Back to Journals » Journal of Inflammation Research » Volume 16
Neutrophil Extracellular Traps Aggravate Contrast-Induced Acute Kidney Injury by Damaging Glomeruli and Peritubular Capillaries
Authors Wang H ![]() , Gao T, Zhang R, Hu J, Gao S, Wang Y, Qi X, Zhou Y, Zheng G, Dong H
, Gao T, Zhang R, Hu J, Gao S, Wang Y, Qi X, Zhou Y, Zheng G, Dong H
Received 30 July 2023
Accepted for publication 17 November 2023
Published 28 November 2023 Volume 2023:16 Pages 5629—5646
DOI https://doi.org/10.2147/JIR.S433110
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Heng Wang,1,* Tingting Gao,1,* Ruijing Zhang,2,* Jie Hu,1 Siqi Gao,1 Yuwen Wang,1 Xiaotong Qi,1 Yun Zhou,3 Guoping Zheng,1,4 Honglin Dong1
1Department of Vascular Surgery, The Second Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2Department of Nephrology, The Second Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 3Shanxi Provincial Integrated TCM and WM Hospital, Taiyuan, People’s Republic of China; 4Centre for Transplant and Renal Research, Westmead Institute for Medical Research, The University of Sydney, Sydney, NSW, Australia
*These authors contributed equally to this work
Correspondence: Honglin Dong, Department of Vascular Surgery, The Second Hospital of Shanxi Medical University, No. 382, Wuyi Road, Xinghualing District, Taiyuan City, Shanxi Province, 030001, People’s Republic of China, Email [email protected] Guoping Zheng, Centre for Transplant and Renal Research, Westmead Institute for Medical Research, The University of Sydney, Sydney, NSW, Australia, Email [email protected]
Background: Contrast-induced acute kidney injury (CI-AKI) is considered to be the third leading cause of hospital-acquired kidney injury. Current studies mostly suggest that contrast agents mainly harm renal tubular epithelial cells, but we hypothesized that the development of CI-AKI should be the result of the interaction of renal vascular and tubular injury.
Methods: First we constructed a CI-AKI mouse model and verified the success of the model by pathological injury and serum creatinine level. Immunohistochemistry, protein quantification and qRT-PCR were used to detect the location and level of expression of neutrophil extracellular traps (NETs) in the kidney. Then, we blocked the in vivo accumulation of NETs using GSK484 and DNase I and detected the expression of NETs and the damage of glomerular and peritubular capillaries.
Results: We first identified the presence of NETs in CI-AKI mice, and NETs were mainly accumulated in glomeruli and peritubular capillaries. The expression of NETs was positively correlated with the severity of CI-AKI kidney. After inhibition of NETs release or promotion of NETs degradation by drugs, renal vascular endothelial cell injury was reduced and renal pathological changes and creatinine levels were reversed in CI-AKI mice. In addition, inhibition of NETs reduced apoptosis and pyroptosis of renal cells and attenuated inflammation in vivo.
Conclusion: These findings suggest that NETs are involved in the development of CI-AKI by damaging glomerular and peritubular capillary endothelial cells. This study will provide a new strategy for clinical prevention and treatment of CI-AKI.
Keywords: contrast-induced acute kidney injury, neutrophils, neutrophil extracellular traps, glomeruli, peritubular capillaries
Introduction
In recent years, contrast agents (CM) have been used with increasing frequency. Contrast-induced acute kidney injury (CI-AKI) occurs in up to 30% of patients receiving iodine contrast and is often considered the third leading cause of hospital-acquired kidney injury.1–3 The Kidney Disease: Improving Global Outcomes (KDIGO) guidelines define CI-AKI as a rise in serum creatinine (SCr) ≥0.5 mg/dL (≥44 μmol/L) or a 25% increase from baseline assessed 48 hours after CM application.4,5 Current therapeutic approaches, such as prophylactic hydration and statins, are not well suited to prevent the development of CI-AKI.6,7 Therefore, it is critical to understand the mechanisms of injury in CI-AKI and to identify potential targets for intervention. There are three main injury mechanisms in CI-AKI, including direct toxic effects, renal vasoconstriction, and overproduction of reactive oxygen species (ROS).8–10 Current studies have mostly concluded that CM mainly harm renal tubular epithelial cells, but we found that the effects of renal vascular injury have been neglected. The development of CI-AKI should be the result of the interaction between renal vascular and tubular injury.
Neutrophils are the most abundant white blood cells in the peripheral circulation and are the first line of defense for the body’s immunity.11 Neutrophils perform their functions through four main mechanisms: phagocytosis, degranulation, release of cytokines, and release of neutrophil extracellular traps (NETs).12 In some cases, neutrophils are activated, releasing NETs and usually accompanied by cell death, a process known as NETosis.13,14 NETs are a reticular structure with DNA as the backbone, which is inlaid with proteins that have bactericidal and permeability-increasing effects, such as citrullinated histone 3 (CitH3), myeloperoxidase (MPO) and elastase of neutrophils (ELANE).15 Recent studies have revealed that NETs not only respond rapidly to pathogen invasion, but also participate in pathophysiological processes such as sterile inflammation and immune diseases, and even damage cells and tissues of the body.16–18
Recently, neutrophils were found to infiltrate the renal interstitium in mice with ischemia-reperfusion (IRI) kidney injury, and necrotic tubular cells induced the formation of NETs, further aggravating kidney injury.19 In addition, NETs can promote vascular injury by activating the complement bypass pathway and occluding microvessels through platelet-neutrophil interactions.20,21 Importantly, in rheumatic diseases such as rheumatoid arthritis, systemic lupus erythematosus and vasculitis, neutrophils play an important role in relation to endothelial cell activation and dysfunction.22 Immune complexes with autoantibodies in rheumatic diseases activate neutrophils to release NETs to induce endothelial damage, and NETs are an important source of immunogenic nucleic acids that maintain inflammation.23–25 However, it is not clear whether NETs are involved in the development of CI-AKI.
Here, we investigated the relationship between the expression of NETs and renal injury in a mouse model of CI-AKI. To investigate whether elimination of NETs could ameliorate renal vascular endothelial cell injury and alleviate CI-AKI, we pretreated in CI-AKI mice using GSK484 and DNase I. We propose new potent strategies for the prevention and treatment of CI-AKI.
Materials and Methods
Animal Model: Contrast-Induced Acute Kidney Injury
After one week of adaptive feeding in the animal room of the cardiology s laboratory of Shanxi Medical University, a mouse CI-AKI model was constructed with reference to a previous study.26 Mice were dehydrated for 16 hours and then injected intraperitoneally with N-nitro-L-arginine methyl ester (L-NAME, 10 mg/kg) after 15 minutes, indomethacin (Indomethacin, 10 mg/kg) after 15 minutes, and iodixanol (Iodixanol, 6.24 g I2/kg) after another 15 minutes. The mice were then fed and watered normally and executed after 24 hours, and blood and kidney tissues were obtained.
Mice were divided into 8 groups for the experiment, with 6 mice in each group. They were Control group, 2 h group, 6 h group, 12 h group, 24 h-L group, 24 h-H group, 48 h group, and 72 h group, respectively. After the representative contrast injection, the mice were executed at different time points. 24 h-L, low-dose iodixanol (6.24 g I2/kg) was injected intraperitoneally. 24 h-H group, high-dose iodixanol (12.48 g I2/kg) was injected intraperitoneally. The above drugs were purchased from MedChemExpress.
Drug Treatment
C57BL/6 mice were divided into four groups: control, CI-AKI, CI-AKI+GSK484, and CI-AKI+DNase I. The CI-AKI group was injected with a low dose of contrast. GSK484 is a potent and reversible inhibitor of peptidylarginine deiminase 4 (PADI4). The intervention was performed one week before CI-AKI mapping until the day of execution with an intraperitoneal dose of 4 mg/kg. DNase I, deoxyribonuclease I, is a nucleic acid endonuclease that can digest single- or double-stranded DNA. Intervention was performed one week prior to CI-AKI modeling until the day of execution at an intraperitoneal dose of 5 mg/kg. The above drugs were purchased from MedChemExpress.
Serum Creatinine Measurement
Blood was obtained using the mouse eye extraction method and placed in a yellow procoagulation tube to promote coagulation. After centrifugation at 3000 rpm for 10 minutes, the upper layer of serum was obtained and stored in a −80°C refrigerator if long-term storage was required. The creatinine oxidase method was used to determine the creatinine level in mouse serum, and the detailed procedure is described in the instruction manual of Nanjing Creatinine (CRE) Assay Kit (C011-2-1).
Determination of Serum Inflammatory Factors
The serum of mice was detected by ELISA, and the detection indexes and kits were as follows: TNF-α (RUIXIN BIOTECH, RXSWSXQ202412M), IL-6 (RUIXIN BIOTECH, RX203049M). Strictly follow manufacturer’s instructions.
Renal Pathology Staining
Kidney tissues were fixed in 10% paraformaldehyde at 4°C for 24 h and embedded in paraffin blocks. The tissues were stained with hematoxylin-eosin (HE) or Periodic Acid-Schiff (PAS) reagents according to standard conventional protocols. Acute kidney injury was scored according to the degree of tubular injury according to the method in the article by Wei et al.27 Where tubular injury was graded 0–4, with 0 representing no injury; 1 representing tubular epithelial cell swelling, vacuolar degeneration, necrosis, and loss of brush border involving <25% of the cortical tubules; and 2, 3, and 4 indicating tubular injury of 25%-50%, 50%-75%, and more than 75% of the cortical tubules. The above pathological assessments were performed independently by two pathology researchers who were not informed of the subgroup.
Immunohistochemical Staining
Paraffin sections were first dewaxed and treated with antigen repair. The sections were then closed at room temperature with goat serum for 30 min. The primary antibody was added dropwise at the appropriate concentration and incubated overnight at 4°C in a wet box. The next day, the tissue was covered with a drop of secondary antibody of the same species as the primary antibody and incubated at room temperature for 1 h. Then, the nuclei were restained with hematoxylin stain. Finally, the slices were sealed by dehydration and placed under a white light microscope for interpretation of the results. The antibodies used were MPO (Proteintech, Cat No. 66177-1) and PV-1 (Servicebio, GB113141).
Immunofluorescence Staining
For the immunofluorescence (IF) co-localization of the two target proteins, we used the following method. Paraffin sections were first dewaxed, treated with antigen repair and serum blocked. Then the first primary antibody was added and incubated overnight at 4°C in a wet box. The corresponding secondary antibody labeled with CY3 was added the next day. The second primary antibody is then added dropwise and incubated overnight at 4°C in a wet box. On the third day, the FITC-labeled corresponding secondary antibody was added and washed after incubation. Then the nuclei were restained with DAPI staining solution. Sealing was done with anti-fluorescence quenching blocker. Finally, image acquisition is performed. If only one antibody is available, the corresponding steps are reduced. The antibodies used in this experiment are: Ly6G (Servicebio, GB11229), CitH3 (Abcam, ab5103), CD31 (Servicebio, GB11063-2), VCAM-1 (Servicebio, GB113376-100), MPO (Proteintech, Cat No. 66177-1), PADI4 (Abcam, ab128086), ELANE (Boster Biotech, PB1114), GSDMD (Servicebio, GB114198), Caspase-1 (Servicebio, GB11383) and IL-1β (Servicebio, GB11113). ImageJ software (NIH, Bethesda, MD, USA) was used for semi-quantitative analyses of the fluorescence intensity.
Transmission Electron Microscope Detection
Firstly, mouse kidneys were pre-fixed with 3% glutaraldehyde and refixed with 1% osmium tetroxide. Then acetone was dehydrated several times step by step. Then infiltration and embedding were done with dehydrating agent and Epon812 embedding agent. Ultrathin sections of approximately 60–90 nm were cut off using an ultrathin sectioning machine and spread onto a copper mesh. Then, the sections were stained with uranyl acetate for 10 min at room temperature, followed by lead citrate for 1 min. Finally, images of the kidney tissue on the copper mesh were acquired by JEM-1400FLASH transmission electron microscopy.
Quantitative Real-Time PCR
The primer sequences (Sangon Biotech Co., Ltd., Shanghai, China) used in this study are listed in Table 1. The purity and concentration of RNA were measured by ultra-micro spectrophotometer. The mRNA was then reverse transcribed to cDNA using Takara’s Reverse Transcription Kit, and the detailed steps are described in the instruction manual of Item No. RR036A. Next, the real-time fluorescence kit was used to detect the expression of the target gene, and the detailed steps are described in the instruction manual of item number MF797. The Ct value of GAPDH was normalized and the relative gene expression level was calculated by the 2−∆∆Ct method.
 |
Table 1 Primers of Mouse qRT-PCR |
Western Blot
Mouse kidney tissue was placed in enhanced RIPA lysate and thoroughly ground using a tissue crusher. Quantification was performed with the BCA protein concentration assay kit pair according to the manufacturer’s instructions. Total proteins were separated by SDS-PAGE gel electrophoresis. Subsequently, the proteins were transferred to PVDF membranes. Closure was performed with TBST buffer containing 5% skim milk powder at room temperature. Then incubated with primary antibody at 4°C overnight. Next, the membrane was incubated with the appropriate secondary antibody at room temperature for 1 h. Finally, the protein signal was developed in the ChemiDoc system using ECL luminescent solution. Protein expression level analysis was performed using Image Lab software. The target proteins were normalized with internal reference proteins. The antibodies used in this experiment were: GAPDH (Servicebio, GB15002), β-actin (Servicebio, GB15001), PADI4 (Abcam, ab128086), CitH3 (Abcam, ab5103), ELANE (Boster Biotech. PB1114) and GSDMD (Servicebio, GB114198).
Statistical Analyses
The data of this experiment were expressed as mean ± standard deviation. If the experimental data were normally distributed, t-test or one-way ANOVA was used. If the experimental data were non-normally distributed, the Mann–Whitney U-test was used. Tukey’s test or Dunn's multiple comparison test was used for the analysis between each experimental group. Pearson’s algorithm was used for correlation tests. All statistical analysis data and graphs were obtained using GraphPad Prism 8.0.2 software. P < 0.05 was considered to be statistically significant.
Results
Induction of CI-AKI Model
We divided the mice into low-dose group (24h-L) and high-dose group (24h-H) according to the contrast agent injection dose. The kidneys of mice were firstly stained with HE and the renal tubular injury score was used as the pathological diagnosis. The results showed that compared with normal mouse kidneys, in the 24h-L group the renal tubular epithelial cells were swollen, vacuolated, necrotic, and lost brush border (Figure 1A and B). And in the 24h-H group, the kidney damage was more severe. Currently, SCr is the preferred criterion for the clinical diagnosis of CI-AKI. Compared to the control group, the SCr level in the 24h-L group had met the diagnostic criteria for CI-AKI, while the elevation was more pronounced in the 24h-H group (Figure 1C). In addition, SCr was detected at different time points in mice injected with low-dose CM. The results showed that the SCr levels in mice injected with CM continued to increase during 24h, while the SCr values dropped back to normal levels at 48h and 72h (Figure 1D). This indicates that for normal mice, CI-AKI is a transient injury with the possibility of normalization.
 |
Figure 1 Successful establishment of CI-AKI model in mice. (A) HE staining of mouse kidney (magnification 400×). (B) Quantification of renal tubular injury scores (n=6). (C) The SCr levels in different contrast dose groups (n=6). (D) The SCr levels in mice at different time periods after contrast injection (n=6). (E) Transmission electron micrographs of kidney tissues in mice. Among them, renal tubular epithelial cells (RTECs), vascular endothelial cells (VECs), nucleus (N), mitochondria (Mi), rough endoplasmic reticulum (RER), peduncle: yellow arrow (↑), mitochondrial swelling: red arrow (↑), basement membrane: green arrow (↑), vacuole: blue arrow (↑), autophagy: purple arrow (↑). Magnification 20,000×. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, indicating statistically significant data between groups; “ns” indicates no statistical significance. |
Meanwhile, we performed transmission electron microscopy on the control group, 24h-L group and 24h-H group (Figure 1E). The glomerular filtration barrier of normal mice had normal morphology, with clear structures of peduncle and endothelial window pores, and the basement membrane was continuous and basically the same thickness. The tubular epithelial cells were intact and clear, and only a few secondary lysosomes and autophagy were found in the cytoplasm. The vascular endothelial cells in the renal tubular interstitium showed no obvious lesions. There was a small amount of autophagy in glomerular vascular endothelial cells in the 24h-L group. Mitochondria in renal tubular epithelial cells were slightly swollen, while autophagy and vacuoles were abundant in the cytoplasm. The morphology of vascular endothelial cells in the renal tubular interstitium was intact, and only slight swelling of mitochondria in the cytoplasm was seen. In the 24h-H group, some regions of the pedicles in the glomerular filtration barrier had fused, and mitochondria in the vascular endothelial cells had swollen (cristae broken and lysed), and a small amount of autophagy was also seen. The morphology of renal tubular epithelial cells was more intact, but a large amount of autophagy was seen in the cytoplasm. The mitochondria of the vascular endothelial cells in the renal tubular interstitium were mildly swollen.
Increased NETs are Associated with the Degree of CI-AKI Progression
Some studies have reported that NETs are observed in human aseptic inflammatory sites, such as ischemia in perfused kidneys. Therefore, it is reasonable to speculate that NETs would also be involved in the mechanism of injury in CI-AKI. Neutrophil markers (Ly6G) and NETs markers (CitH3), were co-localized by IF in the kidney of a mouse CI-AKI model. We found infiltration of neutrophils and release of NETs in the kidneys of CI-AKI mice. We also captured neutrophils undergoing NETosis but not yet ruptured (Unruptured Neutrophils group), neutrophils undergoing NETosis that had ruptured (Ruptured Neutrophils group), and the release of NETs in patches (Patchy Neutrophils group) (Figure 2A). In contrast, no infiltrating neutrophils and NETs markers were shown in healthy mouse kidneys.
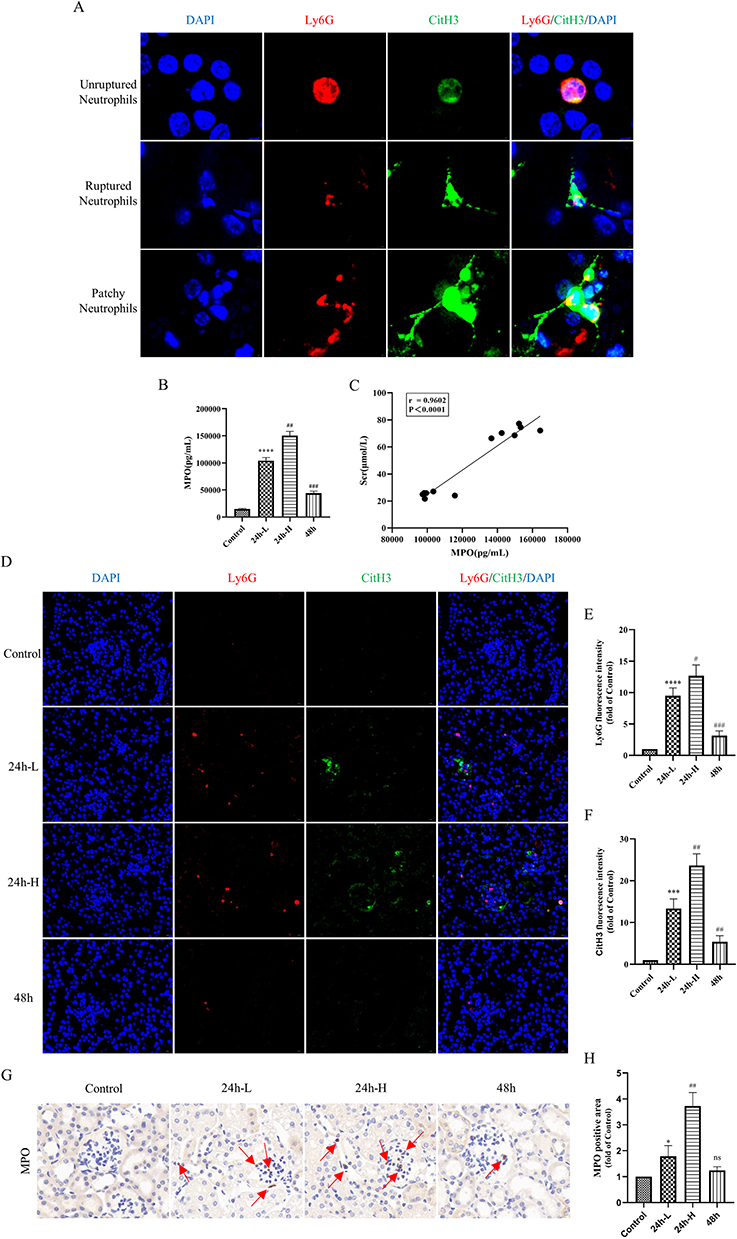 |
Figure 2 NETs are present in the kidneys of CI-AKI mice and correlate with the degree of kidney injury. (A) IF of kidney sections. Among them, blue, DAPI, nucleus; red, Ly6G, mouse neutrophil surface marker; green, CitH3, NETs-specific marker. Magnification 4000×. (B) ELISA method to detect the expression level of serum MPO in mice (n=6). (C) Correlation analysis of serum MPO levels with SCr in CI-AKI mice (n=12). (D–F) IF double staining and semiquantitative analysis of renal paraffin sections (n=6). Among them, blue, DAPI; red, Ly6G; green, CitH3. Magnification 630×. (G and H) Histochemical staining of kidney paraffin sections for MPO (n=6). Magnification 1000×. * VS control group, *P < 0.05, ***P < 0.001, ****P < 0.0001, indicating statistically significant data between groups; # VS 24 h-L group, #P < 0.05, ##P < 0.01, ###P < 0.001, indicating statistically significant data between groups; ns indicates no statistical significance. |
To investigate the relationship between serum NETs levels and SCr in CI-AKI mice, we performed correlation analysis. Among them, the serum levels of NETs markers (MPO) were elevated in CI-AKI mice, and the increase in expression was more significant in the 24h-H group. However, the level of MPO in the 48h group decreased back (Figure 2B). Then, we performed Pearson correlation test on the serum MPO levels and SCr in CI-AKI mice, and there was a positive correlation between them (Figure 2C). Meanwhile, we performed double-label IF staining of Ly6G and CitH3 in the kidneys of each group of mice, and the results showed that the expression of NETs was also significantly higher in the more injured kidneys (Figure 2D–F). And the immunohistochemical (IHC) staining of MPO had the same expression trend (Figure 2G and H).
In addition, in the assay of renal proteins, the more severe the contrast-induced kidney injury in mice, the more protein expression of PADI4 and CitH3 was observed (Figure 3A–C). In the quantitative analysis of renal mRNA, the mRNA expression trends of markers of NETs (PADI4, MPO, ELANE) and specific receptors of NETs (TLR2, TLR4) were also as expected (Figure 3D and E). And the serum inflammatory factors TNF-α and IL-6 in mice also showed similar trends to the levels of NETs (Figure 3F and G). These data suggest that NETs may be involved in the mechanism of injury in CI-AKI and are associated with inflammation.
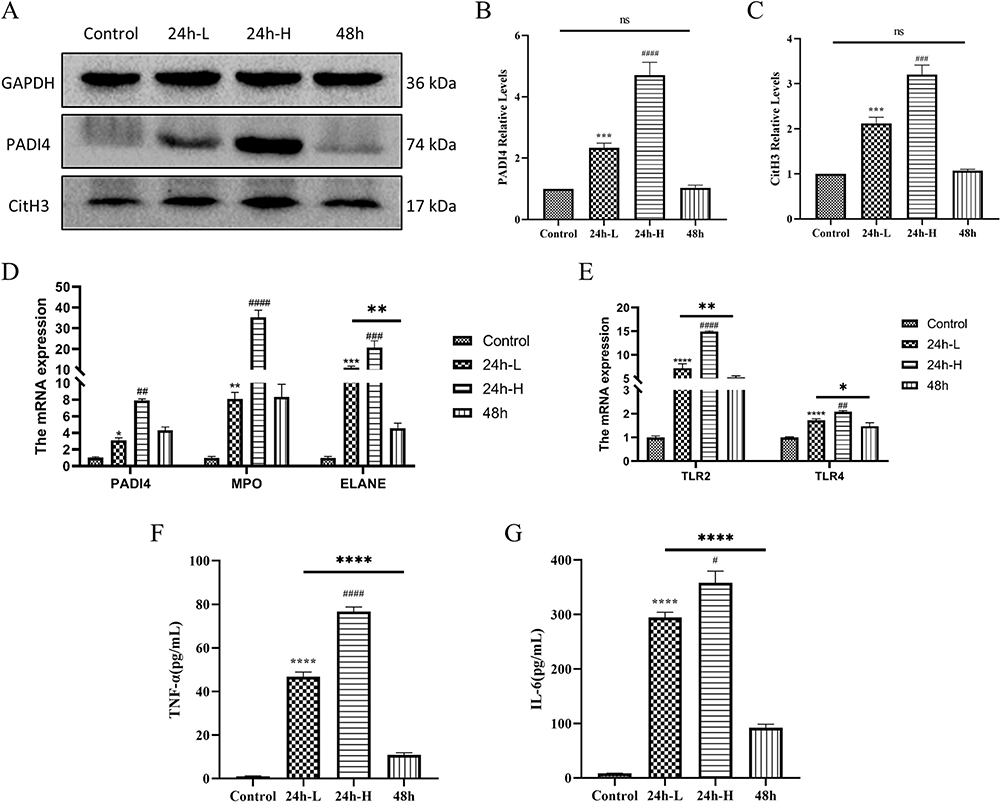 |
Figure 3 Expression of NETs protein and mRNA in the kidney of CI-AKI mice. (A–C) Expression and quantification of PADI4 and CitH3 in mice kidney by Western blot (n=3). (D and E) Detection of PADI4, MPO and ELANE, and mRNA expression of TLR2 and TLR4 by qRT-PCR (n=3). (F and G) Expression levels of inflammatory factors TNF-α and IL-6 in serum of mice (n=3). * VS control group, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, indicating statistically significant data between groups; # VS 24 h-L group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001, indicating statistically significant data between groups; ns indicates no statistical significance. |
NETs Mainly Accumulate in the Microcirculatory System of the Kidney of CI-AKI Mice
The microcirculatory system of the kidney consists mainly of glomeruli and peritubular capillaries (PTC), which are the first barrier to blood and the first line of defense for neutrophils to respond to injury.28,29 Therefore, we found that NETs were mainly deposited within the renal vascular system. We performed IF co-localization of CD31 and MPO in the kidneys of mice (Figure 4A). The results showed that MPO was mainly expressed in the glomeruli and the expression of CD31 was reduced in CI-AKI mice. In addition, we performed double-label IF staining of Ly6G and CitH3, and neutrophils and NETs were deposited in the glomeruli of CI-AKI mice, but not in normal mice (Figure 4B). We also performed serial sections of CI-AKI mouse kidneys for pathologic staining (HE, PAS) and IF staining (CD31+CitH3), respectively (Supplementary Figure 1). This suggests that NETs are deposited in the glomeruli and may damage vascular endothelial cells.
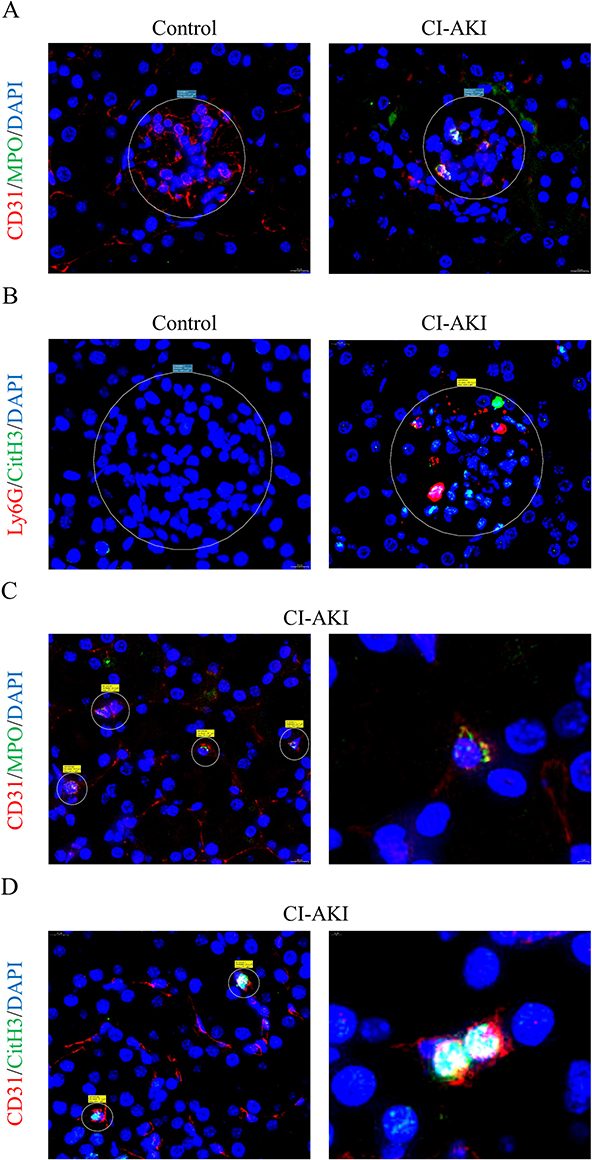 |
Figure 4 NETs accumulate in the glomeruli and PTC of CI-AKI mice. (A) IF co-staining: CD31, red, vascular endothelial cell marker; MPO, green, NETs marker. Magnification 1300×. (B) IF co-staining: Ly6G, red; CitH3, green. Magnification 1300×. (C and D) Neutrophil residency and NETs release in PTC. Left side magnification 1300×, right side magnification 4000×. |
As for PTC, which has been less studied in CI-AKI disease, we also observed the presence of NETs for the first time. In kidney sections from CI-AKI mice, the products of NETs could be seen in the PTC by double-label IF staining with CD31 and MPO, indicating that neutrophils reside in the PTC (Figure 4C). In addition, the presence of CitH3+ cells in PTC was also detected by IF double staining for CD31 and CitH3 (Figure 4D). Since most of the PTC are surrounded by a single layer of vascular endothelial cells, the significance of NETs for its damage may be more important.
In addition, vascular cell adhesion molecule (VCAM)-1 (also known as CD106), which is activated in vascular endothelial cells in response to inflammatory stimuli.30 It promotes adhesion, activation and migration of circulating leukocytes.31 As seen in Supplementary Figure 2A, VCAM-1 was barely expressed in normal mouse kidneys, whereas it was increased in CI-AKI mice. This suggests that endothelial cells in the kidneys of CI-AKI mice promote the recruitment and migration of inflammatory cells. At the same time, we performed TUNEL staining of the kidneys (Supplementary Figure 2B and C). The results showed that glomerular endothelial cells underwent apoptosis.
GSK484 and DNase I Reduce NETs Production in CI-AKI Mice
We divided the mice into four groups: control group, CI-AKI group, CI-AKI+GSK484 group, and CI-AKI+DNase I group. Among them, GSK484 inhibited NETosis by suppressing PADI4 activity, and DNase I could degrade NETs products. We first observed the expression of PADI4 in mice, and both GSK484 and DNase I could reduce its expression (Supplementary Figure 3A and B). In addition, it also reduced the infiltration of neutrophils in the kidney and the expression of NETs markers ELANE, CitH3 and MPO (Supplementary Figure 3C and D; Figure 5A–E). Meanwhile, we detected serum MPO by ELISA, and in the drug intervention group, the serum MPO level of mice was decreased (Figure 5F). WB method was used to detect the protein expression of ELANE and the trend was as expected (Figure 5G and H). This suggests that GSK484 and DNase I pretreatment can successfully reduce neutrophil infiltration in the kidney and decrease the accumulation of NETs.
 |
Figure 5 GSK484 or DNase I reduces the accumulation of NETs in CI-AKI mice. (A–C) IF co-staining results and semi-quantitative analysis of Ly6G and CitH3 in mouse kidney sections (n=6). Magnification 630×. (D and E) Immunohistochemical staining and semi-quantitative analysis of MPO in mouse kidney sections (n=6). Magnification 1000×. (F) The expression of MPO in mouse serum (n=3). (G and H) Quantification of protein in mouse kidney ELANE (n=3). * VS control group, ***P < 0.001, ****P < 0.0001, indicates statistically significant data between groups; # VS CI-AKI group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001, indicates statistically significant data between groups. |
Inhibition of NETs Reduces Pathological Damage in CI-AKI
To evaluate the kidney injury in mice, we first performed HE and PAS staining of the kidneys, and the results showed that the kidney injury was significantly reduced after reducing the accumulation of NETs compared to the CI-AKI group (Figure 6A and B). Next, we examined the level of SCr in mice, and lowering the level of NETs resulted in a decrease in SCr (Figure 6C). Thus, we confirmed that renal pathological changes in CI-AKI mice could be significantly alleviated by inhibiting the accumulation of NETs.
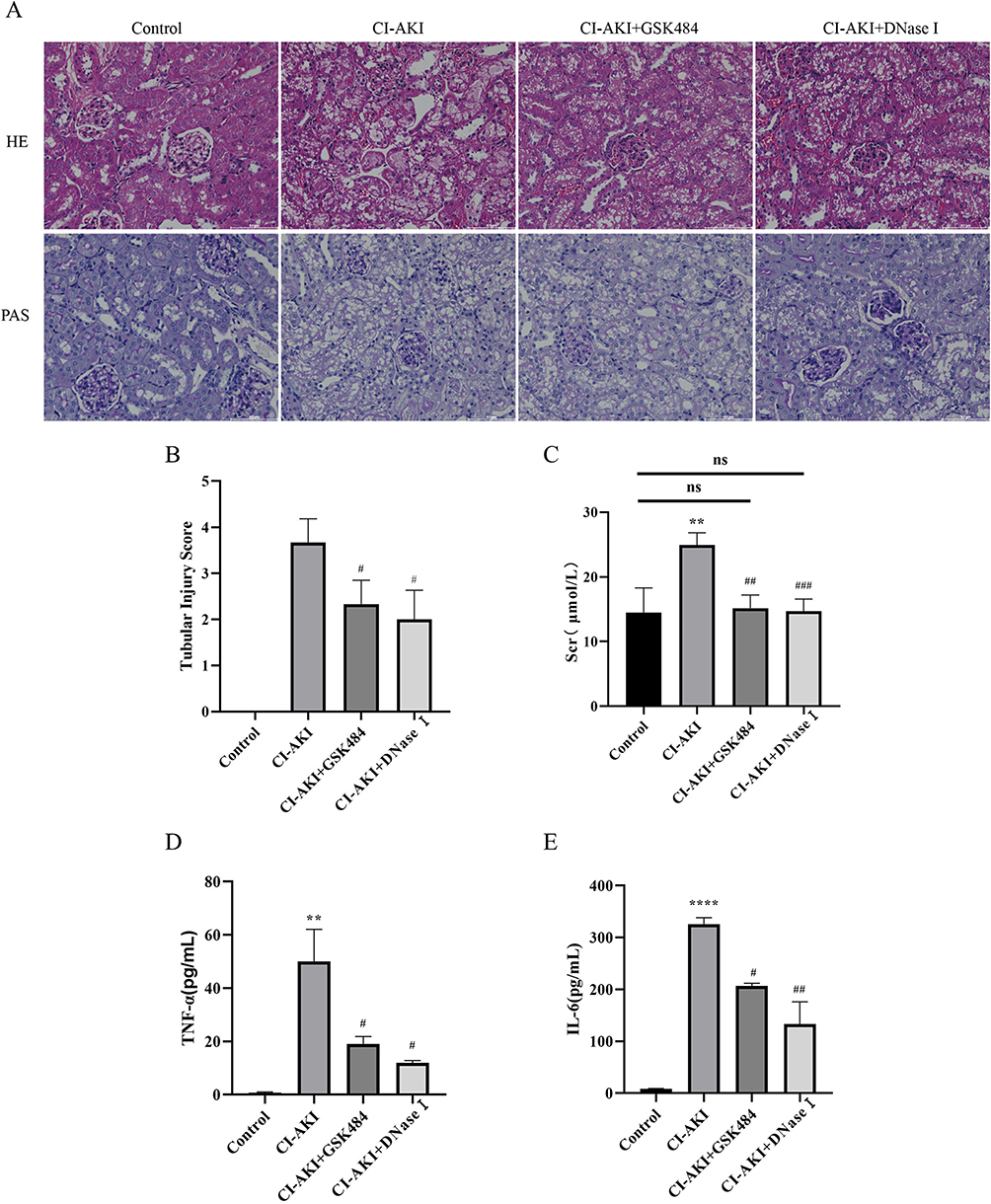 |
Figure 6 GSK484 or DNase I attenuates kidney injury in mice. (A) HE and PAS staining of mouse kidney tissue (magnification 400×). (B) Renal tubular injury score (n=6). (C) Expression of SCr in mice (n=6). (D and E) Expression levels of serum inflammatory factors TNF-α and IL-6 in mice (n=3). * VS control group, **P < 0.01, ****P < 0.0001, indicating statistically significant data between groups; # VS CI-AKI group, #P < 0.05, ##P < 0.01, ###P < 0.001, indicating statistically significant data between groups; ns indicates no statistical significance. |
Neutrophils are the first line of defense of the body and an important factor in the production of inflammation in the body. We wanted to clarify whether the release of NETs promotes the expression of inflammation, and whether inhibition of NETs reduces inflammation. The results showed that either blocking the release of NETs or degrading the products of NETs could reduce inflammation in mice in vivo (Figure 6D and E). However, it is noteworthy that inflammatory factors did not return to normal levels, considering the pro-inflammatory effect of other immune cells such as macrophages.
Inhibition of NETs Reduces Glomerular and Peritubular Capillary Endothelial Cell Injury
We found that NETs mainly accumulate in the renal microcirculatory system and may be involved in disease development by damaging glomeruli and peritubular capillaries. Therefore, we wanted to clarify the protective effects of blocking NETs formation or accelerating NETs degradation on renal vascular endothelial cells in CI-AKI mice. By IF staining of CD31 and CitH3, the expression of vascular endothelial markers was reduced in CI-AKI mice, mainly manifested by diminished CD31 fluorescence intensity and discontinuous color development. In contrast, CD31 expression was significantly higher in the drug intervention group (Figure 7A–C). In addition, IF staining for CD31 and MPO showed a trend consistent with the above (Supplementary Figure 4). This suggests that reducing NETs expression can significantly alleviate glomerular vascular endothelial cell injury in mice.
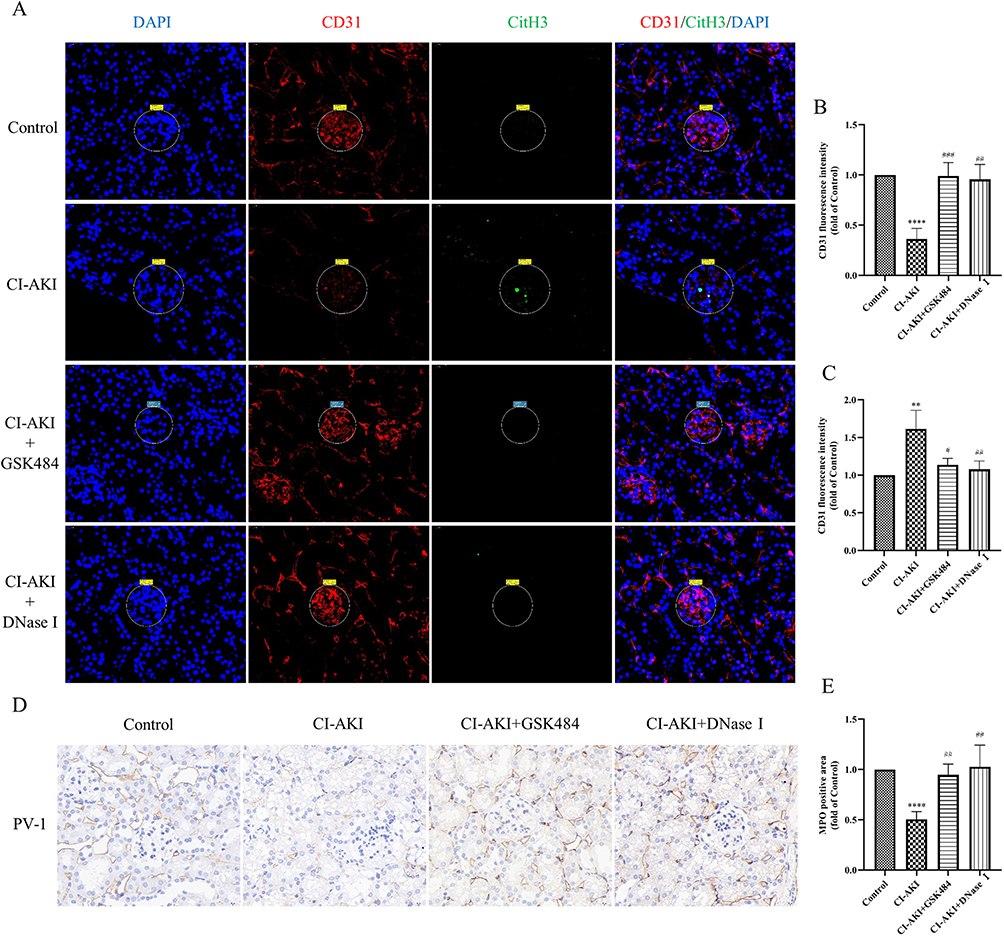 |
Figure 7 Reduction of NETs levels alleviates endothelial cell injury in glomeruli and PTC. (A–C) IF co-staining and semi-quantitative analysis of CD31 and CitH3 in mouse kidney sections (n=6). Magnification 630×. (D and E) Immunohistochemical staining and semi-quantitative analysis of PV-1 in mouse kidney sections (n=6). Magnification 1300×. * VS control group, **P < 0.01, ****P < 0.0001, indicating statistically significant data between groups; # VS CI-AKI group, #P < 0.05, ##P < 0.01, ###P < 0.001, indicating statistically significant data between groups. |
In addition, we also performed immunohistochemical staining for the mouse PTC marker PV-1 (Figure 7D and E). Compared with CD31, PV-1 was a more specific marker of PTC endothelial cells. The results showed that the kidneys of CI-AKI mice had significantly reduced PTC and more extensive damage. In contrast, the drug pretreatment group, by reducing neutrophil infiltration and decreasing the level of NETs, significantly alleviated the damage of PTC. Once again, it was demonstrated that reducing the level of NETs helped to reduce the damage of vascular endothelial cells in mice.
Inhibition of NETs Reduces Apoptosis and Pyroptosis in Kidney Cells
In addition, we explored whether inhibition of NETs attenuated other cell death modalities. TUNEL staining showed that a large number of apoptotic cells were visible in the kidneys of CI-AKI mice, and the number of apoptotic cells was significantly reduced after reducing the accumulation of NETs (Supplementary Figure 5A and B). This suggests that reducing the content of NETs can significantly reduce the occurrence of apoptosis in kidney cells.
Pyroptosis is a form of inflammatory cell death that requires gasdermin D (GSDMD)-mediated cytolysis with the involvement of inflammatory caspases.32 We found that the protein expression of N-GSDMD and GSDMD could be significantly reduced by reducing the content of NETs (Figure 8A–C; Supplementary Figure 5C and D). This suggests that the onset of cellular scorching can be inhibited after reducing the accumulation of NETs in the kidney. In addition, Caspase-1 and IL-1β are also specific markers of cellular pyroptosis. Their expression was reduced after inhibition of NETs accumulation (Figure 8D and E). Interestingly, in CI-AKI mice, cellular pyroptosis occurred mainly in renal tubular epithelial cells, but was not found in vascular endothelial cells (Supplementary Figure 6).
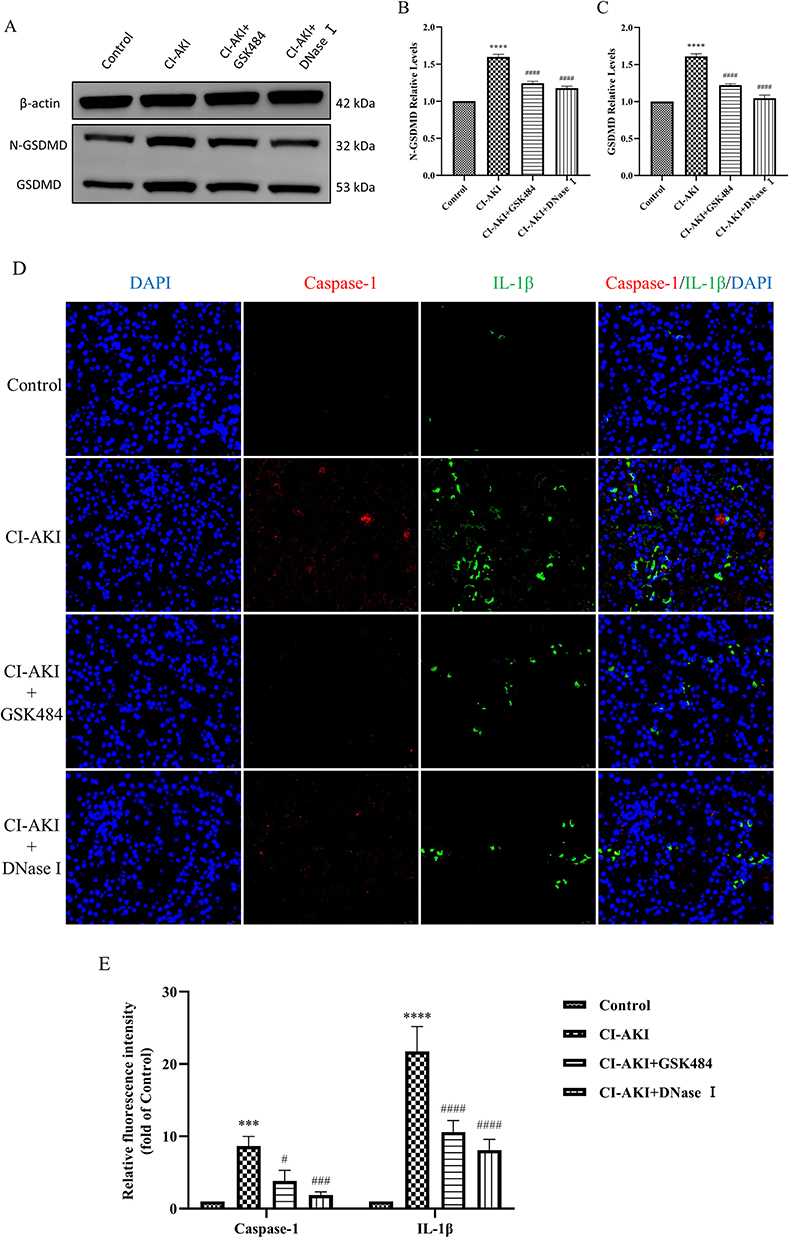 |
Figure 8 Reducing the level of NETs reduces pyroptosis. (A–C) Quantitative analysis of GSDMD and N-GSDMD protein in mouse kidney (n=3). (D and E) IF double staining and semi-quantitative analysis of Caspase-1 and IL-1β in mouse kidney tissue sections (n=3). * VS control group, ***P < 0.001, ****P < 0.0001, indicating statistically significant data between groups; # VS CI-AKI group, #P < 0.05, ###P < 0.001, ####P < 0.0001, indicating statistically significant data between groups. Magnification 630×. |
Discussion
By reviewing the literature, the involvement of NETs in acute kidney injury has been reported.21,33 In the field of CI-AKI research, we reported for the first time that NETs were found in a CI-AKI mouse model. In this study, we found high levels of NETs in serum and kidney tissues of CI-AKI mice, which is consistent with the trend of pathological kidney injury. Interestingly, NETs accumulate mainly in the microcirculatory system of the kidney. In contrast, the glomeruli and peritubular capillaries have been less studied in the exploration of CI-AKI injury mechanisms. Pretreatment of mice with GSK484 or DNase I significantly alleviated renal vascular endothelial cell injury and reversed the pathological changes and creatinine elevation. Therefore, targeting NETs will be an effective strategy to prevent and treat CI-AKI in the future.
The main feature of CI-AKI is a decrease in renal function within a few days after intravascular injection of iodine contrast.34 Continuous measurement of serum creatinine is considered the most practical and sensitive method, and an elevation of 1.0 mg/dL compared to baseline is the accepted diagnostic criterion.35 In 2012, the KDIGO guidelines continued the name of CIN with the diagnostic criteria of renal impairment (increase in serum creatinine more than 25% or 44 μmol/L) within 3 days after intravascular contrast injection and without other etiology.5 In 2018, the European Society of Urogenital Radiology (ESUR) recommended diagnostic criteria is an increase in serum creatinine ≥0.3 mg/dL or ≥1.5–1.9 times the baseline level within 48–72 h of CM use.36
The mechanism of injury of CI-AKI is not fully understood, and there are three main pathophysiological mechanisms, including direct toxic effects, intrarenal vasoconstriction and overproduction of ROS. While the role of contrast agents on renal tubular injury is more studied, the study of renal vascular injury is gradually coming into view. The damage of contrast agents to vascular endothelial cells is multifaceted, and when CM enters the body directly, it acts as a cytotoxic agent through direct contact with the renal vasculature.37 Contrast-induced mitochondrial dysfunction and excessive release of ROS can also damage endothelial cells and cause endothelial dysfunction.38 And the hemodynamic changes triggered by vasoconstriction further reduce the glomerular filtration rate.39 In addition, the operation of contrast agents in the systemic circulatory system reduces the anti-inflammatory effect and anti-thrombotic capacity of the circulatory system, elevating the risk of systemic inflammation.40 Therefore, the role of renal vascular damage in CI-AKI should not be underestimated and may be the key to preventive strategies.
The presence of NETs in AKI and the damaging role they play have been widely reported in recent studies, and this has served as an important inspiration for our research on CI-AKI. In aseptic inflammation, the massive release of damage-associated pattern molecules (DAMPs) triggers neutrophil infiltration and tissue damage.41,42 Among them, the occurrence of NETosis plays a crucial role. When the contrast agent enters the vasculature, neutrophils are the first to be activated, and they are already responding rapidly and being recruited when the kidney is just showing damage or even before it has manifested itself. The occurrence of NETosis indicates that the neutrophil response to pathogens or sterile inflammation is extremely rapid and can be initiated within hours or even minutes.13,43 And this became the basis of our initial hypothesis and the focus of our attention. Furthermore, it has been shown that in an ischemic kidney injury model, secretion of histones by dead renal tubular cells triggers the release of NETs from neutrophils, which in turn exacerbates tubular cell death.19 Therefore, in this study, we can learn that NETs are at the core of kidney injury.
Early occurrence of NETosis in neutrophils facilitates rapid clearance of pathogens, but when this protective mechanism is imbalanced, it poses a threat to the normal organismal environment. Due to the different mechanisms of NETosis occurrence and key components of action in renal diseases, it is necessary to propose targeted inhibition regimens. The protective effect of neutrophils should be maintained as much as possible while mitigating the excessive damage of NETosis.
PADI4, a key enzyme of classical NETosis, is currently a popular target for research in various kidney-related diseases.44 In animal models of renal IRI, PADI4-selective inhibitors such as Cl-amidine, YW3-56 or GSK484 significantly attenuated neutrophil infiltration, inflammatory factor secretion and NETs formation.19,21 In addition, the use of the PADI4 inhibitors BB-Cl-amidine or GSK199 in a mouse model of lupus nephritis (LN) reduced the formation of type 1 IFNs and NETs while protecting blood vessels, kidneys, and skin from lupus-related damage.45,46 Although progress has been made in the production of selective PADI4 inhibitors, no safe and efficient PADI4 inhibitors are currently available for clinical use.47
In addition to inhibiting NETs formation, renal tissue injury can also be attenuated by degrading NETs components, mainly in a variety of autoimmune diseases. In a mouse model of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), MPO or LL37 inhibitors attenuated NETs-mediated glomerular endothelial cell injury.48,49 In contrast, in a rat AAV model, intravenous immunoglobulin significantly reduced ANCA titers and decreased the formation of NETs.50 Exogenous DNase I could degrade antibody complexes in a mouse model of systemic lupus erythematosus (SLE), thereby decreasing the level of NETs in vivo and reducing the accumulation of NETs in the glomerulus.51 In addition, the application of DNase I or histone neutralizing antibodies in mouse IRI models can also achieve the effect of reducing the accumulation of NETs and partially alleviating AKI.21,52
In addition, we found that inhibition of NETs accumulation alleviated apoptosis and pyroptosis. And cell pyroptosis occurred mainly in renal tubular epithelial cells, while it was not found in vascular endothelial cells. GSDMD was cleaved by caspase-1 to generate N-GSDMD fragments, which could perforate the cytosolic or nuclear membrane and increase membrane permeability, leading to macrophage pyroptosis and neutrophil NETosis.53,54 Our results show that the expression of GSDMD and N-GSDMD is reduced after inhibition of NETs, which is equally relevant for attenuating cell pyroptosis.
Despite some of our work, there are still some shortcomings in this experiment. First, the early infiltration of NETs in the kidney of CI-AKI mice was not significant, and many intravascular neutrophils and NETs would be washed away if perfusion was performed before kidney sampling. Secondly, we would also like to perform scanning electron microscopy of the kidney, a technique that allows visual detection of NETs morphology. Finally, we would like to add cellular experiments to our future studies. In particular, the questions of whether CM can induce NETosis independently, the observation of neutrophil and endothelial cell co-culture, and the role of NETs for cellular phenotypic transformation. In our future studies, we will focus on the triggers of NETs during the CI-AKI process, as well as the key components of NETs that play an injurious role.
Conclusion
In conclusion, this experiment demonstrates for the first time that NETs are present in the kidney of CI-AKI mice and participate in the development of the disease by mediating the endothelial cells of the renal microcirculation. Elimination of NETs could significantly reduce the pathological injury and blood creatinine levels in the kidney and play a role in alleviating CI-AKI. To this end, further exploration of the injury mechanisms of NETs will help to propose prevention and treatment strategies targeting CI-AKI.
Data Sharing Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding authors.
Ethics Statement
All animal experiments were conducted in compliance with National Institutes of Health guidelines and were approved by the Ethics Committee of the second Hospital of Shanxi Medical University (No: DW2022012).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The research was backed by three projects, including the National Natural Science Foundation of China (Grant No. 81770695), the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi Province (Grant No. 2022L151) and the Postgraduate Education Innovation Project of Shanxi Province (Grant No. 2021Y38).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that may have influenced the work reported in this paper.
References
1. Fähling M, Seeliger E, Patzak A, Persson PB. Understanding and preventing contrast-induced acute kidney injury. Nat Rev Nephrol. 2017;13(3):169–180. doi:10.1038/nrneph.2016.196
2. Morcos R, Kucharik M, Bansal P, et al. Contrast-induced acute kidney injury: review and practical update. Clin Med Insights Cardiol. 2019;13:1179546819878680. doi:10.1177/1179546819878680
3. Tao SM, Wichmann JL, Schoepf UJ, Fuller SR, Lu GM, Zhang LJ. Contrast-induced nephropathy in CT: incidence, risk factors and strategies for prevention. Eur Radiol. 2016;26(9):3310–3318. doi:10.1007/s00330-015-4155-8
4. Stevens PE, Levin A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158(11):825–830. doi:10.7326/0003-4819-158-11-201306040-00007
5. Lameire N, Kellum JA. Contrast-induced acute kidney injury and renal support for acute kidney injury: a KDIGO summary (Part 2). Crit Care. 2013;17(1):205. doi:10.1186/cc11455
6. Weisbord SD, Gallagher M, Jneid H, et al. Outcomes after angiography with sodium bicarbonate and acetylcysteine. N Engl J Med. 2018;378(7):603–614. doi:10.1056/NEJMoa1710933
7. Zhou X, Dai J, Xu X, et al. Comparative efficacy of statins for prevention of contrast-induced acute kidney injury in patients with chronic kidney disease: a network meta-analysis. Angiology. 2019;70(4):305–316. doi:10.1177/0003319718801246
8. Kusirisin P, Chattipakorn SC, Chattipakorn N. Contrast-induced nephropathy and oxidative stress: mechanistic insights for better interventional approaches. J Transl Med. 2020;18(1):400. doi:10.1186/s12967-020-02574-8
9. Ma C, Chen T, Ti Y, et al. Ranolazine alleviates contrast-associated acute kidney injury through modulation of calcium independent oxidative stress and apoptosis. Life Sci. 2021;267:118920. doi:10.1016/j.lfs.2020.118920
10. Vlachopanos G, Schizas D, Hasemaki N, Georgalis A. Pathophysiology of Contrast-Induced Acute Kidney Injury (CIAKI). Curr Pharm Des. 2019;25(44):4642–4647. doi:10.2174/1381612825666191210152944
11. Altmann DM. The immune regulatory role of neutrophils. Immunology. 2019;156(3):215–216. doi:10.1111/imm.13049
12. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The Neutrophil. Immunity. 2021;54(7):1377–1391. doi:10.1016/j.immuni.2021.06.006
13. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi:10.1083/jcb.200606027
14. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23(3):279–287. doi:10.1038/nm.4294
15. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385
16. Wigerblad G, Kaplan MJ. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol. 2023;23(5):274–288. doi:10.1038/s41577-022-00787-0
17. Zheng F, Ma L, Li X, et al. Neutrophil extracellular traps induce glomerular endothelial cell dysfunction and pyroptosis in diabetic kidney disease. Diabetes. 2022;71(12):2739–2750. doi:10.2337/db22-0153
18. Liu J, Dong Z. Neutrophil extracellular traps in ischemic AKI: new way to kill. Kidney Int. 2018;93(4):1019. doi:10.1016/j.kint.2018.02.002
19. Nakazawa D, Kumar SV, Marschner J, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol. 2017;28(6):1753–1768. doi:10.1681/asn.2016080925
20. Pieterse E, Rother N, Garsen M, et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler Thromb Vasc Biol. 2017;37(7):1371–1379. doi:10.1161/atvbaha.117.309002
21. Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, Reeves WB. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018;93(2):365–374. doi:10.1016/j.kint.2017.08.014
22. Wang L, Luqmani R, Udalova IA. The role of neutrophils in rheumatic disease-associated vascular inflammation. Nat Rev Rheumatol. 2022;18(3):158–170. doi:10.1038/s41584-021-00738-4
23. Gestermann N, Di Domizio J, Lande R, et al. Netting neutrophils activate autoreactive B cells in lupus. J Immunol. 2018;200(10):3364–3371. doi:10.4049/jimmunol.1700778
24. Peng Y, Wu X, Zhang S, et al. The potential roles of type I interferon activated neutrophils and neutrophil extracellular traps (NETs) in the pathogenesis of primary Sjögren’s syndrome. Arthritis Res Ther. 2022;24(1):170. doi:10.1186/s13075-022-02860-4
25. Kronbichler A, Bajema I, Geetha D, Säemann M. Novel aspects in the pathophysiology and diagnosis of glomerular diseases. Ann Rheum Dis. 2023;82(5):585–593. doi:10.1136/ard-2022-222495
26. Zhang Q, Liu X, Li N, Zhang J, Yang J, Bu P. Sirtuin 3 deficiency aggravates contrast-induced acute kidney injury. J Transl Med. 2018;16(1):313. doi:10.1186/s12967-018-1690-5
27. Wei H, Jiang D, Yu B, et al. Nanostructured polyvinylpyrrolidone-curcumin conjugates allowed for kidney-targeted treatment of cisplatin induced acute kidney injury. Bioact Mater. 2023;19:282–291. doi:10.1016/j.bioactmat.2022.04.006
28. Apelt K, Bijkerk R, Lebrin F, Rabelink TJ. Imaging the Renal Microcirculation in Cell Therapy. Cells. 2021;10(5):1087. doi:10.3390/cells10051087
29. Chade AR. Renal vascular structure and rarefaction. Compr Physiol. 2013;3(2):817–831. doi:10.1002/cphy.c120012
30. He Y, Li H, Yao J, et al. HO‑1 knockdown upregulates the expression of VCAM‑1 to induce neutrophil recruitment during renal ischemia‑reperfusion injury. Int J Mol Med. 2021;48(4). doi:10.3892/ijmm.2021.5018
31. Ma YR, Ma YH. MIP-1α enhances Jurkat cell transendothelial migration by up-regulating endothelial adhesion molecules VCAM-1 and ICAM-1. Leuk Res. 2014;38(11):1327–1331. doi:10.1016/j.leukres.2014.08.019
32. Zhong X, Zeng H, Zhou Z, et al. Structural mechanisms for regulation of GSDMB pore-forming activity. Nature. 2023;616(7957):598–605. doi:10.1038/s41586-023-05872-5
33. Wu X, You D, Cui J, et al. Reduced neutrophil extracellular trap formation during ischemia reperfusion injury in C3 KO mice: C3 requirement for NETs release. Front Immunol. 2022;13:781273. doi:10.3389/fimmu.2022.781273
34. Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol. 2011;58(24):e44–e122. doi:10.1016/j.jacc.2011.08.007
35. Berkseth RO, Kjellstrand CM. Radiologic contrast-induced nephropathy. Med Clin North Am. 1984;68(2):351–370. doi:10.1016/s0025-7125(16)31135-x
36. van der Molen AJ, Reimer P, Dekkers IA, et al. Post-contrast acute kidney injury - Part 1: definition, clinical features, incidence, role of contrast medium and risk factors: recommendations for updated ESUR Contrast Medium Safety Committee guidelines. Eur Radiol. 2018;28(7):2845–2855. doi:10.1007/s00330-017-5246-5
37. Ronda N, Potì F, Palmisano A, et al. Effects of the radiocontrast agent iodixanol on endothelial cell morphology and function. Vascul Pharmacol. 2013;58(1–2):39–47. doi:10.1016/j.vph.2012.08.005
38. Guo Y, Li W, Qian M, et al. D-4F ameliorates contrast media-induced oxidative injuries in endothelial cells via the AMPK/PKC pathway. Front Pharmacol. 2020;11:556074. doi:10.3389/fphar.2020.556074
39. Caiazza A, Russo L, Sabbatini M, Russo D. Hemodynamic and tubular changes induced by contrast media. Biomed Res Int. 2014;2014:578974. doi:10.1155/2014/578974
40. Fauser C, Ullisch EV, Kübler W, Haller C. Differential effects of radiocontrast agents on human umbilical vein endothelial cells: cytotoxicity and modulators of thrombogenicity. Eur J Med Res. 2001;6(11):465–472.
41. Watts ER, Walmsley SR. Getting DAMP(s) wets the whistle for neutrophil recruitment. Immunity. 2018;48(5):846–848. doi:10.1016/j.immuni.2018.04.027
42. Yazdani HO, Chen HW, Tohme S, et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. J Hepatol. 2017. doi:10.1016/j.jhep.2017.09.010
43. Li W, Terada Y, Tyurina YY, et al. Necroptosis triggers spatially restricted neutrophil-mediated vascular damage during lung ischemia reperfusion injury. Proc Natl Acad Sci U S A. 2022;119(10):e2111537119. doi:10.1073/pnas.2111537119
44. Du M, Yang L, Gu J, Wu J, Ma Y, Wang T. Inhibition of peptidyl arginine deiminase-4 prevents renal ischemia-reperfusion-induced remote lung injury. Mediators Inflamm. 2020;2020:1724206. doi:10.1155/2020/1724206
45. Liu Y, Lightfoot YL, Seto N, et al. Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight. 2018;3(23):e124729. doi:10.1172/jci.insight.124729
46. Knight JS, Subramanian V, O’Dell AA, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. 2015;74(12):2199–2206. doi:10.1136/annrheumdis-2014-205365
47. Liu X, Arfman T, Wichapong K, Reutelingsperger CPM, Voorberg J, Nicolaes GAF. PAD4 takes charge during neutrophil activation: impact of PAD4 mediated NET formation on immune-mediated disease. J Thromb Haemost. 2021;19(7):1607–1617. doi:10.1111/jth.15313
48. Antonelou M, Michaëlsson E, Evans RDR, et al. Therapeutic myeloperoxidase inhibition attenuates neutrophil activation, ANCA-mediated endothelial damage, and crescentic GN. J Am Soc Nephrol. 2020;31(2):350–364. doi:10.1681/asn.2019060618
49. Lu Y, Jiang H, Li B, et al. Telomere dysfunction promotes small vessel vasculitis via the LL37-NETs-dependent mechanism. Ann Transl Med. 2020;8(6):357. doi:10.21037/atm.2020.02.130
50. Uozumi R, Iguchi R, Masuda S, et al. Pharmaceutical immunoglobulins reduce neutrophil extracellular trap formation and ameliorate the development of MPO-ANCA-associated vasculitis. Mod Rheumatol. 2020;30(3):544–550. doi:10.1080/14397595.2019.1602292
51. Hakkim A, Fürnrohr BG, Amann K, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107(21):9813–9818. doi:10.1073/pnas.0909927107
52. Zhuang S, Xia S, Huang P, et al. Targeting P2RX1 alleviates renal ischemia/reperfusion injury by preserving mitochondrial dynamics. Pharmacol Res. 2021;170:105712. doi:10.1016/j.phrs.2021.105712
53. Karmakar M, Minns M, Greenberg EN, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun. 2020;11(1):2212. doi:10.1038/s41467-020-16043-9
54. Silva CMS, Wanderley CWS, Veras FP, et al. Gasdermin-D activation by SARS-CoV-2 triggers NET and mediate COVID-19 immunopathology. Crit Care. 2022;26(1):206. doi:10.1186/s13054-022-04062-5
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identification and Validation of Hub Genes Related to Neutrophil Extracellular Traps-Mediated Cell Damage During Myocardial Infarction

Ke D, Ni J, Yuan Y, Cao M, Chen S, Zhou H
Journal of Inflammation Research 2024, 17:617-637
Published Date: 1 February 2024
Neutrophil Extracellular Traps Enhance Procoagulant Activity and Predict Poor Prognosis in Patients With Metastatic Breast Cancer
Gong Y, Chen B, Tan Q, Wei W
International Journal of General Medicine 2025, 18:1247-1259
Published Date: 4 March 2025
Mesenchymal Stem Cell-Derived Exosomes in Anti-NET Therapy: Mechanisms, Challenges, and Future Perspectives
Ye Y, Ye Y, Tian M, Zhao Y, Guo Z, Jin C, Duan S, Zheng Y
International Journal of Nanomedicine 2025, 20:14481-14497
Published Date: 4 December 2025
The Role of Neutrophils in Non-Alcoholic Fatty Liver Disease: Mechanisms and Clinical Significance
Zhang F, Li W
Journal of Inflammation Research 2026, 19:585447
Published Date: 4 March 2026
Neutrophil Extracellular Traps in Dry Eye: A Comprehensive Review of Pathogenic Mechanisms and Implications for Targeted Therapy
Guo X, Wang C, Liu W, Sun Y, Su L, Wang H, Liu B
International Journal of General Medicine 2026, 19:583609
Published Date: 23 March 2026