), Singapore; 2Faculty of Life Sciences, University of Manchester, Manchester, UK
Abstract: Inflammasomes are cytosolic multi-protein complexes that regulate the secretion of the proinflammatory cytokines, IL-1β and IL-18, and induce pyroptosis, an inflammatory form of cell death. The NLRP3 inflammasome is the most well-characterized member of this family and functions by sensing intracellular pathogen- and damage-associated molecular patterns and activating caspase-1, which processes the biologically inactive IL-1β and IL-18 precursors into active cytokines. Recent studies have identified an alternative mechanism of inflammasome activation, termed the non-canonical inflammasome, which is triggered by cytosolic sensing of lipopolysaccharide (LPS) derived from bacteria that have escaped phagolysosomes. This pathway is independent of Toll-like receptor 4 (TLR4), the well-known extracellular receptor for LPS, but instead depends on the inflammatory protease, caspase-11. Although our understanding of caspase-11 activation is still in its infancy, it appears to be an essential mediator of septic shock and attenuates intestinal inflammation. In this review, we bring together the latest data on the roles of caspase-11 and the mechanisms underlying caspase-11-mediated activation of the non-canonical inflammasome, and consider the implications of this pathway on TLR4-independent immune responses to LPS.
Keywords: caspase-11, caspase-1, LPS, NLRP3, IL-1β, IL-18
Introduction
The innate arm of the immune system is efficiently equipped with a diverse and highly conserved set of receptors known as pattern recognition receptors (PRRs). These receptors are capable of sensing microbial products (pathogen-associated molecular patterns, or PAMPs), as well as nonmicrobial/host products (danger-associated molecular patterns, or DAMPs). This strategy of primary recognition is one of the first lines of defense against bacterial infections, driving a full-blown innate immune response and subsequently promoting and supporting the adaptive arm of immune response.
The innate immune system encounters pathogens at various sites in the body, namely, epithelial barriers of the skin, gastrointestinal tract, respiratory tract, and urogenital tract. A set of innate sentinel cells, such as dendritic cells, Langerhans cells, and macrophages, collectively known as professional antigen-presenting cells, actively patrol the tissues underneath epithelial barriers, acting as the front line of defense. Antigen-presenting cells express a wide range of PRRs both on the cell surface and intracellularly, including Toll-like receptor (TLR) 4, which detects lipopolysaccharide (LPS), a major component of the outer cell membrane of Gram-negative bacteria. Innate detection of PAMPs, eg, LPS, via these PRRs leads to cellular activation. The activated antigen-presenting cells, in turn, produce proinflammatory and antimicrobial mediators and engulf the invading bacteria by phagocytosis, triggering migration of the cells to the neighboring draining lymph nodes. The antigenic contents of the bacteria are processed into peptides and presented on their cell surface in the context of major histocompatibility complex (MHC) molecules. Recognition of the peptide:MHC complex results in activation of naïve antigen-specific T-cells, which become polarized into different T helper cell subsets in the presence of additional signals from the antigen-presenting cells. Here, the adaptive immune response enters the battle. T-cells migrate to the site of infection, produce a range of cytokines and chemokines to recruit additional immune cells, and activate B-cells to undergo affinity maturation in order for them to produce high-affinity antibodies. Together, cellular and humoral adaptive immune responses act in a harmonized fashion to thoroughly eliminate the infectious agent.
LPS-induced inflammatory responses via the TLR4 pathway
The major prototype of innate immune receptors is the family of germline-encoded TLRs.1 TLRs are expressed on the surface and within the cytosolic compartments of immune cells and, upon sensing their specific ligand, ie, structural motifs from bacteria, fungi, and viruses, can initiate a signaling cascade leading to proinflammatory responses. TLR4 is the most well-characterized member of this family. It was first described as a human homologue of Drosophila Toll protein2 and consists of an extracellular leucine-rich repeat (LRR) domain, a transmembrane domain, and an intracellular Toll/interleukin (IL)-1 receptor (TIR) domain.3 A few years after its discovery, LPS from Gram-negative bacteria was identified as the major ligand for TLR4, as shown by impaired LPS-induced signaling in mice harboring mutated TLR4 and by hyporesponsiveness of TLR4-deficient mice to bacterial LPS.4–6 Myeloid differentiation factor 2 (MD2), first identified as an LPS-binding molecule in the early 1990s,7 was shown to confer LPS responsiveness to TLR4 a decade later by Shimazu et al.8 TLR4 has recently been shown to bind directly to specific high-mobility group box-1 (HMGB1) isoforms to promote TLR4 signaling.9 CD14 was also identified as a co-receptor, facilitating LPS recognition by TLR4–MD2 complexes and promoting TLR4 endocytosis.10–14 Products of respiratory syncytial virus,15 some heat shock proteins,16 β-defensin 2,17 and hyaluronic acid18 have also been indicated as other putative ligands capable of eliciting TLR4-mediated responses.
LPS engagement via the TLR4 receptor complex triggers two distinct signaling pathways (Figure 1). The first of these is dependent on the myeloid differentiation primary response gene 88 (MyD88) protein. The MyD88-dependent pathway involves recruitment of tumor necrosis factor (TNF)-receptor associated factor 6 (TRAF6) and IL-1 receptor-associated kinases (IRAKs), leading to self-ubiquitination and oligomerization of TRAF6. Transforming growth factor beta (TGFβ)-activated kinase 1 binding protein (TAB2) and TAB3 proteins are recruited to the complex, activating TGFβ-activated kinase 1 (TAK1), which, in turn, results in activation of the inhibitor of κB (IκB) complex (IKK), IκBα degradation, and the subsequent activation and nuclear translocation of nuclear transcription factor κB (NF-κB).
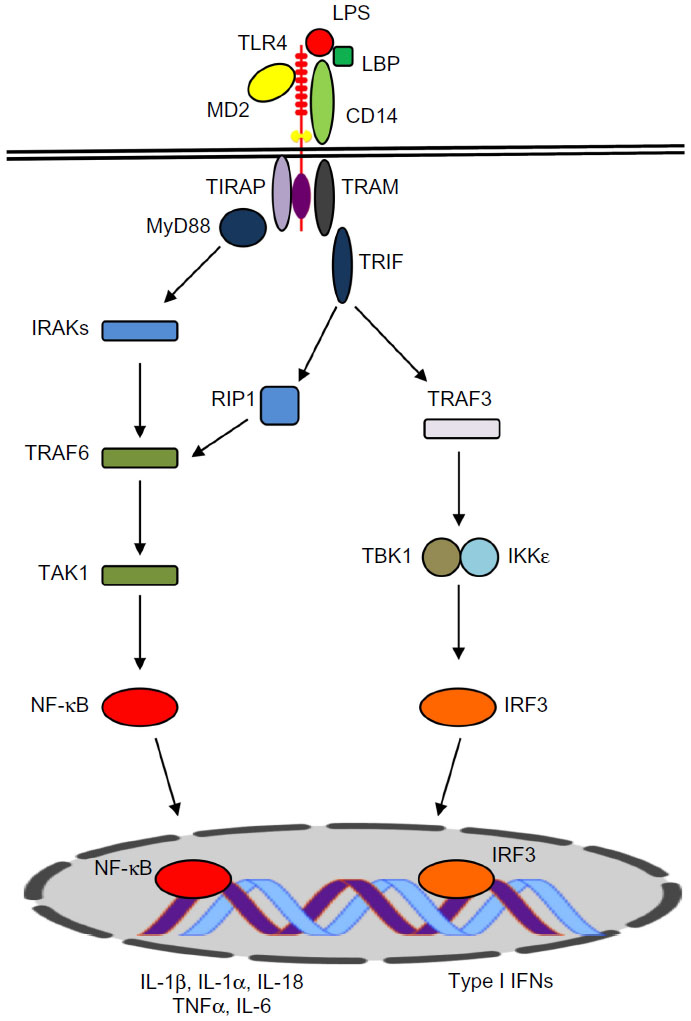 | Figure 1 Schematic view of the TLR4 signaling pathway. |
The MyD88-independent pathway is mediated by the TIR-domain-containing adapter-inducing interferon-β (TRIF), which can also recruit TRAF6 to activate NF-κB and mitogen-activated protein kinase (MAPK) (Figure 1). This pathway may also involve TRIF-related adaptor molecule (TRAM). TRIF–TRAM signaling leads to activation of interferon regulatory factor 3 (IRF3) and the induction of type-I interferon (IFN) responses and IFN-inducible genes.19–21 TLR4 has also been reported to utilize other TIR domain-containing adaptor molecules, including TIR domain-containing adaptor protein (TIRAP) and sterile alpha and heat/armadillo motif-containing protein (SARM).22 Collectively, the activation of these signaling pathways culminates in transcription of proinflammatory genes and a cascade of inflammatory responses that eradicate the microbial insult at the site of inflammation.
Considering the broad role of TLR4 signaling in promoting inflammation, it might be predicted that impaired TLR4 function would result in an imbalance in the immune response in various immunopathological conditions, including infection, autoimmunity, autoinflammation, and cancer. Indeed, polymorphisms in TLR4 genes have been associated with a wide range of inflammatory conditions and susceptibility to various infections. A specific mutation in the TLR4 gene was reported to cause endotoxin hyporesponsiveness in humans,23,24 and TLR4 mutations have been implicated in susceptibility to some infections, including meningococcal25,26 and respiratory syncytial virus infections.27–29 TLR4 variations have also been associated with noninfectious diseases, such as cancers,30,31 autoimmune conditions (eg, systemic lupus erythematosus),32 ulcerative colitis,33 auto-inflammatory familial Mediterranean fever,34 and cardiac diseases.35,36
The canonical inflammasome acts downstream of TLR4 signaling
TLR4/LPS engagement leads to activation of NF-kB and transcription of a set of proinflammatory mediators, including cytokines (eg, TNFα, IL-6, IL-1β, and IL-18), chemokines, and others (eg, prostaglandins). IL-1β, IL-1α, and IL-18 are all members of the IL-1 family and are produced as pro-form proteins. IL-1β and IL-18 are biologically inactive and must be cleaved and catalyzed into their bioactive forms by the enzyme caspase-1. This catalytic event takes place in a high-molecular-weight multi-protein complex known as the inflammasome, which was first described by Martinon et al.37 The NOD-like receptor pyrin-domain containing 3 (NLRP3) inflammasome is the most extensively characterized member of the inflammasome family and consists of NLRP3, a cytosolic sensor from the NOD-like receptor (NLR) family, the apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) adaptor molecule, and activated caspase-1. This pathway represents the canonical inflammasome activation, which requires two signals to be functional (Figure 2). The first signal is provided by TLR-ligand engagement, which primes the cells and results in transcription of certain components of the inflammasome, including NLRP3, and transcription and translation of pro-IL-1β and pro-IL-18. The second signal, which results in the activation of caspase-1 and catalytic processing of pro-IL-1β and pro-IL-18 into their mature forms, is provided by danger/stress signals through mechanisms that have yet to be fully understood (reviewed in Schroder and Tschopp,38 Zambetti et al,39 Latz et al,40 and von Moltke et al41). In contrast to IL-1β and pro-IL-18, pro-IL-1α is constitutively active in resting non-hematopoietic cells, and although it is still cleaved to its mature form, pro-IL-1α does not have a caspase-cleavage site.42 Pan-caspase inhibitors do not affect IL-1α processing, and evidence suggests that pro-IL-1α is instead cleaved via Ca2+-dependent calpain-like proteases. However, the secretion of this cytokine does seem to be dependent on caspase-1 following canonical inflammasome activation.42–44 Although IL-1α release appears to be a product of conventional inflammasome machinery, this cytokine has been shown to be induced through inflammasome-independent mechanisms as well. Freigang et al have recently reported that fatty acids selectively induce IL-1α (but not IL-1β) production by uncoupling mitochondrial respiration. IL-1α release, triggered by cholesterol crystals, is assumed to be the major culprit in atherosclerosis and vascular damage.45 IL-1α also harbors a nuclear localization sequence which prompts it to be translocated into the nucleus and act as a transcription factor.46 Collectively, various reports suggest that IL-1α does not always fit in as a typical product of canonical inflammasome activation.
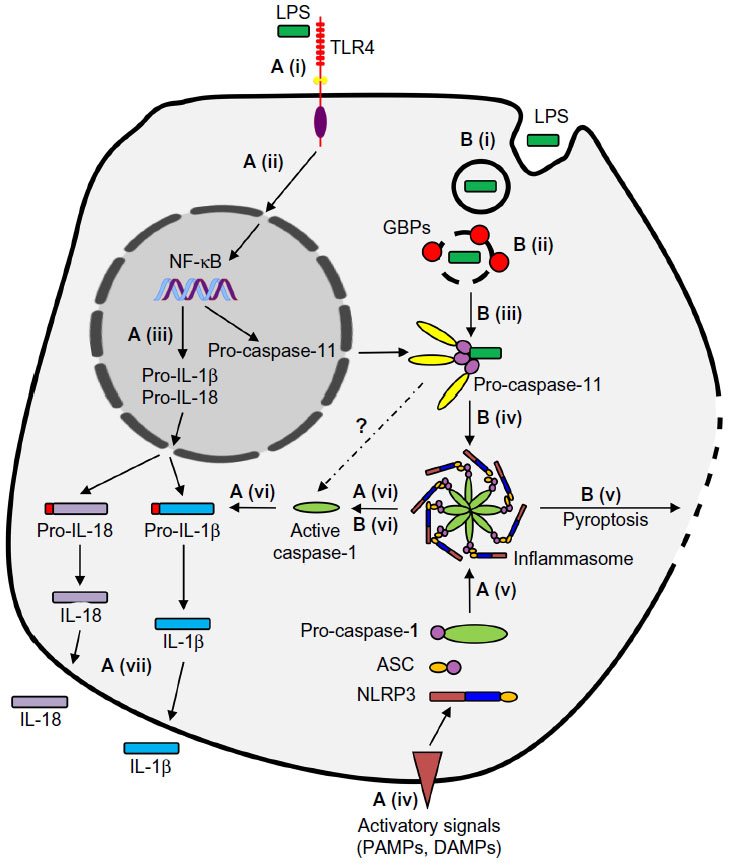 | Figure 2 Canonical and noncanonical pathways of inflammasome activation. |
Evidence for TLR4-independent inflammasome activation by LPS
Since the discovery of the TLR4 and caspase-1-mediated pathway described above, it was assumed that LPS detection and the resulting immune response, including the well-described production and release of IL-1β, IL-18, and other cytokines, were solely mediated through TLR4 signaling.47 Multiple studies showed that TLR4-deficient mice were more susceptible to bacterial disease than those with functional TLR4.48,49 However, evidence began to emerge suggesting that the mechanism behind LPS detection and the regulation of the immune response to pathogens was more complex than at first understood, and that it was highly dependent on the model of infection and bacterial dose.
The first piece of evidence was a report that showed LPS activation was not completely abrogated in TLR4-deficient mice. Indeed, all of the classic transcriptional processes usually instigated by stimulation with LPS were abolished in this model, including the increased expression of proinflammatory cytokines as described above.50,51 However, LPS-stimulated cells from these mice were not defective in nontranscriptional processes, such as phagocytosis, autophagy, and pyroptosis, suggesting an alternative signaling pathway could be instigated following the detection of LPS.52–55 It has been proposed that this alternative pathway is triggered by LPS-containing stimuli from Gram-negative bacteria, which have evaded detection by TLR4 and gained entry into the cytoplasm, possibly by modifying their structure or environment.56,57
This breach of the classical immune defense response requires an alternative pathway for detection of the bacteria and activation of the NLRP3 inflammasome. This is referred to as the non-canonical inflammasome pathway and is mediated by murine caspase-11, or its putative human orthologs caspase-4 and caspase-5.58
The role of caspase-11 in the non-canonical inflammasome pathway
Murine proinflammatory caspase-11 has been shown to have vital roles in the clearance of bacterial infection, including triggering a type of inflammasome-dependent cell death, known as pyroptosis. Unlike caspase-1, which is constitutively expressed, expression of the precursor pro-caspase-11 in macrophages requires an initial inflammatory stimulus provided by TLR agonists, eg, LPS.59
The role of caspase-11 in the activation of the non-canonical pathway first came to light when it was discovered that the conventional caspase-1 knockout model, used to report many of the mechanisms in the inflammasome pathway, was also Casp11-deficient. The loci of Casp1 and Casp11 are in close proximity to each other within the genome, which can result in genes not segregating during recombination, producing a double mutant. This led to differences in the phenotype of inflammasome that was activated by different types of stimuli being masked.60,61 This finding not only caused the reevaluation of previous results gathered from the knockout model, but it also enabled true single Casp11- and Casp1-deficient models to be produced. It was reported that similar levels of IL-1β and IL-18 were released in both Casp1- and Casp11-deficient models compared to wild type, when stimulated with ATP, monosodium urate crystals, and nigericin (these became known as classic canonical stimuli). However, when non-canonical stimuli, such as Escherichia coli and LPS/cholera toxin B (CTB) were used instead, caspase-1 could not be processed in macrophages from Casp11-deficient models, and both IL-1β and IL-18 release was abrogated, demonstrating that all components of the NLRP3 inflammasome were still required.58 Although this demonstrated a clear role for caspase-11 in the release of IL-1β, a further study indicated that direct cleavage of IL-1β by caspase-11 was unlikely, due to the differing crystal structures of caspase-1 and caspase-11.59 Instead, it was suggested that the two caspases work together closely to coordinate the release of IL-1β and IL-18, in order to drive a maximal inflammatory response. A recent study shows this proposal with evidence that caspase-11 can activate caspase-1, possibly through activation of the inflammasome, although the precise mechanism is yet to be confirmed.58 Other inflammatory roles of caspase-11 have also been elucidated, including inflammasome-independent pyroptosis58,62,63 and release of HMGB1 and IL-1α.58,64 In contrast to IL-1β, IL-1α does not require caspase-1 for secretion both in humans and mice in response to non-canonical stimuli. This finding led to the question of how these caspase-11-dependent mechanisms result in IL-1α and HMGB1 release, and pyroptosis.Caspase-4 and caspase-5 have also been shown to share many of the roles of caspase-11; however, there is currently limited evidence confirming this.65,66
Caspase-11 detects cytosolic LPS
The identification of the non-canonical inflammasome pathway led to further investigation into how some bacteria activate caspase-11. Several pathogens, including Salmonella enterica serovar Typhimurium and Legionella pneumophila, are capable of evading the canonical caspase-1 activation pathway, and can cause rapid caspase-11-dependent cell death (<4 hours) in macrophages. These pathogens enter the cytosol of host cells, using type III and type IV secretion systems (T3SS and T4SS), respectively, and activate the non-canonical inflammasome.64,67 Therefore, it was hypothesized that caspase-11 acts as a sensor of intracellular LPS (Figure 2). Recently, two research groups independently found evidence to support this hypothesis, demonstrating that direct transfection of LPS into the intracellular compartments of the cell activates the non-canonical inflammasome pathway.56,57 It was confirmed that only Gram-negative bacteria triggered the non-canonical inflammasome, resulting in pyroptosis of the cell, IL-1β secretion, and caspase-1 processing.56,57 Both studies also indicated that where previous reports had reported that CTB activated the caspase-11 inflammasome, this was in fact due to LPS that was bound to the CTB during those experiments. Therefore, it was suggested that CTB could act as a transporter of LPS into the cytosol. When only CTB was present, it failed to trigger the non-canonical pathway, and only caused activation when bound to LPS from E. coli serotype O111:B4, and not any other LPS O serotypes. Both Hagar et al and Kayagaki et al found that the lipid A moiety of LPS was also able to activate caspase-11.56,57 The evasion of TLR4 detection by modifications to lipid A has previously been described. Indeed, the authors showed that pathogens, including Helicobacter pylori, Yersinia pestis, and Francisella novicida, could evade activation of the non-canonical pathway through deacylation of lipid A.56,57 These groundbreaking studies led to the hypothesis that TLR4 signaling is only required for the detection of extracellular and vacuolar LPS, and for upregulation of transcription of pro-caspase-11, although this latter role can be shared by signaling through other TLRs. Conversely, when LPS is in abundance or has managed to enter the cytosol through other processes, which could include lysis of LPS-containing vacuoles, it is detected by alternative sensors within the cell, and activates a caspase-11-dependent inflammasome pathway. This prevents the immune system from being compromised when bacteria manage to bypass the first line of defense through evasion of TLR4.56,57,63
Proposed model of caspase-11 activation
As described earlier, evidence gained from experimental transfection of LPS directly into the cytosol has revealed details of the mechanisms involved in activation of caspase-11. However, many of the processes involved in caspase-11 activation in response to bacterial infection are still to be elucidated, and the question remains, what causes LPS to become internalized to the cytosol, activating the non-canonical inflammasome pathway? Currently, the majority of evidence suggests that internalization is mediated by type-I IFNs and IFN-inducible GTPases, as described below.
Two studies independently showed that, following detection of bacterial infection, endogenous type-I IFNs were necessary for caspase-11 activation and caspase-11-mediated cell death.67,68 However, a recent study demonstrated that when LPS was directly transfected into the cytosol, upregulation of pro-caspase-11 expression and caspase-11 activation was independent of type-I IFNs.64,69 Therefore, it was deduced that IFN-stimulated genes (ISGs) were required for cytosolic recognition of LPS. Supporting the involvement of IFNs in the activation of the non-canonical pathway, a recent study suggested that the small IFN-inducible GTPases, the guanylate-binding proteins (GBPs), are vital for caspase-11 activation in response to intracellular bacteria.69 Bone marrow-derived macrophages from mice deficient in GBPs on chromosome 3 (GBP1, 2, 3, 5, and 7) were infected with Gram-negative vacuolar-contained bacteria that activate the non-canonical pathway, including wild-type S. Typhimurium. These GBP-deficient models showed significantly reduced cell death and secretion of IL-1β and IL-18, compared to wild-type mice, independent of LPS priming.69 This was also shown using L. pneumophila, where the GBP-deficient macrophages additionally showed reduced caspase-11-dependent pyroptosis.70
Although evidence is building demonstrating the requirement of GBPs in the internalization of Gram-negative bacteria, the mechanism that these GBPs employ to ensure detection of cytosolic pathogens and activate the non-canonical pathway has not yet been confirmed. It has been reported that GBPs can lyse pathogen-containing vacuoles, releasing pathogens into the cytosol, as shown in Figure 2.69,71 Meunier et al indicated that this process allows the recognition of intracellular S. Typhimurium, an essential host defense process for caspase-11 activation of the non-canonical inflammasome.69 A similar mechanism is employed by GBPs mediating host defense to Toxoplasma gondii. GBPs induce lysis, blebbing, and destabilization of the membrane surrounding the parasite, rupturing the vacuole and releasing the parasite into the cytosol where it quickly deteriorates.72 However, it remains to be seen if this defense strategy is induced by all types of bacterial infection, as no vacuolar disruption was observed by Pilla et al using L. pneumophila.70 In this study, the authors suggest that GBPs could instead play a scaffolding role in the oligomerization of caspase-11, heightening the response of caspase-11 following sensing of LPS in the cytosol.70
The importance of GBPs in activating the non-canonical pathway is only just starting to be unraveled. Further clarification is required regarding the particular GBPs involved, the mechanisms of action employed in the presence of different strains of bacteria, and how these different mechanisms of resistance might have evolved to maintain host defense.
Caspase-11 sensing of LPS
Although the above studies elucidated that caspase-11 and the non-canonical inflammasome are activated in response to cytosolic LPS, the mechanism of detection was unknown until recently. Previous studies hypothesized that an unidentified sensor was able to detect cytosolic LPS and activate caspase-11 in response.56,57 However, a recent report has suggested that it is the caspases themselves that directly sense and bind to the LPS.73 Using electroporation of LPS into human monocytic cell lines and immortalized mouse bone marrow-derived macrophages, expressing caspase-4 and caspase-11, respectively, Shi et al demonstrated that pyroptosis occurred and was dependent on caspase-4/11.73 However, when the experiment was repeated using muramyl dipeptide (MDP), a peptidoglycan constituent of both Gram-positive and Gram-negative bacteria, no cell death was induced. The pyroptosis resembled caspase-1-dependent pyroptosis, so the authors used clustered regularly interspaced short palindromic repeats (CRISPR) technology to make a Casp1-deficient model, yet cell death still occurred in these cells. Further investigations prompted siRNA knockdown of caspase-4/11 expression, which caused abrogation of LPS-induced pyroptosis and highlighted the role of caspase-4/11 in the immune response to cytoplasmic LPS. Confirming their findings, recombinant caspase-4/11 appeared as oligomers when purified from E. coli (containing LPS), but only appeared as monomers when purified from insect cells where LPS levels were threefold lower. Additionally, oligomerization of these monomers could be induced when LPS was added. This suggested that direct binding between LPS and caspase-4/11 was taking place. The group went a step further, showing that caspase-4/11 mutants that lacked the caspase-activation and recruitment domain (CARD) did not bind to LPS and oligomerization did not take place. It was also demonstrated that caspase-5 had a similar role in detecting cytoplasmic LPS, by directly binding to LPS.73 This has advanced the field of inflammasome and caspase-4/5/11 research considerably; however, it still remains to be determined how caspase-4/5/11 CARD domains, but not CARD domains of other proteins, are able to directly sense LPS.73
Additional roles of caspase-11
Besides the well-described roles of caspase-11 in cell death and cytokine maturation that have become synonymous with the non-canonical pathway, there are reports of caspase-11 mediating several other host immune defenses.
Although the release of bacteria into the cytosol seems to instigate the caspase-11-dependent non-canonical inflammasome pathway as described above, the lysis of phagosomes also seems to be part of a positive feedback loop that ensures maximum detection of pathogens. There is evidence to suggest that caspase-11 itself causes the release of bacteria from vacuoles into the cytosol, aiding pathogen detection and limiting pathogen growth, by modulating actin polymerization and, therefore, fusion of bacteria-containing vacuoles with lysosomes.74 Caspase-11 appears to regulate cofilin phosphorylation, a crucial event for the process of actin remodeling.75 Interestingly, when nonpathogenic material is contained within the vacuoles, this process does not occur, which gives rise to the question of how caspase-11 discriminates between self and non-self membranes. However, to date, this mechanism has only been reported to occur in L. pneumophila infection.74 Consequently, it seems highly probable that GBPs are required to ensure pathogens are released into the cytosol for detection by caspase-11 in order to induce a maximal immune response.
Role of caspase-11 in vivo
As with many host defense mechanisms, in certain situations the immune response in vivo can be more detrimental than beneficial. Caspase-11 is no exception. In a mouse model of sepsis, in which the components of the non-canonical pathway (apart from caspase-1) were absent, the mice were protected against a lethal dose of LPS, as there was no caspase-11-mediated cell death.58 A further study also showed that hyperactivation of caspase-11, caused by high concentrations of LPS during endotoxemia, was hugely damaging to the host.56 This has major implications in clinical sepsis, where multiple inflammatory cascades may become hyperactivated and result in significant tissue damage, causing great detriment to the patient.
An additional study also described a symbiotic relationship between caspase-1 and caspase-11, in which they balance the effects of each other, as demonstrated by caspase-11 significantly increasing host susceptibility to S. Typhimurium in the absence of caspase-1.62
The role of the canonical NLRP3 and NLRC4 inflammasomes in intestinal inflammation and the development of inflammatory bowel disease (IBD) pathogenesis has been well documented.76 Therefore, several groups have explored the possibility that the non-canonical inflammasome could be similarly involved. Caspase-11 is constitutively expressed at low levels in the colonic mucosa, probably to avoid continuous triggering by the presence of the intestinal microflora, but levels were significantly increased during intestinal inflammation.77,78 In an acute colitis model induced by dextran sodium sulfate (DSS) (2% w/v), Casp11-deficient mice were more susceptible to inflammation (higher weight loss and colon shortening, increased diarrhea and rectal bleeding) as a result of higher colonic tissue damage and epithelial barrier permeability.77–79 A higher DSS dose (4% w/v) induced lethality of Casp11-deficient mice.78,79 Mucosal damage was associated with massive inflammatory infiltrates in the lamina propria of Casp11-deficient colons after DSS-induced colitis,77–79 which seemed to correlate with increased colonic expression of CCL5.78 Adoptive bone marrow transplants indicated that the protective role of caspase-11 was associated with both epithelial cells and hematopoietic cells infiltrating the lamina propria.79
IL-1β and IL-18 are important regulators of colonic inflammation, acting on epithelial cells to promote tissue repair. Therefore, the involvement of caspase-11 in secretion of these cytokines during DSS-induced colitis has been explored, although with contradicting results. Williams et al79 reported that both IL-1β and IL-18 levels were attenuated in colonic homogenates from DSS-treated Casp11-deficient mice, while Oficjalska et al77 reported a reduction in IL-18 only. Administration of recombinant IL-18 ameliorated disease pathogenesis in DSS-treated Casp11-deficient mice.77,79 Contradictive results were reported by Demon et al, who reported that neither IL-1β nor IL-18 was reduced in colon homogenates from Casp11-deficient mice.78 Demon et al have also shown that IL-1β was largely dependent on caspase-1 and not caspase-11.78 These discrepancies could be due to different microbiota composition among mouse facilities or the source of sample used for cytokine measurement (ie, colon homogenates versus organ culture supernatants). The role of IL-1β/IL-18 activated via both the canonical and non-canonical inflammasome pathways during acute intestinal inflammation continues to be debated. Further studies with alternative physiologically relevant models that resemble human IBD more closely will certainly help to clarify these issues.
Caspase-11-mediated protection of acute intestinal inflammation has not been confirmed in mouse models of colitis-associated cancer induced by azoxymethane/DSS treatments. Indeed, the same levels of morbidity or mortality were observed in Casp11-deficient mice and wild-type controls (similar number and size of polyps, and similar levels of hyperplasia and dysplasia).79 Correspondingly, concentrations of IL-18 in the colons of Casp11-deficient mice and wild-type mice were comparable in the context of tumorigenesis.79
The role of the non-canonical inflammasome in gut inflammation in response to enteric bacteria has also been investigated recently.80 The authors showed that murine caspase-11 regulated the release of IL-18 (both the precursor and activated form) from cecal explants following intestinal S. Typhimurium infection, whereas it was dispensable for IL-1β, which required NLRP3, ASC, and caspase-1.80 Moreover, caspase-11 also contributed to reduce bacterial burden in the intestine. Likewise, the human ortholog, caspase-4, regulated IL-18 release and limited bacterial growth in human epithelial cells. Additionally, caspase-4 was shown to increase the sensitivity of human cells to endotoxins, while its absence gave protection against endotoxins.81 Further studies are needed to precisely define the role of caspase-4/5/11 in inflammation and to establish the type of cells (ie, myeloid or epithelial origin) in which it functions.
Future perspectives
Excellent progress has been made recently toward understanding the processes involved in activation of the caspase-11-dependent non-canonical pathway. However, many questions still remain to fully understand the complex mechanisms involved. Although roles have been identified for many of the components known to be involved in this pathway, it is possible that certain components may have multiple functions, as observed for GBPs. In addition, many parallels have been described between caspase-11 and its human orthologs, caspase-4 and caspase-5, but the exact effector functions of the human caspases remain poorly understood.
Given the strong evidence linking the caspase-11-dependent non-canonical pathway with sepsis and intestinal inflammation, translation of the knowledge gained through studying the role of caspase-11 in inflammation will help to identify molecular targets for clinical therapies for inflammatory conditions, including endotoxemia and IBD.
Acknowledgments
This research was funded by Singapore Immunology Network (SIgN, A*STAR). We would like to thank Kerry McLaughlin of Insight Editing London for the critical review of the paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. | |
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. | |
Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A. 1998;95(2):588–593. | |
Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. | |
Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749–3752. | |
Qureshi ST, Larivière L, Leveque G, et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J Exp Med. 1999;189(4):615–625. | |
Kirkland TN, Virca GD, Kuus-Reichel T, et al. Identification of lipopolysaccharide-binding proteins in 70Z/3 cells by photoaffinity cross-linking. J Biol Chem. 1990;265(16):9520–9525. | |
Shimazu R, Akashi S, Ogata H, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189(11):1777–1782. | |
Yang H, Wang H, Ju Z, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212(1):5–14. | |
Schumann RR, Leong SR, Flaggs GW, et al. Structure and function of lipopolysaccharide binding protein. Science. 1990; 249(4975):1429–1431. | |
Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249(4975):1431–1433. | |
Frey EA, Miller DS, Jahr TG, et al. Soluble CD14 participates in the response of cells to lipopolysaccharide. J Exp Med. 1992; 176(6):1665–1671. | |
Jiang Z, Georgel P, Du X, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6(6):565–570. | |
Zanoni I, Ostuni R, Marek LR, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147(4):868–880. | |
Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401. | |
Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164(2):558–561. | |
Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298(5595):1025–1029. | |
Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195(1):99–111. | |
Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2005;7(1):12–19. | |
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. | |
Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6(9):644–658. | |
O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007; 7(5):353–364. | |
Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25(2):187–191. | |
Smirnova I, Hamblin MT, McBride C, Beutler B, Di Rienzo A. Excess of rare amino acid polymorphisms in the Toll-like receptor 4 in humans. Genetics. 2001;158(4):1657–1664. | |
Allen A, Obaro S, Bojang K, et al. Variation in Toll-like receptor 4 and susceptibility to group A meningococcal meningitis in Gambian children. Pediatr Infect Dis J. 2003;22(11):1018–1019. | |
Faber J, Meyer CU, Gemmer C, et al. Human toll-like receptor 4 mutations are associated with susceptibility to invasive meningococcal disease in infancy. Pediatr Infect Dis J. 2006;25(1):80–81. | |
Awomoyi AA, Rallabhandi P, Pollin TI, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. 2007;179(5):3171–3177. | |
Löfgren J, Marttila R, Renko M, Rämet M, Hallman M. Toll-like receptor 4 Asp299Gly polymorphism in respiratory syncytial virus epidemics. Pediatr Pulmonol. 2010;45(7):687–692. | |
Goutaki M, Haidopoulou K, Pappa S, et al. The role of TLR4 and CD14 polymorphisms in the pathogenesis of respiratory syncytial virus bronchiolitis in greek infants. Int J Immunopathol Pharmacol. 2014;27(4):563–572. | |
Zheng SL, Augustsson-Bälter K, Chang B, et al. Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the CAncer Prostate in Sweden Study. Cancer Res. 2004;64(8):2918–2922. | |
Zhou Q, Wang C, Wang X, et al. Association between TLR4 (+896A/G and +1196C/T) polymorphisms and gastric cancer risk: an updated meta-analysis. PLoS One. 2014;9(10):e109605. | |
Rupasree Y, Naushad SM, Rajasekhar L, Uma A, Kutala VK. Association of TLR4 (D299G, T399I), TLR9 -1486T>C, TIRAP S180L and TNF-α promoter (-1031, -863, -857) polymorphisms with risk for systemic lupus erythematosus among South Indians. Lupus. 2015;24(1):50–57. | |
Török HP, Glas J, Tonenchi L, Mussack T, Folwaczny C. Polymorphisms of the lipopolysaccharide-signaling complex in inflammatory bowel disease: association of a mutation in the Toll-like receptor 4 gene with ulcerative colitis. Clin Immunol. 2004;112(1):85–91. | |
Karakose MZ, Yapali S, Salman E, Aksu K, Karakose S, Akarca US. TLR2 and TLR4 gene expression levels and associated factors during acute attack and attack-free periods in familial Mediterranean fever. Clin Rheumatol. 2015;34(4):785–790. | |
Edfeldt K, Bennet AM, Eriksson P, et al. Association of hypo-responsive toll-like receptor 4 variants with risk of myocardial infarction. Eur Heart J. 2004;25(16):1447–1453. | |
Guven M, Ismailoglu Z, Batar B, et al. The effect of genetic polymorphisms of TLR2 and TLR4 in Turkish patients with coronary artery disease. Gene. 2015;558(1):99–102. | |
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. | |
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. | |
Zambetti LP, Laudisi F, Licandro G, Ricciardi-Castagnoli P, Mortellaro A. The rhapsody of NLRPs: master players of inflammation. and a lot more. Immunol Res. 2012;53(1–3):78–90. | |
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. | |
von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013; 31:73–106. | |
Gross O, Yazdi AS, Thomas CJ, et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012; 36(3):388–400. | |
Yazdi AS, Drexler SK. Regulation of interleukin 1α secretion by inflammasomes. Ann Rheum Dis. 2013;72 Suppl 2:ii96–ii99. | |
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–3732. | |
Freigang S, Ampenberger F, Weiss A, et al. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14(10):1045–1053. | |
Werman A, Werman-Venkert R, White R, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci U S A. 2004;101(8):2434–2439. | |
Beutler B. Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol. 2000;12(1):20–26. | |
Malley R, Henneke P, Morse SC, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100(4):1966–1971. | |
Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165(10):5392–5396. | |
Meng J, Gong M, Björkbacka H, Golenbock DT. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J Immunol. 2011;187(7):3683–3693. | |
Triantafilou M, Triantafilou K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 2002;23(6):301–304. | |
Branger J, Leemans JC, Florquin S, Weijer S, Speelman P, Van Der Poll T. Toll-like receptor 4 plays a protective role in pulmonary tuberculosis in mice. Int Immunol. 2004;16(3):509–516. | |
Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304(5673):1014–1018. | |
Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. | |
West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. | |
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. | |
Kayagaki N, Wong MT, Stowe IB, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–1249. | |
Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflamma- some activation targets caspase-11. Nature. 2011;479(7371):117–121. | |
Wang S, Miura M, Jung Yk, et al. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem. 1996;271(34):20580–20587. | |
Kuida K, Lippke JA, Ku G, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267(5206):2000–2003. | |
Li P, Allen H, Banerjee S, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80(3):401–411. | |
Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8(6):471–483. | |
Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16(3):319–326. | |
Casson CN, Copenhaver AM, Zwack EE, et al. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013;9(6):e1003400. | |
Vigano E, Mortellaro A. Caspase-11: the driving factor for noncanonical inflammasomes. Eur J Immunol. 2013;43(9):2240–2245. | |
Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83. | |
Broz P, Ruby T, Belhocine K, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490(7419):288–291. | |
Rathinam VA, Vanaja SK, Waggoner L, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. | |
Meunier E, Dick MS, Dreier RF, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509(7500):366–370. | |
Pilla DM, Hagar JA, Haldar AK, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A. 2014;111(16):6046–6051. | |
MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12(5):367–382. | |
Martens S, Parvanova I, Zerrahn J, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005;1(3):e24. | |
Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. | |
Akhter A, Caution K, Abu Khweek A, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37(1):35–47. | |
Li J, Brieher WM, Scimone ML, et al. Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat Cell Biol. 2007;9(3):276–286. | |
Zambetti LP, Mortellaro A. NLRPs, microbiota, and gut homeostasis: unravelling the connection. J Pathol. 2014;233(4):321–330. | |
Oficjalska K, Raverdeau M, Aviello G, et al. Protective role for caspase-11 during acute experimental murine colitis. J Immunol. 2015;194(3):1252–1260. | |
Demon D, Kuchmiy A, Fossoul A, Zhu Q, Kanneganti TD, Lamkanfi M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 2014;7(6):1480–1491. | |
Williams TM, Leeth RA, Rothschild DE, et al. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am J Physiol Gastrointest Liver Physiol. 2015;308(2):G139–G150. | |
Knodler LA, Crowley SM, Sham HP, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe. 2014;16(2):249–256. | |
Kajiwara Y, Schiff T, Voloudakis G, et al. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol. 2014;193(1):335–343. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.