Back to Journals » ImmunoTargets and Therapy » Volume 12
Novel Immunotherapies for Myasthenia Gravis
Received 8 January 2023
Accepted for publication 3 March 2023
Published 4 April 2023 Volume 2023:12 Pages 25—45
DOI https://doi.org/10.2147/ITT.S377056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Sruthi S Nair,1,2 Saiju Jacob1,3
1Department of Neurology, University Hospitals Birmingham, Birmingham, B15 2TH, UK; 2Department of Neurology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Thiruvananthapuram, Kerala, India; 3Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, B15 2TT, UK
Correspondence: Saiju Jacob, Consultant Neurologist, University Hospitals Birmingham, Edgbaston, Birmingham, B15 2TH, United Kingdom, Tel +44 0121 3716844, Fax +44 0121 4605300, Email [email protected]
Abstract: Myasthenia gravis (MG), a prototype autoimmune neurological disease, had its therapy centred on corticosteroids, non-steroidal broad-spectrum immunotherapy and cholinesterase inhibitors for several decades. Treatment-refractory MG and long-term toxicities of the medications have been major concerns with the conventional therapies. Advances in the immunology and pathogenesis of MG have ushered in an era of newer therapies which are more specific and efficacious. Complement inhibitors and neonatal Fc receptor blockers target disease-specific pathogenic mechanisms linked to myasthenia and have proven their efficacy in pivotal clinical studies. B cell-depleting agents, specifically rituximab, have also emerged as useful for the treatment of severe MG. Many more biologicals are in the pipeline and in diverse stages of development. This review discusses the evidence for the novel therapies and the specific issues related to their clinical use.
Keywords: myasthenia gravis, refractory myasthenia gravis, complement inhibitors, FcRn blocker, B cell depletion
Introduction and Background
Introduction
Myasthenia gravis (MG) is a chronic autoimmune disease of the neuromuscular junction (NMJ) clinically characterized by fatigable weakness of the ocular, limb and bulbar (speech, swallowing and respiratory) muscles. It is the most common acquired disease of the NMJ with an incidence of 0.3–2.8 per 100,000.1 The burden of MG translates not only to disabling symptoms and increased hospitalizations, but also marked economic, social and emotional costs.2
In MG, well-characterized antibodies have been identified against specific targets in the post-synaptic membrane.3 Antibodies against acetylcholine receptor (AChR) are observed in a vast majority (nearly 85%) of generalized MG and antibodies against other components, namely muscle specific kinase (MuSK), lipoprotein related protein 4 (LRP4) and agrin have been reported in 5%, 2% and less than 1%, respectively in the remaining population. The remaining 5–8% with generalized disease have no currently attributable antigenic targets or antibodies and are referred to as seronegative MG.4
The clinical severity of MG can range from mild ocular symptoms to frequent respiratory and bulbar crisis. In general, one-fifth of MG patients manifest only ocular weakness and are referred to as ocular MG whereas the rest, with a more diffuse weakness, are referred to as generalized MG (gMG). Ocular MG is less likely (approximately 50%) to have antibodies compared with gMG.5 Distinctive clinical phenotypes have been recognized for each of the antibody subtypes of MG.3 AChR antibody positive MG are classified into three subtypes: early onset (<50 years of age), late onset (>50 years) and thymoma associated MG (TAMG). Early onset MG has a female preponderance and strong associations with human leukocyte antigen (HLA) DR3-D8 and thymic follicular hyperplasia. In contrast, late onset MG often occurs in males, has no HLA association and may display anti-striational antibodies despite harbouring atrophic thymus.4 Thymoma occurs in 15–30% of MG and is commonly associated with co-existing anti-ryanodine receptor and anti-titin antibodies.6,7 MuSK-MG tends to present with a bulbar-dominant phenotype and usually does not have any significant thymic abnormalities.8
Immunopathology of MG
The origin of the autoimmunity in MG has not been precisely elucidated, though genetic susceptibility and environmental triggers such as viral infections have been implicated. Pathological changes in the thymus are believed to play a pivotal role in the pathogenesis of AChR-MG.9 The most accepted hypothesis centres around the failure of self-tolerance which occurs intrathymically in AChR-MG. Self-tolerance is ensured by a balance between immune cell generation and the timely removal of auto-reactive lymphocytes. The thymus is the primary organ for the maturation and differentiation of T cells. During their maturation, T cells including the T regulatory cells (Tregs) are exposed to thymic myoid cells which express AChR and autoimmune regulatory (AIRE) medullary epithelial cells. The latter play an important role in the clonal deletion of T cells sensitized to auto-antigens during the development of central tolerance. The lymphocytes which display auto-reactivity are removed within the thymus whereas those that escape this culling are suppressed in the periphery by the Treg cells.10
Two important factors which contribute to immune breakdown in MG are thymic pathology and genetics which are vastly different in early and late onset MG. In early onset MG, the thymus shows structural changes in the form of thymic follicular hyperplasia. The germinal centres of the follicles are implicated as the site of origin of autoimmunity in them. The mechanisms include aberrant production of cytokines and imbalance in the function of T effector and Treg cells. There is increase in the pro-inflammatory Th17 and T follicular helper cells (Tfh) which promote B cell activation and generate autoantibodies.10,11 Regulatory B cells which suppress autoimmunity are also functionally abnormal in MG. In TAMG, pathological abnormalities consistent with immune proliferation in the areas adjacent to the tumour have been reported without any follicular hyperplasia. Thymomas express loss of AIRE making the cells susceptible for mounting autoimmune responses.4 Late onset MG is associated with involuted thymus with paucity of the myoid cells and regulatory thymic cells which prevent autoimmunity. No specific thymic pathology has been demonstrated in MuSK-MG.4
In the NMJ, nerve action potential provokes the activation of presynaptic voltage gated calcium channels which initiates a cascade of events culminating in the release of acetyl choline (ACh) into the synaptic cleft. Acetyl choline binds to the AChR in the synaptic folds of the postsynaptic membrane resulting in the opening of the central pore of the receptors and subsequently the voltage gated sodium channels, which then initiate the muscle action potential.12 Normal NMJ function requires clustering of AChR in the crests of the synaptic folds which is achieved by a kinase enzyme, MuSK. Activation of MuSK occurs during the development of NMJ by the interaction of agrin released from the developing motor axons with a post-synaptic protein, LRP4. Binding of this complex is crucial for the dimerization and activation of MuSK. MuSK activation induces a series of phosphorylation reactions recruiting DOK7 and rapsyn and finally inducing clustering and stabilization of AChR.13
The postsynaptic neuromuscular dysfunction in MG is produced by IgG auto-antibodies which act by three mechanisms: (i) functional blockade of the receptors, (ii) receptor internalization and degradation and (iii) complement activation and destruction of the membrane.14 Antibodies against AChR are IgG1 and IgG3 subtypes which are capable of fixing complements. Complement fixation and generation of the membrane attack complex result in damage to AChR and voltage gated sodium channels in the post-synaptic membrane which reduces the safety factor and increases the threshold for excitation. MuSK antibodies which are the IgG4 isotype do not significantly activate the complement pathway. Both IgG4 MuSK and IgG1/G2 LRP4 antibodies prevent the clustering of AChR in the post-synaptic membrane which is essential for normal transmission.15
Current Management of MG
The treatment strategy in MG is guided by the clinical pattern (ocular versus generalized), autoantibody subtype and disease severity. Current therapy of MG revolves around symptomatic treatment (cholinesterase inhibitors), long-term immunomodulatory therapy including thymectomy and management of acute crisis.
Cholinesterase inhibitors such as pyridostigmine can produce rapid relief of symptoms in mild MG and are usually prescribed as the initial therapy. The majority of the patients, however, require suppression of autoantibody production with immunosuppressive treatment which remains the cornerstone of MG therapy. Oral corticosteroids are initially used to induce remission while long-term maintenance is achieved with either low dose oral corticosteroids or non-steroidal immunosuppressants. Azathioprine, methotrexate, ciclosporin, tacrolimus and mycophenolate mofetil are the most frequently used non-steroidal agents. Agents such as cyclophosphamide are usually reserved for poorly responsive patients. In addition, rapidly acting, but short-lasting therapies such as intravenous immunoglobulin (IVIg) or therapeutic plasma exchange (PLEX) are used as rescue therapies in patients with imminent or incident respiratory crisis and severe MG.16 Therapeutic thymectomy is recommended for all thymomatous MG and a subset of non-thymomatous AChR antibody positive gMG patients with an age of onset less than 50 years.16–18 The role of thymectomy in older patients and antibody negative MG has not been convincingly established.16
Therapy of MuSK-MG with IgG4 antibodies differs from AChR-MG. MuSK-MG has limited response to IVIg and may occasionally show paradoxical worsening with cholinesterase inhibitors. No consistent thymic abnormalities have been demonstrated in MuSK MG and hence thymectomy is not recommended.19 The novel complement blocking therapies are also ineffective in MuSK-MG as MuSK antibodies do not usually activate the complement pathway.13
The standard management of MG produces beneficial response in more than 75% of patients.20 Many require long-standing therapy, sometimes lifelong, entailing the high risk of side effects from steroids and the non-specific immunosuppressive treatment.21
Need for Novel Immunotherapeutic Agents
Despite being one of the best studied immune diseases, there are clear unmet needs in MG therapy. Conventional therapies in MG induce remission in 70–80%, but very few attain sustained stable remission off therapy.20,22 This imposes the risk of long-term exposure to immunotherapies with cumulative toxicities including opportunistic infections, malignancies and systemic organ dysfunction. A primary reason for such toxicity is the non-specificity of immune targets for the standard medications.
Need for prolonged therapy with corticosteroids with their myriad metabolic and immunological side effects is necessitated by the delayed onset of action of many of the non-steroidal agents. Patients with serious co-morbidities are often unable to tolerate immunotherapy, steroids in particular.23,24
A subset of MG, approximately 10–15%, may be classified as treatment-refractory, though this entity has been variably defined in different studies. Mantegazza et al defined refractory MG as at least one of inadequate response to standard immunotherapies, relapses or crisis on attempting dose reduction of immunosuppressive therapies, recurrent and frequent crisis on therapy, intolerable side effects of the medications or co-morbidities which limit the therapy.20 Drug refractory patients have recurrent disease exacerbations and need for hospitalizations including ventilatory support, are prone for co-morbidities including end-organ dysfunction and have poor functional quality of life.25,26
Newer Immunotherapies in MG
A plethora of medications have been launched in MG therapeutic trials over the last decade. These predominantly include targeted immunotherapies directed specifically against the various cells and immune pathways implicated in MG pathogenesis (Figure 1). Inhibition of complement pathway and neonatal Fc receptor (FcRn) targeting for hindering IgG recycling are two of the novel and most successful mechanisms identified among them. Inhibition of B and T cells by direct depletion or through cytokines have also been successful. A few of the drugs, namely, eculizumab, ravulizumab and efgartigimod, have obtained approval from regulatory agencies while the majority are in various phases of development.27,28 This review will summarize the new and emerging immunotherapies in MG with focus on the complement inhibitors, FcRn blockers, and B cell depleting therapies (Table 1).
 |
Table 1 Summary of the Newer Molecules for Myasthenia Gravis Therapy |
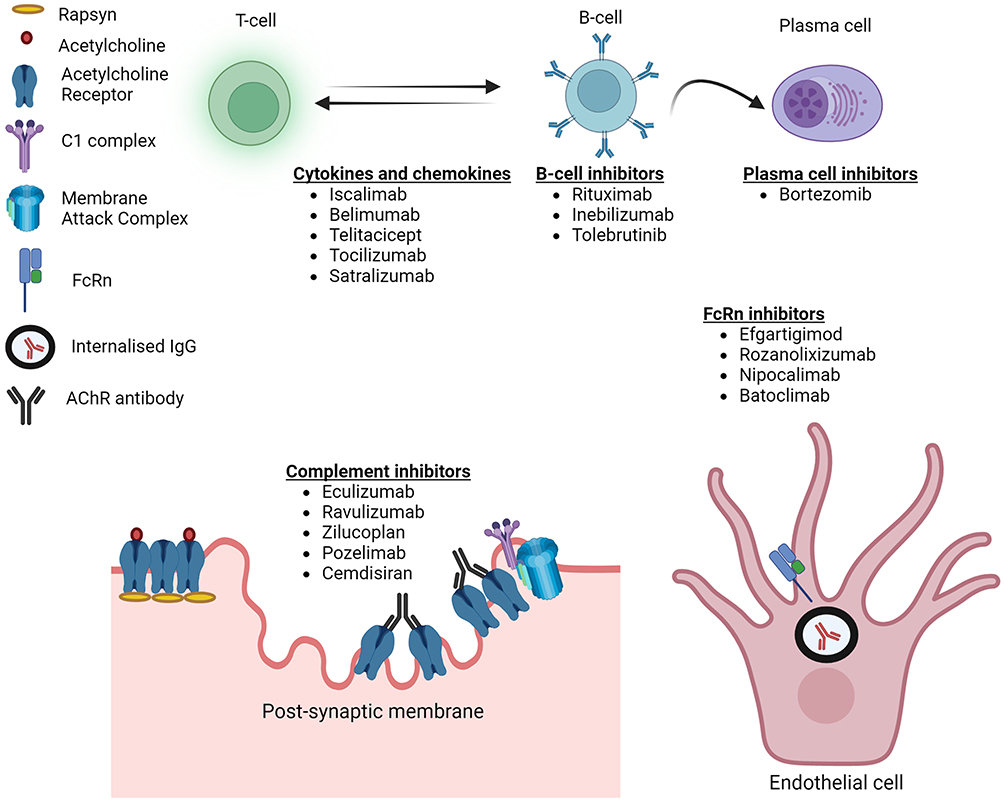 |
Figure 1 Targets of novel therapeutic agents in myasthenia gravis. |
Complement Inhibitors
Complement Pathway and Role in MG
The complement system is an integral part of immune surveillance. It is mediated by a set of proteins which are activated by three separate mechanisms: classical, lectin and alternative pathways.29 The autoantibodies in MG (particularly IgG1 and IgG3 AChR antibodies, but not usually the IgG4 MuSK antibodies) activate the complement system through the classical pathway. The inciting antibody binds to C1q complement protein which sets off a cascade of enzymatic reactions that result in the production of C3 convertase, a key molecule of the complement system. C3 convertase generates and combines with C3b to form C5 convertase which then cleaves C5 into C5a and C5b. C5b further joins with C6, C7, C8 and C9 to form the main effector molecule C5b6789, also referred to as the “membrane attack complex” (MAC) or the “terminal complement component” (TCC). The lysis of the post-synaptic membrane by the MAC is an important pathogenetic mechanism in AChR-MG.30
Animal and human studies have revealed the complement system as a viable and powerful target for therapy in AChR-MG.30 Complement cascade can be disrupted by molecules which inhibit the key players in the cascade, the prime targets being C5, C3 and C1 components.31 Agents against C5, eculizumab, ravulizumab, and zilucoplan, have proven to be the most successful in human clinical trials while many more molecules are in the pipeline.
Eculizumab
Eculizumab is a humanized monoclonal antibody (mAb) directed against C5 complement protein. Binding of eculizumab to C5 prevents its cleavage to C5a and C5b and further prevents the C5a-induced chemotaxis of inflammatory cells and the formation of MAC. It was previously licensed for therapy of paroxysmal nocturnal hemoglobinuria (PNH) and atypical haemolytic uraemic syndrome (HUS)32,33 and more recently for aquaporin IgG positive neuromyelitis optica spectrum disorder (NMOSD).34 It was approved for therapy of gMG in 2019.
The product carries a boxed warning on the risk of meningococcal infection.35 The subjects for the clinical trials were included after mandatory vaccination against Neisseria meningitidis at least 2 weeks prior to the first infusion.26 The other less serious side effects include headache and nasopharyngitis.26
The pilot phase 2 study of eculizumab in AChR antibody positive refractory MG showed encouraging results with rapid and clinically meaningful improvement in the treatment group.36 The phase 3 placebo-controlled randomized REGAIN study (Safety and Efficacy of Eculizumab in AChR positive Refractory Generalized Myasthenia Gravis; NCT01997229) enrolled 125 AChR antibody positive refractory gMG who were assigned to either eculizumab or placebo. The induction dose of eculizumab was 900 mg on day 1, weeks 1, 2 and 3 and 1200 mg in week 4, and thereafter maintenance dose of 1200 mg every second week for 26 weeks. Primary endpoint assigned was the change in Myasthenia Gravis Activity of Daily Living (MG-ADL) score from baseline to week 26 using worst-rank ANCOVA and the secondary endpoints assessed were the change from baseline in the total scores of Quantitative Myasthenia Gravis score (QMG), Myasthenia Gravis Composite (MGC) and Myasthenia Gravis Quality of Life 15 (MG-QOL-15), and the proportion of responders.26
The study failed to attain significance for the primary endpoint (mean rank of 56.6 vs 68.3, p=0.0698). However, the secondary outcomes were significantly better in the intervention group including changes in QMG and MGQOL-15 scores and the responder analysis for MG-ADL and QMG. In the pre-specified sensitivity analysis, significant difference in all scores were noted between the two groups in favour of eculizumab, starting from week 1 and sustained through week 26. A major drawback detected in the trial design and possibly the reason for the negative result for the primary endpoint was the use of the worst rank analysis. This relegated all patients who discontinued therapy to the lowest rank irrespective of the reason for such discontinuation. This was notable in the eculizumab group where three patients who had good therapeutic responses discontinued due to side effects other than myasthenic worsening, namely Moraxella lacunata bacteremia, bowel perforation and prostatic carcinoma.26 In a post-hoc analysis, 25% of refractory MG on eculizumab had attained minimal manifestations status at 26 weeks, which was double that of the placebo group.37
The side effects were mild to moderate, and the most common ones were headache, upper respiratory infection and nasopharyngitis, reported equally in both the groups. Meningococcal infection was not noted. Fewer patients in the eculizumab group had MG exacerbations and need for rescue therapy.26
The participants of the REGAIN trial were enrolled into an open label extension (OLE) phase for a maximum of 4 years. After a blinded induction phase (active drug provided as 1200 mg every 2 weeks for eculizumab group and 900 mg on day 1 and weekly for 3 weeks for the previous placebo group), all patients were continued on 1200 mg once in 2 weeks. A total of 117 participants (56 in eculizumab/eculizumab group and 61 in the placebo/ eculizumab group) entered the study. Primary endpoint was change in mean MG-ADL score over time. Interim analysis showed a reduction of 75% in the episodes of myasthenic worsening compared with the baseline. Infections of specific interest occurred in less than one-fifth of the study group and none had meningococcal meningitis. Improvements in myasthenia scores and quality of life scores were sustained with rapid improvements in the placebo/eculizumab group.38
Various post hoc analyses of the REGAIN trial and OLE have underlined the efficacy and broad-spectrum responses with eculizumab. At the end of the OLE, 84.7% and 71.4% patients were classified as responders based on clinically meaningful improvements in MG-ADL (≥3 points) or QMG scores (≥5 points). While the majority were early responders (response within 12 weeks), new responders continued to emerge with longer term therapy.39 In the REGAIN trial, eculizumab-treated patients were two times more likely to have achieved minimal manifestation post intervention status compared with placebo at week 26, while in the OLE at 130 weeks, a substantial majority (88%) of the patients had attained improved status and 57.3% had reached minimal manifestation status.37 Minimal symptom expression defined as MG-ADL score of 0–1 or MG-QOL-15 score of 0–3 was attained by a significantly higher proportion of eculizumab-treated patients at week 26 of REGAIN.40
Eculizumab was shown to be uniformly beneficial in subgroups in REGAIN and OLE who presumably had the worst spectrum of refractory MG as defined by failed use of chronic IVIg therapy and rituximab. Eculizumab was administered in both these subsets after a sufficient washout period. The 17 patients on chronic IVIg who completed OLE (8 in eculizumab/eculizumab and 9 in placebo/eculizumab groups, respectively) had higher exacerbation rate in the year preceding randomization compared with the total REGAIN cohort. Eculizumab in REGAIN and OLE produced rapid and sustained improvement in the majority and reduced the exacerbation rate by more than two-thirds from 150 exacerbations per 100 patient-years in the pre-treatment year to 47 exacerbations per 100 patient-years during treatment.41 Another subgroup analysis on 14 patients who were previously exposed to rituximab did not show any difference from the unexposed group in terms of efficacy or safety of eculizumab.42
Eculizumab could also produce rapid improvement in two of three ventilator-dependent AChR-MG patients who were previously resistant to other immunotherapies, IVIg and PLEX. While two achieved minimal manifestations status with 4–6 weeks of therapy, the third had partial amelioration of symptoms allowing transition to non-invasive ventilation.43
The RCT and OLE, various subgroup studies and case reports have firmly established the role of eculizumab as a rescue therapy in resistant MG, but its role as a first-line agent and duration of therapy are undefined. The annual cost of therapy which exceeds half a million US dollars has been a major deterrent to the wider use of this drug.44,45
Ravulizumab
Ravulizumab is a long-acting C5 complement inhibitor and has a mechanism of action similar to that of eculizumab. The long half-life of this molecule necessitates fewer intravenous infusions for maintenance. This drug was previously approved for treatment of PNH and is under investigation for atypical HUS and IgA nephropathy.46 It was licensed by regulatory authorities for gMG in 2022.
In the phase 3 randomized placebo-controlled CHAMPION-MG study (NCT03920293), 175 adults with symptomatic AChR antibody positive gMG were recruited to receive ravulizumab infusion versus placebo. The participants were randomized 1:1 to the treatment and control arms. The dosage of ravulizumab was weight-based given as 2400–3000 mg single loading dose on day one followed by maintenance doses of 3000–3600 mg every 8 weeks starting from day 15. The primary efficacy endpoint of significant improvement in MG-ADL and for the secondary outcomes were achieved in the treatment group at 26 weeks. No marked difference in adverse effects was noted between the two groups.47 The open-label extension phase of the study is ongoing.
Zilucoplan
Zilucoplan is a small macrocyclic peptide molecule which prevents the terminal activation of the complement cascade by two mechanisms. It binds to the C5 complement component and prevents its cleavage. It additionally binds to the existing C5b to prevent its attachment to C6. The molecule can be given as a subcutaneous injection. The advantages of this molecule include its small size which ensures good NMJ penetration, ability to concomitantly administer IVIg therapy as this is not an antibody and the potential for self-administration.48
In the phase 2 clinical study in symptomatic adult AChR-MG patients, 44 patients were randomized and received one of the three interventions – once daily subcutaneous injection of zilucoplan at 0.3 mg/kg, once daily zilucoplan at 0.1 mg/kg or placebo. The main efficacy endpoints of changes in MG-ADL and QMG scores were assessed at the end of 12 weeks treatment. High-dose zilucoplan group showed a rapid and statistically significant improvement in the scores compared with placebo. They also had reduced need for rescue therapies. No serious treatment emergent adverse reactions were reported with zilucoplan.49 The phase 3 trial to study the efficacy and tolerability of 0.3 mg/kg zilucoplan versus placebo (RAISE; NCT04115293) has been completed.
Pozelimab with Cemdisiran
Pozelimab is a human mAb against C5 complement whereas cemdisiran is a small synthetic interfering ribonucleic acid (siRNA) which can suppress the hepatic production of C5. Both molecules are given as subcutaneous injection and are generally safe and well-tolerated at different doses. Loading dose of intravenous pozelimab 15 mg/kg followed by four repeat doses of pozelimab 400 mg administered subcutaneously once every week was found to inhibit complement activation in healthy volunteers.50 In animal studies, combination of pozelimab with cemdisiran allowed lower doses and decreased dosing frequency compared with use of the individual agents separately.51 The phase 3 randomized controlled trial of the combination (intravenous pozelimab loading followed by 4-weekly subcutaneous injections along with cemdisiran 200 mg subcutaneous 4-weekly) versus placebo in gMG is ongoing (NCT05070858).
FcRn Blockers
FcRn Pathway in Health
Immunoglobulins are heterodimers made of two heavy and two light chains. Each immunoglobulin molecule has a variable region which houses the Fab (antigen-binding) fragment and a constant region with the complement-binding crystallizable fragment (Fc). The attachment of the Fc fragments to Fc receptors is crucial in regulating their interaction with the cellular immune mechanisms and enhancing the serum levels of IgG. The latter function is modulated by neonatal Fc receptors (FcRn) widely distributed in multiple cell types, particularly endothelial and myeloid cells. FcRn helps in the recycling of IgG and increases its longevity in circulation. The binding of FcRn to IgG is pH dependent and occurs only in an acidic pH. After the uptake of IgG by the cells, it is transported to the acidic environment of endosomes where FcRn binds to IgG and protects it from degradation by the lysosomal enzymes. FcRn transports IgG back to the surface and releases it in the neutral physiological pH.52 This pH-dependent binding is essential for prolonging the half-life of IgG antibodies which typically remain in circulation nearly 4 times longer than the other immunoglobulin subtypes (IgM and IgA). Serum albumin shares the long half-life and FcRn-mediated recycling by binding to a site distinct from that of IgG.53 Serum proteins and IgG molecules which do not bind to FcRn are degraded within the lysosomes.54
FcRn Blocking Agents in MG
Antibodies in MG are proven to be pathogenic and all the different antibody subtypes of seropositive MG belong to the IgG class. Blocking of FcRn can interfere with their recycling and thereby reduce the concentration of the pathogenic IgG autoantibodies.54 Many authors consider this technique similar to a “medical plasma exchange”, given the rapid reduction in IgG levels. While reducing the autoreactive antibodies, these agents keep the concentrations of the other antibody subtypes (IgM and IgA) stable.55 About 25% of the normal IgG response is also retained with these therapies and recovery of IgG levels occur faster compared with B-cell depleting therapies.56 These factors may allow the body to mount an effective immune response on therapy and sustain a lower risk of infection.
Novel therapeutic agents which target FcRn to reduce IgG levels are classified under the umbrella term ‘antibodies which enhance the degradation of IgGs’ or ‘Abdegs’.52 The various molecules include engineered Fc fragments (efgartigimod), monoclonal antibodies against FcRn (rozanolixizumab, nipocalimab, batoclimab and orilanolimab), and peptide fragments. Efgartigimod was approved for therapy of gMG in 2021, while the others are in various phases of development. By reducing the pathogenic autoantibodies, they prevent all the terminal effects of the antibodies including complement activation, apoptosis by internalization and blocking.54 An unintentional class effect noted in healthy volunteers and patients is a mild to moderate reduction of serum albumin levels, possibly resulting from a spill-over blockade of albumin turnover.53 This can potentially influence the blood levels of highly albumin-bound medications including concomitant anti-myasthenic therapies and adversely affect the lipid levels.53
Efgartigimod
Efgartigimod is an engineered Fc domain of human IgG1 with increased affinity for FcRn receptors than the endogenous IgG, thereby competitively inhibiting the latter’s recycling. Unlike FcRn mAbs, efgartigimod retains the pH-dependent binding. The intact pH-dependent binding allows the recycling of efgartigimod, prolonging its half-life in circulation.54
In the exploratory phase 2 study, 24 confirmed AChR antibody positive gMG patients were randomized to receive four 10 mg/kg weekly doses of efgartigimod or placebo. The primary endpoints were safety and tolerability. The treatment-related adverse effects were mild and generally comparable between the two groups. Headache and reduction in lymphocyte and monocyte counts were more commonly noted in the intervention arm. One patient in the efgartigimod group developed herpes zoster in the infusion arm. A clear positive therapeutic effect was discerned in the treatment group as early as 1 week after the first infusion with about three-fourths of the treatment group showing rapid improvement. The study was not powered to assess efficacy.57
The phase 3 multicentric randomized controlled ADAPT trial of efgartigimod proved its efficacy against placebo as an add-on therapy for AChR antibody positive gMG. This study included adults with gMG irrespective of the antibody status on a stable dose of anti-myasthenic medicines and an MG-ADL score of ≥5. Efgartigimod 10 mg/kg/dose was administered as a weekly infusion for 4 weeks and the cycle was repeated after a variable interval of 8 weeks or more depending on the re-emergence of symptoms and tolerability. The total follow-up period was 26 weeks. Among the167 patients (84 efgartigimod and 83 placebo) enrolled, 77% were AChR antibody positive. Compared with placebo, efgartigimod-treated AChR-MG had significant improvement in MG-ADL score at 4 weeks (primary outcome). The other efficacy scores (QMG, MGC and MGQoL15 revised) showed a similar pattern with the maximum improvement noted by 4−5 weeks and sustained for 7 weeks. The drug was tolerated well and adverse effects including serious ones were no more common than in placebo. Headache was similar in the two groups whereas mild to moderate infections were more common in the intervention group compared with placebo (46% vs 37%).58
Efgartigimod produced about 60–70% reduction in the levels of total IgG and all IgG subtypes commencing in the first week of the initial dose, the levels inversely corresponding with clinical improvement.57,58 Levels of anti-AChR antibody also fell by 40–70% in most of the patients with recovery by 12 weeks after the first infusion. The resurgence in antibody and IgG levels occurred prior to or paralleled symptomatic worsening.54,57,58
Nipocalimab
Nipocalimab is a fully human aglycosylated IgG1 monoclonal Ab against FcRn which strongly binds to FcRn independent of pH. Multiple intravenous doses of nipocalimab in 50 healthy volunteers produced a marked reduction of serum IgG levels by 85%, and the low levels were maintained for 24 days. The drug exhibited a satisfactory safety and tolerability profile.56
In the phase 2 study (NCT03772587), 68 patients with gMG were divided into five treatment arms to receive either of 4 dosages of nipocalimab or placebo as intravenous infusions over 8 weeks. A dose-dependent reduction in IgG and anti-AChR antibody levels was noted which correlated well with clinical improvement.59 Results are awaited from the ongoing phase 3 randomized controlled study in adults (NCT04951622) and phase 2/3 open-label study in children (NCT05265273).
Rozanolixizumab
Rozanolixizumab is a high affinity humanized IgG4 mAb against FcRn which is administered subcutaneously. This drug completed a phase 2 trial in 43 patients with moderate to severe gMG who were positive for either of AChR or MuSK antibodies. The study was conducted in 2 periods separated by 2 weeks. In period 1, patients were randomized to receive either rozanolixizumab subcutaneous infusions 7 mg/kg once a week for 3 weeks or placebo and then re-randomized in period 2 to receive 3 doses of either 7 mg/kg or 4 mg/kg rozanolixizumab. This model enabled the investigators to assess the sustainability of the drug effect, adverse effects on longer use, and efficacy of a lower dose. The drug was generally well-tolerated with headache being the most frequent side effect, more common in the treatment arm. The primary efficacy outcome of improvement in QMG score from day 1 to 29 was not significant but overall data including antibody measurements suggested potential efficacy for the drug in moderate to severe gMG.60 The phase 3 study (NCT03971422) to confirm efficacy is ongoing.
B Cell Depleting Therapies
Role of B Cells in MG Pathogenesis
The pathogenesis of MG is believed to be initiated by T cell dysregulation, but autoreactive B cells play a major role in driving and propagating the dysfunction. In the presence of follicular dendritic cells, pro-inflammatory T helper cells and cytokines, there is proliferation of the autoreactive B cells within the germinal centres and in the periphery. The final differentiation into the plasma cells which secrete the pathogenic antibodies is crucial in the pathogenesis.61,62
Targeting B Cells in MG Therapy
B cells can be targeted directly by depleting or inhibiting cells of the B cell lineage or indirectly by targeting their facilitators such as cytokines or other immune cells.63 B cell-based therapies are an attractive option as the pathogenic antibodies are derived from plasma cells. The distinct subtype of plasma cells which produce the antibodies may influence the therapeutic response. MuSK antibodies are secreted by short-living plasma cells which are highly susceptible to depletion by anti CD19/20 agents whereas the IgG1 and IgG3-secreting long-living plasma cells lack these surface markers.64,65 In this section, we will discuss the direct B cell and plasma cell depleting therapies.
Rituximab
Rituximab is a chimeric mouse/human antiCD20 mAb which has gained wide acceptance as a therapy for severe and refractory MG in the last few years. Rituximab rapidly depletes the mature and memory B cells in the peripheral blood while largely sparing the pre-B cells and plasma cells located in the bone marrow and secondary lymphoid organs. This depletion is achieved by antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and apoptosis of the target cells. The effects persist for about 6 months before the circulating B cells are repopulated from the bone marrow.66
The pathogenic antibodies in MG are produced by plasma cells which reside in the bone marrow. Rituximab spares the long-lived plasma cells which do not express CD20 while it does diminish the short-lived plasma cells and B regulatory cells (interleukin-10 producing cells) which have CD20 expression.65 In general, the suppression of antibody production is transient in MG.15 Anti-AChR antibodies are produced by the long-lived plasma cells whereas the MuSK antibodies are thought to be secreted from the short-living plasma cell blasts.64 Theoretically, this suggests that MuSK-MG may be more responsive to rituximab therapy.
To date, there has been a single phase 2 randomized controlled trial for rituximab in MG.67 The bulk of the efficacy and safety information for this drug exists as case series and reports and systematic reviews (Table 2).67–78 The most commonly used rituximab regimens include 2 doses of 1 g 2 weeks apart or 375 mg/m2 infusions repeated weekly for 4 weeks. Some authors have used low dose regimens of 375 mg/m2 or 500 mg twice given 2 weeks apart or 600 mg single dose inductions. Reinfusions are usually guided by clinical symptoms or B cell repopulation.69
 |
Table 2 Summary of Systematic Reviews, Randomized Trials, and Prospective Studies of Rituximab in Myasthenia Gravis |
The phase 2 B-cell Targeted Treatment in MG (BeatMG) study published in 2021 was a placebo controlled randomized study in adults with mild to moderate gMG with positive AChR antibody. The study required the patients to be on a minimum dose of 15 mg of prednisolone per day. Fifty-two patients were recruited to receive either two cycles of rituximab at 6-month intervals with 4 doses of 375 mg/m2 in each cycle, or matched placebo. A futility design was employed, and the primary endpoint was a reduced requirement for steroid 4 weeks prior to week 52. The study failed to attain significance in the primary steroid sparing outcome (60% with rituximab versus 56% with placebo). Secondary efficacy and other exploratory outcomes were also not different between the two groups though the placebo group had thrice the requirement for rescue therapy for myasthenia exacerbations compared with the study group. No specific safety concerns were recorded with rituximab. The inclusion of patients with a low disease activity (as suggested by good placebo response) and the prerequisite of high prednisolone dose were probable contributors to the negative results.67
Among the two early systematic reviews which compared the efficacy of rituximab in the different antibody subgroups of MG, divergent outcomes were recorded. While both reviews recorded a positive therapeutic outcome with rituximab, only one study showed a significant difference between AChR-MG and MuSK-MG.68,69 Tandan et al, recorded a higher frequency of positive response with MuSK-MG than AChR-MG patients. Minimal manifestation status was attained in 72% and 30%, respectively and pharmacological or complete stable remission in 47% and 16%, respectively for MuSK-MG and AChR-MG in this analysis. A more recent study of rituximab in refractory MG reiterated this observation.72 A multicentric prospective study which compared rituximab therapy with conventional immunotherapy in MuSK-MG found marked clinical improvement and lower corticosteroid requirement in the rituximab-treated group.74
Therapeutic efficacy of rituximab in AChR-MG was specifically examined in two systematic reviews where unequivocal clinical improvement was noted in 68–77% of the subjects.70,71 One of these reviews compared routine and low dose rituximab (171 and 89 subjects, respectively) in refractory AChR-MG and failed to record any significant difference in the therapeutic outcomes or side effects. Low dose rituximab protocol as monotherapy or add-on immunotherapy was also shown to have excellent efficacy and safety profile in new-onset and early MG in a few retrospective series.78–80 However, none of these studies had a comparison arm with routine immunotherapy.
Rituximab has also been reported to be efficacious in juvenile MG. A retrospective multicentre study from France included 27 paediatric MG patients treated with rituximab as first-line agent and 37 on standard therapy. Rituximab therapy was associated with superior outcomes in terms of clinical improvement, tapering of glucocorticoid dose and cessation of oral immunosuppressant therapy compared with conventional first-line therapies. This study did not report any adverse effects in the rituximab group.81 Similar observations were noted with smaller series of refractory juvenile MG patients.82,83
Adverse effects have been documented in 4.2–26.1% of patients,68,69,72,80 the most important ones being infusion reactions, cytopenias, infections including opportunistic infections and prolonged hypogammaglobulinaemia.69,72 Although most of the infections have been mild to moderate, serious infections and sepsis have resulted in deaths in many series and merit meticulous follow up. Increasing age is a risk factor for serious infections.84 Two confirmed cases of progressive multifocal leukoencephalopathy in rituximab-treated MG patients who were on concomitant therapy with multiple immunosuppressants have been reported.84,85
Inebiluzumab
Inebilizumab is a humanized mAb targeting CD19 expressed in the B lineage cells. The expression of CD19 marker covers a broader group of cells than CD20. CD20 marker is restricted to pre-B cells in the bone marrow and the circulating naïve, mature and memory B cells whereas CD19 is additionally expressed on the early pro-B cells and the majority of plasma cells in blood and secondary lymphoid organs and about half of the plasma cells in the bone marrow.86 The predominant mechanism of B cell depletion is through antibody dependent cell-mediated cytotoxicity. The drug was approved for treatment of aquaporin-4 IgG positive NMOSD in June 2020 and is under evaluation for immunotherapy for kidney transplant recipients and IgG4 disease.34,87 In the trials for NMOSD, the drug was well-tolerated with mild to moderate side effects in line with other B cell depleting agents.87
Inebilizumab is currently undergoing phase 3 clinical studies in MG (NCT04524273). The Myasthenia gravis Inebilizumab Trial (MINT) study is a randomized placebo-controlled trial of intravenous inebilizumab (300 mg given on Day 1, 15 and 183) in antibody positive (either anti AChR or MuSK antibody) gMG as an add-on therapy.
Bortezomib
Bortezomib, a potent and reversible proteasome inhibitor, produces immune effects by targeting the plasma cells. Proteasomes are ubiquitous intracellular protein complexes which regulate the cell turnover and mediate the degradation of pro-apoptotic factors, thereby playing a central role in protein homeostasis. Misfolded and dysfunctional proteins are tagged by the proteasomes and marked as substrates for enzyme-mediated proteolysis. Bortezomib binds and induces apoptosis of the 26S proteasome, a cardinal player in nuclear and cytosolic protein degradation pathways. This results in the build-up of non-functional proteins including pro-apoptotic factors within the cells.88
Plasma cells are non-dividing cells which secrete antibodies, including the pathogenic autoantibodies in MG. Broadly, there are two groups of plasma cells designated as short and long living plasma cells, the latter being particularly important in chronic dysimmune diseases. As they are terminally differentiated and non-dividing, they are usually resistant to radiotherapy, glucocorticoids, standard oral immunosuppressants, and CD19/20 inhibitors. However, the rapid intracellular synthesis of antibodies in these cells renders them highly susceptible to mechanisms which deter the protein degradation pathways such as proteasome inhibitors. In addition, bortezomib also targets the nuclear factor κB (NF- κB) signalling pathway which has important anti-apoptotic functions and is often upregulated in inflammatory diseases.88
Bortezomib was primarily approved for use in multiple myeloma and mantle-cell lymphoma, but is a potential therapeutic option in a variety of autoimmune conditions resistant to standard therapy including gMG.89 Its utility in MG was supported by experimental data in culture of thymic cells from patients with early onset MG where bortezomib demonstrably depleted the resident autoreactive long-lived plasma cells with in vitro reduction in anti-AChR antibody production from them.90
A single case report has suggested clinical benefit for bortezomib in MG. One MuSK-MG patient who was poorly responsive to glucocorticoids, IVIg, PLEX, immunoadsorption and rituximab was treated with bortezomib and achieved a speedy and sustained improvement in symptoms.91 The drug was administered subcutaneously at a dose of 1.3 mg/m2 body surface, four doses given within 2 weeks. A single-centre open-label study of bortezomib in antibody-mediated autoimmune diseases (TAVAB, NCT02102594) including MG was terminated due to recruitment difficulties.89
The main limiting side effect is bortezomib-induced polyneuropathy. This neurotoxicity is dose-dependent and perpetuated by repeated cycles. The propensity for neuropathy is diminished by restricting the dose to a single cycle of bortezomib which appears sufficient to induce remission in immunological diseases and by adopting subcutaneous rather than intravenous route of administration.92
Tolebrutinib
Tolebrutinib belongs a class of molecules referred to as Bruton’s Tyrosine Kinase (BTK) inhibitors. The facilitatory role of BTK in innate and adaptive immunity, in particular, its influence on the proliferation and maturation of B cells, has made BTK inhibitors an attractive therapeutic option for autoimmune diseases. BTK belongs to the protein kinase family and is important in the signalling pathways involved in B cell proliferation and function including production of antibodies and cytokines, and antigen presentation. It is also expressed in myeloid cells (components of innate immunity) and influences the production of inflammatory cytokines and adhesion molecules which promote inflammation.93 The suppression of these functions by BTK inhibitors results in their efficacy in autoimmune diseases and B cell malignancies. Tolebrutinib, an orally administered irreversible covalent BTK inhibitor, is currently being evaluated in a phase 3 placebo-controlled randomized trial in gMG (NCT05132569).
Cytokine and Chemokine Targeting Therapies
Cytokines and Chemokines in MG
Cytokines are small signalling molecules which help in the coordination of immune system function and communication between the various immune and inflammatory cells. These are secreted by stimulated cells (chiefly T helper cells and macrophages) and act on a variety of target cells which harbour their specific receptors. Chemokines are a specific subgroup of cytokines with chemoattractant properties that attract leukocytes to sites of inflammation.94
The targeting of cytokines can impact either or both of B and T cell mediated mechanisms of pathogenesis. The viable targets include the following.63
- CD40-CD154 pathway: This is important for the activation of B cells and differentiation of plasma cells through other immune cells.
- B cell activating factor (BAFF) pathway: The pathway has a key role in B cell maturation and survival.
- CXCL13 signalling: CXCL13 chemokine and its receptor CXCR5, expressed on B cells and Tfh cells play a major role in the migration of B cells to secondary lymphoid organs and for the development of germinal centres.
- Interleukin 17 (IL-17) pathway: IL-17 is a cytokine secreted by the pro-inflammatory Th17 cells and is overexpressed in MG.
- Interferon-I pathway: This pathway is overactivated in TAMG and Early onset MG and could be responsible for the chronic inflammatory changes.
- Interleukin 6 (IL-6) pathway: IL-6 has pro-inflammatory activity and influences both T and B cells. It induces the conversion of T helper to Th17 proinflammatory cells and promotes the maturation and survival of plasma cells.
Belimumab
Belimumab is a human recombinant neutralizing mAb against a cytokine of the tumour necrosis factor (TNF) family called the B-cell activating factor (BAFF), also known as B Lymphocyte Stimulator (BLyS). Receptors for BAFF are located in all circulating B cells and BAFF plays key roles in B cell survival, maturation and immune function.95 Overexpression of BAFF promotes the survival of autoreactive B cells and is correlated with autoimmune diseases including MG. Suppression of BAFF by belimumab causes the depletion of the B cell lineage cells dependent on it for survival and homeostasis.96
Clinical efficacy of belimumab was shown in systemic lupus erythematosus for which it is an approved therapy. However, the drug’s performance in MG has been disappointing, notwithstanding the fact that some of the commonly used MG therapies indirectly affect BAFF activity. A 24-week phase 2 study of intravenous belimumab in MG did not find any benefit for the therapy over placebo.97
Iscalimab
Iscalimab is a fully human non-depleting antiCD40 mAb which targets the activation and signalling pathways mediated by CD40. CD40 is a co-stimulatory molecule expressed in B cells and other antigen presenting cells which interacts with its ligand CD154 (CD40L) located on activated T cells. The CD40-CD154 co-stimulatory pathway is essential for the T cell dependent antibody responses, and the differentiation, survival and activation of memory B cells, dendritic cells and macrophages.98 A phase 2 placebo-controlled randomized trial of iscalimab (CFZ533) as an add-on therapy in moderate to severe MG (NCT02565576) is currently underway.
Interleukin 6 Antagonists
Interleukin 6 (IL-6) is a soluble cytokine which mediates a wide variety of functions including inflammation, immune response and haematological functions. In the acute phase of inflammation, IL-6 facilitates the production of a subgroup of acute phase reactants and augments the immune function by stimulating haematopoiesis. However, it plays a major role in the transition of acute neutrophilic inflammation to chronic mononuclear infiltrates paving the way for potentially harmful chronic inflammatory response.99 Its role in AChR-MG was illustrated by the presence of elevated IL-6 in treatment-naïve patients and the subsequent suppression of the levels with immunotherapy.100 Antibodies against IL-6 could downregulate the pro-inflammatory Th-17 response and suppress the symptoms in experimental autoimmune MG models.101
Tocilizumab and satralizumab are mAbs against IL-6 receptors which have been approved for other chronic autoimmune diseases. Subcutaneously administered satralizumab was effective in reducing relapses of NMOSD both as monotherapy and add-on immunotherapy and has been recently approved for this indication.102,103 Jonsson et al, described two patients with refractory MG who responded to intravenous tocilizumab after failure of classical immunotherapy, thymectomy and repeated doses of IVIg and rituximab.104 Both the drugs are under development as therapy for gMG (NCT05067348 and NCT04963270).
Subcutaneous Immunoglobulin
Chronic IVIg administration is a therapeutic option in refractory gMG. Subcutaneously administered immunoglobulin is an attractive alternative for IVIg as it avoids hospital admission, reduces utilization of health resources, preserves patient autonomy and portends better quality of life. Subcutaneous immunoglobulin (SCIg) was also associated with more persistent blood levels of the drug and fewer systemic adverse effects.105,106
In a prospective open-label study, SCIg (2 g/kg) was administered over 4 weeks in 23 adults with worsening mild or moderate MG. The primary outcome of significant reduction in QMG scores at 6 weeks was achieved compared with baseline at weeks 2, 4 and 6. Headache and infusion reactions were the common side effects which occurred in 74% and 61% of patients, respectively.107 The magnitude of improvement in the QMG scores in this study was comparable to the pivotal study with IVIg.108 This study provided class IV data for the non-inferiority of SCIg over IVIg.107 However, with a global shortage of immunoglobulin products, it is unlikely that this will be useful in large number of MG patients.
Chimeric Antigen Receptor (CAR) and Chimeric Autoantibody Receptor (CAAR) T Cell Therapy
Chimeric antigen receptor (CAR) T cell therapy is a cell-based therapy which has shown marked success in the treatment of B cell malignancies over the last 6 years. The technique involves harvesting of T cells from the patient and genetically modifying them by attaching an artificially engineered receptor referred to as a chimeric antigen receptor (CAR). When the CAR T cells are infused into the circulation, the modified receptors bind to a specific antigen on the target cells (e.g., cancerous cells in B cell malignancies) and destroy them. The effect of CAR T cells is sustained long-term by their in vivo multiplication.109,110
More recently, the application of CAR T cell therapy have been extended beyond cancer treatment to autoimmune diseases, fibrosis, degenerative diseases and infections. In autoimmune diseases where a specific pathogenic antibody is identified, the preferred approach is using an engineered T cell which can bind the specific autoreactive B-cell. This technique is referred to as Chimeric autoantibody receptor (CAAR) T cell therapy and by specifically targeting B cells which express the autoantibody, a general depletion of the B cell lineage cells is avoided. In most of the autoimmune diseases, however, the pathogenic antibody is not well-delineated. Here, using a CAR T-cell model targeting the specific B cell antigen (e.g., CD 19-specific CAR) ensures a more thorough suppression of the abnormal cell clones compared with mAbs aiming at the same antigens.110 The limiting side effects include cytokine release syndrome, neurological side effects, infusion reactions and cytopenias.109
The ongoing studies in MG include engineered T cells directed against B cell Maturation Antigen (BCMA) (NCT04146051) and MuSK antibody (NCT05451212).
Haematopoietic Stem Cell Transplantation
Autologous haematopoietic stem cell transplantation (HSCT) has emerged as a technique to attenuate or even completely terminate disease activity in many autoimmune neurological diseases including MG. The procedure involves stimulating the production of haematopoietic cells, harvesting them from circulation, ablating and resetting the immune system and then re-infusing the treated cells from the patient. The evidence for HSCT in MG is limited to a handful of patients. Bryant et al, conducted a retrospective analysis of 7 patients with severe MG who had received autologous HSCT (one for co-existent follicular lymphoma). All had attained stable remission off therapy after a median follow up of 40 months.111 Similar outcomes were reported in other single case reports.112,113 The high propensity for early and late debilitating side effects including infections, secondary autoimmune diseases, and neoplasms have relegated HSCT to a lower position among the treatment options. Myasthenia gravis has been reported rarely as a chronic graft-versus-host reaction in recipients of HSCT.114
Limitations and Challenges of the Novel Agents
The novel therapies have brought with them some formidable challenges which limit their liberal use. The primary and immediate concern is the exorbitantly high cost, particularly for the recently approved eculizumab and efgartigimod. A cost-effectiveness analysis demonstrated that the addition of these drugs to conventional therapy increased the expenses 7- to 9-fold and discouraged the use of these medicines as benchmarks for costing decisions for emerging drugs.45 While acknowledging their undeniable benefits in refractory MG, the report of the analysis also suggested restricting the use of these medicines to a limited subset of AChR-positive MG patients.45
The use of these medicines is also limited by many unanswered questions. There is little understanding of their role in early MG and impact on reduction of doses of other immunosuppressant therapy in refractory MG. The optimal redosing schedule for efgartigimod and duration of therapy for all the novel therapies are not well-understood. Of particular concern is the unquantified risk of long-term toxicities including opportunistic infections and neoplasms which may emerge with extended use.
Conclusions
The newer biologicals herald an era where MG therapies are targeted to the specific pathomechanisms of the disease. Complement inhibitors, FcRn blockers and B cell depleters have established their position in MG management protocols. These medicines come with concerns pertaining to long-term safety, optimal usage and prohibitive cost. This necessitates the judicious use of these drugs in the correct clinical scenario for the correct patient. With the current evidence, they are maximally useful in patients with refractory MG, though cautious use in early severe disease may bring down the disease burden for the patient. Accumulation of real-world data will help in the logical placing of the drugs in treatment protocols. Research directed towards safer therapies and curative treatment for MG are essential additions to this rapidly expanding spectrum.
Disclosure
Prof. Dr. Saiju Jacob reports personal fees from and is part of the Advisory Board for ArgenX Ltd and UCB Pharma; speaker fees from Terumo BCT; personal fees from Regeneron and Immunovant, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010;10:1–9. doi:10.1186/1471-2377-10-46
2. Lehnerer S, Jacobi J, Schilling R, et al, Burden of disease in myasthenia gravis: taking the patient’s perspective. J Neurol. 2022;269(6):3050–3063. doi:10.1007/s00415-021-10891-1
3. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023–1036. doi:10.1016/S1474-4422(15)00145-3
4. Huijbers MG, Marx A, Plomp JJ, Le Panse R, Phillips WD. Advances in the understanding of disease mechanisms of autoimmune neuromuscular junction disorders. Lancet Neurol. 2022;21(2):163–175. doi:10.1016/S1474-4422(21)00357-4
5. Evoli A, Iorio R. Controversies in ocular myasthenia gravis. Front Neurol. 2020;11. doi:10.3389/fneur.2020.605902
6. Lovelace RE, Younger DS. Myasthenia gravis with thymoma. Neurology. 1997;48(Suppl5):76S–81S. doi:10.1212/WNL.48.Suppl_5.76S
7. Romi F. Thymoma in myasthenia gravis: from diagnosis to treatment. Autoimmune Dis. 2011;2011:1–5. doi:10.4061/2011/474512
8. Guptill JT, Sanders DB, Evoli A. Anti-musk antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. 2011;44(1):36–40. doi:10.1002/mus.22006
9. Levinson AI, Song D, Gaulton G, Zheng Y. The intrathymic pathogenesis of myasthenia gravis. Clin Dev Immunol. 2004;11(3–4):215–220. doi:10.1080/17402520400001769
10. Alahgholi-Hajibehzad M, Kasapoglu P, Jafari R, Rezaei N. The role of T regulatory cells in immunopathogenesis of myasthenia gravis: implications for therapeutics. Expert Rev Clin Immunol. 2015;11(7):859–870. doi:10.1586/1744666X.2015.1047345
11. Leite MI, Jones M, Ströbel P, et al. Myasthenia gravis thymus. Am J Pathol. 2007;171(3):893–905. doi:10.2353/ajpath.2007.070240
12. Rodríguez Cruz PM, Cossins J, Beeson D, Vincent A. The neuromuscular junction in health and disease: molecular mechanisms governing synaptic formation and homeostasis. Front Mol Neurosci. 2020;13:610964. doi:10.3389/fnmol.2020.610964
13. Borges LS, Richman DP. Muscle-specific kinase myasthenia gravis. Front Immunol. 2020;11:707. doi:10.3389/fimmu.2020.00707
14. Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Res. 2016;5:1513. doi:10.12688/f1000research.8206.1
15. Huda R. New approaches to targeting B cells for myasthenia gravis therapy. Front Immunol. 2020;11:240. doi:10.3389/fimmu.2020.00240
16. Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96(3):114–122. doi:10.1212/WNL.0000000000011124
17. Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375(6):511–522. doi:10.1056/NEJMoa1602489
18. Wolfe GI, Kaminski HJ, Aban IB, et al. Long-term effect of thymectomy plus prednisone versus prednisone alone in patients with non-thymomatous myasthenia gravis: 2-year extension of the MGTX randomised trial. Lancet Neurol. 2019;18(3):259–268. doi:10.1016/S1474-4422(18)30392-2
19. Clifford KM, Hobson-Webb LD, Benatar M, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis. Muscle Nerve. 2019;59(4):404–410. doi:10.1002/mus.26404
20. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord. 2018;11:175628561774913. doi:10.1177/1756285617749134
21. Khadilkar S, Chaudhari C, Patil T, Desai N, Jagiasi K, Bhutada A. Once myasthenic, always myasthenic? Observations on the behavior and prognosis of myasthenia gravis in a cohort of 100 patients. Neurol India. 2014;62(5):492. doi:10.4103/0028-3886.144438
22. Schneider-Gold C, Hagenacker T, Melzer N, Ruck T. Understanding the burden of refractory myasthenia gravis. Ther Adv Neurol Disord. 2019;12:175628641983224. doi:10.1177/1756286419832242
23. Bacci ED, Coyne KS, Poon JL, Harris L, Boscoe AN. Understanding side effects of therapy for myasthenia gravis and their impact on daily life. BMC Neurol. 2019;19(1):335. doi:10.1186/s12883-019-1573-2
24. Menon D, Bril V. Pharmacotherapy of generalized myasthenia gravis with special emphasis on newer biologicals. Drugs. 2022;82(8):865–887. doi:10.1007/s40265-022-01726-y
25. Harris L, Graham S, MacLachlan S, Exuzides A, Jacob S. A retrospective longitudinal cohort study of the clinical burden in myasthenia gravis. BMC Neurol. 2022;22(1):172. doi:10.1186/s12883-022-02692-4
26. Howard JF, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16(12):976–986. doi:10.1016/S1474-4422(17)30369-1
27. Menon D, Barnett C, Bril V. Novel treatments in myasthenia gravis. Front Neurol. 2020;11:538. doi:10.3389/fneur.2020.00538
28. Schneider-Gold C, Gilhus NE. Advances and challenges in the treatment of myasthenia gravis. Ther Adv Neurol Disord. 2021;14:175628642110654. doi:10.1177/17562864211065406
29. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. 2015;6. doi:10.3389/fimmu.2015.00262
30. Albazli K, Kaminski HJ, Howard JF. Complement inhibitor therapy for myasthenia gravis. Front Immunol. 2020;11:917. doi:10.3389/fimmu.2020.00917
31. Dalakas MC. Role of complement, anti-complement therapeutics, and other targeted immunotherapies in myasthenia gravis. Expert Rev Clin Immunol. 2022;18(7):691–701. doi:10.1080/1744666X.2022.2082946
32. Zhou S, Dong X, Chen C. Efficacy and safety of eculizumab for paroxysmal nocturnal hemoglobinuria: a systematic review and meta-analysis. J Pediatr Hematol Oncol. 2021;43(6):203–210. doi:10.1097/MPH.0000000000002178
33. Mahat U, Matar RB, Rotz SJ. Use of complement monoclonal antibody eculizumab in Shiga toxin producing Escherichia coli associated hemolytic uremic syndrome: a review of current evidence. Pediatr Blood Cancer. 2019;66(11):e27913. doi:10.1002/pbc.27913
34. Levy M, Fujihara K, Palace J. New therapies for neuromyelitis optica spectrum disorder. Lancet Neurol. 2021;20(1):60–67. doi:10.1016/S1474-4422(20)30392-6
35. Benamu E, Montoya JG. Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis. Curr Opin Infect Dis. 2016;29(4):319–329. doi:10.1097/QCO.0000000000000279
36. Howard JF, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled Phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48(1):76–84. doi:10.1002/mus.23839
37. Mantegazza R, Wolfe GI, Muppidi S, et al. Post-intervention status in patients with refractory myasthenia gravis treated with eculizumab during REGAIN and its open-label extension. Neurology. 2021;96(4):e610–e618. doi:10.1212/WNL.0000000000011207
38. Muppidi S, Utsugisawa K, Benatar M, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. 2019;60(1):14–24. doi:10.1002/mus.26447
39. Howard JF, Karam C, Yountz M, O’Brien FL, Mozaffar T; For the REGAIN Study Group. Long‐term efficacy of eculizumab in refractory generalized myasthenia gravis: responder analyses. Ann Clin Transl Neurol. 2021;8(7):1398–1407. doi:10.1002/acn3.51376
40. Vissing J, Jacob S, Fujita KP, et al. ‘Minimal symptom expression’ in patients with acetylcholine receptor antibody-positive refractory generalized myasthenia gravis treated with eculizumab. J Neurol. 2020;267(7):1991–2001. doi:10.1007/s00415-020-09770-y
41. Jacob S, Murai H, Utsugisawa K, et al. Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study. Ther Adv Neurol Disord. 2020;13:175628642091178. doi:10.1177/1756286420911784
42. Siddiqi ZA, Nowak RJ, Mozaffar T, et al. Eculizumab in refractory generalized myasthenia gravis previously treated with rituximab: subgroup analysis of
43. Usman U, Chrisman C, Houston D, Haws CC, Wang A, Muley S. The use of eculizumab in ventilator‐dependent myasthenia gravis patients. Muscle Nerve. 2021;64(2):212–215. doi:10.1002/mus.27326
44. Mantegazza R, Cavalcante P. Eculizumab for the treatment of myasthenia gravis. Expert Opin Biol Ther. 2020;20(9):991–998. doi:10.1080/14712598.2020.1786530
45. Tice JA, Touchette DR, Lien PW, Agboola F, Nikitin D, Pearson SD. The effectiveness and value of eculizumab and efgartigimod for generalized myasthenia gravis: a summary from the institute for clinical and economic review’s New England comparative effectiveness public advisory council. JMCP. 2022;28(1):119–124. doi:10.18553/jmcp.2022.28.1.119
46. McKeage K. Ravulizumab: first global approval. Drugs. 2019;79(3):347–352. doi:10.1007/s40265-019-01068-2
47. Vu T, Meisel A, Mantegazza R, et al. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evidence. 2022;1(5):EVIDoa2100066. doi:10.1056/EVIDoa2100066
48. Howard JF, Vissing J, Gilhus NE, et al. Zilucoplan: an investigational complement C5 inhibitor for the treatment of acetylcholine receptor autoantibody–positive generalized myasthenia gravis. Expert Opin Investig Drugs. 2021;30(5):483–493. doi:10.1080/13543784.2021.1897567
49. Howard JF, Nowak RJ, Wolfe GI, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 2020;77(5):582–592. doi:10.1001/jamaneurol.2019.5125
50. Weyne J, Ni Y, DelGizzi R, et al. A randomized, double-blind, placebo-controlled phase 1 study of the pharmacokinetics and pharmacodynamics of REGN3918, a human antibody against complement factor C5, in healthy volunteers. Blood. 2018;132(Supplement1):1039. doi:10.1182/blood-2018-99-112262
51. Devalaraja-Narashimha K, Huang C, Cao M, et al. Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination with cemdisiran in non-human primates. PLoS One. 2022;17(6):e0269749. doi:10.1371/journal.pone.0269749
52. Lünemann JD. Getting specific: targeting Fc receptors in myasthenia gravis. Nat Rev Neurol. 2021;17(10):597–598. doi:10.1038/s41582-021-00547-z
53. Ward ES, Gelinas D, Dreesen E, et al. Clinical significance of serum albumin and implications of FcRn inhibitor treatment in IgG-mediated autoimmune disorders. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.892534
54. Wolfe GI, Ward ES, de Haard H, et al. IgG regulation through FcRn blocking: a novel mechanism for the treatment of myasthenia gravis. J Neurol Sci. 2021;430:118074. doi:10.1016/j.jns.2021.118074
55. Ulrichts P, Guglietta A, Dreier T, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018;128(10):4372–4386. doi:10.1172/JCI97911
56. Ling LE, Hillson JL, Tiessen RG, et al. M281, an Anti‐FcRn antibody: pharmacodynamics, pharmacokinetics, and safety across the full range of IgG reduction in a first‐in‐human study. Clin Pharmacol Ther. 2019;105(4):1031–1039. doi:10.1002/cpt.1276
57. Howard JF, Bril V, Burns TM, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. 2019;92(23):e2661–e2673. doi:10.1212/WNL.0000000000007600
58. Howard JF, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526–536. doi:10.1016/S1474-4422(21)00159-9
59. Guptill J, Antozzi C, Bril V, et al. Vivacity-MG: a phase 2, multicenter, randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, efficacy, pharmacokinetics, pharmacodynamics, and immunogenicity of nipocalimab administered to adults with generalized myasthenia gravis Neurology ; 2021;96 (15 Supplement):2157.
60. Bril V, Benatar M, Andersen H, et al. Efficacy and safety of rozanolixizumab in moderate-to-severe generalised myasthenia gravis: a phase 2 RCT. Neurology. 2021;96(6):e853e865. doi:;96(6):e853e865. doi:10.1212/WNL.0000000000011108
61. Stathopoulos P, Kumar A, Heiden JAV, Pascual-Goñi E, Nowak RJ, O’Connor KC. Mechanisms underlying B cell immune dysregulation and autoantibody production in MuSK myasthenia gravis: B cell abnormalities in MuSK myasthenia gravis. Ann NY Acad Sci. 2018;1412(1):154–165. doi:10.1111/nyas.13535
62. Yi JS, Guptill JT, Stathopoulos P, Nowak RJ, O’Connor KC. B cells in the pathophysiology of myasthenia gravis:. Muscle Nerve. 2018;57(2):172–184. doi:10.1002/mus.25973
63. Behin A, Le Panse R. New pathways and therapeutic targets in autoimmune myasthenia gravis. JND. 2018;5(3):265–277. doi:10.3233/JND-170294
64. Fichtner ML, Jiang R, Bourke A, Nowak RJ, O’Connor KC. Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front Immunol. 2020;11:776. doi:10.3389/fimmu.2020.00776
65. Zografou C, Vakrakou AG, Stathopoulos P. Short- and long-lived autoantibody-secreting cells in autoimmune neurological disorders. Front Immunol. 2021;12:686466. doi:10.3389/fimmu.2021.686466
66. Stathopoulos P, Dalakas MC. Evolution of anti-B cell therapeutics in autoimmune neurological diseases. Neurotherapeutics. 2022;19(3):691–710. doi:10.1007/s13311-022-01196-w
67. Nowak RJ, Coffey CS, Goldstein JM, et al. Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: the BeatMG study. Neurology. 2022;98(4):e376–e389. doi:10.1212/WNL.0000000000013121
68. Iorio R, Damato V, Alboini P, Evoli A. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol. 2014;262. doi:10.1007/s00415-014-7532-3
69. Tandan R, Hehir MK, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve. 2017;56(2):185–196. doi:10.1002/mus.25597
70. Di Stefano V, Lupica A, Rispoli MG, Di Muzio A, Brighina F, Rodolico C. Rituximab in AChR subtype of myasthenia gravis: systematic review. J Neurol Neurosurg Psychiatry. 2020;91(4):392–395. doi:10.1136/jnnp-2019-322606
71. Li T, Zhang GQ, Li Y, et al. Efficacy and safety of different dosages of rituximab for refractory generalized AChR myasthenia gravis: a meta-analysis. J Clin Neurosci. 2021;85:6–12. doi:10.1016/j.jocn.2020.11.043
72. Zhao C, Pu M, Chen D, et al. Effectiveness and safety of rituximab for refractory myasthenia gravis: a systematic review and single-arm meta-analysis. Front Neurol. 2021;12:736190. doi:10.3389/fneur.2021.736190
73. Anderson D, Phan C, Johnston WS, Siddiqi ZA. Rituximab in refractory myasthenia gravis: a prospective, open‐label study with long‐term follow‐up. Ann Clin Transl Neurol. 2016;3(7):552–555. doi:10.1002/acn3.314
74. Hehir MK, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology. 2017;89(10):1069–1077. doi:10.1212/WNL.0000000000004341
75. Jing S, Song Y, Song J, et al. Responsiveness to low-dose rituximab in refractory generalized myasthenia gravis. J Neuroimmunol. 2017;311:14–21. doi:10.1016/j.jneuroim.2017.05.021
76. Beecher G, Anderson D, Siddiqi ZA. Rituximab in refractory myasthenia gravis: extended prospective study results: rituximab in Refractory MG. Muscle Nerve. 2018;58(3):452–455. doi:10.1002/mus.26156
77. Landon-Cardinal O, Friedman D, Guiguet M, et al. Efficacy of rituximab in refractory generalized anti-achr myasthenia gravis. J Neuromuscul Dis. 2018;5(2):241–249. doi:10.3233/JND-180300
78. Du Y, Li C, Hao Y, et al. Individualized regimen of low-dose rituximab monotherapy for new-onset AChR-positive generalized myasthenia gravis. J Neurol. 2022;269(8):4229–4240. doi:10.1007/s00415-022-11048-4
79. Brauner S, Eriksson-Dufva A, Hietala MA, Frisell T, Press R, Piehl F. Comparison between rituximab treatment for new-onset generalized myasthenia gravis and refractory generalized myasthenia gravis. JAMA Neurol. 2020;77(8):974. doi:10.1001/jamaneurol.2020.0851
80. Li H, Huang Z, Jia D, et al. Low-dose rituximab treatment for new-onset generalized myasthenia gravis. J Neuroimmunol. 2021;354:577528. doi:10.1016/j.jneuroim.2021.577528
81. Molimard A, Gitiaux C, Barnerias C, et al. Rituximab therapy in the treatment of juvenile Myasthenia gravis: the French experience. Neurology. 2022;98(23):e2368–e2376. doi:10.1212/WNL.0000000000200288
82. Zingariello CD, Elder ME, Kang PB. Rituximab as adjunct maintenance therapy for refractory juvenile myasthenia gravis. Pediatr Neurol. 2020;111:40–43. doi:10.1016/j.pediatrneurol.2020.07.002
83. Ramdas S, Della Marina A, Ryan MM, et al. Rituximab in juvenile myasthenia gravis-an international cohort study and literature review. Eur J Paediatr Neurol. 2022;40:5–10. doi:10.1016/j.ejpn.2022.06.009
84. Caballero-ávila M, Álvarez-Velasco R, Moga E, et al. Rituximab in myasthenia gravis: efficacy, associated infections and risk of induced hypogammaglobulinemia. Neuromuscular Disorders. 2022;32(8):664–671. doi:10.1016/j.nmd.2022.06.006
85. Afanasiev V, Demeret S, Bolgert F, Eymard B, Laforêt P, Benveniste O. Resistant myasthenia gravis and rituximab: a monocentric retrospective study of 28 patients. Neuromuscular Disorders. 2017;27(3):251–258. doi:10.1016/j.nmd.2016.12.004
86. Chen D, Gallagher S, Monson NL, Herbst R, Wang Y. Inebilizumab, a B cell-depleting anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med. 2016;5(12):107. doi:10.3390/jcm5120107
87. Frampton JE. Inebilizumab: first approval. Drugs. 2020;80(12):1259–1264. doi:10.1007/s40265-020-01370-4
88. Mao Y. Structure, dynamics and function of the 26s proteasome. In: Harris JR, Marles-Wright J, editors. Macromolecular Protein Complexes III: Structure and Function. Subcellular Biochemistry. Springer International Publishing; 2021:1–151. doi:10.1007/978-3-030-58971-4_1
89. Kohler S, Märschenz S, Grittner U, Alexander T, Hiepe F, Meisel A. Bortezomib in antibody-mediated autoimmune diseases (TAVAB): study protocol for a unicentric, non-randomised, non-placebo controlled trial. BMJ Open. 2019;9(1):e024523. doi:10.1136/bmjopen-2018-024523
90. Gomez AM, Willcox N, Vrolix K, et al. Proteasome inhibition with bortezomib depletes plasma cells and specific autoantibody production in primary thymic cell cultures from early-onset myasthenia gravis patients. J Immunol. 2014;193(3):1055–1063. doi:10.4049/jimmunol.1301555
91. Schneider-Gold C, Reinacher-Schick A, Ellrichmann G, Gold R. Bortezomib in severe MuSK-antibody positive myasthenia gravis: first clinical experience. Ther Adv Neurol Disord. 2017;10(10):339–341. doi:10.1177/1756285617721093
92. Klimas R, Sgodzai M, Motte J, et al. Dose-dependent immunomodulatory effects of bortezomib in experimental autoimmune neuritis. Brain Communications. 2021;3(4):fcab238. doi:10.1093/braincomms/fcab238
93. Robak E, Robak T. Bruton’s kinase inhibitors for the treatment of immunological diseases: current status and perspectives. J Clin Med. 2022;11(10):2807. doi:10.3390/jcm11102807
94. Delves PJ. The immune system. Adv Immunol. 2000;34:37.
95. Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. 2002;2(7):465–475. doi:10.1038/nri844
96. Ragheb S, Lisak RP. B-cell-activating factor and autoimmune myasthenia gravis. Autoimmune Dis. 2011;2011:1–10. doi:10.4061/2011/939520
97. Hewett K, Sanders DB, Grove RA, et al. Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology. 2018;90(16):e1425–e1434. doi:10.1212/WNL.0000000000005323
98. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229(1):152–172. doi:10.1111/j.1600-065X.2009.00782.x
99. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(2):S3. doi:10.1186/ar1917
100. Uzawa A, Kuwabara S, Suzuki S, et al. Roles of cytokines and T cells in the pathogenesis of myasthenia gravis. Clin Exp Immunol. 2021;203(3):366–374. doi:10.1111/cei.13546
101. Aricha R, Mizrachi K, Fuchs S, Souroujon MC. Blocking of IL-6 suppresses experimental autoimmune myasthenia gravis. J Autoimmun. 2011;36(2):135–141. doi:10.1016/j.jaut.2010.12.001
102. Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114–2124. doi:10.1056/NEJMoa1901747
103. Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19(5):402–412. doi:10.1016/S1474-4422(20)30078-8
104. Jonsson DI, Pirskanen R, Piehl F. Beneficial effect of tocilizumab in myasthenia gravis refractory to rituximab. Neuromuscular Disorders. 2017;27(6):565–568. doi:10.1016/j.nmd.2017.03.007
105. Abolhassani H, Sadaghiani MS, Aghamohammadi A, Ochs HD, Rezaei N. Home-based subcutaneous immunoglobulin versus hospital-based intravenous immunoglobulin in treatment of primary antibody deficiencies: systematic review and meta analysis. J Clin Immunol. 2012;32(6):1180–1192. doi:10.1007/s10875-012-9720-1
106. Hadden RDM, Marreno F. Switch from intravenous to subcutaneous immunoglobulin in CIDP and MMN: improved tolerability and patient satisfaction. Ther Adv Neurol Disord. 2015;8(1):14–19. doi:10.1177/1756285614563056
107. Beecher G, Anderson D, Siddiqi ZA. Subcutaneous immunoglobulin in myasthenia gravis exacerbation: a prospective, open-label trial. Neurology. 2017;89(11):1135–1141. doi:10.1212/WNL.0000000000004365
108. Barth D, Nouri MN, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. 2011;76(23):2017–2023. doi:10.1212/WNL.0b013e31821e5505
109. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. doi:10.1056/NEJMra1706169
110. Aghajanian H, Rurik JG, Epstein JA. CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nat Metab. 2022;4(2):163–169. doi:10.1038/s42255-022-00537-5
111. Bryant A, Atkins H, Pringle CE, et al. Myasthenia gravis treated with autologous hematopoietic stem cell transplantation. JAMA Neurol. 2016;73(6):652. doi:10.1001/jamaneurol.2016.0113
112. Sharrack B, Saccardi R, Alexander T, et al. Autologous haematopoietic stem cell transplantation and other cellular therapy in multiple sclerosis and immune-mediated neurological diseases: updated guidelines and recommendations from the EBMT Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of EBMT and ISCT (JACIE). Bone Marrow Transplant. 2020;55(2):283–306. doi:10.1038/s41409-019-0684-0
113. Das J, Sharrack B, Snowden JA. Autologous hematopoietic stem-cell transplantation in neurological disorders: current approach and future directions. Expert Rev Neurother. 2020;20(12):1299–1313. doi:10.1080/14737175.2020.1820325
114. Tsutsumi Y, Kamiishi T, Kikuchi R, Ito S, Matsuoka S, Teshima T. Myasthenia gravis after allogeneic bone marrow transplantation: a case report and literature review. Hematol Oncol Stem Cell Ther. 2019;12(2):110–114. doi:10.1016/j.hemonc.2017.04.001
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.