Back to Archived Journals » Journal of Neurorestoratology » Volume 5
Neuroprotection of hyperbaric oxygen treatment for traumatic brain injury involving autophagy pathway in rats
Authors Liu WC, Wen L, Wang H, Gong JB, Zhan TX, Meng YY, Yang XF
Received 5 August 2016
Accepted for publication 10 January 2017
Published 7 April 2017 Volume 2017:5 Pages 85—91
DOI https://doi.org/10.2147/JN.S119037
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Hongyun Huang
Wen-Chao Liu, Liang Wen, Hao Wang, Jiang-Biao Gong, Tian-Xiang Zhan, Yuan-Yuan Meng, Xiao-Feng Yang
Department of Neurosurgery, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
Abstract: In the present study, we evaluated the changes in autophagy after hyperbaric oxygen (HBO) treatment for traumatic brain injury (TBI) and investigated whether autophagy takes part in the neuroprotection after HBO treatment. Male Sprague Dawley rats were assigned to four groups randomly: sham injury, sham injury and HBO, TBI, and TBI and HBO. The HBO rats received HBO treatment for 100 min immediately after injury. Rats were sacrificed at 24 h after the brain injury and the levels of cleaved caspase-3 and the number of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells in the injured cortex were measured to determine cell death. The expression levels of autophagy-associated proteins were measured by immunohistochemistry and Western blotting to assess changes in autophagy. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cell density and cleaved caspase-3 expression were increased 24 h after TBI. These increases were suppressed by post-TBI HBO treatment. Immunohistochemistry and Western blotting of autophagy-associated proteins showed that TBI can induce autophagy and that HBO treatment can further upregulate the expression of autophagy makers, as shown by an increase in LC3, ATG-5, and Beclin-1 expression and reduction in P62 expression. In conclusion, HBO treatment can reduce apoptosis and further upregulate autophagy in the injured cortex after brain injury, and the autophagy pathway may take part in the neuroprotection provided by HBO treatment for TBI.
Keywords: autophagy, hyperbaric oxygen treatment, traumatic brain injury, apoptosis, neuroprotective effect
Introduction
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity in children and young adults worldwide.1 Brain injury initiates progressive tissue injury through a series of events that lead to neuronal death, brain edema, and impaired cognitive functions.2–4 As more and more data regarding the pathologic changes associated with TBI are obtained, it appears that TBI is a complicated secondary neurodegenerative disease. Primary injuries, such as traumatic axonal injury and cerebral contusions, can cause irreversible damage, and secondary injuries cause cerebral ischemia, oxidative stress, inflammation, and neural apoptosis.5 Recently, the autophagy pathway was shown to be involved in the pathologic processes of both injuries of TBI.6–9
Autophagy is a metabolic process that has complex relationship with apoptosis.10 This process conduces to sustaining the balance of synthesis and degradation, which is necessary for cell homeostasis.11 Autophagy has been showed to take part in the pathogenic mechanism of many diseases, including cerebral ischemia, cerebral hemorrhage, and TBI.7,9,12–15 There is increasing evidence that autophagy can promote neuron survival, and activation of autophagy may serve as a protective effect post-TBI.7,8,16 However, the concrete role of autophagy pathway in brain injury remains controversial and unclear.17
Several studies have indicated the protective effects of hyperbaric oxygen (HBO) treatment for TBI, including reduction of cerebral edema, inhibition of inflammatory response, protecting the blood brain barrier, and improved neurologic recovery.18–21 In this study, we examine the changes of autophagy markers in the cortex after HBO treatment in TBI rats and investigate the effect of the autophagy in this process.
Materials and methods
Animals and HBO treatment
All animal procedures were approved by and conducted in strict accordance with the ethical guidelines of Zhejiang University Animal Experimentation Committee. Every attempt was made to minimize the discomfort and pain of the animals. Male Sprague Dawley rats (350–380 g; Shanghai Laboratory Animal Center) were assigned to four groups randomly: sham injury (n=20), HBO (n=20), TBI (n=20), and TBI and HBO (n=20). For HBO administration, rats were placed in the hyperbaric chamber (100% oxygen at 2.5 atm absolute) for 100 min immediately after brain injury. The rats were returned to their cages after HBO treatment.
Traumatic brain injury
We used the lateral fluid-percussion model to induce rat TBI, as described previously.22 Briefly, 24 h before brain injury, the rats were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneal injection). A craniotomy of 4.8 mm diameter was performed using a trephine drill, 2.5 mm lateral to the median line, maintaining the exposed dura intact. A female Luer lock was secured on the craniotomy. Then, the rats were returned to their cages to recover overnight. The next day, the skin incision was opened and the rat was connected to the fluid percussion brain injury device (Custom Design and Fabrication, Richmond, VA, USA). A moderate TBI (1.8–2.0 atm) was performed by releasing the pendulum onto the cylinder. Immediately after brain injury, the hub assembly was removed, the wound was cleaned, and the incision was sutured. The animals were then returned to their cages. Sham injury group rats received craniotomies without exposure to trauma. These animals were killed 24 h after the operation.
Immunohistochemistry and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) method
At 24 h after sham or TBI procedures, the rats were anesthetized by 10% chloral hydrate and transcardially perfused with phosphate-buffered saline (PBS; 100 mL) and 4% paraformaldehyde solution. The brains were removed and the brain tissues immersed in 30% sucrose overnight at 4°C. Post-fixed brain tissues were sectioned on a sliding microtome.
The TUNEL method was performed in accordance with the directions of In Situ Cell Death Detection Kit (Hoffman-La Roche Ltd., Basel, Switzerland). Briefly, sections were immersed in 0.1% Triton-X and washed with PBS three times for a total of 30 min. Subsequently, they were exposed to 0.3% H2O2 at room temperature for 30 min and then washed with PBS. The sections were immersed in 50 μL of reaction mixture at 37°C for 1 h in the dark. Next, the sections were rinsed thrice in PBS and blocked for 30 min, followed by diaminobenzidine coloration. Light microscopy was performed for observation using an Olympus BX51 optical microscope in bright field; ten fields per section were randomly chosen to perform the counting of TUNEL-positive cells. There were six rats in each of the four groups.
Sections 4 μm thick were used for immunofluorescence assay of Beclin-1, ATG-5, and LC3. The sections were blocked with 5% bovine serum albumin at room temperature for 1 h and then incubated with primary antibodies (1:200) overnight. The following day, after incubation, the sections were washed for 15 min with PBS and then incubated with secondary antibody: Alexa Fluor 488 goat anti-rabbit IgG (1:300; Thermo Fisher Scientific, Waltham, MA, USA; Cat. No. A-24922). The sections were then incubated in 4′,6-diamidino-2-phenylindole (1 ng/μL; Sigma-Aldrich Co., St Louis, MO, USA; D9542) for nuclear counterstaining at room temperature for 5 min. Laser confocal scanning microscope (Olympus, FV1000) was used to detect the fluorescent signals. Ten fields per section were randomly selected to perform the counting of Beclin-1-, ATG-5-, and LC3-positive cells. There were six rats in each of the four groups.
Western blotting
Twenty four hours after TBI, the brains were rapidly removed and the injured cortex was quickly dissected. The injured cortical tissues were homogenized with lysis solution. After centrifugation at 12,000 ×g for 30 min, the protein concentrations were measured with bicinchoninic acid protein assay kit (Beyotime Biotechnology). An equal quality of protein (50 µg/lane) was separated by 10% sodium dodecylsulfate polyacrylamide gel electrophoresis and then transferred onto a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). The membranes were then blocked in 5% skimmed milk for 2 h and incubated with primary antibodies at 4°C overnight. The following primary antibodies were used: P62 (1:1000; Abcam, Cambridge, UK), ATG-5 (1:1000; Abcam), Beclin-1 (1:1000; Cell Signaling Technology, Danvers, MA, USA), LC3 (1:10,000; Cell Signaling Technology), caspase-3 (1:10,000; Cell Signaling Technology), and GAPDH antibody (1:10,000; Cell Signaling Technology). After the membranes had been washed three times in tris-buffered saline with Tween, they were incubated in secondary antibodies for 2 h. Then the membranes were washed with tris-buffered saline with Tween. Blotted protein bands were exposed to X-ray film, and the results were quantified with Quantity One Software (Bio-Rad). There were eight rats in each of the four groups.
Statistical analyses
Statistical Package for the Social Sciences (SPSS) 20.0 (IBM Corporation, Armonk, NY, USA) was used for statistical analysis. All data are presented as mean ± standard error of mean. One-way analysis of variance and Tukey’s test were used for comparisons among groups. p Values <0.05 were considered statistically significant.
Results
HBO treatment decreased apoptosis in the injured cortex after TBI
HBO treatment decreased the number of TUNEL-positive cells in the cortex after TBI
To elucidate the protective mechanism of HBO in TBI, we examined the results of TUNEL staining to explore apoptosis in the injured cortex. There were few TUNEL-positive cells in the cortex of the sham injury group (Figure 1A, B). Twenty-four hours after TBI in the injured cortex, the TUNEL-positive cells were markedly increased in the TBI group compared with the sham group (31.7%±3.1%; p<0.001; Figure 1C and 1E). However, the number of TUNEL-positive cells was significantly decreased in the post-TBI HBO group compared to the TBI group (15.6%±3.5%; p<0.001; Figure 1D and 1E). Quantitative analysis indicated that HBO treatment clearly had a protective effect on delayed neuronal cell death in the cortex of TBI rats.
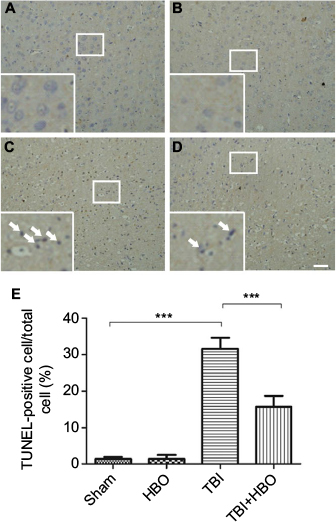 | Figure 1 TUNEL immunohistochemistry staining of the injured cortex: (A) sham injury, (B) HBO, (C) TBI, (D) TBI + HBO, and (E) Quantitative analysis of TUNEL-positive cells. Notes: The number of TUNEL-positive cells in the total number of cells was evaluated in ten microscopic fields per section. HBO treatment could significantly decrease the number of TUNEL-positive cells after TBI. Arrows indicate TUNEL-positive cells. ***p<0.001. Magnification, 10×; scale bar =100 μm Abbreviations: HBO, hyperbaric oxygen; TBI, traumatic brain injury; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling. |
HBO treatment downregulated the expression of cleaved caspase-3 in the cortex after TBI
Twenty-four hours after brain injury, the expression levels of cleaved caspase-3 (assayed by Western blotting) of the TBI and TBI + HBO groups were increased by 1.67±0.13 and 1.16±0.08 compared with the sham group, respectively (Figure 2A, B). The expression level of cleaved caspase-3 was significantly increased in the injured cortex after TBI (p<0.05, TBI versus sham) and this increase was suppressed by post-TBI HBO treatment (p<0.05, TBI versus TBI + HBO).
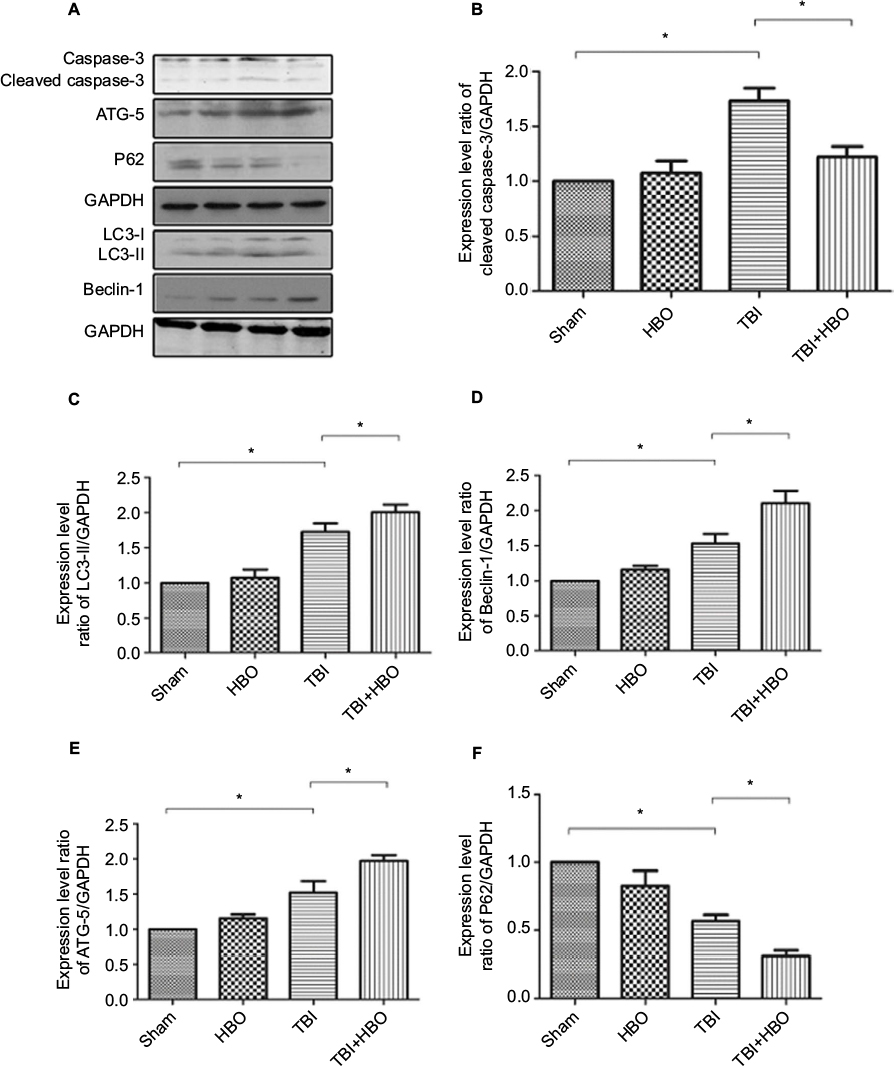 | Figure 2 Western blotting of cleaved caspase-3, P62, ATG-5, Beclin-1, and LC3 in rats’ injured cortex after TBI. Notes: GAPDH was used as a load control. Values are the mean ± SEM. The expression levels of cleaved caspase-3, ATG-5, Beclin-1, and LC3 significantly increased and the expression level of P62 significantly decreased in the injured cortex 24 h after TBI. Post-TBI HBO treatment further upregulated the levels of ATG-5, Beclin-1, and LC3 and downregulated the levels of cleaved caspase-3 and P62, compared with rats in the TBI group. *p<0.05. Magnification, 20×. Abbreviations: HBO, hyperbaric oxygen; SEM, standard error of the mean; TBI, traumatic brain injury. |
HBO treatment significantly increased autophagy in the injured cortex 24 h after TBI
Western blotting
The expression level of LC3-II was upregulated in the injured cortex 24 h after the brain injury, compared to the sham group (1.75±0.38, p<0.05). Post-TBI HBO treatment could further increase LC3-II level, compared to rats in the TBI group (2.02±0.11, p<0.05; Figure 2A, C).
The expression level of Beclin-1 significantly increased in the injured cortex 24 h after the injury, compared to the sham group (1.55±0.08, p<0.05). Post-TBI HBO treatment could efficiently increase Beclin-1 level 24 h after the injury, compared to the TBI group (2.16±0.09, p<0.05; Figure 2A, D).
The expression level of ATG-5 significantly increased 24 h after brain injury, compared to the sham group (1.52±0.35, p<0.05). Post-TBI HBO treatment could further increase ATG-5 level 24 h after the injury, compared to the TBI group (1.97±0.07, p<0.05; Figure 2A, E).
The expression level of P62 significantly decreased at 24 h after brain injury in the injured cortex, compared with the sham group (0.58±0.07, p<0.05). Post-TBI HBO treatment could further decrease P62 level 24 h after the injury, compared to the TBI group (0.31±0.04, p<0.05; Figure 2A, F).
Immunofluorescence
At 24 h after injury, the number of LC3-positive cells was increased in the injured cortex, compared to rats in the sham injury group (18.9%±4.6%, p<0.05). Post-TBI HBO treatment further increased the number of LC3-positive cells, compared to rats in the TBI group (26.1%±5.4%, p<0.05; Figure 3).
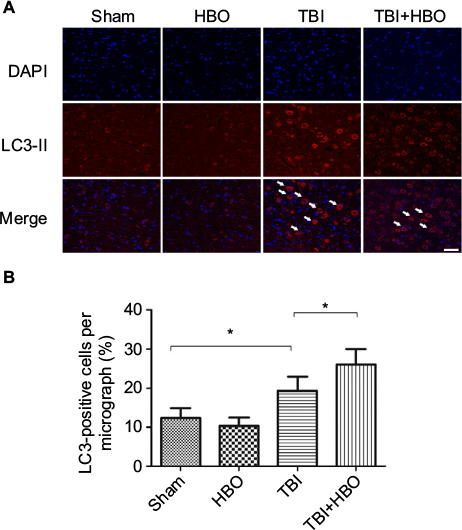 | Figure 3 Immunofluorescence analysis of LC3 expression in the cortex. Notes: The average number of LC3-positive cells in the total number of cells was evaluated in ten microscopic fields. Arrows indicate LC3-positive cells. Post-TBI HBO treatment significantly increased the LC3-positive cells. *p<0.05. Magnification, 20×; scale bar =100 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; HBO, hyperbaric oxygen; TBI, traumatic brain injury. |
At 24 h after injury, the number of Beclin-1-positive cells was increased in the injured cortex, compared to rats in the sham injury group (21.5%±4.3%, p<0.05). Post-TBI HBO treatment further increased the number of Beclin-1-positive cells, compared to rats in the TBI group (32.7%±3.9%, p<0.05; Figure 4).
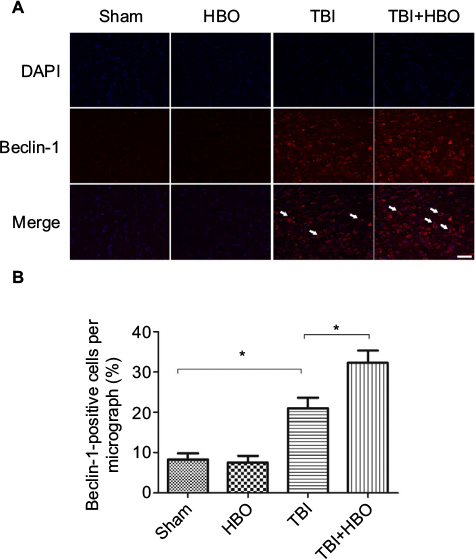 | Figure 4 Immunofluorescence analysis of Beclin-1 expression in the cortex. Notes: The average number of Beclin-1-positive cells in the total number of cells was evaluated in ten microscopic fields. Arrows indicate Beclin-1-positive cells. Post-TBI HBO treatment significantly increased the Beclin-1-positive cells. *p<0.05. Magnification, 20×; scale bar =100 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; HBO, hyperbaric oxygen; TBI, traumatic brain injury. |
At 24 h after brain injury, the number of ATG-5-positive cells was significantly increased, compared to rats in the sham injury group (21.5%±4.1%, p<0.05). Post-TBI HBO treatment further increased the number of ATG-5-positive cells, compared to rats in the TBI group (40.1±5.9, p<0.05; Figure 5).
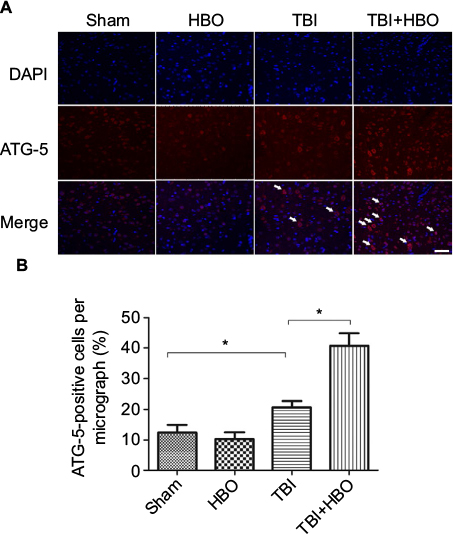 | Figure 5 Immunofluorescence analysis of ATG-5 expression in the cortex. Notes: The average number of ATG-5-positive cells in the total number of cells was evaluated in ten microscopic fields. Arrows indicate ATG-5-positive cells. Post-TBI HBO treatment significantly increased the ATG-5-positive cells. *p<0.05. Magnification, 20×; scale bar =100 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; HBO, hyperbaric oxygen; TBI, traumatic brain injury. |
Discussion
In the present study, we found that the TUNEL-positive cells and the expression of cleaved caspase-3 in the injured cortex were significantly increased 24 h after brain injury, and HBO treatment could attenuate cortical cell death and brain injury. We also observed that TBI activated autophagy in the injured cortex at 24 h after TBI, and HBO treatment could further upregulate the level of autophagy after TBI. These results suggest that activation of autophagy may be involved in the neuroprotection provided by HBO treatment after TBI.
Apoptosis plays a crucial role in the pathogenesis of brain injury. Apoptosis of neurons and glia is a key signaling pathway that leads to brain injury and contributes to the overall pathology of TBI.23,24 In several experimental models of TBI, activation of proapoptotic molecules was observed in the early period and in tissues away from the injured site, weeks after brain trauma.23,25,26 Several studies have demonstrated that the balance between proapoptotic and antiapoptotic factors is a key mechanism of apoptosis.27–29 Activation of death-inducing factors, such as Bax, p53, and tumor-suppressor gene, induces apoptosis. In accordance with previous studies, we verified that TBI could induce significant apoptosis of injured cortex, which was evaluated by activation of caspase-3 and TUNEL assay.
In addition to apoptosis, autophagy can also be activated by TBI.7,8,16,17,30,31 Increased autophagy after TBI was first reported by Diskin; it was found that the expression level of Beclin-1 was significantly increased in the closed-head injury models of mice.30 Also, Lai et al first found ultrastructural authentication of autophagosomes in controlled cortical impact brain injury in mice. It was demonstrated that the number of autophagosomes and autolysosomes was increased 2 h after TBI and lasted for an extended period in the injured cortex.32 Autophagy is essential for the maintenance of cell survival and cellular homeostasis. In the present study, autophagic activity was assessed by the levels of proteins Beclin-1, LC3, and ATG-5, which are biomarkers for detecting changes in autophagy. Beclin-1, a novel Bcl-2-homology (BH)-3 domain protein, plays an important role in autophagy. The proautophagic activity of Beclin-1 can be attenuated by Bcl-2, and therefore, Beclin-1–Bcl-2 complex is thought to regulate the switch between autophagy and apoptosis.33 LC3, a mammalian ortholog of yeast Atg8, is one of the most reliable markers in the study of autophagy induction.33 When autophagy is activated, LC3-I is conjugated with phosphatidylethanolamine to form LC3-II, and LC3-II accumulation reflects autophagic flux.34,35
The role of autophagy after TBI has been studied with rapamycin, which activates autophagy by inhibition of mammalian target of rapamycin. It was found that autophagy might play a beneficial role and maintained cellular homeostasis after brain injury.36 However, the concrete role of autophagy is not yet clear. Several studies demonstrated that autophagy pathway participates in the neuropathology of brain injury.15,37,38 In this study, we found that HBO treatment could increase autophagy and attenuate injured cortex cell death. Together with previous studies, we suggest autophagy pathway may be involved in the neuroprotection provided by HBO treatment for TBI.
Previous researches have confirmed that HBO treatment reduces neuronal apoptosis by reducing p53 and increasing bcl-2 expression.39–43 Bcl-2 proteins were previously shown to exert a neuroprotective effect by reducing the release of cytochrome c and blocking caspase activation.44 Bcl-2 is not only involved in the pathway of apoptosis, but also participates in the regulation of the autophagy signaling pathways.45,46 Our results further suggest that HBO could protect against brain injury by increasing autophagy. This upregulation of autophagy may be partially caused by the increased Bcl-2 after HBO. In addition, the two signaling pathways interact with each other and jointly play an important role in the neuroprotection provided by HBO treatment.
In conclusion, TBI induced autophagy in the injured cortex 24 h after the injury. HBO treatment could attenuate apoptosis and further upregulate autophagy. Therefore, we propose that autophagy pathway participates in the neuroprotection provided by HBO treatment in TBI. Further studies are required to provide concrete evidence of the mechanisms of the activation of autophagy by HBO.
Acknowledgment
This project was supported by grants from the National Natural Science Foundation of China (81470052 and 81200955).
Disclosure
The authors report no conflicts of interest in this work.
References
Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129(4):1021–1029. | ||
Cernak I, Noble-Haeusslein LJ. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab. 2010;30(2):255–266. | ||
Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. | ||
Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4–9. | ||
Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7(8):728–741. | ||
Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2008;4(1):88–90. | ||
Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28(4):674–683. | ||
Luo CL, Li BX, Li QQ, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. | ||
Jin Y, Lin Y, Feng JF, Jia F, Gao GY, Jiang JY. Moderate hypothermia significantly decreases hippocampal cell death involving autophagy pathway after moderate traumatic brain injury. J Neurotrauma. 2015;32(14):1090–1100. | ||
Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012;3(1):17–27. | ||
Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. | ||
Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144–153. | ||
Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med. 2012;25 (Suppl 1):30–34. | ||
Carloni S, Buonocore G, Longini M, Proietti F, Balduini W. Inhibition of rapamycin-induced autophagy causes necrotic cell death associated with Bax/Bad mitochondrial translocation. Neuroscience. 2012;203:160–169. | ||
Son JH, Shim JH, Kim KH, Ha JY, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. 2012;44(2):89–98. | ||
Zhang YB, Li SX, Chen XP, et al. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull. 2008;24(3):143–149. | ||
Lai Y, Hickey RW, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28(3):540–550. | ||
Huang L, Obenaus A. Hyperbaric oxygen therapy for traumatic brain injury. Med Gas Res. 2011;1:21. | ||
Zhang Y, Yang Y, Tang H, et al. Hyperbaric oxygen therapy ameliorates local brain metabolism, brain edema and inflammatory response in a blast-induced traumatic brain injury model in rabbits. Neurochem Res. 2014;39(5):950–960. | ||
Geng F, Ma Y, Xing T, Zhuang X, Zhu J, Yao L. Effects of hyperbaric oxygen therapy on inflammasome signaling after traumatic brain injury. Neuroimmunomodulation. 2016;23(2):122–129. | ||
Hadanny A, Efrati S. The efficacy and safety of hyperbaric oxygen therapy in traumatic brain injury. Expert Rev Neurother. 2016;16(4):359–360. | ||
McIntosh TK, Vink R, Noble L, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28(1):233–244. | ||
Clark RS, Kochanek PM, Chen M, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13(8):813–821. | ||
Tashlykov V, Katz Y, Gazit V, Zohar O, Schreiber S, Pick CG. Apoptotic changes in the cortex and hippocampus following minimal brain trauma in mice. Brain Res. 2007;1130(1):197–205. | ||
Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147(6):1575–1583. | ||
Newcomb JK, Zhao X, Pike BR, Hayes RL. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp Neurol. 1999;158(1):76–88. | ||
Slemmer JE, Zhu C, Landshamer S, et al. Causal role of apoptosis-inducing factor for neuronal cell death following traumatic brain injury. Am J Pathol. 2008;173(6):1795–1805. | ||
Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17(10):927–938. | ||
Raghupathi R, Conti AC, Graham DI, et al. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in cellular Bcl-2 immunoreactivity. Neuroscience. 2002;110(4):605–616. | ||
Diskin T, Tal-Or P, Erlich S, et al. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma. 2005;22(7):750–762. | ||
Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17(8):467–484. | ||
Lai YC, Hickey RW, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cerebr Blood F Met. 2008;28(3):540–550. | ||
Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007;581(11):2156–2161. | ||
Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152(4):657–668. | ||
Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36(12):2503–2518. | ||
Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26(1):86–93. | ||
Feng Y, Gao J, Cui Y, et al. Neuroprotective effects of resatorvid against traumatic brain injury in rat: involvement of neuronal autophagy and TLR4 signaling pathway. Cell Mol Neurobiol. 2017;37(1):155–168. | ||
Yao J, Zheng K, Zhang X. Rosiglitazone exerts neuroprotective effects via the suppression of neuronal autophagy and apoptosis in the cortex following traumatic brain injury. Mol Med Rep. 2015;12(5):6591–6597. | ||
Zhang QX, Chang Q, Cox RA, Gong XM, Gould LJ. Hyperbaric oxygen attenuates apoptosis and decreases inflammation in an ischemic wound model. J Invest Dermatol. 2008;128(8):2102–2112. | ||
Liu Z, Jiao QF, You C, Che YJ, Su FZ. Effect of hyperbaric oxygen on cytochrome C, Bcl-2 and Bax expression after experimental traumatic brain injury in rats. Chin J Traumatol. 2006;9(3):168–174. | ||
Palzur E, Zaaroor M, Vlodausky E, Milman F, Soustiel JF. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008;1221:126–133. | ||
Weber SU, Koch A, Kankeleit J, et al. Hyperbaric oxygen induces apoptosis via a mitochondrial mechanism. Apoptosis. 2009;14(1):97–107. | ||
Chen X, Duan XS, Xu LJ, et al. Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience. 2014;266:235–243. | ||
Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. | ||
Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009;5(5):720–722. | ||
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.