Back to Journals » The Application of Clinical Genetics » Volume 15
Novel Compound Heterozygous Variants in the CDC6 Gene in a Russian Patient with Meier-Gorlin Syndrome
Authors Zabnenkova V ![]() , Shchagina O
, Shchagina O ![]() , Makienko O
, Makienko O ![]() , Matyushchenko G, Ryzhkova O
, Matyushchenko G, Ryzhkova O ![]()
Received 12 October 2021
Accepted for publication 22 December 2021
Published 6 January 2022 Volume 2022:15 Pages 1—10
DOI https://doi.org/10.2147/TACG.S342804
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Viktoriia Zabnenkova,1 Olga Shchagina,1 Olga Makienko,2 Galina Matyushchenko,2 Oxana Ryzhkova1
1Molecular Genetics Laboratory № 3 The Shared Resource Centre “Genome”, Federal State Budgetary Scientific Institution Research Centre for Medical Genetics named after Academician N.P. Bochkov, Moscow, Russian Federation; 2Counselling Unit, Federal State Budgetary Scientific Institution Research Centre for Medical Genetics named after Academician N.P. Bochkov, Moscow, Russian Federation
Correspondence: Viktoriia Zabnenkova
Molecular Genetics Laboratory № 3 The Shared Resource Centre “Genome”, Federal State Budgetary Scientific Institution Research Centre for Medical Genetics named after Academician N.P. Bochkov, ul. Moskvorechye, 1, Moscow, 115522, Russian Federation
Tel +7 903 200 92 85
Fax +7 499 324 81 10
Email [email protected]
Background: Meier-Gorlin syndrome (MGS) is a rare genetic syndrome inherited in an autosomal dominant or autosomal recessive manner. The disorder is characterized by bilateral microtia, absence or hypoplasia of the patella, and an intrauterine growth retardation as well as a number of other characteristic features. The cause of the disease is mutations in genes encoding proteins involved in the regulation of the cell cycle (ORC1, ORC4, ORC6, CDT1, CDC6, GMNN, CDC45L, MCM3, MCM5, MCM7, GINS2, and DONSON). Meier-Gorlin syndrome 5 due to mutations in the CDC6 gene is difficult to diagnose, and few clinical data have been described to date. Only one patient (male) with a missense mutation in a homozygous state has been previously reported. This report describes a new clinical case of Meier-Gorlin syndrome 5. This is also the first report of a Russian patient with Meier-Gorlin syndrome.
Case Presentation: The patient, a female, had extremely low physical development, neonatal progeroid appearance, lipodystrophy, thin skin, partial alopecia, cyanosis of the face, triangular face, microgenia, arachnodactyly, delayed bone age, hepatomegaly, hypoplasia of the labia majora, and hypertrophy of the clitoris in addition to known clinical signs. Differential diagnosis was performed with chromosomal abnormalities and Hutchinson-Gilford progeria. According to the results of sequencing of the clinical exome, the patient had two previously undescribed variants in the CDC6 gene, c.230A>G (p.(Lys77Arg)) and c.232C>T (p.(Gln78Ter)), NM_001254.3, in a compound heterozygous state.
Conclusion: This case allows us to learn more about the clinical features and nature of MGS 5 and improve the speed of diagnostics and quality of genetic counseling for such families.
Keywords: MGS 5, ear-patella-short stature syndrome, CDC6 gene, case report
Introduction
Meier-Gorlin syndrome (MGS) or ear-patella-short stature syndrome is a rare disorder characterized by a diagnostic triad: bilateral microtia, aplasia, or hypoplasia of the patella and severe pre- and postnatal growth retardation. However, two signs are sufficient to suggest that the patient has Meier-Gorlin syndrome. In addition, one clinical case was described in which, of the triad, the patient had only short stature.1
In addition to the triad, the disorder has a wide spectrum of clinical variability complicating the diagnosis of Meier-Gorlin syndrome.
Immediately after birth, not only are marked deficiencies in height and weight noticed but also frequent respiratory problems requiring respiratory support. Some patients have pulmonary emphysema. External signs include microcephaly and facial dysmorphias: triangular shape of the face, micrognathia, and microstomia. X-rays show significant bone growth retardation. The chest is hypoplastic. The skin is thin, and the vasculature (mainly the head) is pronounced. Genital involvement may include cryptorchidism in boys. In girls - clitoris hypertrophy and hypoplasia of the labia majora or minora. Three cases of cardiovascular involvement have been described.1,2 Intelligence is usually preserved, but motor and/or language development is delayed.1,3–6
Meier-Gorlin syndrome is a genetically heterogeneous disease. There are 12 known genes (ORC1, ORC4, ORC6, CDT1, CDC6, GMNN, CDC45L, MCM3, MCM5, MCM7, GINS2, and DONSON) that are responsible for the development of the disease.5,7–9 (Supplementary Table 1)MGS can be inherited in either a dominant or recessive manner. The causative genes encode proteins involved in regulation of DNA replication during the cell cycle. Thus, the syndrome is thought to be based on impaired replication mechanisms, resulting in a specific patient phenotype.6
During DNA replication in eukaryotes, multiple replication origins are first recognized and bound in the early G1 phase of the cell cycle by the origin recognition complex (ORC), composed of ORC1-6 proteins. The CDC6, CDT1, and complex MCM2-7 helicase proteins then sequentially complete each ORC-linked replication initiation site to form a prereplication complex, which allows DNA replication to begin. During the S phase, other factors are involved in the prereplication complexes to initiate DNA synthesis.10
CDC6 protein belongs to the AAA+ ATPase family and is necessary to form a prereplication complex, helping to load MCM proteins onto replication origins. It also regulates control points that ensure completion of DNA replication prior to mitosis. CDC6 may also activate CDK2 to promote the progression of phase S and the phase transition from G1 to S.11–16
The only one clinical case of MGS due the CDC6 gene mutations had been described previously.6 Herein, we report a Meier-Gorlin syndrome Russian patient with two novel compound-heterozygous variants in the CDC6 gene.
Materials and Methods
Clinical data was obtained during consultation of the patient with geneticists of Federal State Budgetary Scientific Institution Research Center for Medical Genetics.
The patient’s and parental DNA was isolated from peripheral blood using the Qiagen reagent kit according to the manufacturer’s protocol.
The patient’s DNA analysis was performed on a next generation Illumina NextSeq 500 sequencer using SeqCap EZ HyperCap Workflow reagents. The HGVS nomenclature was applied to the name of the variants identified (http://varnomen.hgvs.org/recommendations/DNA).17 The sequencing data was processed using the standard automated algorithm offered by Illumina for data analysis presented at https://basespace.illumina.com.18 Sample projects of “1000 genomes”, ESP6500, gnomAD (v.2.1.1),19 and RuExac (the database of variants obtained by exome sequencing (WES, CES) in the Research Center for Medical Genetics) were used to estimate the population frequencies of the identified variants.
Exome sequencing data verification was performed by Sanger automatic sequencing on the Applied Biosystems 3500 xL genetic analyzer. The reference sequence of the CDC6 gene used in the work is available in the RefSeq database:20 CDC6 NM_001254.3.
Case Presentation
The proband, female of 1 year and 3 months old, was referred to the Research Centre for Medical Genetics because of severe failure to thrive. She was born from a first pregnancy. Parents were nonconsanguineous and had normal growth. The threat of miscarriage took place at 27 weeks. Delivery was emergent at 38 weeks and 5 days by cesarean section. The condition at birth was severe. Respiratory failure was increasing. The child was on a ventilator for five days. In total, respiratory support was required for five months. The patient was diagnosed with pulmonary hypertension.
Body weight at birth was 1490 g (Z-score −4.50), 40 cm in length (Z-score −4.54). Head circumference was 32.5 cm (Z-score −2.59). Chest circumference was 24 cm. She had hydrocephalic head, short palpebral fissures, ocular and nipple hypertelorism, depressed nasal bridge, epicanthus, microstomy, high-arched palate, hypoplastic low-set ears, short neck, low hair line, and arachnodactyly. The baby had a pronounced progeroid appearance. The subcutaneous fat layer was not pronounced. An external examination of the genitals revealed hypoplasia of the labia majora, and the clitoris was hypertrophied. The liver protruded 3 cm below the edge of the right costal arch and 1 cm below the edge of the left costal arch. Motor activity was reduced.
Upon screening at 9 months, the patient’s condition was severe, but her consciousness was clear, and her posture as active. The baby was responsive to communication. She had a body weight of 3.3 kg (Z-score −6.54), a height of 54 cm (Z-score −6.61), and a body mass index of 11.32 (Z-score −4.67). Her head circumference was 43.5 cm (Z-score −0.76), and her chest circumference was 30 cm. The patient’s physical development was extremely low, disharmonic. Pseudohydrocephalus, high forehead, triangular face, exophthalmos, narrow nasal dorsum, smooth philtrum, microgenia, microtia, and arachnodactyly were observed in the child during the examination. The skin was clean and dry. The girl had cyanosis of the face and upper half of the trunk when crying. The subcutaneous fat was underdeveloped. The patient’s chest was barrel-shaped. Hepatomegaly was observed. The child could not support her head but tried to turn her body from prone to supine. She had a pronounced interest in toys. Her speech consisted of babbling, with chains of syllables. X-ray showed delay in bone age – less than three months vs the patient’s actual age of 9 months. The bone structure was sparse. Valgus deformity of the knee joints was observed. Data on the condition of the patella were not provided because these bones are transparent on X-ray scans in young children, and ultrasonography was not performed. EEG recordings contained no epileptiform or focal pathological activity. Echocardiography showed no signs of heart defects. Patent foramen ovale and diagonal trabecula in the left ventricular cavity were observed.
Another examination was performed at 1 year and 3 months was performed. The girl had a height of 58 cm (Z-score −7.06), a weight of 4 kg (Z-score −6.44) and a head circumference of 45 cm (Z-score −0.5). Hydrocephalic head, high forehead, triangular face, exophthalmos, narrow nasal dorsum, microgenia, microtia, severe hypotrophy, and areas of alopecia on the head were observed. Subcutaneous fat was not developed. The baby did not sit independently and could not steadily support herself on her feet, but she was able to stand with support. She played with toys, held objects in her hands, collected pyramids, and transferred small objects.
The last time the patient was examined when she was 1 year 11 months old. Short stature (63 cm, Z-score −7.04) and growth retardation (5 kg, Z-score −6.16) were noted. Clinical examination revealed dysmorphic facial features, microtia. Absent patella was noted by palpation. (Figure 1).
 |
Figure 1 A girl with Meier-Gorlin syndrome: (A) – Failure to thrive, high forehead, short palpebral fissures, micrognathia; (B) - Bilateral microtia; (C) - Knees with absent patellae. |
Initially, chromosomal abnormality was suspected in the child. Cytogenetic testing was performed. Karyotype 46, XX was detected. Hutchinson-Gilford progeria was assumed. A corresponding study of the LMNA gene was performed. No pathogenic changes were observed. Clinical exome sequencing was ordered, identifying two variants in the CDC6 gene. (Supplementary Table 2; Figure 2A) Parental examination revealed that they were heterozygous carriers of relevant changes in the nucleotide sequence.
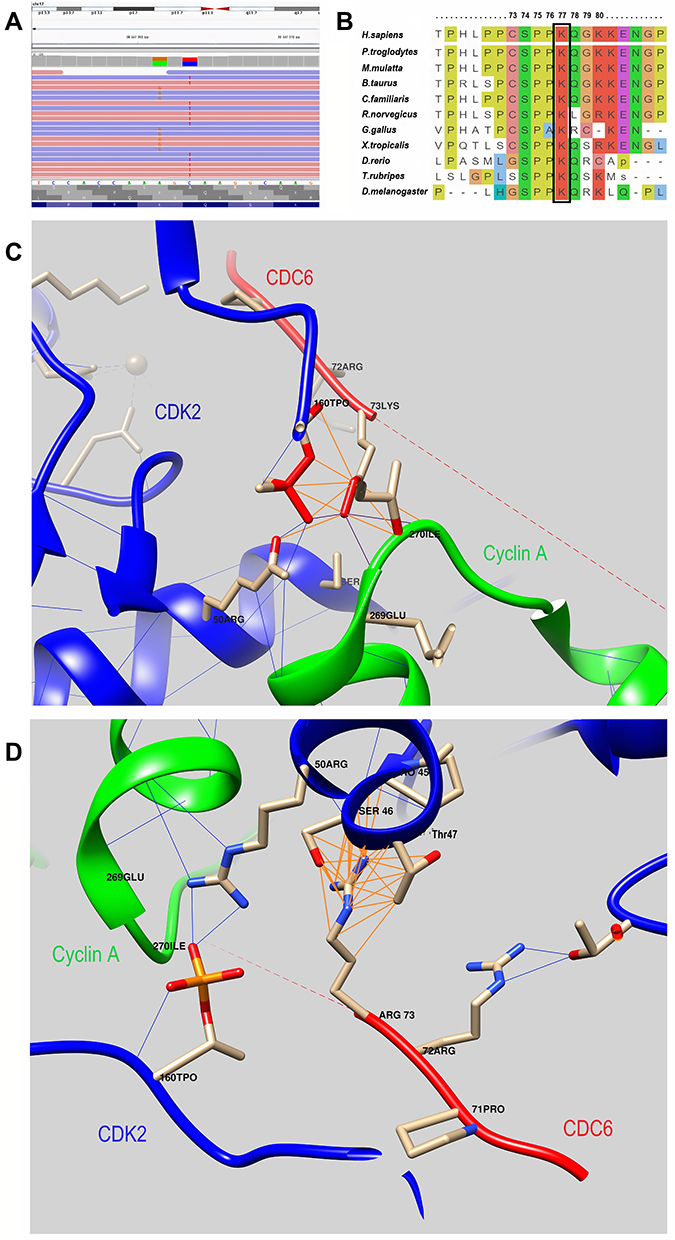 |
Figure 2 (A) - Molecular genetic results: the visualization of variants - c.230A>G, p.(Lys77Arg) and c.232C>T, p.(Gln78Ter) - with the Integrative Genomics Viewer (IGV). (B) - An amino acid sequence of the CDC6 gene in the region of the identified variants in different species. Lys77 is highly conserved across species. It is highlighted in the frame. (C) - 3D model of the interaction of the CDC6 protein with the CDK2/Cyclin A complex (PDB ID 2CCI): wild-type CDC6 interaction. (D) – 3D model of the interaction of the CDC6 protein with the CDK2/Cyclin A complex (PDB ID 2CCI): mutant protein CDC6 interaction. Color code: green chain – Cyclin A, red chain – CDC6, blue chain – CDK2; blue lines – hydrogen bonds, dark Orange lines – contacts. |
Neither variant had been registered in either the GnomAD database (v.2.1.1),19 the clinical databases (Clinvar,21 HGMD Professional (v.2021.1)22), or the database of variants obtained by exome sequencing (WES, CES) in the Research Center for Medical Genetics – RuExac (1337 samples). The exon 3 nucleotide sequence variant of the CDC6 gene (chr17:38447363C>T) results in a premature translation termination site (p.(Gln78Ter), NM_001254.3). According to the pathogenicity criteria, ACMG23 is classified as pathogenic (PVS1, PS3, PM2). The criterium PS3 was used due to the previously functional study24 that demonstrated pathogenicity of LoF variants in the CDC6 gene. A variant in exon 3 of the CDC6 gene (chr17:38447361A>G), resulting in a missense replacement of (p.(Lys77Arg), NM_001254.3), is classified as a variant with uncertain clinical significance (ACMG: PM2, PM3).
Discussion
A patient with Meier-Gorlin syndrome 5 with a p.Thr323Arg homozygous missense mutation (c.968C>G, NM_001254.3) in the CDC6 gene has been previously reported.6 This variant is localized in the functional ATP binding domain of the CDC6 protein. The biological model shows its pathogenicity.24
Our patient’s variants are located in the adjacent codons of the CDC6 gene, outside the functional domain. Since one of the variants is the one terminating translation, p.(Gln78Ter), the presence of functional homozygosity for missense replacement of p.(Lys77Arg) could be hypothesized. Pathogenicity prediction programs provide a contradictory assessment of the variant c.232C>T (p.(Lys77Arg)). The polar amino acid Lys77 is conservative (Figure 2B). It is replaced with more polar amino acid Arg77. DynaMut225 predicted the change in stability (ΔΔGStability) of −0.353 kcal/mol. Lys77 is located at the +3 position of the canonical phosphorylation site for the CDK2/Cyclin A complex: tPHLPPCs(74)PPK(77)QGKK.24,26–28 Cytoplasmic translocation is sufficient to control CDC6 protein activity after initiation during the S phase; this event also prevents repeated replication in eukaryotic cells. The process is phosphorylation-dependent. Phosphorylation of cyclin-dependent kinase 2 at three serine residues, Ser54, Ser74, and Ser106, is thought to be responsible for the cytoplasmic translocation process in the CDC6 protein.
It was determined that Ser74 phosphorylation provides the primary contribution to the cytoplasmic translocation of the CDC6 protein, whereas Ser54 phosphorylation provides a minor contribution, and Ser106 phosphorylation does not affect CDC6 nuclear export.29
Phosphorylation of CDC6 by CDK2/Cyclin A requires sequential build-up of the CDK2/Cyclin A-CDC6 complex: a) binding of Cyclin A to CDK2, b) phosphorylation of CDK2 on Thr160, c) phosphorylation of CDC6 by the CDK2/Cyclin A complex, phosphorylated on CDK2.30
The role of Lys+3 in substrate binding is consistent with the crystal structure of CDK2-cyclin A, which shows hydrogen bonding between the enzyme phosphoryl group at Thr160 and the side chain of Lys+3 in a peptide substrate. CDK2-cyclin A has a well-defined consensus target site ((S/T)PX(K/R)) that prioritizes substrates containing a lysine at the +3 position. Substitutions of the basic residue at the +3 position had the greatest effects on phosphorylation efficiencies.31
Cheng et al evaluated the substrate specificity of [TPO160]CDK2/Cyclin A at the +3 position using a panel of GST-KSPRX substrates: KSPRK, KSPRR and KSPRY. KSPRK was considered the “wild-type”. It was shown that at the +3 position the best substrate for CDK2/Cyclin A is KSPRK. Comparison of the relative phosphorylation efficiencies of the indicated substrates by [TPO160]CDK2/Cyclin A demonstrated that Lys+3 exceeds Arg+3 in 12 times in this parameter.26
Also, the visualization of missense replacement of p. (Lys77Arg) using DynaMut2 and UCSF Chimera32 tools (PDB ID 2CCI33) shows the presence of spatial change and a change of the bonds that are formed by both the replaced and the replacing amino acid. (Figure 2C and D) Thus, in the native protein Lys77 (in the 2CCI model, Lys73) interacts with both phosphothreonine TPO160 (hydrogen bonds) and Arg50 in the CDK2 molecule and with Glu269 and Ile270 in the Cyclin A molecule. In the mutant protein Arg77 (for the 2CCI model, Arg73) forms additional bonds with the CDK2 molecule (Ser46 and Thr47), while there is no interaction with the Cyclin A molecule and no hydrogen bonds with TPO160.
Thus, replacing lysine with arginine at position 77 results in a protein with reduced function. Compound heterozygous LoF mutations are not described for Meier-Gorlin syndrome genes, it is thought to indicate the lethality of these genotypes. Specifically, studies in D. rerio genetic models have been conducted for the CDC6 gene. Fish with LoF variants died during the embryonic stage, while hypomorphic homozygous animals developed during embryogenesis but later had marked developmental delays, with a smaller weight and length in adulthood compared to wild-type animals.24 Hypomorphic mutations in combination with the LoF variant result in a more severe phenotype than true homozygosity.34
These data may suggest the pathogenicity of the genotype identified in the patient.
When comparing the average clinical data from different types of Meier-Gorlin syndrome (Table 1), we noted extremely low physical development in our patient—absence of subcutaneous fat, thin skin, and partial alopecia. There was also a pronounced delay in her motor development. The presence of hepatomegaly, arachnodactyly, and exophthalmos was not previously mentioned in patients with MGS. Furthermore, due to the severity of the clinical course, the minor abnormalities detected in heart development may be important. In addition to these phenotypic features, our patient’s lack of a cleft palate sets her apart from the first reported patient with Meier-Gorlin syndrome 5. It is interesting to note the phenotypic similarity of our patient in the neonatal period with the patient described earlier with Meyer-Gorlin syndrome, caused by mutations in the MCM7 gene. They both had a progeroid appearance and lipodystrophy.7
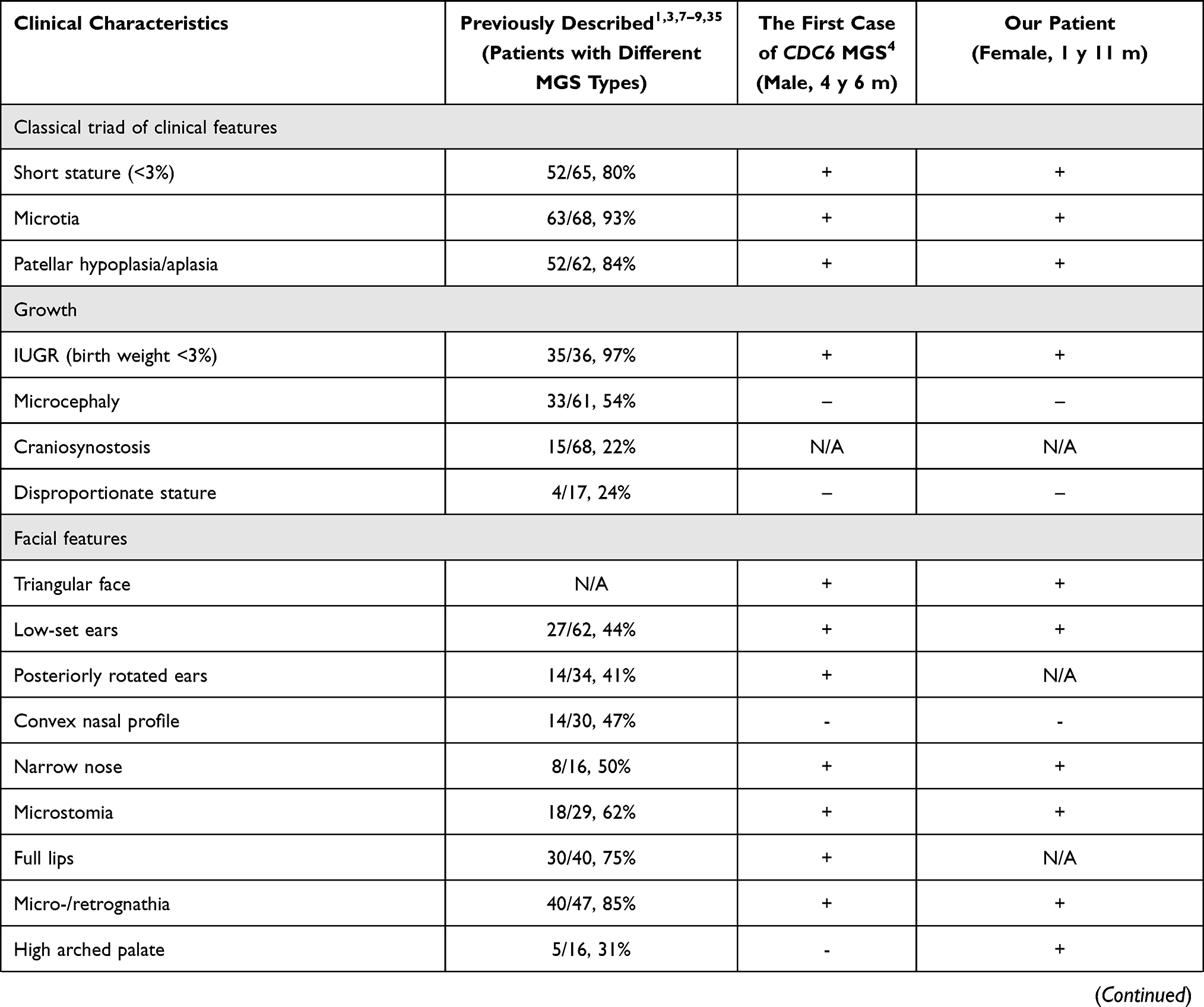 | 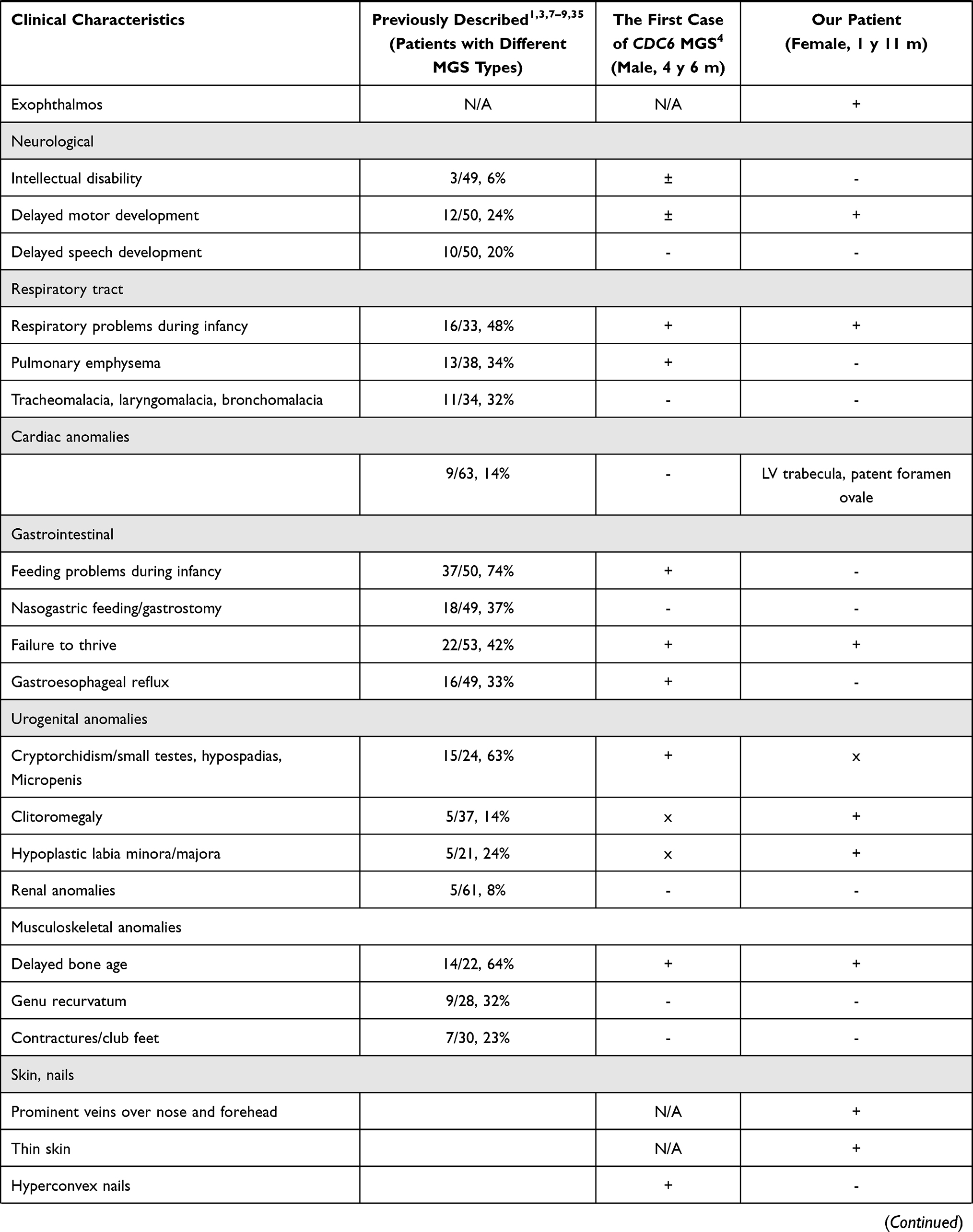 | 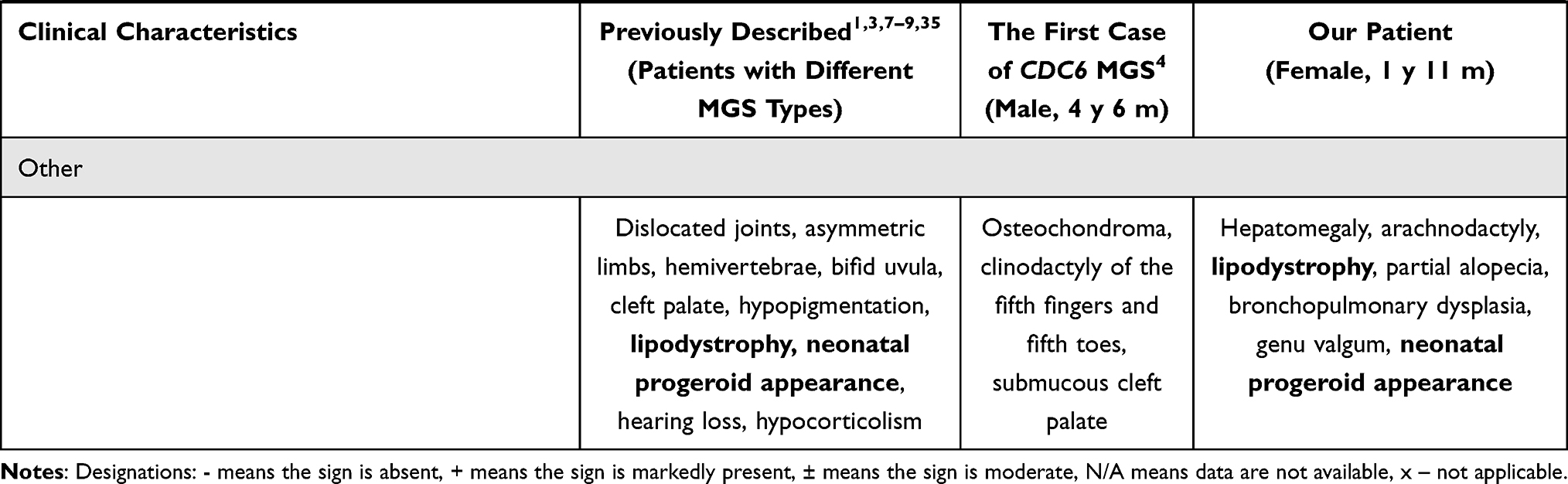 |
Table 1 Clinical Characteristics of Patients with MGS |
Conclusion
For the first time we report a Russian patient with severe form of Meier-Gorlin syndrome, including intrauterine and postnatal growth delay, absent patellae, microtia, neonatal progeroid appearance, lipodystrophy. The girl has two novel compound heterozygous LoF and missense variants in the CDC6 gene. Phenotypic variations within Meier-Gorlin syndrome 5 were identified. In this case, the use of exome sequencing was an integral part of the diagnosis given the nonspecific nature of the manifestation and underestimation of this rare genetic syndrome. This highlights the importance of CES/WES in shortening the diagnostic odyssey in patients with rare genetic disorders.
Abbreviations
AAA+ ATPase family, ATPases associated with various cellular activities; ACMG, The American College of Medical Genetics and Genomics; CDC45L, Cell division cycle 45-like; CDC6, Cell division cycle 6; CDK2, Cyclin-dependent kinase 2; CDT1, Chromatin licensing and DNA replication factor 1; CES, Clinical exome sequencing; EEG, Electroencephalography; GMNN, Geminin DNA replication inhibitor; LMNA, Lamin A/C; LoF, Loss of function; MCM2-7, Minichromosome maintenance 2–7; MGS, Meier-Gorlin syndrome; PM2, PM3, Pathogenic moderate 2 and 3, respectively; PVS1, Pathogenic very strong 1; WES, Whole-exome sequencing.
Data Sharing Statement
All datasets generated or analyzed during the current study are included in this article and are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The Ethics Committee of the Federal State Budgetary Scientific Institution Research Center for Medical Genetics approved this study. All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Written consent was obtained from the parents of the proband for the collection of samples and for DNA sequencing.
Consent for Publication
Written informed consent was obtained from the patient’s parents for publication of this case report. A copy of the written consent is available for review by the Editor of this journal.
Acknowledgments
We would like to thank the specialists at the Department of Early Childhood (The Research and Clinical Institute for Pediatrics named after academician Yuri Veltischev of the Pirogov Russian National Research Medical University of the Russian Ministry of Health) and the Department of Pathology of Newborns and Premature Babies (The Budgetary Healthcare Institute of the Vologda Region, “Vologda Regional Children’s Hospital No. 2”), and geneticist of Research Centre for Medical Genetics Bessonova L.A. for providing the data from the instrumental examination of the patient.
Funding
No funds, grants, or other forms of support were received.
Disclosure
The authors declare that they have no competing interests.
References
1. de Munnik SA, Bicknell LS, Aftimos S, et al. Meier-Gorlin syndrome genotype-phenotype studies: 35 individuals with pre-replication complex gene mutations and 10 without molecular diagnosis. Eur J Human Genet. 2012;20(6):598–606. doi:10.1038/ejhg.2011.269
2. Shawky RM, Elabd HS, Gamal R, Mohammad SA, Gad S. Meier-Gorlin syndrome: report of an additional patient with congenital heart disease. Egypt J Med Human Gen. 2014;15(4):393–398. doi:10.1016/j.ejmhg.2014.04.003
3. de Munnik SA, Hoefsloot EH, Roukema J, et al. Meier-Gorlin syndrome clinical genetics and genomics. Orphanet J Rare Dis. 2015;10(1). doi:10.1186/s13023-015-0322-x
4. Bongers EM, Opitz JM, Fryer A, et al. Meier-Gorlin syndrome: report of eight additional cases and review. Am J Med Genet. 2001;102:115–124. doi:10.1002/ajmg.1452
5. Ting CY, Bhatia NS, Lim JY, et al. Further delineation of CDC45-related Meier-Gorlin syndrome with craniosynostosis and review of literature. Eur J Med Genet. 2020;63(2):103652. doi:10.1016/j.ejmg.2019.04.009
6. Bicknell LS, Bongers EM, Leitch A, et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet. 2011;43(4):356–360. doi:10.1038/ng.775
7. Knapp KM, Jenkins DE, Sullivan R, et al. MCM complex members MCM3 and MCM7 are associated with a phenotypic spectrum from Meier-Gorlin syndrome to lipodystrophy and adrenal insufficiency. Eur J Human Genet. 2021;29(7):1110–1120. doi:10.1038/s41431-021-00839-4
8. Knapp KM, Sullivan R, Murray J, et al. Linked-read genome sequencing identifies biallelic pathogenic variants in DONSON as a novel cause of Meier-Gorlin syndrome. J Med Genet. 2020;57(3):195–202. doi:10.1136/jmedgenet-2019-106396
9. Nabais Sá MJ, Miller KA, McQuaid M, et al. Biallelic GINS2 variant p.(Arg114Leu) causes Meier-Gorlin syndrome with craniosynostosis. J Med Genet. 2021;
10. Li N, Lam WH, Zhai Y, et al. Structure of the origin recognition complex bound to DNA replication origin. Nature. 2018;559(7713):217–222. doi:10.1038/s41586-018-0293-x
11. Shen Z, Chakraborty A, Jain A, et al. Dynamic association of ORCA with prereplicative complex components regulates DNA replication initiation. Mol Cell Biol. 2012;32(15):3107–3120. doi:10.1128/mcb.00362-12
12. Petersen BO, Wagener C, Marinoni F, et al. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000;14(18):2330–2343. doi:10.1101/gad.832500
13. Yuan Z, Schneider S, Dodd T, et al. Structural mechanism of helicase loading onto replication origin DNA by ORC-Cdc6. Proc Natl Acad Sci USA. 2020;117(30):17747–17756. doi:10.1073/pnas.2006231117
14. Mailand N, Diffley JFX. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122(6):915–926. doi:10.1016/j.cell.2005.08.013
15. Diffley JFX. DNA replication: building the perfect switch. Curr Biol. 2001;11:R367–70. doi:10.1016/S0960-9822(01)00196-8
16. Borlado LR, Méndez J. CDC6: from DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis. 2008;29(2):237–243. doi:10.1093/carcin/bgm268
17. HGVS. Available from: https://varnomen.hgvs.org/.
18. BaseSpace apps. Available from: https://basespace.illumina.com.
19. GnomAD. Available from: https://gnomad.broadinstitute.org/.
20. RefSeq database. Available from: https://www.ncbi.nlm.nih.gov/refseq/.
21. ClinVar. Available from: https://www.ncbi.nlm.nih.gov/clinvar/.
22. HGMD. Available from: http://www.hgmd.cf.ac.uk/.
23. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
24. Yao L, Chen J, Wu X, Jia S, Meng A. Zebrafish cdc6 hypomorphic mutation causes Meier-Gorlin syndrome-like phenotype. Hum Mol Genet. 2017;26(21):4168–4180. doi:10.1093/hmg/ddx305
25. DynaMut2. Available from: http://biosig.unimelb.edu.au/dynamut2/.
26. Cheng KY, Noble MEM, Skamnaki V, et al. The role of the phospho-CDK2/cyclin A recruitment site in substrate recognition. J Biol Chem. 2006;281(32):23167–23179. doi:10.1074/jbc.M600480200
27. Chi Y, Carter JH, Swanger J, Mazin AV, Moritz RL, Clurman BE. A novel landscape of nuclear human CDK2 substrates revealed by in situ phosphorylation. Sci Adv. 2020;6:eaaz9899. doi:10.1126/sciadv.aaz9899
28. Takeda DY, Wohlschlegel JA, Dutta A. A bipartite substrate recognition motif for cyclin-dependent kinases. J Biol Chem. 2001;276(3):1993–1997. doi:10.1074/jbc.M005719200
29. Yim H, Park JW, Woo SU, et al. Phosphorylation of Cdc6 at serine 74, but not at serine 106, drives translocation of Cdc6 to the cytoplasm. J Cell Physiol. 2013;228(6):1221–1228. doi:10.1002/jcp.24275
30. van Roey K, Orchard S, Kerrien S, et al. Capturing cooperative interactions with the PSI-MI format. Database. 2013;2013. doi:10.1093/database/bat066
31. Stevenson-Lindert LM, Fowler P, Lew J. Substrate specificity of CDK2-cyclin A: what is optimal? J Biol Chem. 2003;278(51):50956–50960. doi:10.1074/jbc.M306546200
32. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
33. PDB. Available from: https://www.rcsb.org/structure/2CCI.
34. Wilkie AO. The molecular basis of genetic dominance. J Med Genet. 1994;31(2):89–98. doi:10.1136/jmg.31.2.89
35. Fenwick AL, Kliszczak M, Cooper F, et al. Mutations in CDC45, encoding an essential component of the pre-initiation complex, cause Meier-Gorlin syndrome and craniosynostosis. Am J Hum Genet. 2016;99(1):125–138. doi:10.1016/j.ajhg.2016.05.019
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.