Back to Journals » Journal of Inflammation Research » Volume 19
Neutrophil Extracellular Traps in Heart Failure: From Pathophysiological Mechanisms to Therapeutic Targets
Authors Ye S ![]() , Dong Z, Jiang X, Li X, Wang X
, Dong Z, Jiang X, Li X, Wang X ![]() , Li B, Yu R, Fan G, Wang Y, Zhu M, Wei J
, Li B, Yu R, Fan G, Wang Y, Zhu M, Wei J ![]()
Received 12 February 2026
Accepted for publication 13 May 2026
Published 23 May 2026 Volume 2026:19 603127
DOI https://doi.org/10.2147/JIR.S603127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qing Lin
Shengbo Ye,1,2,* Zhengwei Dong,1,* Xiaorui Jiang,1,* Xingyuan Li,1 Xinlu Wang,1,3 Bin Li,1,4 Rui Yu,1 Genhao Fan,1 Yongxia Wang,1 Mingjun Zhu,1 Jingjing Wei1
1Department of Cardiovascular Disease, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, Henan, 450000, People’s Republic of China; 2Collaborative Innovation Center of Prevention and Treatment of Major Diseases by Chinese and Western Medicine, Henan University of Chinese Medicine, Zhengzhou, Henan, 450000, People’s Republic of China; 3Prevention and Control Center for Chronic Disease, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, Henan, 450000, People’s Republic of China; 4Henan Evidence-Based Medicine Center of Chinese Medicine, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, Henan, 450000, People’s Republic of China
*These authors have contributed equally to this work
Correspondence: Jingjing Wei, Department of Cardiovascular Disease, The First Affiliated Hospital of Henan University of Chinese Medicine, Renmin Road 19, Jinshui District, Zhengzhou, 450000, People’s Republic of China, Email [email protected] Mingjun Zhu, Department of Cardiovascular Disease, The First Affiliated Hospital of Henan University of Chinese Medicine, Renmin Road 19, Jinshui District, Zhengzhou, 450000, People’s Republic of China, Email [email protected]
Abstract: Heart failure (HF) is a clinical syndrome characterized by myocardial remodeling, pathological fibrosis, and chronic sterile inflammation. Within the pathophysiological network of HF and its complex comorbidities, neutrophil extracellular traps (NETs) have been identified as key pathological mediators driving tissue damage. This review aims to explore the regulatory mechanisms of NETs in the progression of heart failure (HF) and their cross-organ pathological effects, and to summarize the latest research advances in NET-targeted interventions for the treatment of HF. NETs are reticular structures composed of antimicrobial proteins, such as myeloperoxidase (MPO) and histones, attached to a decondensed DNA scaffold. Under pathological conditions, excessive NET release and impaired clearance trigger inflammatory cascades. Aberrant NETs formation significantly promotes cardiac remodeling and pathological fibrosis by mediating immunothrombosis, inducing cardiomyocyte apoptosis, and activating fibroblasts.NETs serve as a critical pathophysiological link connecting HF to systemic comorbidities. Therapeutic strategies targeting NETs primarily include inhibiting PAD4 or associated signaling axes to block NETs formation, utilizing enzymatic reactions to facilitate NETs clearance, uncovering the novel potential of established clinical agents, and employing nanotechnology-based precision delivery systems. These findings offer new avenues for precision immunotherapy in HF and highlight considerable potential for clinical translation.
Keywords: heart failure, neutrophil extracellular traps, pathology, inflammation, therapeutic targets
Introduction
Over 37.7 million individuals worldwide are afflicted by heart failure (HF), a cardiovascular condition with a rising global burden.1 Current epidemiological evidence indicates that despite therapeutic advances, the five-year mortality rate of heart failure remains as high as 50%, underscoring the urgent need for novel intervention targets.2,3
Neutrophils act as the immune system’s first line of defense against invading pathogens.4 By phagocytosing pathogens, degranulating, generating reactive oxygen species (ROS), and secreting chemokines and cytokines to recruit additional immune cells, they orchestrate the host’s immune response.5 Accumulating evidence suggests that neutrophils release reticular structures called NETs, which encapsulate nuclear DNA and related nuclear and cytoplasmic components to carry out their immune defense duties.6 However, NETs contribute to the pathogenesis of HF through mechanisms such as generating inflammation, promoting prothrombotic states, and facilitating the creation of atherosclerotic plaque, in addition to their critical function in fighting infections.7 NETs are positioned as a unique therapeutic target due to their dual role as a pathogenic element in disease and an essential host defense mechanism.8
Pathologically, HF is characterized by myocardial remodeling, cardiomyocyte hypertrophy, fibrosis, cell apoptosis, and chronic low-grade sterile inflammation.9 HF frequently manifests as a shared endpoint of several systemic comorbidities rather than occurring in isolation. Clinically, concomitant conditions such as diabetes, hypertension, chronic kidney disease (CKD), and autoimmune diseases are commonly seen in HF patients.8,10 These diseases not only increase the likelihood of HF but also hasten its pathological development. Recent research reveals that NETs may serve as a crucial mediator linking these systemic diseases to heart damage.11 To better understand the complex systemic pathophysiology of HF and develop more focused therapy approaches, it is essential to comprehend the mediating role of NETs in cardiac comorbidities.
Currently, while the medications recommended in standard guidelines have optimized the clinical management of heart failure, they remain unable to effectively curb the ongoing damage caused by systemic inflammation.12,13 Furthermore, while the role of NETs in cardiac injury has been previously explored, a systematic elucidation of NETs as a central hub integrating between heart failure and its multisystemic comorbidities remains lacking.8,10 This review distinguishes itself by shifting the focus from localized cardiac damage to a comprehensive integration of the interactive mechanisms by which NETs drive HF progression through systemic complications. Finally, we summarize the latest advances in targeting NETs, aiming to offer novel perspectives for precision immunotherapy in heart failure.
In order to provide new theoretical underpinnings and intervention targets for the clinical prevention and treatment of heart failure, this narrative review, based on a comprehensive literature search of multiple databases, aims to clarify the pathophysiological mechanisms of NETs in HF, as well as possible therapeutic approaches targeting NETs (Detailed search methods can be found in the Supplementary Material 1).
Formation and Regulatory Mechanisms of NETs
Neutrophils release complex reticular structures called NETs in response to specific stimuli.14 Decondensed DNA, histones (H2A, H2B, H3, and H4), neutrophil proteins (myeloperoxidase MPO, neutrophil elastase NE, calmodulin, or calgranin), and antimicrobial proteins (azurophilin 1, cathelicidin, and lysozyme C) comprise their primary structural components.15–18 These structural components empower NETs to execute their fundamental physiological role in innate immunity. In the context of normal cell function, neutrophils rely on physiological oxidative function, which is a regulated process governed by O2 metabolism. During physiological inflammation, this oxygen metabolism facilitates a controlled “oxidative burst” that triggers NET deployment, enabling the localized entrapment and neutralization of invading pathogens.19 Importantly, the safety of this defense mechanism depends strictly on the cellular redox balance. If a deficiency of cellular antioxidants occurs, this physiological oxidative process can generate excessive oxidative stress,20 ultimately leading to cell damage and marking the critical transition of NETs from protective immune responders to pathological drivers in cardiovascular diseases.21
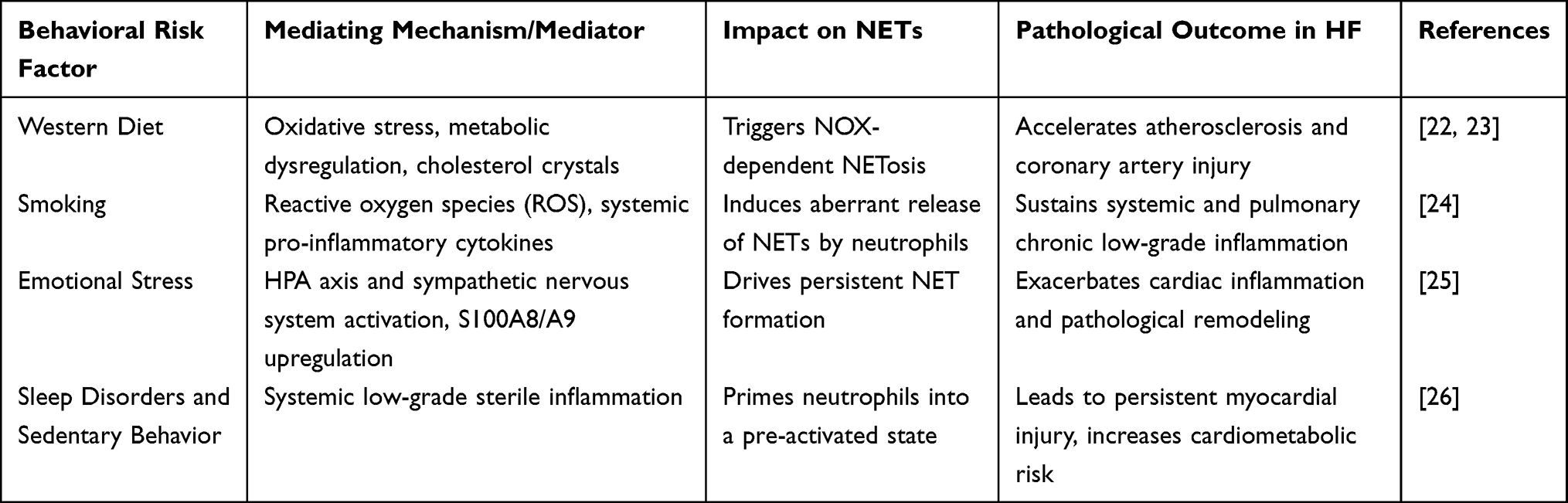 |
Table 1 Impact of Behavioral Risk Factors on NETs and Heart Failure Progression |
The process by which NETs develop and are released into the extracellular space is termed NETosis.27 NETosis is often initiated by diverse microorganisms and pro-inflammatory mediators, including potent activators such as phorbol 12,13-dimyristate 13-acetate (PMA), lipopolysaccharide (LPS), bacteria, viruses, and environmental factors, including cigarette smoke. It may also be induced by cytokines, chemokines, immune complexes, and other physiological signals.15,28,29 Additionally, chronic behavioral risk factors, including Western diets, alcohol consumption, and psychological stress, have been identified as significant contributors to aberrant NET formation. These factors, alongside sleep deprivation and sedentary behavior, act as chronic stressors that trigger profound neuroendocrine dysregulation, primarily characterized by the overactivation of the sympathetic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis. This neuroendocrine stress synergizes with low-grade systemic inflammation to prime neutrophils for persistent NET release and exacerbate myocardial injury.25,26,30 The specific molecular mechanisms by which these behavioral stressors orchestrate the neuroendocrine-immune axis to drive NETosis and their subsequent clinical implications are summarized in Table 1. The formation process (NETosis) is conventionally categorized into two types: NOX2-dependent and NOX2-independent.31 NOX2-dependent NETosis is primarily characterized by the activation of signaling proteins, including PKC (protein kinase C) and the NADPH oxidase complex, in response to stimuli such as antibodies, microbes, cholesterol, and PMA. This leads to elevated intracellular calcium levels and NOX activation, ultimately generating significant reactive oxygen species (ROS).28,32 By oxidatively modifying lipids or proteins on azurophilic granule membranes, ROS destabilizes membrane structures and facilitates the dissociation of NE from MPO on the granule membrane and its release into the cytoplasm. The dissociated NE and MPO then translocate into the nucleus, driven by ROS. Within the nucleus, NE synergistically degrades histone H4 with the calcium-dependent protease peptidyl arginine deiminase 4 (PAD4), while PAD4 simultaneously catalyzes histone H3 citrullination (citH3), resulting in chromatin decondensation. Meanwhile, MPO catalyzes the interaction of H2O2 with halides (eg, Cl−) in acidic conditions to form potent oxidants such as hypochlorous acid (HOCl), further promoting chromatin decondensation18,27,28 The NOX2-independent NETosis process occurs more rapidly (5–60 minutes) 33 This process is initiated by Staphylococcus aureus or calcium ionophores such as ionomycin, which activate small conductance calcium-potassium channels (SK3 channels) on neutrophils to induce mitochondrial reactive oxygen species (mtROS) generation. MPO and NE are released when mtROS disrupts neutrophil granules.15,33–35 Vesicles derived from the nucleolar membrane then bleb from the inner and outer nuclear membranes. Nuclear DNA-containing vesicles are released intact into the extracellular environment, where they eventually rupture, releasing chromatin and forming NETs15,33 (Figure 1).
 |
Figure 1 Formation and Regulatory Mechanisms of NETs. Two core mechanisms of NETs formation. The classical NOX2-dependent pathway, where ROS accumulation drives chromatin decondensation, ultimately leading to plasma membrane rupture and NETs release. The NOX2-independent pathway, primarily driven by mitochondrial ROS (mtROS), releases NETs extracellularly via nuclear budding and vesicular transport. Abbreviations: Ca2⁺, calcium ion; CXCR1/2, C-X-C motif chemokine receptor 1/2; IL-8, interleukin-8; LPS, lipopolysaccharide; MPO, myeloperoxidase; mtROS, mitochondrial reactive oxygen species; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NOX2, NADPH oxidase 2; PAD4, protein-arginine deiminase 4; PKC, protein kinase C; PMA, phorbol 12,13-dimyristate 13-acetate; ROS, reactive oxygen species; SK3, small conductance calcium-activated potassium channel 3; SOCE, store-operated calcium entry; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor-alpha; TNFR, tumor necrosis factor receptor. |
Pathophysiological Role of NETs in Heart Failure and Comorbidities
The final stage of many cardiovascular diseases is represented by HF. NETs are secretions triggered by the innate immune response to pathological stimuli.14 They also act as pivotal mediators mediating sterile inflammation, thrombosis, and tissue fibrosis.7 Although NETs facilitate the removal of bacteria, their overproduction and impaired clearance are factors in HF. This pathological influence manifests predominantly through inflammatory responses, thrombosis, and ventricular remodeling.
Inflammatory Cascade: NETs as Drivers and Amplifiers of Inflammation
NETs serve as both the source and amplifier of sterile inflammation in chronic HF.7,9 The DNA and histones that NETs release function as damage-associated molecular patterns (DAMPs), which can bind to a variety of pattern recognition receptors (PRRs) and increase inflammation.36–38 This inflammatory response differs across different HF subgroups. The inflammatory profiles of heart failure diverge fundamentally between the reduced (HFrEF) and preserved (HFpEF) ejection fraction phenotypes. In HFrEF, inflammation typically emerges as a secondary, localized response to facilitate tissue repair following acute myocardial loss. Conversely, the pathology of HFpEF is driven by a systemic metabolic milieu, sustained by comorbidities such as obesity and diabetes, that fosters a persistent pro-inflammatory state. This chronic systemic stress keeps coronary microvascular endothelial cells in an activated state, which orchestrates neutrophil recruitment and culminates in a massive surge of NET release.39,40 Transcriptomic research demonstrated that patients with HF with preserved ejection fraction (HFpEF) had significantly heightened activation of NETs-related pathways, such as neutrophil elastase NE and defensin alpha 4, than patients with HF with reduced ejection fraction (HFrEF). This indicates a particularly pronounced connection between NETs and microvascular inflammation associated with endothelial dysfunction in HFpEF.41
Furthermore, epicardial adipose tissue (EAT) has pronounced pro-inflammatory secretory properties in HF secondary to atrial fibrillation (AF).36,37 Myeloperoxidase (MPO) and NETs are abundant in the vesicles secreted by EAT from AF patients, which induces increased local inflammation. MPO expression peaks in individuals with persistent AF.42 Reactive oxygen species (ROS) and oxidative stress are readily induced by hyperglycemic diabetes, and they act synergistically with chronic inflammatory mediators such as TNF-α and IL-6 to activate neutrophil signaling pathways.43 Myocardial interstitial fibrosis and increased stiffness are driven by NETs-mediated chronic immune-inflammatory responses, which impair heart contractility and diastolic function. This is the primary mechanism by which inflammatory cardiomyopathy culminates in HF.
HF is frequently induced by sterile inflammation, myocarditis secondary to various infectious agents, including Coxsackievirus B3 and Influenza A virus, while end-stage HF may result from persistent autoimmune reactions.44 These pathogens, much like SARS-CoV-2, are present throughout the clinical course of SARS-CoV-2 infection, and patients with COVID-19 complicated by fulminant myocarditis had elevated circulating levels of NET biomarkers (free DNA, MPO-DNA, Cit-H3).45,46 The study also revealed that the 13-kD cytokine mediator (MK) interacts with low-density lipoprotein receptor-related protein 1 (LRP1) to increase neutrophil infiltration into the heart and induce NETosis. This suggests that NET formation is mediated by the MK-LRP1 pathway, which exacerbates cardiac fibrosis and inflammation.47
Finally, it is important to consider the long-term low-grade inflammatory response induced by a hyperglycemic environment. Under such conditions, the body produces significant amounts of reactive oxygen species (ROS) due to elevated oxidative stress levels. ROS function as second messengers, stimulating neutrophil signaling pathways and promoting the formation of NETs.
Chronic low-grade inflammation is present in diabetic patients, and elevated levels of inflammatory cytokines (including TNF-α and IL-6) further trigger the release of extracellular traps (NETs) derived from neutrophils.22 The aberrant metabolic status in diabetic patients also contributes to the accumulation of cholesterol crystals within atherosclerotic plaques. These crystals not only act as stimulants for NETosis but also serve as amplification signals for inflammasome activation.23 Ultimately, metabolic abnormalities promote the persistence of NETs in plaques. This drives atherothrombosis and cardiac injury, eventually culminating in HF (Figure 2).
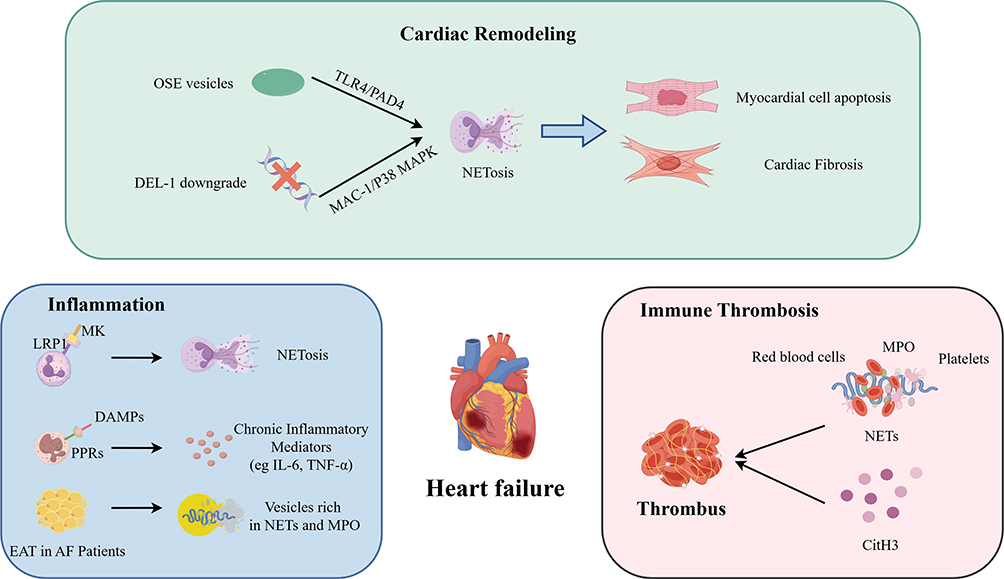 |
Figure 2 Pathophysiological mechanisms of NETs in the development and progression of HF. During cardiac remodeling, downregulation of OSE vesicles or DEL-1 triggers NETosis via the TLR4 and MAC-1 signaling axes, respectively, leading to cardiomyocyte apoptosis and interstitial fibrosis. During inflammation DAMPs bind to numerous pattern recognition receptors (PRRs), exacerbating cardiac inflammation and fibrosis. EAT in AF patients secretes numerous vesicles rich in MPO and NETs, promoting local inflammation. MK binds to LDL receptor-related protein 1 (LRP1) to facilitate neutrophil infiltration into the myocardium and induce NETosis. In the prothrombotic cascade, NETs act as physical scaffolds to recruit platelets and Erythrocytes, promoting Immunothrombosis. Elevated expression of NETs markers (particularly CitH3) also promotes fibrin clot compaction and inhibits thrombolysis. These pathological processes collectively drive the onset and progression of HF. Abbreviations: AF, atrial fibrillation; CitH3, citrullinated histone H3; DAMPs, damage-associated molecular patterns; DEL-1, developmental endothelial locus-1; EAT, epicardial adipose tissue; LRP1, LDL receptor-related protein 1; MAC-1, macrophage-1 antigen; MAPK, mitogen-activated protein kinase; MK, midkine; MPO, myeloperoxidase; NETs, neutrophil extracellular traps; OSE, oxidation-specific epitopes; PAD4, protein-arginine deiminase 4; PRRs, pattern recognition receptors; TLR4, Toll-like receptor 4. |
Immune Thrombosis: NET-Mediated Coagulation Activation and Microcirculatory Dysfunction
Thrombosis directly compromises the prognosis of ischemic HF.48 NETs’ histone components directly promote coagulation, and they provide a physical scaffold for platelet and red blood cell aggregation to generate thrombi.49 Furthermore, elevated expression of NETs markers—particularly CitH3—is closely associated with fibrin clot densification and enhanced fibrinolysis inhibition.50 This mechanistically explains why, following acute myocardial infarction, left ventricular thrombus (LVT) persists and proves resistant to resolution in certain patients despite anticoagulant therapy.51
At the molecular level, LNK (SH2B3) works as an inhibitor of cytokine signaling in hematopoietic cells, and LNK deficiency leads to hematopoietic stem cell proliferation.52 Studies have demonstrated that in LNK-deficient animals, oxidized phospholipids (OXPLs) enhance NETosis by activating platelet-activating factor receptor (PAFR) on neutrophil membranes, accelerating atherosclerosis and thrombosis.53 However, plaque rupture is associated with the immune-related GTPase family M protein (IRGM). Studies have demonstrated that IRGM activates the engagement of MAPK family members (including p38, JNK, and ERK) and downstream effectors of MAPK-activated cytoplasmic phospholipase A2 (cPLA2), further promoting NET formation and resulting in the pro-inflammatory phenotype of thrombi.54 Additionally, von Willebrand factor (VWF) has been reported to positively regulate NET production by interacting with the SLC44A2 receptor, thereby causing cardiomyocyte death through NET-mediated mitochondrial malfunction.55 Concurrently, the protective transcription factor KLF2 is downregulated in HF, releasing its inhibitory impact on NETosis and encouraging microvascular thrombosis and cardiac hypertrophy.56,57 These dysregulated molecular switches collectively drive the pathological course of HF.
Additionally, excessive NETs release due to chronic inflammation in patients with chronic kidney disease (CKD) contributes to elevated thrombosis rates, with NETs showing a significant association with vascular access thrombosis risk.58 Similarly, overexpression of platelet P-selectin induces PAD4-dependent NET production in chronic thromboembolic pulmonary hypertension (CTEPH).59 It will impair thrombolysis and exacerbate right HF.NETs are released in primary hypertension when angiotensin II activates NADPH oxidase to produce reactive oxygen species (ROS), which act synergistically with PAD4.60 NETs induce intravascular thrombosis, which occludes blood flow in small and large coronary arteries. The number of functional myocardial contractile units is decreased as a result of myocardial ischemia and necrosis, which is a critical driver in the progression of ischemic heart disease and HF (Figure 2).
Cardiac Remodeling: From Direct Cardiotoxicity to Pathological Fibrosis
Cardiomyocytes can be directly injured by NETs and their constituent parts, which can also induce pathological remodeling.61 Extracellular vesicles containing oxidation-specific epitopes (OSE) are released from the damaged myocardium during acute myocardial infarction (AMI), activating NETs via TLR4- and PAD4-dependent pathways. By binding to Annexin A5 to form a complex that potentiates calcium influx into neutrophils, S100A12 plays a pivotal role in this process. Elevated intracellular calcium levels activate calcium-dependent signaling pathways for NETosis, hence exacerbating cardiac damage following ischemia-reperfusion.62
By causing autophagic apoptosis and cell death in cardiomyocytes, NETs exacerbate cardiac dysfunction in atrial fibrillation-induced HF. The numerous cytoplasmic and granular proteins within NETs trigger autophagic signaling pathways within cardiomyocytes. Conversely, stimuli such as mtDNA released from cardiomyocytes during rapid pacing further promote neutrophils to develop NETs. Changes in ventricular wall structure, architecture, and function result from this vicious cycle between NETs and cardiomyocytes, which also induces cardiomyocyte loss and fibrosis. This evolution moves the disease from myocardial damage toward irreversible HF.63 Additionally, studies have demonstrated that a deficiency in aldehyde dehydrogenase 2 (ALDH2) exacerbates myocardial ischemia-reperfusion injury by promoting NETosis through activation of the endoplasmic reticulum stress/MGST2/LTC4/NOX2 pathway, thereby providing novel therapeutic targets for individuals with ALDH2 gene mutations and HF.64
Studies on the endogenous inhibitor developmental endothelial locus-1 (DEL-1, also known as EDIL3; commonly referred to as DEL-1) suggest that the involvement of NETs in cardiac remodeling may be multifaceted. In non-ischemic pressure-overloaded HF, NETs predominantly induce pathological damage. According to Zhao et al, when cardiac endothelial DEL-1 is downregulated, the neutrophil MAC-1/P38 MAPK pathway is no longer inhibited, which induces an excessive release of NETs that exacerbates myocardial hypertrophy and fibrosis65 However, during the healing phase of ischemic myocardial infarction, NETs demonstrate a protective role. Wei et al revealed that whereas DEL-1 deficiency similarly increased NETs, in a milieu without acute thrombosis, the DNA component of NETs induced polarization toward the Mertk MHC-IIlo-int phenotype by activating the TLR9 signaling pathway in macrophages This promotes polarization toward a Mertk MHC-IIlo-int phenotype, which attenuates adverse remodeling by increasing phagocytosis of cellular debris.66 The pleiotropic effects of NETs during tissue remodeling present a substantial challenge for determining the optimal timing of therapeutic interventions. Crucially, the impact of NETs on cardiac remodeling is highly context-dependent, differing fundamentally between acute ischemic injury and chronic metabolic progression. In the acute phase of post-infarction repair, transient NET release facilitates physiological repair and necessary debris clearance; thus, early suppression of NETs in this specific acute setting can actually hinder macrophage-dependent debris clearance (Figure 2). This interference impairs physiological scar formation and ultimately elevates the vulnerability to cardiac rupture.67 However, under chronic stress (such as in the early stages of metabolic or hypertensive heart disease), the persistent presence of NETs directly drives pathological fibrosis by continually activating fibroblasts and inducing cardiomyocyte apoptosis, which leads to ventricular stiffening and diastolic dysfunction.68 Establishing specific therapeutic windows and disease-specific contexts is thus essential to avoid these severe complications while pursuing NET-targeted therapies.
Systemic Comorbidity: NETs as the Pivotal Hub Linking Systemic Diseases to HF
Through NET-mediated shared pathophysiological pathways, a number of systemic disorders eventually compromise cardiac function, resulting in HF.10 Diabetes and HF have a strong association.43 Significant amounts of ROS are produced by high oxidative stress levels in a hyperglycemic setting. ROS function as second messengers, triggering neutrophil signaling pathways that promote the formation of NETs. Diabetic patients exhibit persistent chronic low-grade inflammation, and elevated levels of inflammatory cytokines (including TNF-α and IL-6) further trigger neutrophil production of NETs.22 The accumulation of cholesterol crystals within atherosclerotic plaques is another consequence of the aberrant metabolic state in diabetic individuals. These crystals function as supplementary signals for inflammasome activation in addition to acting as NETosis stimuli.23 Ultimately, NETs in atherosclerotic plaques are resistant to degradation, which facilitates their persistence and has pro-inflammatory effects.
Cardiovascular disease and thrombosis are more prevalent in patients with chronic kidney disease (CKD).69 Prior research has shown that the severity of chronic kidney disease (CKD) increases the incidence and types of arterial thrombotic events.70 Patients with chronic kidney disease frequently have low-grade inflammation. Neutrophils are activated by inflammatory mediators, including IL-8 and IL-6, which promote the release of NETs. In CKD patients, this persistent inflammatory state may result in increased NET production, raising the risk of thrombosis.71
Abnormally high pulmonary artery pressure is the hallmark of pulmonary arterial hypertension (PAH), which can induce right ventricular failure, progressive dyspnea, and fatal outcomes.38 NETs are involved in the pathophysiology of pulmonary arterial smooth muscle cells (PASMCs) proliferation in pulmonary arterial hypertension (PAH). NETs trigger the ILK/β-parvin/RAC1 signaling cascade by activating the CCDC25 receptor on the PASMC surface. This induces morphological and cytoskeletal remodeling in PASMCs, which eventually results in luminal stenosis and hypertrophy of the pulmonary artery wall.72 According to additional research, NETs can also induce angiogenesis and inflammation by activating endothelial cells through the MPO/H2O2-dependent TLR4/NFκB signaling pathway.73
Increased cardiac workload and deterioration of the cardiac microenvironment mediated by NETs are the ultimate consequences of these systemic pathologies, driven by increased right ventricular afterload (PAH) or elevated levels of metabolic stressors (such as in diabetes or chronic kidney disease)10 (Figure 3).
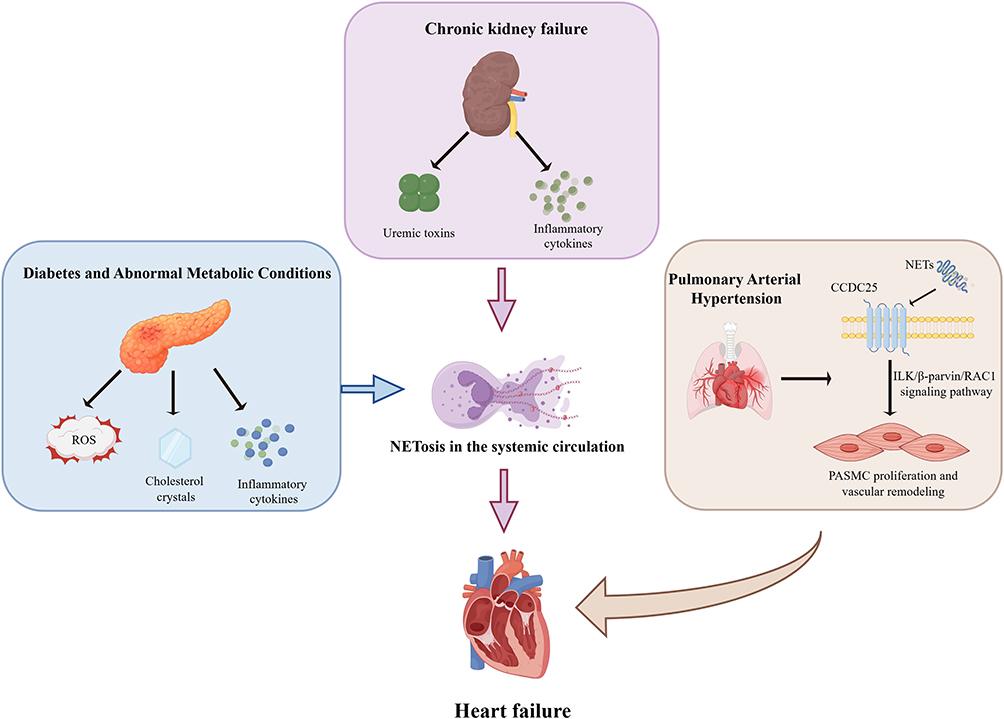 |
Figure 3 NETs as a Pathophysiological Bridge Linking Systemic Comorbidities to HF. Diabetes and metabolic abnormalities trigger systemic NETosis via reactive oxygen species (ROS) generated by high oxidative stress, cholesterol crystals, and pro-inflammatory cytokines. In chronic kidney disease, uremic toxins and inflammatory mediators, such as IL-6 and IL-8, synergistically promote NET release. Within the pulmonary vasculature, NETs bind to CCDC25 receptors on pulmonary arterial smooth muscle cells (PASMCs), activating the ILK/β-parvin/RAC1 signaling axis to induce luminal stenosis and vascular hypertrophy. These multi-organ pathophysiological processes ultimately converge to exacerbate HF progression by increasing right ventricular afterload or intensifying systemic metabolic stress. Abbreviations: CCDC25, coiled-coil domain-containing protein 25; ILK, integrin-linked kinase; NETs, neutrophil extracellular traps; PASMCs, pulmonary arterial smooth muscle cells; RAC1, Ras-related C3 botulinum toxin substrate 1; ROS, reactive oxygen species. |
In summary, the progression of heart failure is classically driven by five intertwined pathophysiological mechanisms, including persistent inflammation, pathological remodeling, cardiomyocyte death, microvascular thrombosis, and neurohormonal or hemodynamic stress. Remarkably, as discussed throughout this section, recent evidence identifies NETs not merely as a byproduct but as critical active mediators participating in every single one of these pathogenic cascades. This multidimensional crosstalk is summarized in Table 2, highlighting why targeting NETs offers a unique opportunity to simultaneously block multiple detrimental pathways in HF.
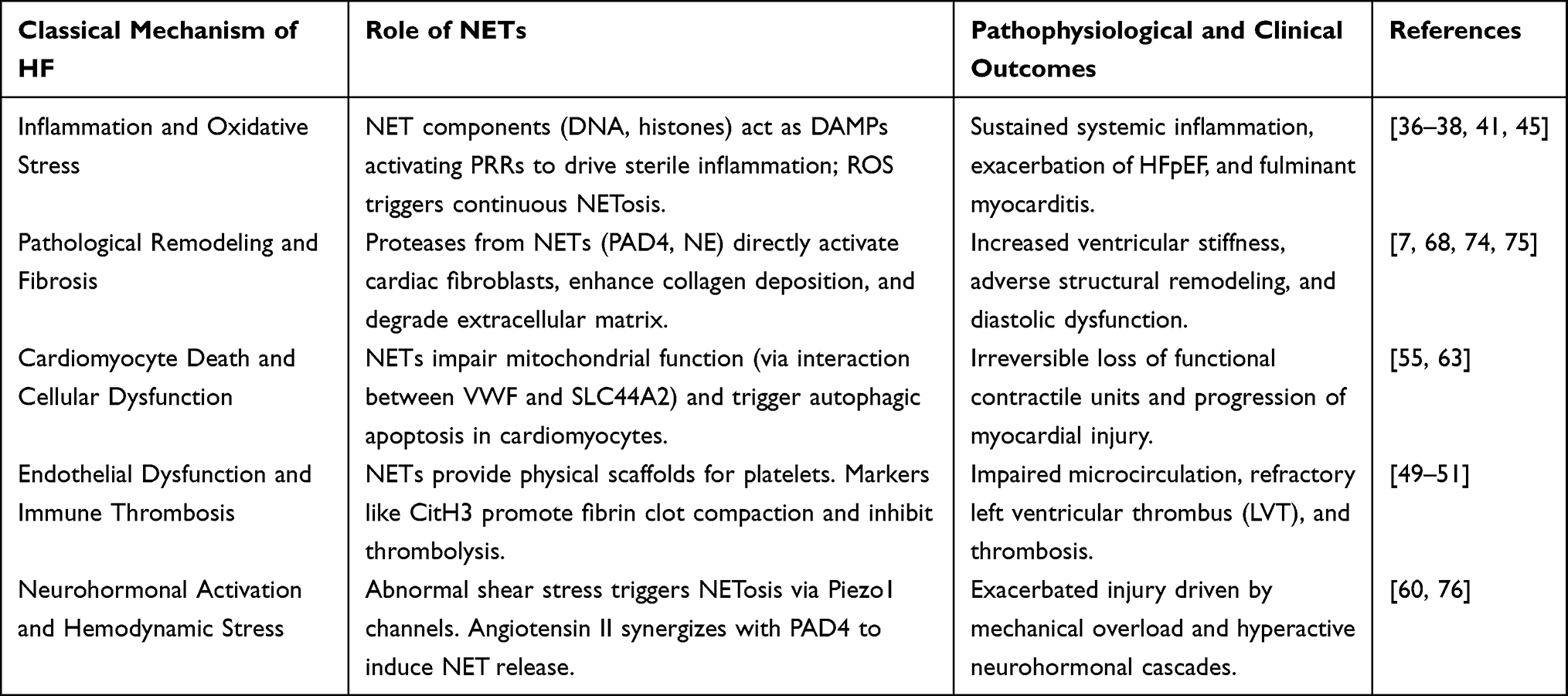 |
Table 2 Crosstalk Between the Five Classical Mechanisms of Heart Failure and NETs |
Furthermore, to enhance the characterization of early subclinical states, contemporary research paradigms have refined the progression of heart failure into six distinct stages. Evaluating the pathological trajectory of NETs within this framework highlights their evolving, stage-dependent roles: during the early phases from behavioral or biological risks to subclinical pre-heart failure (Stages 1–3), metabolic stress stemming from comorbidities triggers a low-level release of NETs, which initiates fibroblast activation and subclinical remodeling. Conversely, in the advanced phases spanning symptomatic progression to refractory heart failure (Stages 4–6), a massive surge in NETs acts as a primary amplifier of immunothrombosis and irreversible myocardial injury.77 Although large-scale quantitative evidence across all six specific stages remains limited, the progressive nature of NETs alongside disease evolution underscores their significant translational potential for the clinical grading of heart failure severity. Consequently, targeted modulation or inhibition of NETs during these early phases (Stages 1 and 2) presents a critical therapeutic window, holding immense preventive promise for halting the transition from subclinical risk factors to overt cardiac dysfunction.
To provide a more intuitive overview of the scientific foundation discussed above, the key clinical and experimental studies that have shaped our understanding of NETs in HF are systematically summarized in Table 3.
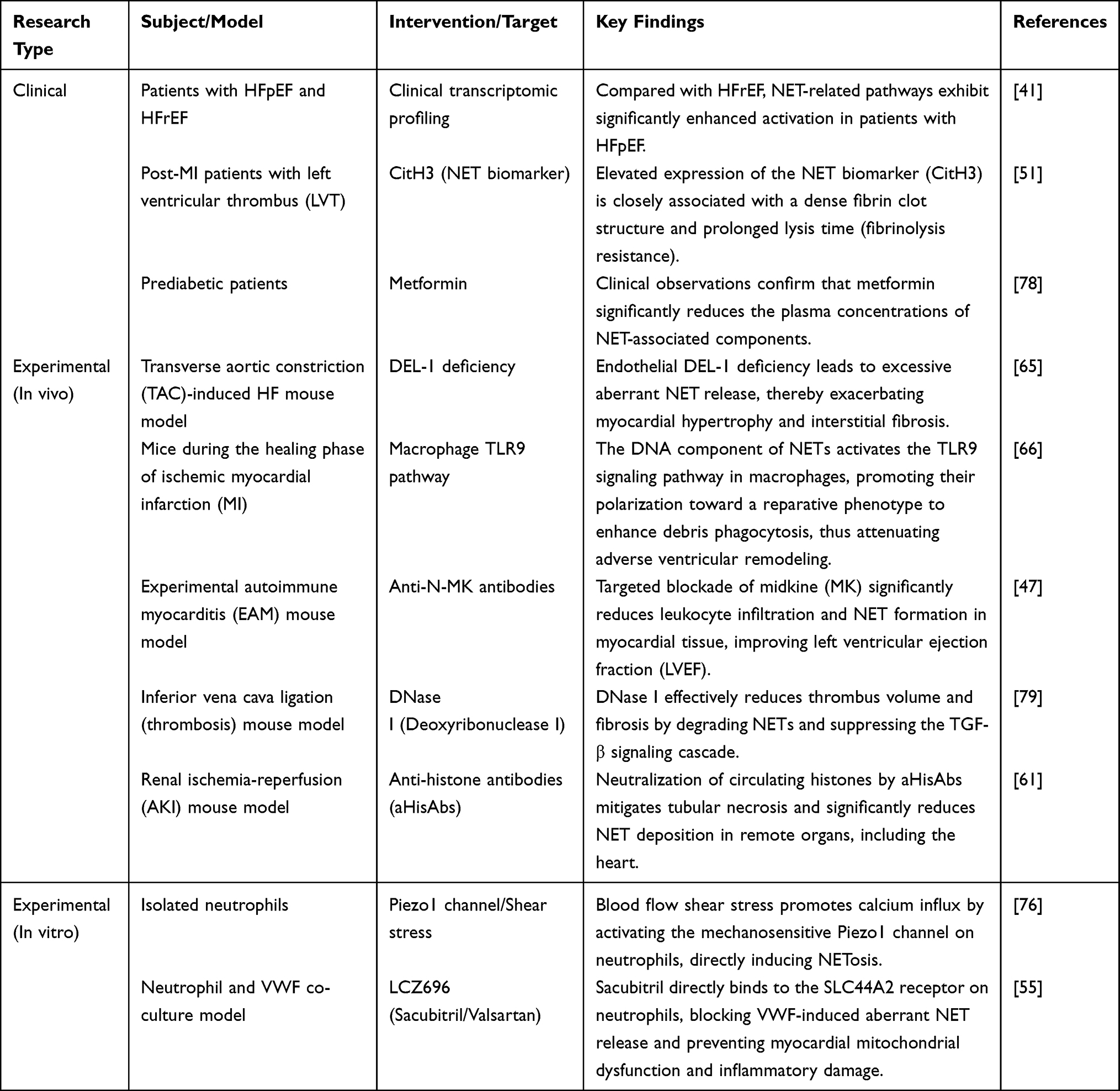 |
Table 3 Summary of Key Clinical and Experimental Studies Targeting NETs in Heart Failure |
NETs as Potential Therapeutic Targets for HF and Comorbidities
Cardiovascular System: Directly Targeting Cardiac and Vascular Pathologies
One of the main mechanisms in the pathological course of HF is immune thrombosis driven by NETs, which exacerbates myocardial ischemia and functional failure by impairing microcirculation. NETs not only function as physical scaffolds to trap platelets and red blood cells,80 but also interact with coagulation factors such as von Willebrand factor (VWF) to promote the formation of dense, refractory thrombus formations. When left ventricular thrombi persist, and ischemic HF is present, this condition is pronounced.55 Even with standardized anticoagulant therapy clinically, some individuals with post-MI HF still have persistent left ventricular thrombus (LVT). Mróz et al revealed that elevated expression of the NETs biomarker (CitH3) was substantially linked with a dense fibrin clot structure and extended lysis time51 These results highlight the limitations of monotherapy anticoagulants against NET-rich thrombi, indicating that when treating HF aggravated by refractory thrombus, NET degradation agents should be used in combination. Thus, multifaceted therapies that target NET-mediated prothrombotic pathways have great potential to improve microcirculation and prognosis in patients with HF.
DNA can be degraded by an enzyme called deoxyribonuclease 1 (DNase1). Breaking down the DNA components of NETs, it attenuates their biological activity, which mitigates thrombus fibrosis and promotes thrombus breakdown and absorption.79,81 Smriti Sharma et al demonstrated in an in vivo mouse inferior vena cava ligation model that DNase1 efficiently lowers thrombus size and fibrosis by degrading NETs and consequently suppressing the TGF-β signaling cascade79 Neutrophils are activated by cardiac damage and hemodynamic changes during HF, which results in the large release of NET.76,80 Given its capacity to lower NET levels, DNase1 holds promise as a potential therapeutic strategy for HF. Furthermore, DNase1 has exhibited excellent safety profiles in clinical applications for cystic fibrosis patients, providing a clinical safety reference for its potential application in HF.82
A novel polysaccharide hydrogel nanoparticle was created by de La Taille et al This nanoparticle can precisely target and bind to P-selectin on thrombus surfaces functionalized with fucoidan. Importantly, the particles concurrently loaded rtPA (fibrinolytic enzyme) and DNase I. These dual-drug-loaded nanoparticles demonstrated a notable synergistic thrombolytic effect within thrombi rich in NETs, according to in vitro and in vivo preclinical experimental results. This strategy reduced the effective dose of rtPA, which decreased the risk of bleeding, while simultaneously significantly increasing thrombus dissolution rates.83 This presents a highly promising translational strategy for precision anti-thrombotic therapy in HF.
Previous investigations have established that hemodynamic shear stress can activate mechanosensitive neutrophils. However, the molecular mechanisms by which shear stress induces NETosis through mechanotransduction mechanisms remain unknown.84 In vitro research by Sara Baratchi’s team confirmed that blood flow shear stress directly promotes calcium influx by activating the mechanosensitive channel Piezo1 on the neutrophil surface. Piezo1 inhibitors (GsMTx4, ruthenium red) or calcium chelators (EGTA) completely abrogate this action.76 In addition to providing a theoretical foundation for the development of anti-thrombotic medications such as Piezo1 inhibitors, this finding provides novel insights into the functional regulation of neutrophils within mechanical microenvironments.
Chronic low-grade sterile inflammation is the main pathogenic mechanism underpinning the onset and progression of HF, with NETs playing a major amplifying role as essential mediators of inflammatory development.7 By preventing important steps in NET release or limiting the function of their main enzymes, it is possible to abrogate the inflammatory process at its source, giving a potential treatment method to delay the course of HF.
For specific HF subtypes, interfering in upstream signaling pathways can have significant anti-inflammatory and protective benefits. Midkine (MK), a heparin-binding growth factor rich in basic amino acids, exacerbates inflammatory reactions by attracting and activating immune cells such as neutrophils and monocytes.85 MK exacerbates cardiac inflammation in myocarditis by promoting polymorphonuclear neutrophil (PMN) migration and infiltration through low-density lipoprotein receptor-related protein 1 (LRP1).86 Anti-N-MK antibody treatment significantly decreased leukocyte infiltration in the heart tissue of experimental autoimmune myocarditis (EAM) mice in vivo, according to Weckbach et al Concurrently, heart contractile performance improved, with significantly preserved left ventricular ejection fraction (LVEF) compared to untreated controls According to these findings, MK targeting reduces NETosis, which mitigates cardiac inflammation and damage.47 This discovery suggests a novel therapeutic target for certain types of HF, such as post-viral myocarditis HF.
LTC4, an inflammatory mediator, is released excessively when ALDH2 deficiency induces endoplasmic reticulum stress. This, in turn, induces accelerated NETosis and exacerbates myocardial damage, ultimately resulting in an adverse HF prognosis. Based on in vitro studies using patient-derived samples, inhibiting LTC4 receptors with pranlukast can prevent endoplasmic reticulum stress-induced NETosis in patients with ALDH2 gene abnormalities. Pranlukast significantly decreased neutrophil infiltration and NET deposition, decreased infarct size, and enhanced cardiac function in an ALDH2-knockout mouse myocardial I/R model in vivo, as Yang K et al demonstrated.64
One essential drug for treating HF is sacubitril/valsartan (LCZ696).87 Through combined in silico molecular docking and in vitro thermostability tests, Mang et al demonstrated that sacubitril, the active component in LCZ696, can directly target and bind to the SLC44A2 receptor on the surface of neutrophils By successfully preventing upstream von Willebrand factor (VWF) signaling from activating SLC44A2, this binding prevents the aberrant release of NETs downstream, which in turn prevents myocardial mitochondrial dysfunction and inflammatory damage.55 This finding implies that LCZ696 produces an additional, additive cardioprotective effect via suppressing NETosis. This not only explains the molecular basis for its pleiotropic effects but also provides theoretical justification for clinically targeting SLC44A2 in combination therapy for HF.
A major pathological alteration that induces ventricular wall stiffness, decreased compliance, and compromised contractile performance is Cardiac fibrosis, which is also a defining characteristic of the development of HF.9 NETs not only function as physical scaffolds, but the active components and important enzymes they release (such as PAD4) can directly activate fibroblasts, enabling collagen deposition and pathological remodeling.7,74 A key enzyme in the production of NETs, PAD4 catalyzes the citrullination of histone H3, which induces chromatin decondensation and the release of NETs.15 PAD4 was previously found to be a key factor in age-related organ fibrosis by Martinod et al Further in vivo research on a cardiac aging model by Van Bruggen et al confirmed that, while maintaining cardiac contraction and relaxation capabilities, selective deletion of the PAD4 gene in neutrophils significantly decreased collagen deposition in the hearts of aged mice According to experimental findings, fibrosis-induced HF associated with aging can be effectively countered by targeting PAD4. Cl-amidine is a pan-PAD inhibitor, demonstrating particularly significant inhibitory effects against PAD4 activity.88 In vitro biochemical and structural studies indicate that Cl-amidine competitively binds to the active site of PAD4, thereby irreversibly inactivating its enzymatic activity.89
Blocking the Lung-Heart Inflammation Axis: Intervening in Respiratory Disease-Driven HF Progression
NETs act as key mediators driving the progression from respiratory disorders to heart failure. Neutrophil elastase (NE) is a serine protease released by neutrophils. As a key component of NETs, NE can degrade extracellular matrix (ECM) components such as collagen and elastic fibers alongside other proteases, thereby disrupting tissue structural integrity.18 Studies have demonstrated that NE promotes fibroblast proliferation by degrading insulin receptor substrate-1 (IRS-1) to activate the PI3K/Akt pathway.75 Gregory et al found that administering the NE inhibitor ONO-5046 to mice with pulmonary fibrosis in vivo significantly improved fibrosis severity, despite having no apparent effect on inflammatory infiltration Furthermore, in vitro experimental results demonstrated that NE inhibitors suppress NE’s direct proliferation and differentiation effects on fibroblasts by blocking its protease activity.90 Although more research is needed to determine whether NE inhibitors are effective in treating HF, these experimental results offer compelling support for them as a treatment approach for pulmonary fibrosis. The possible use of NE inhibitors in HF with a pronounced fibrotic component may be investigated in future studies. Suzuki et al injected Cl-amidine intraperitoneally into mice that had lung fibrosis induced by bleomycin (BLM) According to these in vivo experimental findings, Cl-amidine reduces fibrosis by blocking NETs.91 Therefore, a major potential therapeutic target for HF associated with aging and inflammation may be the selective suppression of PAD4 activity by experimental inhibitors such as Cl-amidine. However, the clinical translation of pan-NET inhibitors like Cl-amidine requires careful evaluation. Since NETs are fundamental to innate host defense, systemic and non-selective suppression of NETosis inherently carries the risk of severe immunosuppression and increased susceptibility to opportunistic infections, which must be carefully managed in vulnerable HF patients.
A macrolide antibiotic with broad-spectrum antibacterial, anti-inflammatory, and immunomodulatory properties is erythromycin.92 In addition to being a major contributor to cardiovascular disorders such as HF, smoking is a major risk factor for COPD. Erythromycin pretreatment significantly decreased ROS levels produced by neutrophils activated with cigarette smoke extract (CSE), according to in vitro research by Hui Zhang et al on neutrophils in COPD patients Additionally, it inhibited the development of NETs in vitro by significantly reducing the inflammatory mediators (IL-1β, IL-12, and TNF-α) generated by monocyte-derived dendritic cells (mDCs) activated by NETs.24 HF and COPD are linked by similar risk factors and reciprocal pathophysiological mechanisms.93 This work highlights erythromycin’s potential to prevent NET formation, providing a novel therapeutic approach for the concurrent treatment of HF and COPD to enhance patient outcomes. Furthermore, the clinical application of erythromycin in HF management necessitates extreme caution. As a macrolide antibiotic, it is well-documented to induce QT interval prolongation, which can trigger fatal ventricular arrhythmias in patients with pre-existing structurally abnormal hearts. Therefore, its application must be carefully weighed against these arrhythmogenic risks.
Alleviating Cardio-Renal Syndrome: Mitigating Secondary Cardiac Damage Induced by Acute Kidney Injury
Damaged kidneys release DAMPs, which travel through the circulation and have an impact on the heart. NETs are essential in this process because they connect the pathophysiological systems of the kidneys and heart. One of the main constituents of NETs, histones, can function as damage-associated molecular patterns (DAMPs). They exacerbate inflammation by triggering the release of inflammatory cytokines such as TNF-α and IL-6 through the activation of Toll-like receptors (TLR2/4).61,94 Free histones also have a significant positive charge that can directly damage cell membrane structures and result in cell death.95 In AKI, antibodies that can specifically recognize and bind to histones are known as anti-histone antibodies (aHisAbs). Histones’ direct cellular harm and pro-inflammatory effects are mitigated when aHisAbs bind to them, neutralize their charge toxicity, and stop them from interacting with receptors.96 In an in vivo mouse model of renal ischemia-reperfusion injury (IRI), Nakazawa et al demonstrated that pretreatment with aHisAbs improved renal function, decreased tubular cell necrosis and expression of inflammatory mediators (eg, TNF-α, IL-6), and significantly reduced NET deposition in renal tissue and other affected organs61 These results show that by reducing circulating histone levels, aHisAbs can mitigate inflammatory damage to several organs, including the heart. Renal IRI and myocardial ischemia-reperfusion injury (such as post-infarction HF) share DAMP-driven pathogenic pathways,97 indicating that aHisAbs may have protective effects in such HF conditions.
Regulating Metabolic Dysregulation: Targeting Immune Mechanisms in Diabetes-Related HF
Metabolic diseases are high-risk factors for HF, and NETs are the connection between metabolic stress and vascular damage. Current evidence suggests that the widely prescribed antidiabetic medication metformin has potent anti-inflammatory properties, especially when it comes to inhibiting NETs formation.98 Metformin significantly decreased the plasma concentrations of neutrophil extracellular trap (NET)-associated components in prediabetic individuals, according to clinical observations by Menegazzo et al. Metformin inhibits the protein kinase C (PKC)-NADPH oxidase pathway, which prevents NET formation, according to additional in vitro research.78 These results offer a theoretical foundation for metformin’s possible ability to prevent diabetic heart complications. Despite these theoretical benefits, metformin must be used judiciously in clinical settings. Its application carries a risk of fatal lactic acidosis, making it strictly contraindicated in patients presenting with acute decompensated HF or concurrent severe renal impairment. The potential therapeutic agents and their clinical safety considerations are summarized in Table 4.
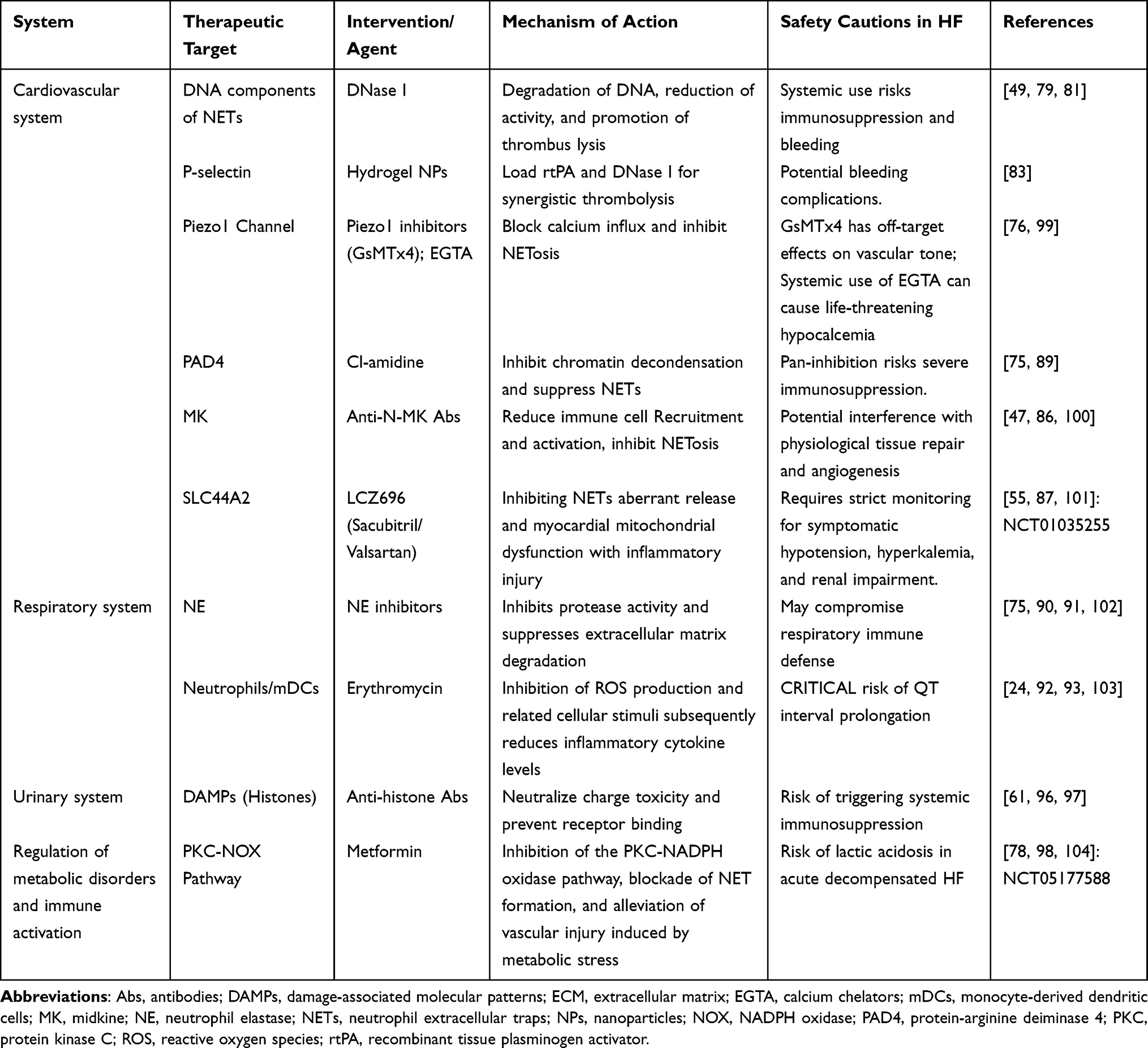 |
Table 4 Summary of Strategies for NETs as Potential Therapeutic Targets |
Conclusion
The role of NETs in the pathophysiological network of HF is described in this article. On the one hand, NETs release DAMPs, which activate immune pathways and maintain a chronic low-grade inflammatory state in the heart; on the other hand, NETs act as physical scaffolds that promote immune thrombogenesis, which impairs microcirculation. NETs promote cardiomyocyte mortality and mitochondrial malfunction, which leads to cardiac remodeling, the direct pathophysiological induction of structural alterations in the heart. Furthermore, NETs represent a crucial molecular hub connecting behavioral risk factors (eg., unhealthy lifestyle and chronic stress) and systemic comorbidities (diabetes, hypertension, chronic kidney disease, respiratory diseases) to cardiac damage across various systems.
Active research is also being done on NET-targeting interventions to prevent HF and associated conditions. Current strategies cover a variety of phases, from increasing NETs clearance (eg, utilizing DNase1) to preventing NETs production at the source (eg, targeting the PAD4 enzyme or the VWF-SLC44A2 signaling axis) Notably, research on drug repurposing has shown that established pharmaceuticals such as metformin and erythromycin, as well as therapeutically available drugs such as sacubitril/valsartan, have extra cardioprotective benefits via blocking NETosis This offers practical translational approaches for clinical intervention. Although these intervention strategies have demonstrated significant potential in preclinical studies, they currently lack support from clinical trials. Therefore, further validation of the efficacy and safety of these targets remains a core challenge in achieving precision medicine translation.
Targeted treatment for NETs still has difficulties, though. Future studies should also aim to reconcile the preservation of neutrophils’ natural immune response with the efficient targeting of pathological NETosis, while carefully selecting safe therapeutic windows to avoid fatal side effects such as severe immunosuppression or bleeding. Attention should also be paid to the intricate mechanisms governing interactions between NETs and other innate or adaptive immune cells. Simultaneously, exploring the stage-dependent evolution of NETs across the spectrum of HF, from subclinical risk phases to advanced stages, holds significant promise for early prevention and clinical grading. More thorough research into the heterogeneity of NETs across various comorbidities and HF phenotypes will provide more accurate precision medicine strategies for successfully treating HF.
Abbreviations
Abs, antibodies; AF, atrial fibrillation; AKI, acute kidney injury; AMI, acute myocardial infarction; CCDC25, coiled-coil domain-containing protein 25; CitH3, citrullinated histone H3; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; DAMPs, damage-associated molecular patterns; DEL-1, developmental endothelial locus-1; DNase1, deoxyribonuclease 1; EAT, epicardial adipose tissue; ECM, extracellular matrix; EGTA, ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; ILK, integrin-linked kinase; LRP1, LDL receptor-related protein 1; LVT, left ventricular thrombus; MAC-1, macrophage-1 antigen; mDCs, monocyte-derived dendritic cells; MK, midkine; MPO, myeloperoxidase; mtROS, mitochondrial reactive oxygen species; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NOX, NADPH oxidase; NPs, nanoparticles; OSE, oxidation-specific epitopes; PAD4, protein-arginine deiminase 4; PAH, pulmonary arterial hypertension; PASMCs, pulmonary arterial smooth muscle cells; PKC, protein kinase C; PMA, phorbol 12,13-dimyristate 13-acetate; PRRs, pattern recognition receptors; RAC1, Ras-related C3 botulinum toxin substrate 1; ROS, reactive oxygen species; rtPA, recombinant tissue plasminogen activator; SGLT2, sodium-glucose cotransporter 2; SLC44A2, solute carrier family 44 member 2; TLR, Toll-like receptor; TNF-α, tumor necrosis factor-alpha; VWF, von Willebrand factor.
Data Sharing Statement
Data that support the findings of this study are available from the corresponding author, upon reasonable request.
Author Contributions
S.Y.: Visualization, Writing – original draft, Writing – review and editing. Z.D.: Investigation, Data curation, Writing – review and editing. X.J.: Investigation, Data curation, Writing – review and editing. X.L.: Validation, Supervision, Writing – review and editing. X.W.: Methodology, Validation, Supervision, Writing – review and editing. B.L.: Methodology, Validation, Supervision, Writing – review and editing. R.Y.: Validation, Supervision, Writing – review and editing. G.F.: Validation, Supervision, Writing – review and editing. Y.W.: Validation, Supervision, Writing – review and editing. M.Z.: Conceptualization, Project administration, Writing – review and editing. J.W.: Conceptualization, Funding acquisition, Writing – review and editing. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Henan Province Science and Technology Research Projects (Grant No. 252102311270), the Youth Talent Support Project of the China Association of Chinese Medicine (2025-QNRC2-B01), the National Natural Science Foundation of China (Grant No. 82505477), the Major and Difficult Diseases Clinical Collaboration Project of Integrated Traditional Chinese and Western Medicine for Heart Failure (Grant No. ZDYN-2024-A-090), and the Noncommunicable Chronic Diseases-National Science and Technology Major Project (Grant No. 2024ZD0522000, 2024ZD0522001).
Disclosure
All authors of this study claim there is no conflict of interest.
References
1. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):368–18. doi:10.1038/nrcardio.2016.25
2. Jones NR, Roalfe AK, Adoki I, Hobbs FDR, Taylor CJ. Survival of patients with chronic heart failure in the community: a systematic review and meta-analysis. Eur J Heart Fail. 2019;21(11):1306–1325. doi:10.1002/ejhf.1594
3. Zhang SY. Chinese Guidelines for the Diagnosis and Treatment of Heart Failure 2024. J Geriatr Cardiol. 2025;22(3):277–331. doi:10.26599/1671-5411.2025.03.002
4. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. doi:10.1083/jcb.201006052
5. Mutua V, Gershwin LJ. A Review of Neutrophil Extracellular Traps (NETs) in Disease: potential Anti-NETs Therapeutics. Clin Rev Allergy Immunol. 2021;61(2):194–211. doi:10.1007/s12016-020-08804-7
6. Hasler P, Giaglis S, Hahn S. Neutrophil extracellular traps in health and disease. Swiss Med Wkly. 2016;146:w14352. doi:10.4414/smw.2016.14352
7. Sorvillo N, Cherpokova D, Martinod K, Wagner DD. Extracellular DNA NET-Works With Dire Consequences for Health. Circ Res. 2019;125(4):470–488. doi:10.1161/circresaha.119.314581
8. Geng X, Wang DW, Li H. The pivotal role of neutrophil extracellular traps in cardiovascular diseases: mechanisms and therapeutic implications. Biomed Pharmacother. 2024;179:117289. doi:10.1016/j.biopha.2024.117289
9. Kostin S, Krizanic F, Kelesidis T, Pagonas N. The role of NETosis in heart failure. Heart Fail Rev. 2024;29(5):1097–1106. doi:10.1007/s10741-024-10421-x
10. Ling S, Xu JW. NETosis as a Pathogenic Factor for Heart Failure. Oxid Med Cell Longev. 2021;2021:6687096. doi:10.1155/2021/6687096
11. Shirakawa K, Sano M. Neutrophils and Neutrophil Extracellular Traps in Cardiovascular Disease: an Overview and Potential Therapeutic Approaches. Biomedicines. 2022;10(8):1850. doi:10.3390/biomedicines10081850
12. Liori S, Kapelios CJ, Savarese G, Filippatos G. Heart failure evidence update 2026. Heart Fail Rev. 2026;31(1):1. doi:10.1007/s10741-026-10609-3
13. Kallash M, Frishman WH. The Potential Role of Anti-Inflammatory Therapy in Heart Failure Treatment. Cardiol Rev. 2026;2026:1160. doi:10.1097/crd.0000000000001160
14. Hidalgo A, Libby P, Soehnlein O, Aramburu IV, Papayannopoulos V, Silvestre-Roig C. Neutrophil extracellular traps: from physiology to pathology. Cardiovasc Res. 2022;118(13):2737–2753. doi:10.1093/cvr/cvab329
15. Wang Y, Du C, Zhang Y, Zhu L. Composition and Function of Neutrophil Extracellular Traps. Biomolecules. 2024;14(4):416. doi:10.3390/biom14040416
16. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385
17. Ruiz-Limón P, Ortega R, Arias de la Rosa I, et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl Res. 2017;183:87–103. doi:10.1016/j.trsl.2016.12.003
18. Burgener SS, Schroder K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb Perspect Biol. 2020;12(7):37028. doi:10.1101/cshperspect.a037028
19. Ibrahim N, Eilenberg W, Neumayer C, Brostjan C. Neutrophil Extracellular Traps in Cardiovascular and Aortic Disease: a Narrative Review on Molecular Mechanisms and Therapeutic Targeting. Int J Mol Sci. 2024;25(7):3983. doi:10.3390/ijms25073983
20. Natorska J, Ząbczyk M, Undas A. Neutrophil extracellular traps (NETs) in cardiovascular diseases: from molecular mechanisms to therapeutic interventions. Kardiol Pol. 2023;81(12):1205–1216. doi:10.33963/v.kp.98520
21. Szymańska Z, Staniewski A, Karpiński M, et al. The Role of Neutrophil Extracellular Networks in Cardiovascular Pathology. Cells. 2025;14(19):1562. doi:10.3390/cells14191562
22. Zhu Y, Xia X, He Q, et al. Diabetes-associated neutrophil NETosis: pathogenesis and interventional target of diabetic complications. Front Endocrinol. 2023;14:1202463. doi:10.3389/fendo.2023.1202463
23. Josefs T, Barrett TJ, Brown EJ, et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight. 2020;5(7):134796. doi:10.1172/jci.insight.134796
24. Zhang H, Qiu SL, Tang QY, et al. Erythromycin suppresses neutrophil extracellular traps in smoking-related chronic pulmonary inflammation. Cell Death Dis. 2019;10(9):678. doi:10.1038/s41419-019-1909-2
25. Meng S, Huang T, Zhou Z, Yu L, Wang H. Chronic mild stress exacerbates atrial fibrillation and neutrophil extracellular traps formation through S100A8/A9 signaling. Signal Transduct Target Ther. 2025;108:2.
26. Pérez-Olivares L, Soehnlein O. Contemporary Lifestyle and Neutrophil Extracellular Traps: an Emerging Link in Atherosclerosis Disease. Cells. 2021;2021(8):1985. doi:10.3390/cells10081985
27. Dąbrowska D, Jabłońska E, Garley M, Ratajczak-Wrona W, Iwaniuk A. New Aspects of the Biology of Neutrophil Extracellular Traps. Scand J Immunol. 2016;84(6):317–322. doi:10.1111/sji.12494
28. Wang H, Kim SJ, Lei Y, et al. Neutrophil extracellular traps in homeostasis and disease. Signal Transduct Target Ther. 2024;9(1):235. doi:10.1038/s41392-024-01933-x
29. Frade-Sosa B, Sanmartí R. Neutrophils, neutrophil extracellular traps, and rheumatoid arthritis: an updated review for clinicians. Reumatol Clin. 2023;19(9):515–526. doi:10.1016/j.reumae.2023.10.002
30. Liu Y, Guo Y, Wu X, Yan P, Wei Y. Neutrophil Extracellular Traps: potential Therapeutic Targets of Traditional Chinese Medicine and Natural Products for Cardiovascular Diseases. Pharmaceuticals. 2026;19(1):183. doi:10.3390/ph19010183
31. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi:10.1083/jcb.200606027
32. Kenny EF, Herzig A, Krüger R, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 2017;6:24437. doi:10.7554/eLife.24437
33. Pilsczek FH, Salina D, Poon KK, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185(12):7413–7425. doi:10.4049/jimmunol.1000675
34. Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:38. doi:10.3389/fimmu.2013.00038
35. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. 2015;112(9):2817–2822. doi:10.1073/pnas.1414055112
36. Shaihov-Teper O, Ram E, Ballan N, et al. Extracellular Vesicles From Epicardial Fat Facilitate Atrial Fibrillation. Circulation. 2021;143(25):2475–2493. doi:10.1161/circulationaha.120.052009
37. Gaeta M, Bandera F, Tassinari F, et al. Is epicardial fat depot associated with atrial fibrillation? A systematic review and meta-analysis. Europace. 2017;19(5):747–752. doi:10.1093/europace/euw398
38. Maron BA, Humbert M. Finding Pulmonary Arterial Hypertension-Switching to Offense to Mitigate Disease Burden. JAMA Cardiol. 2022;7(4):369–370. doi:10.1001/jamacardio.2022.0011
39. Halade GV, Bäck M, Kain V. Inflammation-resolution signalling in cardiac repair, remodelling, and heart failure. Eur Heart J Open. 2025;5(6):oeaf157. doi:10.1093/ehjopen/oeaf157
40. Peikert A, Vacca A, Norata GD, Schiattarella GG, Osto E. Cellular Interactions and Immunometabolic Mechanisms in Heart Failure With Preserved Ejection Fraction: from Molecular Mechanisms to Clinical Evidence. Circ Heart Fail. 2026;19(3):e012674. doi:10.1161/circheartfailure.125.012674
41. VANE BJ, Tromp J, Gevaert AB, et al. Activation of Neutrophil Extracellular Trap Formation in Patients with Heart Failure and a Preserved Ejection Fraction. J Card Fail. 2025;2025:1. doi:10.1016/j.cardfail.2025.02.015
42. Meulendijks ER, Al-Shama RFM, Kawasaki M, et al. Atrial epicardial adipose tissue abundantly secretes myeloperoxidase and activates atrial fibroblasts in patients with atrial fibrillation. J Transl Med. 2023;21(1):366. doi:10.1186/s12967-023-04231-2
43. Vecchié A, Montecucco F, Carbone F, Dallegri F, Bonaventura A. Diabetes and Vascular Disease: is It All About Glycemia? Curr Pharm Des. 2019;25(29):3112–3127. doi:10.2174/1381612825666190830181944
44. Zeng JH, Liu YX, Yuan J, et al. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. 2020;48(5):773–777. doi:10.1007/s15010-020-01424-5
45. Nicolai L, Leunig A, Brambs S, et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation. 2020;142(12):1176–1189. doi:10.1161/circulationaha.120.048488
46. Hammond ME, Christensen ED, Belenky M, Snow GL, Shah K, Hammond MEH. Evidence of autoinflammation as a principal mechanism of myocardial injury in SARS-CoV-2 PCR-positive medical examiner cases. Diagn Pathol. 2023;18(1):114. doi:10.1186/s13000-023-01397-7
47. Weckbach LT, Grabmaier U, Uhl A, et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. J Exp Med. 2019;216(2):350–368. doi:10.1084/jem.20181102
48. Shantsila E, Lip GY. Preventing Thrombosis to Improve Outcomes in Heart Failure Patients. Prog Cardiovasc Dis. 2016;58(4):386–392. doi:10.1016/j.pcad.2015.09.005
49. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–15885. doi:10.1073/pnas.1005743107
50. Ząbczyk M, Ariëns RAS, Undas A. Fibrin clot properties in cardiovascular disease: from basic mechanisms to clinical practice. Cardiovasc Res. 2023;119(1):94–111. doi:10.1093/cvr/cvad017
51. Mróz K, Paszek E, Polak M, Undas A. Prothrombotic fibrin clot phenotype as a risk factor for persistent left ventricular thrombus following acute myocardial infarction. J Thromb Haemost. 2025;23(9):2903–2912. doi:10.1016/j.jtha.2025.06.003
52. Tong W, Lodish HF. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med. 2004;200(5):569–580. doi:10.1084/jem.20040762
53. Dou H, Kotini A, Liu W, et al. Oxidized Phospholipids Promote NETosis and Arterial Thrombosis in LNK(SH2B3) Deficiency. Circulation. 2021;144(24):1940–1954. doi:10.1161/circulationaha.121.056414
54. Sun S, Zou X, Wang D, et al. IRGM/Irgm1 deficiency inhibits neutrophil-platelet interactions and thrombosis in experimental atherosclerosis and arterial injury. Biomed Pharmacother. 2023;158:114152. doi:10.1016/j.biopha.2022.114152
55. Mang G, Chen J, Sun P, et al. Von Willebrand factor exacerbates heart failure through formation of neutrophil extracellular traps. Eur Heart J. 2024;45(37):3853–3867. doi:10.1093/eurheartj/ehae517
56. Tang X, Wang P, Zhang R, et al. KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression. J Clin Invest. 2022;132(3):147191. doi:10.1172/jci147191
57. Riascos-Bernal DF, Sibinga NE. Neutrophil extracellular traps in cardiac hypertrophy: a KLF2 perspective. J Clin Invest. 2022;132(3):156453. doi:10.1172/jci156453
58. Lee HW, An JN, Lee HS, et al. Neutrophil extracellular traps and heparin-induced antibodies contribute to vascular access thrombosis in hemodialysis patients. Kidney Res Clin Pract. 2021;40(4):712–723. doi:10.23876/j.krcp.21.080
59. Sun L, Li H, Li Y, et al. Proteomic insights into platelet dysregulation and pathogenic mechanisms of chronic thromboembolic pulmonary hypertension. J Transl Med. 2025;23(1):1074. doi:10.1186/s12967-025-06891-8
60. Yu F, Chen J, Zhang X, Ma Z, Wang J, Wu Q. Role of Neutrophil Extracellular Traps in Hypertension and Their Impact on Target Organs. J Clin Hypertens. 2025;27(1):e14942. doi:10.1111/jch.14942
61. Nakazawa D, Kumar SV, Marschner J, et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J Am Soc Nephrol. 2017;28(6):1753–1768. doi:10.1681/asn.2016080925
62. Ichimura S, Misaka T, Ogawara R, et al. Neutrophil Extracellular Traps in Myocardial Tissue Drive Cardiac Dysfunction and Adverse Outcomes in Patients With Heart Failure With Dilated Cardiomyopathy. Circ Heart Fail. 2024;17(6):e011057. doi:10.1161/circheartfailure.123.011057
63. He L, Liu R, Yue H, et al. Interaction between neutrophil extracellular traps and cardiomyocytes contributes to atrial fibrillation progression. Signal Transduct Target Ther. 2023;8(1):279. doi:10.1038/s41392-023-01497-2
64. Yang K, Gao R, Chen H, et al. Myocardial reperfusion injury exacerbation due to ALDH2 deficiency is mediated by neutrophil extracellular traps and prevented by leukotriene C4 inhibition. Eur Heart J. 2024;45(18):1662–1680. doi:10.1093/eurheartj/ehae205
65. Zhao M, Zheng Z, Yin Z, et al. DEL-1 deficiency aggravates pressure overload-induced heart failure by promoting neutrophil infiltration and neutrophil extracellular traps formation. Biochem Pharmacol. 2023;218:115912. doi:10.1016/j.bcp.2023.115912
66. Wei X, Zou S, Xie Z, et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc Res. 2022;118(9):2179–2195. doi:10.1093/cvr/cvab269
67. Horckmans M, Ring L, Duchene J, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38(3):187–197. doi:10.1093/eurheartj/ehw002
68. Antipenko S, Mayfield N, Jinno M, et al. Neutrophils are indispensable for adverse cardiac remodeling in heart failure. J Mol Cell Cardiol. 2024;189:1–11. doi:10.1016/j.yjmcc.2024.02.005
69. Thakur M, Junho CVC, Bernhard SM, Schindewolf M, Noels H, Döring Y. NETs-Induced Thrombosis Impacts on Cardiovascular and Chronic Kidney Disease. Circ Res. 2023;132(8):933–949. doi:10.1161/circresaha.123.321750
70. Wetmore JB, Kou C, Khan NF, et al. Major adverse thrombotic events and bleeding in stage 4 and 5 chronic kidney disease and dialysis-dependent end-stage kidney disease. J Thromb Haemost. 2025;23(12):3920–3931. doi:10.1016/j.jtha.2025.08.025
71. Ebert T, Pawelzik SC, Witasp A, et al. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins. 2020;12(4):227. doi:10.3390/toxins12040227
72. Sun H, Du Z, Zhang X, et al. Neutrophil extracellular traps promote proliferation of pulmonary smooth muscle cells mediated by CCDC25 in pulmonary arterial hypertension. Respir Res. 2024;25(1):183. doi:10.1186/s12931-024-02813-2
73. Aldabbous L, Abdul-Salam V, McKinnon T, et al. Neutrophil Extracellular Traps Promote Angiogenesis: evidence From Vascular Pathology in Pulmonary Hypertension. Arterioscler Thromb Vasc Biol. 2016;36(10):2078–2087. doi:10.1161/atvbaha.116.307634
74. Heger LA, Schommer N, Fukui S, et al. Inhibition of protein arginine deiminase 4 prevents inflammation-mediated heart failure in arthritis. Life Sci Alliance. 2023;6(10):4. doi:10.26508/lsa.202302055
75. Houghton AM, Rzymkiewicz DM, Ji H, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16(2):219–223. doi:10.1038/nm.2084
76. Baratchi S, Danish H, Chheang C, et al. Piezo1 expression in neutrophils regulates shear-induced NETosis. Nat Commun. 2024;15(1):7023. doi:10.1038/s41467-024-51211-1
77. Jankajova M, Singh RB, Hristova K, et al. Identification of Pre-Heart Failure in Early Stages: the Role of Six Stages of Heart Failure. Diagnostics. 2024;14(23). doi:10.3390/diagnostics14232618
78. Menegazzo L, Scattolini V, Cappellari R, et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018;55(6):593–601. doi:10.1007/s00592-018-1129-8
79. Sharma S, Hofbauer TM, Ondracek AS, et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood. 2021;137(8):1104–1116. doi:10.1182/blood.2020005861
80. Döring Y, Libby P, Soehnlein O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: recent Experimental and Clinical Insights. Circ Res. 2020;126(9):1228–1241. doi:10.1161/circresaha.120.315931
81. Han DSC, Ni M, Chan RWY, et al. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am J Hum Genet. 2020;106(2):202–214. doi:10.1016/j.ajhg.2020.01.008
82. Delfino D, Mori G, Rivetti C, et al. Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease. Biomolecules. 2021;11(3):410. doi:10.3390/biom11030410
83. de La Taille T, Sarfati P, Aid R, et al. Microemulsion-Inspired Polysaccharide Nanoparticles for an Advanced Targeted Thrombolytic Treatment. ACS Nano. 2025;19(2):2944–2960. doi:10.1021/acsnano.4c17049
84. Fukuda S, Yasu T, Predescu DN, Schmid-Schönbein GW. Mechanisms for regulation of fluid shear stress response in circulating leukocytes. Circ Res. 2000;86(1):E13. doi:10.1161/01.res.86.1.e13
85. Yan P, Jimenez ER, Li Z, et al. Midkine as a driver of age-related changes and increase in mammary tumorigenesis. Cancer Cell. 2024;42(11):1936–1954.e9. doi:10.1016/j.ccell.2024.09.002
86. Majaj M, Weckbach LT. Midkine-A novel player in cardiovascular diseases. Front Cardiovasc Med. 2022;9:1003104. doi:10.3389/fcvm.2022.1003104
87. McMurray JJ, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11):993–1004. doi:10.1056/NEJMoa1409077
88. Shi G, Liu L, Cao Y, et al. Inhibition of neutrophil extracellular trap formation ameliorates neuroinflammation and neuronal apoptosis via STING-dependent IRE1α/ASK1/JNK signaling pathway in mice with traumatic brain injury. J Neuroinflammation. 2023;20(1):222. doi:10.1186/s12974-023-02903-w
89. Mondal S, Thompson PR. Protein Arginine Deiminases (PADs): biochemistry and Chemical Biology of Protein Citrullination. Acc Chem Res. 2019;52(3):818–832. doi:10.1021/acs.accounts.9b00024
90. Gregory AD, Kliment CR, Metz HE, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. 2015;98(2):143–152. doi:10.1189/jlb.3HI1014-493R
91. Suzuki M, Ikari J, Anazawa R, et al. PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am J Respir Cell Mol Biol. 2020;63(6):806–818. doi:10.1165/rcmb.2019-0433OC
92. Undheim K. Scaffold Modifications in Erythromycin Macrolide Antibiotics. A Chemical Minireview. Molecules. 2020;25(17):3941. doi:10.3390/molecules25173941
93. Frantz S, Hundertmark MJ, Schulz-Menger J, Bengel FM, Bauersachs J. Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies. Eur Heart J. 2022;43(27):2549–2561. doi:10.1093/eurheartj/ehac223
94. Li X, Ye Y, Peng K, Zeng Z, Chen L, Zeng Y. Histones: the critical players in innate immunity. Front Immunol. 2022;13:1030610. doi:10.3389/fimmu.2022.1030610
95. Lorch Y, Kornberg RD, Maier-Davis B. Role of the histone tails in histone octamer transfer. Nucleic Acids Res. 2023;51(8):3671–3678. doi:10.1093/nar/gkad079
96. Ma W, Zhu J, Bai L, Zhao P, Li F, Zhang S. The role of neutrophil extracellular traps and proinflammatory damage-associated molecular patterns in idiopathic inflammatory myopathies. Clin Exp Immunol. 2023;213(2):202–208. doi:10.1093/cei/uxad059
97. Francisco J, Del Re DP. Inflammation in Myocardial Ischemia/Reperfusion Injury: underlying Mechanisms and Therapeutic Potential. Antioxidants. 2023;12(11):1944. doi:10.3390/antiox12111944
98. Zhou Y, Tao W, Shen F, Du W, Xu Z, Liu Z. The Emerging Role of Neutrophil Extracellular Traps in Arterial, Venous and Cancer-Associated Thrombosis. Front Cardiovasc Med. 2021;8:786387. doi:10.3389/fcvm.2021.786387
99. Ducrocq GP, Anselmi L, Brandt K, Wang J, Velasco VR, Kaufman MP. GsMTx-4 inhibits the exercise pressor reflex and the muscle mechanoreflex primarily through TRPC inhibition. J Physiol. 2025;603(19):5333–5350. doi:10.1113/jp289092
100. Tsai SL, Baselga-Garriga C, Melton DA. Midkine is a dual regulator of wound epidermis development and inflammation during the initiation of limb regeneration. eLife. 2020;9:50765. doi:10.7554/eLife.50765
101. Nouhravesh N, Cyr D, Hernandez AF, et al. In-Hospital or Out-of-Hospital Initiation of Sacubitril/Valsartan Versus Valsartan in Patients With Mildly Reduced or Preserved Ejection Fraction After A Worsening Heart Failure Event: the PARAGLIDE-HF Trial. J Am Heart Assoc. 2025;14(5):e037899. doi:10.1161/jaha.124.037899
102. Santos Bonilha C, Protasio Veras F. Mapping benefit, risk, and opportunity in PAD4 inhibition. Front Immunol. 2026;17:1769421. doi:10.3389/fimmu.2026.1769421
103. Kono Y, Niimura T, Goda M, et al. Cardiovascular Toxicity Profile of Macrolides Investigated Using VigiBase Data: a Pharmacovigilance Study. Cardiovasc Toxicol. 2025;25(3):498–506. doi:10.1007/s12012-025-09970-w
104. Dunlay SM, Givertz MM, Aguilar D, et al. Type 2 Diabetes Mellitus and Heart Failure: a Scientific Statement From the American Heart Association and the Heart Failure Society of America: this statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation. 2019;140(7):e294–e324. doi:10.1161/cir.0000000000000691
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Role of Intestinal Flora and Its Metabolites in Heart Failure
Guan X, Sun Z
Infection and Drug Resistance 2023, 16:51-64
Published Date: 5 January 2023
Mitochondrial Dysfunction in Diabetic Periodontitis: Mechanisms and Therapeutic Potential
Meng L, Wen W
Journal of Inflammation Research 2025, 18:115-126
Published Date: 10 January 2025
Therapeutic Targeting of Signaling Pathways in Abdominal Aortic Aneurysm: From Pathogenesis to Precision Medicine
Shaikh II, Singh S, Feng Y, Shahzad KA, Wang J, Zhou Q, Wang S, Zeng C, Shao C
Drug Design, Development and Therapy 2025, 19:12077-12111
Published Date: 31 December 2025
Neutrophil Extracellular Traps in Diabetic Kidney Disease: Mechanisms of Pathogenesis and Emerging Therapeutic Strategies
Wang B, Zhang R, Liu X, Shang Y, Jin T, Gao C, Yang N, Jin J, He Q
Drug Design, Development and Therapy 2026, 20:583077
Published Date: 21 February 2026