Back to Journals » ImmunoTargets and Therapy » Volume 10
NHS-IL12, a Tumor-Targeting Immunocytokine
Authors Greiner JW, Morillon II YM, Schlom J ![]()
Received 17 February 2021
Accepted for publication 26 March 2021
Published 27 May 2021 Volume 2021:10 Pages 155—169
DOI https://doi.org/10.2147/ITT.S306150
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
John W Greiner, Y Maurice Morillon II, Jeffrey Schlom
Laboratory of Tumor Immunology and Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Correspondence: Jeffrey Schlom
Laboratory of Tumor Immunology and Biology, Center for Cancer Research, National Cancer Institute, 10 Center Drive, Room 8B09, Bethesda, MD, 20892, USA
Tel +1 240-858-3463
Fax +1 240-541-4558
Email [email protected]
Abstract: NHS-IL12 is a novel immunocytokine designed for delivery of IL-12 to the tumor microenvironment (TME). NHS-IL12 consists of two molecules of IL-12 fused to a human IgG1 (NHS76) recognizing DNA/histone complexes, which are often exposed in the necrotic portions of tumors. Preclinical studies demonstrated the tumor-targeting ability and longer plasma half-life for NHS-IL12 when compared with recombinant IL-12 (rIL-12). NHS-IL12 outperformed rIL-12 in enhancing the proliferation and activation of immune as well as antigen-presenting cells, resulting in a more robust primary immune response. NHS-IL12 also reduced the number and function of suppressive myeloid cells (myeloid derived suppressor cells/macrophages) within the TME. In a murine bladder tumor model, NHS-IL12 administration led to a coordinated increase in host immunity with a reduction of immunosuppressive myeloid cells in the TME resulting in substantial reduction in tumor growth. Several preclinical studies have demonstrated increased overall anti-tumor efficacy when NHS-IL12 was combined with either immune-based therapeutics or chemotherapeutic approaches.
Keywords: interleukin-12, NHS-IL12, immunotherapy, cancer, immuno-oncology, review
Introduction
Interleukin (IL)-12 is a potent proinflammatory cytokine composed of the proteins p35 (35 kDa) alpha (α) chain and the p40 (40 kDa) beta (β) chain, covalently bonded to form the heterodimer IL-12 p70 protein.1,2 IL-12 is produced locally by activated dendritic cells (DCs), macrophages, and neutrophils during T-cell priming3 and acts directly on cytotoxic immune effector cells, including natural killer (NK) cells, natural killer T (NKT) cells, and CD8+ T cells, to stimulate their proliferation and increase their cytotoxic functions.4 Besides regulating the transition from innate to adaptive immunity, IL-12 also induces the differentiation of naive T helper cells toward a Th1 phenotype and the production of cytokines (most notably IFNγ) that promotes cell-mediated immunity.5,6 Additionally, IL-12 mediates effector T cells for optimal production of effector memory T cells and T follicular helper cells.7,8 Those IL-12‒mediated changes in the cellular immune compartments along with suppression of angiogenesis and increased immune (ie, T-cell) trafficking all contributed to the profound antitumor effects observed in preclinical mouse tumor models.1,9,10 Preclinical studies showed not only the ability of systemic recombinant IL-12 to substantially reduce large tumor burdens, but also the induction of immune memory to protect mice against tumor challenge. The excitement generated by these preclinical findings underscored IL-12 being ranked third in a comprehensive list of immunotherapeutic agents with high potential for use in treating cancer.11
Subsequent clinical failures negatively impacted the ongoing therapeutic development of rIL-12.12,13 The reported schedule-dependent toxicities and disappointing clinical responses were explained, in part, by the inability of intravenous (i.v.) or subcutaneous (s.c.) administered rIL-12 at the maximum tolerated dose (MTD) to achieve biologically relevant concentrations of rIL-12 in the tumor microenvironment (TME). Despite those setbacks, investigators designed approaches for paracrine/local IL-12 delivery that could optimize IL-12 levels in the TME, while limiting systemic exposure and the associated toxicity. Other strategies, in addition to NHS-IL12, which maximize IL-12 delivery to the TME have been the subject of a separate review.14
In support of that hypothesis, intratumoral rIL-12 injections were found to trigger a potent antitumor CD8+ cytotoxic T lymphocyte (CTL) response that led to complete regression of large established tumors in several murine models.15 More recently, using an orthotopic murine bladder model, local (intravesical) administration of rIL-12 admixed with chitosan, a mucoadhesive biopolymer,16 also resulted in significant tumor regression and subsequent survival advantage.17 Those and other findings argued that IL-12 targeting to the TME could enhance its significant effects on host immunity, which should mitigate toxicity by minimizing systemic exposure. One such tumor-targeting approach, NHS-IL12, an immunocytokine, is the subject of this review.
Components and Characteristics of the Fusion Protein NHS-IL12
NHS-IL12 is a recombinant fusion protein consisting of the human monoclonal IgG1 antibody NHS76 fused at each CH3 C-terminus to human IL-12.18 Figure 1A shows a ribbon diagram that illustrates the immunocytokine NHS-IL12, which consists of the tumor necrosis-targeting NHS7618 antibody fused to two molecules of the human IL-12 heterodimer. NHS76 is a second-generation TNT antibody that has been clinically validated for its ability to target lung tumors following systemic administration (Figure 1B, ref.19). NHS76 targets exposed DNA/histone sites,18,20 thereby directing the proinflammatory cytokine IL-12 to intratumoral necrotic regions.20,21 Because human IL-12 is not cross-reactive with the murine IL-12 receptor, the chimeric surrogate immunocytokine, designated NHS-muIL12, which consists of murine IL-12 fused to the same human NHS76 antibody, was generated and used in preclinical studies with immunocompetent mice.10
 |
Figure 1 NHS-IL12: Structure and concept: (A) Ribbon diagram of NHS-IL12, an immunocytokine consisting of the NHS76 antibody fused to two IL-12p70 heterodimers. (B) Image shows biodistribution of 131I-chTNT, a mouse/human chimeric antibody, in a patient with lung cancer. Arrows identify a tumor lesion. (C) IFNγ production by human PBMCs - NHS-huIL12 (blue) or rHuIL-12 (red). (D) Serum IFNγ levels in cynomolgus macaques (N=2) treated with 40 µg/kg NHS-huIL12 s.c. (green), 40 µg/kg NHS-huIL12 (i.v., pink), or 4 µg/kg rHuIL-12 i.v. (red). Panel B: Reproduced with permission from Yu L, Ju DW, Chen W,Li T, Xu Z, Jiang C, Chen S, Tao Q, Ye D, Hu P, Khawli LA, Taylor CR, Epstein AL. 131I-chTNT radioimmunotherapy of 43 patients with advanced lung cancer. Cancer Biother Radiopharm. 2006;21(1):5–14.19 Copyright © 2006, Mary Ann Liebert, Inc. All rights reserved. Panels A, C, and D: Reproduced from Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget. 2014;5(7):1869–1884.19 Copyright © 2014 Fallon et al. Creative Commons Attribution 3.0 License (https://creativecommons.org/licenses/bssy/3.0/legalcode). Abbreviations: i.v., intravenous; s.c., subcutaneous. |
Functional Differences Between NHS-IL12 and rIL-12
In vitro studies using PHA-stimulated human peripheral blood mononuclear cells (PBMCs) (Figure 1C) or human NK-92 cells revealed that IFNγ production was attenuated following the addition of the immunocytokine. Since IFNγ production has been linked with rHuIL-12 toxicity,12,13 these findings suggest that delivering IL-12 via the immunocytokine might be better tolerated and a safer therapeutic. In subsequent in vivo studies, single s.c. or i.v. injections of human NHS-huIL12 (40 µg/kg) or rHuIL-12 (4 µg/kg) were administered to cynomolgus macaques to examine IFNγ serum levels as well as comparative pharmacokinetics.22 Consistent with the previous in vitro assays, administration of NHS-huIL12 induced much lower serum IFNγ levels than did rHuIL-12 (Figure 1C; ref.22). Pharmacokinetic analyses clearly showed the sustained presence of plasma NHS-huIL12 levels when injected by either route,22 as compared with an i.v. injection of rHuIL-12 (Figure 1D). Thus, NHS-huIL12 was well tolerated as evidenced by (a) an absence of any measurable increase in body temperature, (b) a minor reduction in platelet counts, and (c) no significant injection site reactions. Thus, NHS76 fused to human IL-12 had a longer half-life in vivo but induced lower levels of IFNγ release by immune cells both in vitro and in vivo than recombinant IL-12. The sustained NHS-IL12 plasma levels, but with lower serum IFNγ, can be hypothesized by a lowering of the bioactivity of IL-12 as an immunocytokine compared with the recombinant IL-12 protein and the potential ability of NHS-IL12 to bind to tumor. As mentioned above, NHS is a human immunoglobulin that would have a greater clearance rate in mice vs humans (Figure 1C).
In vivo fluorescence imaging assessed the ability of NHS-muIL12 to target subcutaneous Lewis lung carcinoma (LLC) tumors in syngeneic mice22 (Figure 2A). Localization of fluorescent-labeled NHS-muIL12 within tumors was compared to BC1-muIL12, a human monoclonal antibody (MAb) immunocytokine analogous in structure to NHS-muIL12 but lacking a targeting component.23,24 Following subcutaneous delivery at a distal site, NHS-muIL12 accumulated within tumors to a higher level than did BC1-muIL12 (Figure 2A). To visualize the subcellular localization of NHS-muIL12, tumors from mice administered NHS-muIL12 were isolated and immunohistochemical detection was used to identify the NHS76 antibody (human IgG1). Strong, distinct staining in the nuclei of tumor cells was observed (Figure 2B), which supports the hypothesis of the proposed DNA/histone-binding mechanism in the TME.
 |
Figure 2 (A) Targeting of fluorescence-conjugated NHS-muIL12 or BC1-muIL12 to s.c. LLC tumors in athymic mice. Circles represent the area of the tumor. (B) IHC illustrating NHS-muIL12 binding to exposed cell nuclei within the tumor necrotic regions: arrows indicate NHS-muIL12 deposition. Circles represent the area of the tumor. (C) Antitumor effects of NHS-muIL12 versus rmuIL-12 against s.c. LLC, MC38, and B16 tumors. Average tumor volumes ± SE are shown. Asterisks indicate statistically significant differences between the NHS-muIL12 treatment group versus the control or rMuIL-12 treatment groups (2-way ANOVA followed by Bonferroni’s post-test; *p < 0.05; **p < 0.01; ***p <0.001). Reproduced from Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget. 2014;5(7):1869-1884.22 Copyright © 2014 Fallon et al. Creative Commons Attribution 3.0 License (https://creativecommons.org/licenses/by/3.0/legalcode). Abbreviations: IHC, immunohistochemistry; LLC, Lewis lung carcinoma; MDSCs, myeloid derived suppressor cells; s.c., subcutaneous. |
Subsequent studies using equimolar doses of NHS-muIL12 (10 µg) and rmuIL-12 (5.4 µg) were carried out to directly compare their antitumor potencies in different murine tumor models (Figure 2C). In three different syngeneic subcutaneous tumor models (LLC; MC38 colon carcinoma; B16 melanoma), treatment with NHS-muIL12 was clearly more potent as an antitumor agent when compared with an equimolar dose of rmuIL-1222 (Figure 2C). Employing the intravesical bladder tumor model, the differences between NHS-muIL12 and rmuIL-12 were even more striking. In additional studies, NHS-muIL12 treatment of mice bearing MB49luc bladder tumors resulted in a dose-dependent reduction of tumor growth with complete tumor regression achieved at 0.4 µg NHS-muIL12.25,26 One can surmise that the enhanced antitumor efficacy of NHS-muIL12 stems from its more favorable pharmacokinetic properties combined with its ability to recognize exposed DNA-histone adjuncts and localize to necrotic sites within the tumor. Additionally, large macromolecules are known to selectively accumulate in tumors through a passive process termed the “enhanced permeabilization and retention effect,” which may also apply to immunocytokines.27
Mechanisms of Action of NHS-muIL12
A series of preclinical studies looked at the soluble and cellular factors that accompanied NHS-muIL12 treatment to determine those that might have contributed to in vivo antitumor effects. A hallmark of IL-12 administration is the release of IFNγ, so it was not surprising that NHS-muIL12 induced a dose-dependent serum IFNγ response in vivo.22 NHS-muIL12 also induced the maturation of splenic DCs,28 as determined by upregulation of MHC class I expression and the activation of splenic NK and CD8+ T cells and tumor-infiltrating NK and CD8+ T cells (Table 1). Changes were dose-dependent,22 and NHS-muIL12 was more effective at activating the overall immune response than an equimolar dose of rmuIL-12 (Table 1). P15E is a tumor-associated endogenous retroviral antigen; functional immune studies also detected a high frequency of p15E-specific T cells in NHS-muIL12–treated mice bearing either MC38 or B16 tumors.22 Immune cell depletion studies established the absolute requirement for CD8+ T cells and a contributing role for NK cells to elicit significant antitumor effects in different murine tumor models.22
 |
Table 1 Comparison of NHS-muIL12 vs rmuIL-12 Effects on Markers of Immune Activation |
Mechanistic studies illustrating the cellular changes that contribute to the antitumor effects of NHS-muIL12 in the bladder tumor model revealed another facet of the immunocytokine. Known characteristics of the MB49luc bladder tumor model include early tumor necrosis and an immunosuppressive (Th2 polarized) TME governed by IL-10 signaling.29 Contributing to the immunosuppressive MB49luc bladder TME were large numbers of myeloid-derived cells, primarily CD11b+Gr1+F4/80− myeloid derived suppressor cells (MDSCs) and CD11b+Gr1−F4/80+ macrophages (M2), which suppressed CD4+ and CD8+ T-cell activation; this, in turn, blunted the ability of NHS-muIL12 to control MB49luc bladder tumor growth. A temporal study identified changes occurring within the MB49luc bladder TME during the time of maximum tumor regression, ie, between 72 and 144 hours after the final NHS-muIL12 administration (ref.25; Figure 3A). There was excellent concordance between the phenotypic and functional changes within the MB49luc bladder TME and the antitumor responses to systemic NHS-muIL12. At the earlier time point, MB49luc bladder tumors contained a preponderance of MDSCs and M2 macrophages, eliciting an ongoing immunosuppressive TME and no discernible reduction in MB49luc bladder tumor burden (Figure 3B). However, there were cellular changes in the MB49luc bladder TME following NHS-muIL12 treatment, including (a) a decrease in the number of Ly6C+ and Ly6G+ MDSCs, (b) an increase in M1 macrophages, and (c) an increase in CD4+IFNγ+ cells.26 By 144 hours after the final NHS-muIL12 treatment, the cellular makeup of the MB49luc bladder TME had dramatically changed with the number of MDSCs and macrophages significantly reduced (Figure 4A and B); this, in turn, increased the ratios of CD4+ and CD8+ cells to MDSCs and M2 macrophages.25 When MDSCs were further divided into monocytic and granulocytic, NHS-muIL12 kept a low number of monocytic MDSCs while virtually eliminating granulocytic MDSCs within the MB49luc bladder TME.25 These data are consistent with a previous finding that a robust increase in intratumoral CD8+ T-cell activation and proliferation resulted after targeted depletion of granulocytic MDSCs.30 Changes within the myeloid cell compartment provided compelling evidence for a reversal of cellular immunosuppression within the MB49luc bladder TME elicited by NHS-muIL12 treatment resulting in significant tumor regression. An overall reduction in TME cellular specific potential was demonstrated ex vivo, which may ultimately contribute to a more effective tumor clearance by cytotoxic immune effector cells. Phenotypic/functional changes within the MB49luc bladder TME affirm the tumor-targeting ability of NHS-muIL12 and offer a plausible sequence of events that accompany the significant reductions observed in MB49luc bladder tumors. In addition, NHS-muIL12 targeted actions are multifaceted and include cells that support host antitumor immunity (NK and T cells) as well as those that impede immunotherapy (MDSCs and macrophages). These and other preclinical findings formed the underpinnings for a first-in-human Phase 1 clinical trial of NHS-IL12.
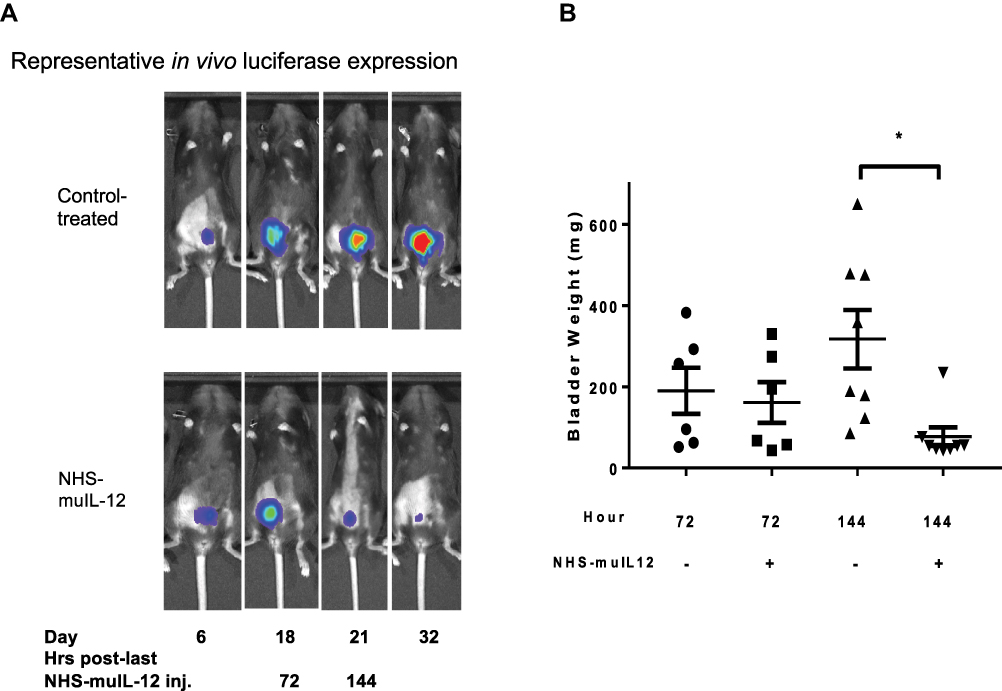 |
Figure 3 Antitumor effects of NHS-muIL12 on MB49luc bladder tumors. (A) Representative in vivo luciferase expression images for control Ig–treated (top row) and NHS-muIL12–treated (bottom row) mice. (B) Individual bladder weights from control Ig–treated (circles, triangles) and NHS-muIL12–treated (squares, inverted triangles) mice. Horizontal lines represent the average bladder weight; error bars represent mean ± SEM, Student’s t-test; *p < 0.05. Reproduced from Morillon YM II, Su Z, Schlom J, Greiner JW. Temporal changes within the (bladder) tumor microenvironment that accompany the therapeutic effects of the immunocytokine NHS-IL12. J Immunother Cancer. 2019;7(1):150.25 Copyright © The Authors 2019. Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/) |
 |
Figure 4 Changes in the myeloid cell populations and immune suppressive activity in the MB49luc bladder TME of control Ig–treated (black bars) and NHS-muIL12–treated (grey bars) mice. Absolute number of each cell type/mg bladder weight for (A) MDSCs and (B) macrophages. Error bars (panels A and B) represent mean + SEM, Student’s test; *p < 0.05. Reproduced from Morillon YM II, Su Z, Schlom J, Greiner JW. Temporal changes within the (bladder) tumor microenvironment that accompany the therapeutic effects of the immunocytokine NHS-IL12. J Immunother Cancer. 2019;7(1):150.25 Copyright © The Authors 2019. Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/). Abbreviation: MDSCs, myeloid derived suppressor cells. |
First-in-Human Phase 1 Trial of NHS-IL12
The phase 1 study of NHS-IL12 in patients with metastatic or locally advanced solid epithelial or mesenchymal tumors31 evaluated the safety, tolerability, pharmacokinetics, and biological and clinical activity of the agent. The trial also analyzed the effects on immune cell subsets, and T-cell receptor (TCR) clonality of NHS-IL12. The single- and multiple-dose escalation study also established the MTD of NHS-IL12 monotherapy given s.c. as a single dose or every 4 weeks. Patients were enrolled in cohorts of three to six using a standard 3x3 approach until MTD was reached. In the single-dose cohort, patients received one dose of NHS-IL12 and were followed for 28 days. In the multiple-dose escalation cohort, patients received NHS-IL12 every 4 weeks at 2, 4, 8, 12, 16.8, and 21.8 µg/kg. Pharmacodynamic data from this trial at 16.8 µg/kg revealed an expected time-dependent rise in serum IFNγ (Figure 5) and IL-10.31 Despite a similar rise in serum IL-12, most multiple-dose subjects had a diminished rise in IFNγ and IL-10 on the second dose, which coincided with the second and subsequent doses of NHS-IL12 being better tolerated (Figure 5, ref.31). Cellular changes in the peripheral blood (ie, PBMCs) at 1 week post‒NHS-IL12 infusion included increased (a) activated (Tim3+) and mature (CD16+/CD56dim) NK cells, and (b) PD-1+ NKT cells, consistent with the known ability of IL-12 to enhance both proliferation and lytic capacity of NK cells.9,32 Reductions in the levels of terminally differentiated CD4+ T cells and plasmacytoid DCs were also present at that time point. At 2 months post-treatment, only a sustained elevation of NKT cells remained. Sequencing of TCR CDR3 (TCRseq) regions on tumor biopsies and matching PBMCs showed that patients with high/medium increase in serum IFNγ levels also had an increase in tumor-infiltrating lymphocyte (TIL) density and TCR diversity in the TME.31 Broadening the tumor-associated TCR diversity suggested T cells trafficking consistent with other actions of NHS-IL12.33,34 NHS-IL12 was well tolerated up to a dose of 16.8 µg/kg, the recommended dose for subsequent clinical study. The study underscored the rationale that further studies should be designed in which NHS-IL12 is combined with other cancer therapeutics; this is addressed in the following section.
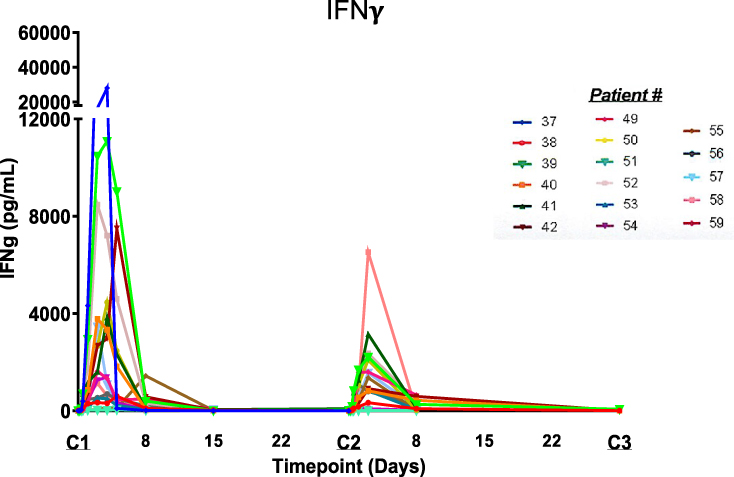 |
Figure 5 Temporal-dependent increases in serum IFNγ levels in individual patients given NHS-IL12 dosed at the MTD of 16.8 mcg/kg every 4 weeks. Reproduced from Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, McMahon S, Lamping E, Marté JL, Madan RA, Bilusic M, Silver MR, Bertotti E, Schlom J, Gulley JL. First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res. 2019;25(1):99–109.31 Copyright © 2019, American Association for Cancer Research. All rights reserved. Abbreviation: MTD, maximum tolerated dose. |
Combination Therapy with NHS-muIL12
In different preclinical murine tumor models, NHS-muIL12 was found to be promiscuous in that when combined with a wide range of agents, additive and/or synergistic antitumor effects resulted.22 For example, when combined with the vaccine tecemotide,35 which contains a 25-mer peptide from the VNTR region of the MUC1 tumor antigen, the combination delayed the growth of subcutaneous MC38/MUC1+ or orthotopic pancreatic (PancO2/MUC1+) tumors in MUC.Tg (transgenic) mice. Reduction in tumor growth was associated with the generation of a potent peptide-specific CD4+ T-cell proliferative response. In other studies, NHS-muIL12 was combined with approved standard-of-care agents/modalities. Radiation induced immune-associated changes in tumor cells, including upregulation of MHC class I molecules and diversification of the intracellular antigenic peptide pool, resulting in better anti-tumor immunity.36 In mice bearing either LLC or MC38 tumors, the combined treatment of NHS-muIL12 with localized, fractionated radiotherapy resulted in additive tumor growth inhibition.22,37 Sunitinib, a receptor tyrosine kinase inhibitor used to treat late-stage kidney cancer and gastrointestinal stromal tumor, can also modulate host immunity.38 Once again, the combined treatment of sunitinib with NHS-muIL12 resulted in an additive improvement in the growth inhibition of Renca renal tumors. Other cancer therapeutics, such as gemcitabine, a nucleoside analog, and docetaxel, an anti-mitotic taxane, have been successfully combined with NHS-muIL12, resulting in either additive or synergistic antitumor effects.39,40 Of interest were studies in which NHS-muIL12 was administered alone or combined with docetaxel to mice bearing large, well-established MC38 tumors. In the first study, NHS-muIL12, not docetaxel, induced significant regression of MC38 tumors when administered at a time point when tumor burden was approximately 150 mm3 (Figure 6A). The ability of NHS-muIL12 to impact MC38 tumor growth was lost when those tumors reached a volume in excess of 500 mm3 (Figure 6B). However, when mice bearing larger MC38 tumors (>500 mm3) were initially treated with docetaxel followed by NHS-muIL12, that sequential therapy induced significant tumor regression and prolonged survival (Figure 6B). Those findings support the hypothesis that initial docetaxel treatment, while not impacting overall tumor growth, enhances tumor necrosis, thus potentiating NHS-muIL12 tumor targeting that culminates in significant reduction in the growth of large, well-established MC38 tumors.
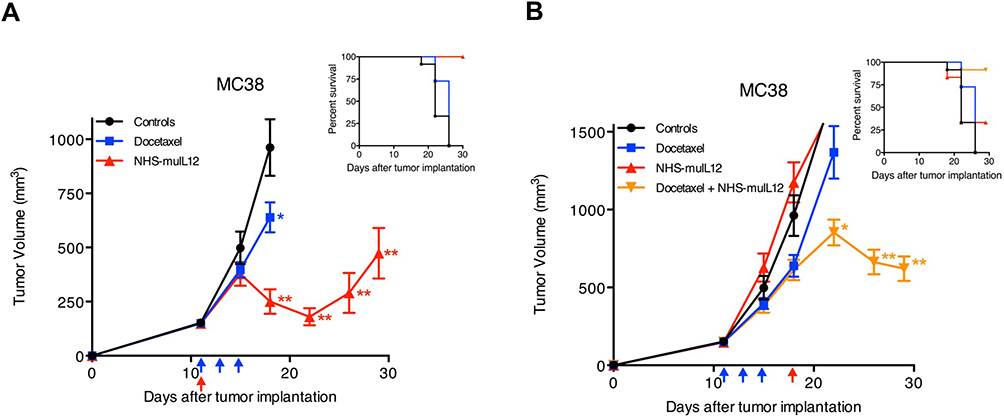 |
Figure 6 Docetaxel and NHS-muIL12 are effective as monotherapies and combined therapies in mice bearing well-established MC38 tumors. (A) Mice bearing well-established MC38 tumors were treated with saline (closed circles), 0.5 µg docetaxel (closed squares; blue arrows), or 50 µg NHS-muIL12 (closed triangles; red arrow). Docetaxel and NHS-muIL12 each significantly delayed tumor growth. (B) To study the combined efficacy of these therapeutic agents, mice bearing MC38 tumors were treated with saline (closed circles), 0.5 mg docetaxel (closed squares; blue arrows), 50 µg NHS-muIL12 (closed triangles; red arrows), or the combination (closed inverted triangles). Graphs show mean tumor volume ± SE. Asterisks indicate statistically significant differences between the sequential treatment combination versus saline-treated controls (1-way ANOVA with Tukey’s post-test; *p < 0.05; **p < 0.0001). Both inserts indicate percent survival of each treatment group. Reproduced from Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget. 2014;5(7):1869–1884.22 Copyright © 2014 Fallon et al. Creative Commons Attribution 3.0 License (https://creativecommons.org/licenses/by/3.0/legalcode). |
The advantage of combining NHS-muIL12 with the anti-PD-L1 MAb avelumab was demonstrated in multiple murine tumor models.10,41 In one study, mice bearing MC38 s.c. tumors received either avelumab or NHS-muIL12 as monotherapies and each was approximately equipotent in delaying tumor growth. The combined treatment, however, reduced MC38 tumor growth by >80% versus control-treated mice (p < 0.0001; 1-way ANOVA with Tukey’s post-test). In a second murine tumor model, MB49, the murine transitional bladder carcinoma cells, significant tumor reduction was achieved only in mice treated with the combination of NHS-muIL12/avelumab (p < 0.01; 1-way ANOVA with Tukey’s post-test). The combined NHS-muIL12/avelumab therapy was particularly effective in treating mice bearing MB49luc intravesical bladder tumors.25,26 Mice were administered avelumab and/or NHS-muIL12 alone or in combination. Tracking bladder tumor involvement with intravital imaging showed substantial reductions in mice treated with the combined therapy. That was verified with subsequent analyses of individual tumor burdens, determined by bladder weights. Only with the combination of NHS-muIL12/avelumab was statistically significant regression of tumor growth achieved in mice bearing MB49luc bladder tumors. The combined treatment induced complete, durable (> 2 months) tumor regression in a majority of mice and those mice were protected from subsequent tumor challenge. These findings indicated that the combination therapy with NHS-muIL12/avelumab generated a potent antigen-specific immune response that led to resolution of primary MB49luc bladder tumors and also generated powerful host immune memory.25,26 In the fourth preclinical model, in BALB/c mice bearing orthotopic EMT-6 breast tumors (> 100 mm3), monotherapy consisted of a single s.c. dose of NHS-muIL12 (2 µg) alone or in combination with avelumab (200 µg x 3), neither of which impacted EMT-6 tumor growth (Figure 7A). Once again, the NHS-muIL12/avelumab combination elicited a synergistic antitumor effect and complete tumor resolution in most mice (88%) (Figure 7A and B), prolonged survival (Figure 7C) and protected mice from tumor-specific rechallenge (Figure 7D).
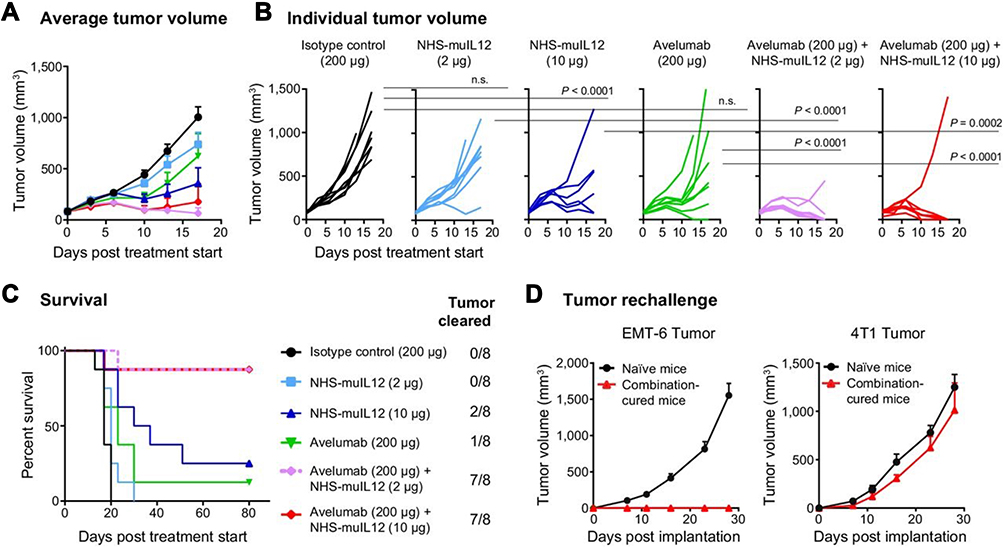 |
Figure 7 NHS-muIL12/avelumab combination treatment had a synergistic antitumor effect and induced long-term protective immunity in EMT-6 tumor-bearing mice. (A–C) EMT-6 tumor-bearing BALB/c mice were treated with: (i) isotype control (200 µg), (ii) NHS-muIL12 (2 µg), (iii) NHS-muIL12 (10 µg), (iv) avelumab (200 µg), (v) NHS-muIL12 (2 µg) + avelumab (200 µg), or (vi) NHS-muIL12 (10 µg) + avelumab (200 µg). (A) Average tumor volumes. (B) Individual tumor volumes. P values were calculated by 2-way ANOVA followed by Bonferroni’s post-test. (C) Kaplan-Meier survival curve and proportion of tumor clearance. (D) Mice in complete remission and naive BALB/c mice were challenged with EMT-6 or 4T1 cells. Error bars represent SEM. Reproduced with permission from Xu C, Zhang Y, Rolfe PA, Hernàndez VM, Guzman W, Kradjian G, Marelli B, Qin G, Qi J, Wang H,Yu H, Tighe R, Lo K-M, English JM, Radvanyi L, Lan Y. Combination therapy with NHS-muIL12 and avelumab (anti-PD-L1) enhances antitumor efficacy in preclinical cancer models. Clin Cancer Res. 2017;23(19):5869–5880.10 Copyright © 2017, American Association for Cancer Research. All rights reserved. |
Mechanisms Involved with Combining NHS-muIL12 and Avelumab
With the profound therapeutic advantage gained by combining NHS-muIL12 with avelumab, additional studies were carried out to examine the underlying mechanisms. Initial studies focused on immune-associated gene expression (NanoString PanCancer Immune Profiling Panels) in EMT-6 tumors of mice treated with NHS-muIL12 and/or avelumab. The combination of NHS-muIL12 (10 µg) and avelumab (200 µg) resulted in differential expression (defined as p < 0.05 and log fold-change >1) of 138 of 770 evaluated genes. At the lower NHS-muIL12 dose (2 µg) and avelumab, the combination resulted in nearly as many differentially expressed genes (120 of 770) relative to EMT-6 tumors from control-treated mice.10 A direct comparison of the differential gene expression profiles between monotherapies and combined therapies revealed profound differences. NHS-muIL12 monotherapy (2 or 10 µg) resulted in differential upregulation of 7 and 45 genes, respectively, while only a single gene was down-regulated following avelumab monotherapy.42 Subsequent studies examined the functional advantages gained by the NHS-muIL12/avelumab combined treatment versus each being administered as a monotherapy.
Upregulation of Serum Cytokines
One of the hallmarks of IL-12 treatment, whether rIL-12 or NHS-muIL12, is a substantial increase in serum IFNγ levels.1,5,6 Indeed, a short course of NHS-muIL12 upregulated serum IFNγ levels which, in turn, enhanced PD-L1 expression levels on MC38 or MB49 s.c. tumors.22 When that same experiment was carried out in IFNγ knockout mice, mPD-L1 expression levels remained unchanged, establishing the absolute need for IFNγ. The addition of avelumab to NHS-muIL12 treatment further increased PD-L1 expression levels on MC38 or MB49 tumors. The ability of the combined NHS-muIL12/avelumab treatment to enhance serum IFNγ was also present in Balb/c mice bearing EMT6 breast tumors. Also increased relative to NHS-muIL12 was the proinflammatory cytokine TNFα, which mediates antitumor effects through damaging tumor vasculature and promoting T-cell and NK-cell cytotoxicity (3.1-fold increase; p < 0.0001; refs.43,44). The combination therapy also increased levels of Th2 cytokines, including IL-5, IL-6, and IL-10, which have been associated with protumor and immunosuppressive responses,33 relative to NHS-muIL12 alone (8.1-, 1.8-, and 1.8-fold increases; p < 0.0001, p < 0.0369, and p < 0.0001, respectively).
Functional Immune Cell Changes with Combined NHS-muIL12/Avelumab Treatment
An in vitro assay confirmed the direct effects of NHS-muIL12 and avelumab on CD8+ T-cell activation.22 Naïve splenic CD8+ T cells from F5 TCR.Tg mice were stimulated with the cognate NP68 or control peptide-pulsed bone marrow-derived dendritic cells (BMDCs), which are known to express mPD-L1. BMDCs were added in the presence of increasing amounts (10–1000 pg/mL) of NHS-muIL12 and/or avelumab (10 µg/mL). The combination of NHS-muIL12/avelumab boosted IFNγ production, suggesting additive effects of the combination on antigen-specific T-cell activation.
To further delineate the in vivo activation status of different immune cell subsets following treatment with NHS-muIL12/avelumab, immune phenotypic signatures were determined via flow cytometry. In the EMT-6 murine breast tumor model, NHS-muIL12 monotherapy enhanced splenic NK and CD8+ T-cell proliferation, which was further enhanced with the addition of avelumab.10 In MC38 tumor-bearing mice, the combined treatment of NHS-muIL12/avelumab increased the percentage of proliferating CD8+ T cells in the TME. In both tumor models, the combined NHS-muIL12/avelumab also increased splenic effector (TEM) and central (TCM) memory T cells when compared with either monotherapy.10
Changes Within the Tumor Microenvironment with Combined NHS-muIL12/Avelumab
NanoString Immune Profiling also identified several serum chemokines involved in tumor inflammation that were upregulated in mice following NHS-muIL12/avelumab combined treatment. CXCL9 and CXCL10, both induced by IL-12,45 were involved in the chemotactic recruitment of CTLs and could support the IL-12 antitumor effects through inhibition of tumor neovascularization.46 The combination of NHS-muIL12/avelumab significantly increased both CXCL9 and CXCL10 serum levels relative to NHS-muIL12 monotherapy (1.4- and 1.8-fold increase; p < 0.0406 and p < 0.0001, respectively). Coupling the chemokine changes with the increase in splenic CD8+ T-cell proliferation,10 it was of interest to examine whether the combined treatment would increase CD8+ T-cell infiltration in the TME. Immunohistochemistry analysis of EMT-6 orthotopic breast tumors confirmed a dose-dependent increase in CD8+ T-cell infiltration with the combination of NHS-muIL12/avelumab (Figure 8A and B). TILs were identified by their small, distinctive nuclear morphologies, and TIL “hotspots” were present within well-defined areas of focal necrosis, identified by the absence of stained tumor cell nuclei. Both NHS-muIL12 and avelumab monotherapies increased the necrotic fraction and the combined treatment enhanced regression of EMT-6 tumors.10 In other studies, immune cell depletion41 established the absolute requirement for CD8+ T cells in order for NHS-muIL12/avelumab to elicit antitumor effects.22 In the MC38 tumor model it was of interest to examine the correlation between antitumor efficacy and the presence of a CD8+ T-cell response to p15E, an endogenous retroviral antigen,46 and a possible tumor rejection antigen. The combined NHS-muIL12/avelumab resulted in a synergistic reduction in MC38 tumor volume and additional analyses revealed a significantly higher frequency of p15E-specific CD8+ splenocytes (ELISpot assay) as compared with those from mice treated with either agent alone.
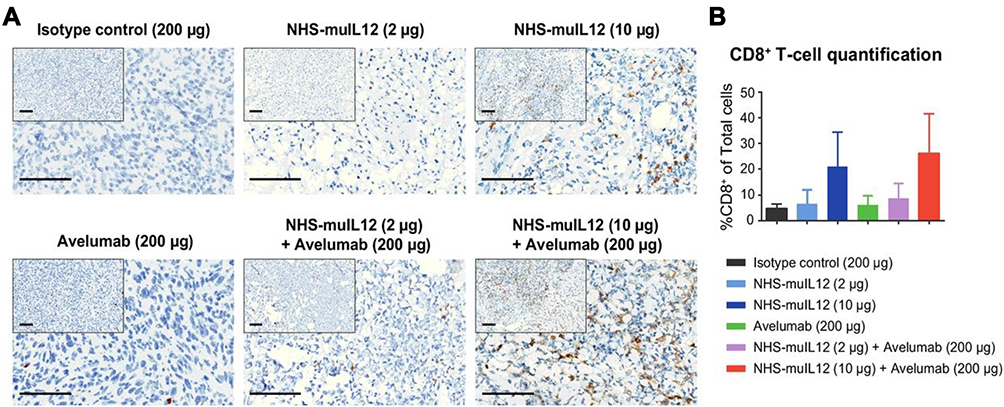 |
Figure 8 NHS-muIL12 treatment enhanced CD8+ T-cell infiltration in EMT-6 tumors. EMT-6 tumor-bearing mice were treated with: (i) isotype control, (ii) NHS-muIL12, (iii) NHS-muIL12, (iv) avelumab, (v) NHS-muIL12 + avelumab, or (vi) NHS-muIL12 + avelumab. Tumors were stained with an anti-mouse CD8 antibody. (A) Representative images of anti-CD8 IHC. Bars, 100 mm. (B) Average percentage of CD8+ cells/total cells in the TME. Error bars represent SEM.Reproduced with permission from Xu C, Zhang Y, Rolfe PA, Hernàndez VM, Guzman W, Kradjian G, Marelli B, Qin G, Qi J, Wang H,Yu H, Tighe R, Lo K-M, English JM, Radvanyi L, Lan Y. Combination therapy with NHS-muIL12 and avelumab (anti-PD-L1) enhances antitumor efficacy in preclinical cancer models. Clin Cancer Res. 2017;23(19):5869–5880.10 Copyright © 2017, American Association for Cancer Research. All rights reserved. Abbreviations: IHC, immunohistochemistry; TME, tumor microenvironment. |
Looking into the Future for NHS-IL12
One can envision NHS-IL12 being an integral part of a combined immunotherapeutic approach to cancer treatment. One particular aspect of NHS-IL12 is its ability to promote antigen presentation through the enhancement of DC maturation.47 Preclinically, murine splenic DCs upregulated MHC class I in an NHS-IL12 dose-dependent manner,22 thus inducing more potent T-cell priming. In a bladder tumor model, infiltrating macrophages were rapidly polarized to an M1 phenotype following NHS-muIL12 administration, consistent with a heightened ability to initiate a tumor-specific T-cell response.25 In a Phase I clinical study,31 NHS-IL12 reduced the frequency of circulating plasmacytoid DCs, again consistent with DC maturation and possible trafficking to lymphatics. The ability of NHS-IL12 to enhance the antigen presentation machinery is one aspect of the immunocytokine leading to enhanced antitumor effects in ongoing preclinical models utilizing peptide-based vaccines.48,49
In one study,49 NHS-muIL12 was one component of a triad of immunotherapeutics, including an HPV-specific vaccine, PDS0101, and a bifunctional checkpoint inhibitor, bintrafusp alfa. Mice bearing a murine lung carcinoma transformed with HPV16 E6 and E7, TC1, were treated with each agent alone and in the various double and triple combinations (Figure 9A). The triple combination, consisting of the PDS0101 vaccine, bintrafusp alfa and NHS-muIL12, was the most effective in reducing tumor growth and subsequently tumor weight (Figure 9B, p < 0.001 vs PBS control), thus improving overall antitumor therapy. Subsequent analyses showed that the triple combination enhanced (a) CD4+ and CD8+ T-cell TME infiltration, (b) proliferation/activation of TILs and HPV-specific T cells, and (c) TCR clonality.49
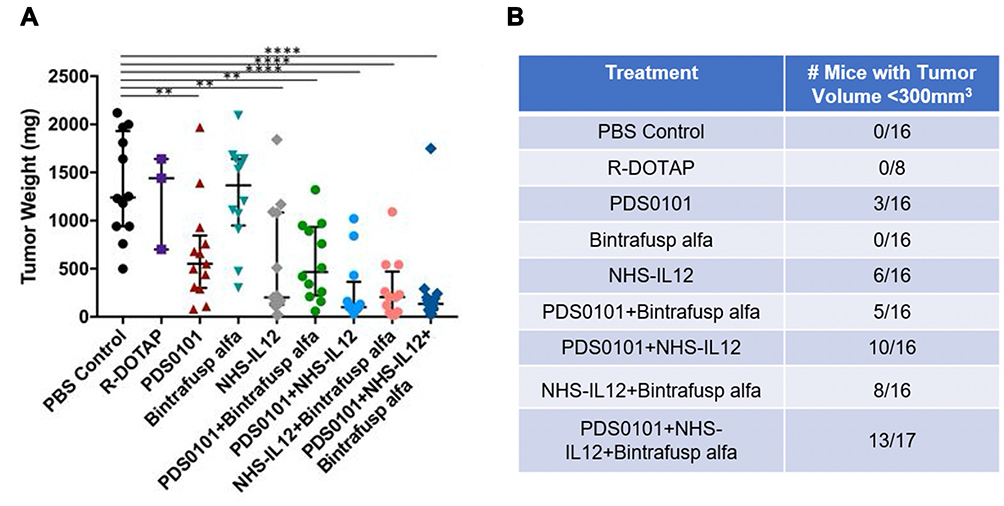 |
Figure 9 Triple combination of PDS0101 vaccine, bintrafusp alfa and NHS-muIL12 leads to significant control of TC-1 tumor growth. B6 mice received PBS, R-DOTAP, PDS0101 vaccine, bintrafusp alfa, or NHS-muIL12 alone or in different combinations. (A) Tumor weights at study end. (B) Number of mice in each treatment group with tumors <300 mm3 at study end. A meta-analysis of two independent experiments is shown. **p <0.01, ****p < 0.0001. Reproduced from Smalley Rumfield C, Pellom ST, Morillon YM II, Schlom J, Jochems C. Immunomodulation to enhance the efficacy of an HPV therapeutic vaccine. J Immunother Cancer. 2020;8(1):e000612.49 © Authors (or their employer(s)) 2020. Creative Commons Attribution Non Commercial (CC BY-NC 4.0) (https://creativecommons.org/licenses/by-nc/4.0/). With permission from BMJ Publishing Group Ltd. |
In another preclinical tumor model,48 NHS-muIL12 was combined with other immuno-oncology agents: (a) a neoepitope-targeted vaccine, (b) an IL-15 superagonist, and (c) an anti-PD-L1 (Figure 10A). That multifaceted treatment regimen promoted the influx of CD8+ TILs and enhanced the expression of transcripts relating to T-cell activation/effector function (Figure 10B). The inclusion of NHS-muIL12 also increased the clonality of the T-cell repertoire in the TME (Figure 10C), which led to a dramatic increase in tumor growth control (Figure 10D, ref.48). The neoepitope vaccine used also resulted in the in situ expansion of T cells reactive against other novel epitopes expressed by the tumor but not part of the vaccine.49
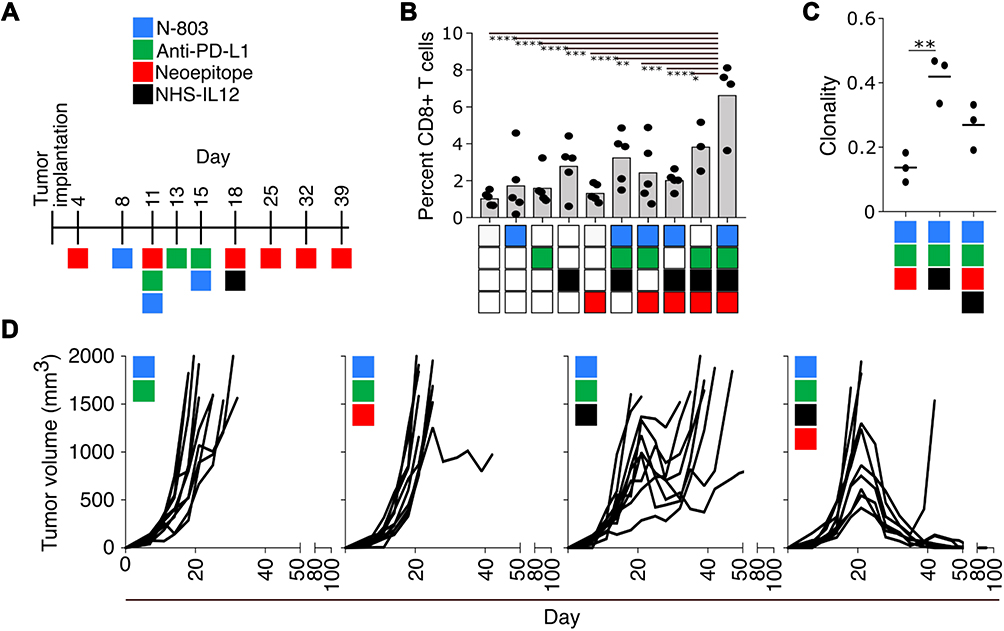 |
Figure 10 Combination immunotherapy: a 9-mer neoepitope vaccine, N-803, anti-PD-L1, and NHS-muIL12. (A) Treatment schedule. (B) Percent CD8+ cells in immunofluorescent tumor sections. (C) Clonality of TCRβ chains detected in tumor infiltrates. (D) Tumor growth curves of mice treated as indicated. *p<0.05; **p <0.01; ***p <0.001; ****p <0.0001 (n=5). Reproduced from Lee KL, Benz SC, Hicks KC, Nguyen A, Gameiro SR, Palena C, Sanborn JZ, Su Z, Ordentlich P, Rohlin L, Lee JH, Rabizadeh S, Soon-Shiong P, Niazi K, Schlom J, Hamilton DH. Efficient tumor clearance and diversified immunity through neoepitope vaccines and combinatorial immunotherapy. Cancer Immunol Res. 2019;7(8):1359–1370.48 Copyright © 2019, American Association for Cancer Research. All rights reserved. Abbreviation: TCR, T-cell receptor. |
NHS-IL12 is being paired with avelumab in an ongoing phase 1b, open-label, dose-escalation study in patients diagnosed with advanced solid tumors.50 The rationale for that particular combination came, for the most part, from preclinical studies. Avelumab was an effective immunotherapeutic in both s.c. and the intravesical MB49luc bladder tumor models.50 In each case, the tumors were known to harbor known rejection antigens (eg, p15E, HY, etc.) that were intrinsically immunogenic, which accounted for immune cells at the tumor/normal tissue interfaces (ie, “hot” tumors). Yet, the murine tumors grew at a rapid pace. One explanation was that tumor growth was a result of an immunosuppressive TME and one component was the PD-1/PD-L1 axis; disruption of that axis with avelumab led to effective immunotherapy. Immune suppression within the TME also seemed to be tied to myeloid cells, particularly MDSCs and subsets of macrophages and those cells seemed to be the primary target for NHS-muIL12.41 Tumor-targeting of NHS-muIL12 depleted granulocytic MDSCs and M2 macrophages and offered a plausible sequence of events leading to tumor resolution. Along with the presence of immune cells within the TME were large numbers of immunosuppressive MDSCs and macrophages, which enabled uncontrolled tumor growth. In both cases with avelumab and NHS-muIL12, the ability to modulate different components of immunosuppression within the TME seemed to reignite resident CD4+ and CD8+ T cells, leading to the significant cytolysis and tumor regression. Those findings also argued that the resident T cells within the TME were not defective (ie, exhausted, etc.), but when unshackled from immunosuppressive components (eg, PD1/PD-L1 axis, myeloid cells) elicited a robust antitumor response. Those findings also underscored the importance of selecting agents that target different components within the TME or host immune machinery, thereby resulting in a synergistic therapeutic effect. It is crucial that as different immune-based agents are combined and tested in clinical settings, the rationale for those efforts continues to be based on sound preclinical data.
Conclusion
The extensive preclinical and early clinical data form a very compelling argument for the continued development of NHS-IL12. Preclinical studies demonstrated that NHS-muIL12 can impact both immune activation targets and immune suppressive events. Targeting those changes within the confines of the TME underscores a “cold-to-hot” immune-based switch that precedes significant antitumor actions. The promise of immune checkpoint inhibitors has created significant enthusiasm for cancer immunotherapy, yet only a subset of patients benefits. NHS-IL12 presents a novel strategy that delivers a powerful Th1 cytokine that can function locally as an immune adjuvant and, when combined, can augment other immunotherapies.
Abbreviations
BMDC, bone marrow-derived dendritic cell; CTL; cytotoxic T lymphocyte; DC, dendritic cell; i.v., intravenous; LLC, Lewis lung carcinoma; MAb, monoclonal antibody; MDSC, myeloid derived suppressor cell; MTD, maximum tolerated dose; mu, murine; NK, natural killer; NKT, natural killer T cells; PBMC, peripheral blood mononuclear cell; s.c., subcutaneous; TCM, central memory T cell; TCR, T-cell receptor; TEM, effector memory T cell; Tg, transgenic; TIL, tumor-infiltrating lymphocyte; TME, tumor microenvironment; Treg, regulatory T cells.
Ethics Statement
Animal care was in compliance with the recommendations of The Guide for Care and Use of Laboratory Animals (National Research Council).
Acknowledgments
The authors thank Debra Weingarten for her editorial assistance in the preparation of this manuscript.
Funding
This study was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):155–168. doi:10.1016/s1359-6101(01)00032-6
2. Lu X. Impact of IL-12 in cancer. Curr Cancer Drug Targets. 2017;17(8):682–697. doi:10.2174/1568009617666170427102729
3. Del Vecchio M, Bajetta E, Canova S, et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13(16):4677–4685. doi:10.1158/1078-0432.Ccr-07-0776
4. Brunda MJ, Luistro L, Rumennik L, et al. Antitumor activity of interleukin 12 in preclinical models. Cancer Chemother Pharmacol. 1996;38(Suppl):S16–S21. doi:10.1007/s002800051031
5. Nakajima C, Uekusa Y, Iwasaki M, et al. A role of interferon-gamma (IFN-gamma) in tumor immunity: t cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice. Cancer Res. 2001;61(8):3399–3405.
6. Nastala CL, Edington HD, McKinney TG, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol. 1994;153(4):1697–1706.
7. Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood. 2011;118(14):3890–3900. doi:10.1182/blood-2011-05-357111
8. Schmitt N, Bustamante J, Bourdery L, et al. IL-12 receptor β1 deficiency alters in vivo T follicular helper cell response in humans. Blood. 2013;121(17):3375–3385. doi:10.1182/blood-2012-08-448902
9. Aste-Amezaga M, D’Andrea A, Kubin M, Trinchieri G. Cooperation of natural killer cell stimulatory factor/interleukin-12 with other stimuli in the induction of cytokines and cytotoxic cell-associated molecules in human T and NK cells. Cell Immunol. 1994;156(2):480–492. doi:10.1006/cimm.1994.1192
10. Xu C, Zhang Y, Rolfe PA, et al. Combination therapy with NHS-muIL12 and avelumab (anti-PD-L1) enhances antitumor efficacy in preclinical cancer models. Clin Cancer Res. 2017;23(19):5869–5880. doi:10.1158/1078-0432.Ccr-17-0483
11. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357–368. doi:10.1111/j.1600-065X.2008.00604.x
12. Haicheur N, Escudier B, Dorval T, et al. Cytokines and soluble cytokine receptor induction after IL-12 administration in cancer patients. Clin Exp Immunol. 2000;119(1):28–37. doi:10.1046/j.1365-2249.2000.01112.x
13. Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–2548.
14. Nguyen KG, Vrabel MR, Mantooth SM, et al. Localized Interleukin-12 for cancer immunotherapy. Front Immunol. 2020;11:575597. doi:10.3389/fimmu.2020.575597
15. Zaharoff DA, Hance KW, Rogers CJ, Schlom J, Greiner JW. Intratumoral immunotherapy of established solid tumors with chitosan/IL-12. J Immunother. 2010;33(7):697–705. doi:10.1097/CJI.0b013e3181eb826d
16. Grabnar I, Bogataj M, Mrhar A. Influence of chitosan and polycarbophil on permeation of a model hydrophilic drug into the urinary bladder wall. Int J Pharm. 2003;256(1–2):167–173. doi:10.1016/s0378-5173(03)00074-7
17. Zaharoff DA, Hoffman BS, Hooper HB, et al. Intravesical immunotherapy of superficial bladder cancer with chitosan/interleukin-12. Cancer Res. 2009;69(15):6192–6199. doi:10.1158/0008-5472.Can-09-1114
18. Gillies SD, Lan Y, Wesolowski JS, et al. Antibody-IL-12 fusion proteins are effective in SCID mouse models of prostate and colon carcinoma metastases. J Immunol. 1998;160(12):6195–6203.
19. Yu L, Ju DW, Chen W, et al. 131I-chTNT radioimmunotherapy of 43 patients with advanced lung cancer. Cancer Biother Radiopharm. 2006;21(1):5–14. doi:10.1089/cbr.2006.21.5
20. Sharifi J, Khawli LA, Hu P, King S, Epstein AL. Characterization of a phage display-derived human monoclonal antibody (NHS76) counterpart to chimeric TNT-1 directed against necrotic regions of solid tumors. Hybrid Hybridomics. 2001;20(5–6):305–312. doi:10.1089/15368590152740707
21. Eckert F, Schmitt J, Zips D, et al. Enhanced binding of necrosis-targeting immunocytokine NHS-IL12 after local tumour irradiation in murine xenograft models. Cancer Immunol Immunother. 2016;65(8):1003–1013. doi:10.1007/s00262-016-1863-0
22. Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget. 2014;5(7):1869–1884.
23. Mariani G, Lasku A, Balza E, et al. Tumor targeting potential of the monoclonal antibody BC-1 against oncofetal fibronectin in nude mice bearing human tumor implants. Cancer. 1997;80(12 Suppl):2378–2384.
24. Rudman SM, Jameson MB, McKeage MJ, et al. A phase 1 study of AS1409, a novel antibody-cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin Cancer Res. 2011;17(7):1998–2005. doi:10.1158/1078-0432.Ccr-10-2490
25. Morillon YMII, Su Z, Schlom J, Greiner JW. Temporal changes within the (bladder) tumor microenvironment that accompany the therapeutic effects of the immunocytokine NHS-IL12. J Immunother Cancer. 2019;7(1):150. doi:10.1186/s40425-019-0620-2
26. Vandeveer AJ, Schlom J, Greiner JW. Systemic immunotherapeutic efficacy of an immunocytokine, NHS-muIL12, in a superficial murine orthotopic bladder cancer model (abstr 1480). Cancer Res. 2016;76(14 Suppl):. doi:10.1158/1538-7445.AM2016-1480
27. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi:10.1016/s0168-3659(99)00248-5
28. Macatonia SE, Hosken NA, Litton M, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154(10):5071–5079.
29. Newton MR, Askeland EJ, Andresen ED, et al. Anti-interleukin-10R1 monoclonal antibody in combination with bacillus Calmette–Guérin is protective against bladder cancer metastasis in a murine orthotopic tumour model and demonstrates systemic specific anti-tumour immunity. Clin Exp Immunol. 2014;177(1):261–268. doi:10.1111/cei.12315
30. Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–1781. doi:10.1136/gutjnl-2013-306271
31. Strauss J, Heery CR, Kim JW, et al. First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res. 2019;25(1):99–109. doi:10.1158/1078-0432.Ccr-18-1512
32. Gumperz JE. CD1d-restricted “NKT” cells and myeloid IL-12 production: an immunological crossroads leading to promotion or suppression of effective anti-tumor immune responses? J Leukoc Biol. 2004;76(2):307–313. doi:10.1189/jlb.0104038
33. Cha E, Fong L. Shuffling the deck with CTLA-4 therapy: deep sequencing of rearranged TCRB genes demonstrates T cell repertoire remodeling in cancer patients. Oncoimmunology. 2018;7(4):e956016. doi:10.4161/21624011.2014.956016
34. Sheikh N, Cham J, Zhang L, et al. Clonotypic diversification of intratumoral T cells following sipuleucel-T treatment in prostate cancer subjects. Cancer Res. 2016;76(13):3711–3718. doi:10.1158/0008-5472.Can-15-3173
35. Samuel J, Budzynski WA, Reddish MA, et al. Immunogenicity and antitumor activity of a liposomal MUC1 peptide-based vaccine. Int J Cancer. 1998;75(2):295–302. doi:10.1002/(sici)1097-0215(19980119)75:2<295::aid-ijc20>3.0.co;2-b
36. Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–1271. doi:10.1084/jem.20052494
37. Eckert F, Jelas I, Oehme M, et al. Tumor-targeted IL-12 combined with local irradiation leads to systemic tumor control via abscopal effects in vivo. Oncoimmunology. 2017;6(6):e1323161. doi:10.1080/2162402x.2017.1323161
38. Farsaci B, Higgins JP, Hodge JW. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int J Cancer. 2012;130(8):1948–1959. doi:10.1002/ijc.26219
39. Burris HA
40. Hodge JW, Garnett CT, Farsaci B, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. 2013;133(3):624–636. doi:10.1002/ijc.28070
41. Fallon JK, Vandeveer AJ, Schlom J, Greiner JW. Enhanced antitumor effects by combining an IL-12/anti-DNA fusion protein with avelumab, an anti-PD-L1 antibody. Oncotarget. 2017;8(13):20558–20571. doi:10.18632/oncotarget.16137
42. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
43. Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002;13(2):135–141. doi:10.1016/s1359-6101(01)00020-x
44. Roberts NJ, Zhou S, Diaz LA, Holdhoff M, Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget. 2011;2(10):739–751. doi:10.18632/oncotarget.344
45. Kanegane C, Sgadari C, Kanegane H, et al. Contribution of the CXC chemokines IP-10 and Mig to the antitumor effects of IL-12. J Leukoc Biol. 1998;64(3):384–392. doi:10.1002/jlb.64.3.384
46. Yang JC, Perry-Lalley D. The envelope protein of an endogenous murine retrovirus is a tumor-associated T-cell antigen for multiple murine tumors. J Immunother. 2000;23(2):177–183. doi:10.1097/00002371-200003000-00001
47. Bianchi R, Grohmann U, Vacca C, Belladonna ML, Fioretti MC, Puccetti P. Autocrine IL-12 is involved in dendritic cell modulation via CD40 ligation. J Immunol. 1999;163(5):2517–2521.
48. Lee KL, Benz SC, Hicks KC, et al. Efficient tumor clearance and diversified immunity through neoepitope vaccines and combinatorial immunotherapy. Cancer Immunol Res. 2019;7(8):1359–1370. doi:10.1158/2326-6066.Cir-18-0620
49. Smalley Rumfield C, Pellom ST, Morillon IYM, Schlom J, Jochems C. Immunomodulation to enhance the efficacy of an HPV therapeutic vaccine. J Immunother Cancer. 2020;8(1):e000612. doi:10.1136/jitc-2020-000612
50. Strauss J, Vugmeyster Y, Sznol M, et al. Phase 1b, open-label, dose-escalation study of M9241 (NHS-IL12) plus avelumab in patients (pts) with advanced solid tumors (abstr 4062). Ann Oncol. 2019;30(suppl_5):v475–v532. doi:10.1093/annonc/mdz253
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.