Back to Journals » Cancer Management and Research » Volume 11
Negative regulators of STAT3 signaling pathway in cancers
Authors Wu M, Song D, Li H, Yang Y, Ma X, Deng S, Ren C, Shu X ![]()
Received 21 February 2019
Accepted for publication 17 April 2019
Published 29 May 2019 Volume 2019:11 Pages 4957—4969
DOI https://doi.org/10.2147/CMAR.S206175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Moli Wu,1,2,* Danyang Song,1,* Hui Li,1 Yang Yang,1 Xiaodong Ma,1 Sa Deng,1 Changle Ren,3 Xiaohong Shu1
1College of Pharmacy, Dalian Medical University, Dalian 116044, People’s Republic of China; 2College of Basic Medical Sciences, Dalian Medical University, Dalian 116044, People’s Republic of China; 3Surgery Department of Dalian Municipal Central Hospital, Dalian Medical University, Dalian 116033, People’s Republic of China
*These authors contributed equally to this work
Abstract: STAT3 is the most ubiquitous member of the STAT family and involved in many biological processes, such as cell proliferation, differentiation, and apoptosis. Mounting evidence has revealed that STAT3 is aberrantly activated in many malignant tumors and plays a critical role in cancer progression. STAT3 is usually regarded as an effective molecular target for cancer treatment, and abolishing the STAT3 activity may diminish tumor growth and metastasis. Recent studies have shown that negative regulators of STAT3 signaling such as PIAS, SOCS, and PTP, can effectively retard tumor progression. However, PIAS, SOCS, and PTP have also been reported to correlate with tumor malignancy, and their biological function in tumorigenesis and antitumor therapy are somewhat controversial. In this review, we summarize actual knowledge on the negative regulators of STAT3 in tumors, and focus on the potential role of PIAS, SOCS, and PTP in cancer treatment. Furthermore, we also outline the STAT3 inhibitors that have entered clinical trials. Targeting STAT3 seems to be a promising strategy in cancer therapy.
Keywords: cancer, STAT3 signaling, PIAS, SOCS, PTP, negative regulators
Introduction
STAT protein family members, including STAT1, STAT2, STAT3, STAT4, STAT5 (STAT5A and STAT5B), and STAT6, are important transducers of many cytokines and growth factors.1 Of the seven members of the STAT protein family, STAT3 is the most common, and is constitutively activated or overexpressed in approximately 70% of human solid and hematological tumors compared with normal tissue.2 STAT3 is activated by phosphorylation to form a homodimer and then translocates to the nucleus. The nuclear homodimer recognizes and binds to STAT3-specific DNA-binding elements that can regulate the expression of target genes associated with cell growth, proliferation, differentiation, apoptosis, and immunoresponse.3 Aberrant activation of STAT3 can induce malignant cell transformation and is associated with poor prognosis of some tumors.4–10 It has been reported that disruption of constitutively activated STAT3 can suppress tumor-cell growth and promote cell apoptosis, and the STAT3-signaling pathway has become an attractive target for cancer therapy.4–7,11 Therefore, great efforts have been made to discover new selective inhibitors targeting STAT3 signaling. Recent studies have shown that several negative regulators of STAT3 signaling, including PIAS, SOCS, and PTP, can effectively prevent cancer progression (Figure 1).12–17 However, these negative regulators have also been reported to correlate with tumor malignancy, and their biological functions in tumorigenesis and antitumor therapy are somewhat controversial. Here, we review actual knowledge on PIAS, SOCS, and PTP in cancer and summarize the STAT3 inhibitors that have entered clinical trials, in order to evaluate the role of targeting STAT3 in cancer therapy.
 | Figure 1 The negative regulators of JAK-STAT signaling. Binding of the ligand to cytokine receptor induces receptor dimerization and activation of receptor associated JAK kinase, which in turn phosphorylates STAT proteins. After forming a homodimer, STAT proteins translocate to the nucleus to control gene expression. Negative regulation of the JAK-STAT pathway is provided by PTPs, SOCS and PIAS proteins. |
Protein inhibitor of activated STAT (PIAS) family
There are four PIAS genes in mammals: PIAS1, PIAS2, PIAS3, and PIAS4. These genes share 40% of sequence homology and a similar domain organization, and the protein corresponding to each gene has a Zn-binding ring-finger domain in the central portion (Figure 2).18 PIAS proteins were initially identified as inhibitors of STAT transcription factors (Figure 1),19,20 but in fact they can regulate a broader range of biological processes, including nuclear trafficking and DNA-damage repair by interacting with other transcription factors, such as NF-κB and p53.21 PIAS proteins may regulate transcription through several mechanisms (Figure 1), eg, blocking the DNA-binding activity of transcription factors, recruiting transcriptional co-repressors, and promoting protein SUMOylation.22 It has been reported that PIAS proteins can bind to activated STAT dimers and prevent them from binding DNA (Figure 1). PIAS1 and PIAS3 bind to STAT1 and STAT3, respectively, and inhibit transcriptional activity of STAT1 and STAT3.19,20
 | Figure 2 Structure characteristics of STATs, PIAS and SOCS. (A) Structure of STAT. ND, N domain; CCD, coiled coil domain; DBD, DNA-binding domain; LD, linker domain; SH2, Src homology 2 domain; TAD, transactivation domain. (B) The domain structure of PIAS proteins. SAP/LXXLL, SAF (scaffold attachment factor A and B) -A/B, Acinus and PIAS domain (it can recognize and bind to AT-rich DNA sequences), Within the SAP domain, there is a conserved LXXLL (it mediates interactions between nuclear receptors and their co-regulators) signature motif; PINIT, Pro-Ile-Asn-Ile-Thr motif (a highly conserved region of PIAS proteins and it may be involved in the nuclear retention of PIAS3); RLD, RING finger-like zinc binding domain; AD/SIM, acidic domain (AD), Within the AD, there is a putative SUMO1 interaction motif (SIM) in all PIASs except PIASy; S/T, serine-threonine rich region (in all PIASs except PIASy). (C) Structure of SOCS proteins. All eight members are characterized by their N-terminal region with variable length and limited homology, an extended SH2 domain (ESS), a central SH2 domain and a conserved SOCS box at the C-terminus. |
A basal amount of PIAS3 has been shown to exist in the nucleus in most normal human epithelial and endothelial cells.23 Due to the inhibitory effect of PIAS3 on STAT3 activation, downregulation of PIAS3 expression may play a critical role in cancer development. As a matter of fact, many studies have demonstrated that PIAS3 expression is reduced in various cancers.24–27 For example, PIAS3 mRNA is undetectable in most lymphoma cells, and absence of PIAS3 partly contributes to the high levels of activated STAT3 in these cells.24 The PIAS3 protein is also absent in most lung squamous-cell carcinoma (SCC).12 Moreover, the association of low PIAS3 expression with increased STAT3 activation has been found in malignant mesothelioma.27 Lastly, it has been shown that there is reduced PIAS3 expression in glioblastoma tissue that can promote glioblastoma-cell proliferation.25,26
In contrast, a lot of studies have demonstrated that upregulation of PIAS3 expression can inhibit cell proliferation and increase drug chemosensitivity in various tumors. Inhibition of constitutively activated STAT3 by curcumin attenuates tumor-cell growth by upregulating PIAS3 in ovarian and endometrial cancer cells.13 Overexpression of PIAS3 contributes to suppression of lung cancer–cell growth and restores drug chemosensitivity.28–30 In prostate cancer, overexpression of PIAS3 induces cancer-cell apoptosis both in vitro and in nude mice.31 In addition, PIAS3 overexpression can reduce STAT3 transcription and inhibit glioblastoma-cell proliferation.26 All these findings indicate that PIAS3 may be an attractive candidate for targeting the JAK–STAT signaling pathway and restoring sensitivity to chemotherapeutic drugs in cancer therapy.
However, overexpression of PIAS genes has also been observed in some cancers.23,32 PIAS1 is overexpressed in human prostate cancer, and enhances cancer-cell growth through inhibition of p21.33,34 High PIAS1 expression is associated with adverse patient outcomes in multiple myeloma.35 Additionally, PIAS3 is overexpressed in colorectal cancer.36 The mechanism of the PIAS proteins promoting tumorigenesis may be related to their SUMO-ligase activity. Through SUMOylation, PIAS proteins can interact with several tumor suppressors and oncogenes including TP53, PML, AKT, MYC, and FAK.36 This field needs to be further explored in the near future.
In conclusion, the biological function of PIAS in tumorigenesis and antitumor therapy is somewhat controversial. Therefore, much more detailed genetic and functional analyses of PIAS should be performed to clarify the inconsistencies and thus better to understand the role of PIAS in cancer therapy.
Suppressor of cytokine–signaling proteins
The mammalian SOCS family consists of eight members: SOCS1–7 and the cytokine-inducible SH2-containing protein. They all are negative-feedback regulators of the JAK–STAT signaling pathway.37 Structures of SOCS family members are characterized by an N-terminal region of variable length and limited homology, a central SH2 domain, and a conserved SOCS box at the C-terminus (Figure 2).38 SOCS proteins inhibit JAK–STAT signaling by mechanisms (Figure 1) of blocking STAT recruitment to the cytokine receptor by shielding the STAT-binding sites of the receptor, binding to JAKs and inhibiting their kinase activities, and targeting receptor proteins or JAKs for proteosomal degradation via ubiquitination.39,40 A positive correlation has been proven between SOCS dysregulation and tumor progression. Several members of the SOCS family have been identified as tumor suppressors, and dysregulation of their biological roles in controlling cytokine and growth-factor signaling may contribute to the development of many human cancers.41,42
SOCS1, SOCS2, and tumors
Hypermethylation and silencing of SOCS1 have been commonly reported in various kinds of tumors, including cervical cancer, esophageal SCC (ESCC), hepatocellular carcinoma, breast cancer, ovarian cancer, glioblastoma multiforme, acute myeloid leukemia, and chronic myeloid leukemia.43–49 Methylation of SOCS1-promoter CpG islands contributes to the transformation of liver cirrhosis to hepatocellular carcinoma.50 The SOCS1 gene has been found to be frequently mutated in both classical Hodgkin's lymphoma and primary mediastinal B-cell lymphoma.51,52 Restoration of SOCS1 gene expression suppresses cell growth in acute myeloid leukemia,53 breast cancer,54 ovarian cancer, and hepatocellular carcinoma.46,55 Hypermethylation of SOCS1 is reversed to an unmethylated state during chronic myeloid leukemia patients’ remission phase.49 In gastric cancer, loss of the SOCS1 protein is involved in tumor progression and lymph-node metastasis.56 Spontaneous colorectal cancer is also found in SOCS1-knockout mice.57 In addition, SOCS1 expression is correlated with the clinical stages of some tumors. The SOCS1 level at stages II-IV is lower than at stage I in colorectal tumors. Meanwhile, the SOCS1 protein is highly expressed in well-differentiated adenocarcinomas.58 High mRNA levels of SOCS1 are also associated with early tumor stages, and can improve clinical outcomes in breast cancer.59 Breast cancer patients with positive SOCS1 expression exhibit decreased incidence of detectable circulating tumor cells in peripheral blood.60 In glioblastoma multiforme, hypermethylation-mediated silencing of SOCS1 enhances tumor radioresistance.47
In light of these findings, SOCS1 displays a role as a tumor suppressor in most tumors through inhibiting tumor proliferation and invasion, as well as reducing the sensitivity of tumor cells to cytokines or hormones. Molecular mechanisms underlying the antiproliferative effect of SOCS1 on tumor cells are inhibition of JAK–STAT3 and other signaling pathways. In non-small-cell lung cancer, SOCS1 presents its potent antiproliferative effects through blockage of the JAK–STAT signaling and FAK-dependent signaling pathways.61 SOCS1 also exerts its growth-inhibitory function through downregulation of cyclin D1, CDK2, and CDK4 in prostate cancer.62 In addition, SOCS1 has been reported to inhibit the invasion and migration of colorectal cancer by preventing epithelial–mesenchymal transition and promotes mesenchymal–epithelial transition by increasing E-cadherin and decreasing ZEB1 observed in cell cultures and mouse-xenograft models.63
Similarly, hypermethylation of SOCS2 has been detected in ovarian cancer.46 SOCS2 CpG islands were found to be hypermethylated in 14% of primary ovarian cancers, but not in normal tissue. Furthermore, high SOCS2 expression is closely associated with favorable prognosis in primary breast cancer, and survival time also shows an evident positive correlation with SOCS2 expression in breast cancer patients.64
SOCS3 and tumors
In various human cancers, reduced expression or silencing of SOCS3 is associated with constitutive STAT3 activation,15 and hyperactivation of STAT3 can contribute to tumorigenesis by inducing multiple tumor-promoting genes.65 Hypermethylation of SOCS3 is mostly found in head-and-neck cancer,66 lung cancer,67 glioma,68 cholangiocarcinoma,69 prostate cancer (but not in benign prostate hyperplasia),70 Barrett esophagus carcinoma, and ulcerative colitis–related colorectal cancer.71,72 Reduced SOCS3 expression has been detected in human malignant melanoma.73 In hepatocellular carcinoma, level of SOCS3 expression is inversely correlated with STAT3 activation.74 Loss of SOCS3 activates STAT3, promotes cell proliferation, and leads to enhanced hepatitis-induced hepatocarcinogenesis.75 Moreover, restoration or upregulation of SOCS3 expression can suppress tumor growth and metastasis in some malignancies.76–78 For example, exogenous SOCS3 can inhibit cell growth and enhance cell sensitivity to radiotherapy in human non-small-cell lung cancer.79 The antitumor mechanism of SOCS3 may involve its negative regulation of the JAK–STAT and other signaling pathways.80–82 In prostate cancer, SOCS3 antagonizes the proliferative and migratory effects of FGF2 by inhibiting p44/p42 MAPK signaling.80 Other studies have also demonstrated that SOCS3 can inhibit the proliferation of mesothelioma cells via multiple signaling pathways, including JAK–STAT3, ERK, Fak, and p53.81 In addition, SOCS3 has also been found to inhibit inflammation-associated tumorigenesis in the colon through both STAT3 and NFκB pathways.82
SOCS4, SOCS5, and tumors
Some studies have proven that SOCS4 can suppress tumor growth.59,83–85 In human breast cancer, SOCS4 expression is inversely associated with TNM stage, and high SOCS4 expression predicts a favorable prognosis.59 Meanwhile, an inverse relationship between SOCS4 and EGFR expression has also been found in aggressive hepatocellular carcinoma.83 Compared with noncancerous gastric tissues, gastric cancer elicits much lower SOCS4 expression, accompanied by hypermethylation of SOCS4-promoter CpG sites.84 Moreover, in vivo studies using several mouse models have demonstrated that SOCS4 is able to suppress tumors derived from epithelial cells and that RUNX1 mediates repression of the SOCS4 promoter to reduce SOCS4 level and increase STAT3 activity, thereby promoting tumor development.85
Similarly to SOCS4, SOCS5 is also able to suppress tumor development.43,86,87 SOCS5 expression is higher in normal human cervical tissue than in neighboring cervical tumors.43 Significant reduction of SOCS5 is detected in the thyroid-gland cancer tissue than normal tissue,86 and exogenous SOCS5 in the highly aggressive anaplastic thyroid cancer cells can reduce or abolish phosphorylation of the STAT3 protein and activation of the PI3K–AKT pathway, which can cause an altered balance between proapoptotic and antiapoptotic molecules and increase sensitivity to chemotherapeutic drugs.87
SOCS6, SOCS7, and tumors
SOCS6 has also been reported to be downregulated in many cancers.88–92 Moreover, exogenous or upregulated SOCS6 can inhibit cancer-cell growth in gastric cancer, prostate cancer, medulloblastoma, glioblastoma, and cervical cancer.88,90,93,94 The role of SOCS6 in tumor suppression is associated with cKit (SCF receptor). The abnormality of SCF–cKit signaling is closely related to certain tumors.95 SOCS6 can interact with cKit via its SH2 domain, which suppresses cKit-dependent pathways.96 Overexpression of SOCS6 in a Ba/F3-Kit cell line causes a decrease in SCF-dependent cell proliferation and a parallel reduction in ERK1, ERK2, and p38 signaling.97
Few data are currently available with regard to the tumor-suppression activity of SOCS7. SOCS7 is downregulated in colon cancer.98 On the other hand, increased expression of SOCS7 can reduce aggressive ability of prostate cancer cells by blocking activation of the JAK–STAT3 pathway.99 Importantly, high levels of SOCS7 predict good disease-free survival and overall survival in breast cancers.59
Upregulated expression of SOCSs in tumors
Many SOCS types are considered tumor suppressors, and upregulation or activation of these proteins is associated with the inhibition or suppression of many malignant tumors. However, SOCSs are also upregulated in some tumors. Constitutive expression of SOCS1 has been detected in chronic myeloid leukemia and correlates with poor response to IFN treatment.100 Expression of SOCS2 is significantly higher in papillary thyroid cancer than in patients with benign disease.101 Additionally, expression of SOCS3 is significantly elevated in human breast cancer.102 In the human melanoma cell line 1286, constitutive SOCS3 can stimulate tumor-cell growth.103 Increased SOCS in tumors can contribute to tumor development, and that is possibly mediated by its negative control of other SOCSs that normally suppress tumor development. For instance, in patients with active acromegaly and colonic polyps, SOCS2 is significantly increased, which causes a reduction in SOCS1 expression and leads to elevated STAT5B levels and consequently exaggerated GH-mediated proliferation of colonic epithelial cells.104
As such, it is suggested that increased expression of SOCS proteins may be a consequence rather than a cause of their antitumor activities. Tumor cells are sustained by several cytokines that can activate JAK–STAT signaling and other molecules to support cell proliferation and survival within the tumor microenvironment. Dysregulation of oncogene expression and function, as well as any other loss of function changes in negative regulation of the JAK–STAT pathway, may overwhelm the capacity of SOCS proteins to inhibit JAK–STAT signaling activation. Therefore, the inhibitory action of SOCS proteins may not have a significant impact on proliferation and survival of tumor cells, even if they are overexpressed.
Protein tyrosine phosphatases
Protein phosphorylation and dephosphorylation occur mainly at tyrosine residues and are catalyzed by PTKs and PTPs, respectively. Tyrosine phosphorylation of STAT3 by specific PTKs is critical for its activation. Therefore, PTPs are very important in the negative regulation of STAT3 activity. There are seven types of PTPs targeting STAT3 (Figure 1): PTPRD, PTPRT, PTPRK, SHP1, SHP2, PTPN9, and TC-PTP.2
Protein tyrosine phosphatase receptor-type D (PTPRD) and tumors
PTPRD belongs to the highly conserved family of receptor PTPs. Capable of interacting directly with STAT3, it negatively regulates STAT3-mediated signaling and thus functions as a tumor suppressor.105–107 The PTPRD gene is frequently inactivated in a number of cancers, such as glioblastoma multiforme, colon cancer, breast cancer, neuroblastoma, lung cancer, and SCC.105,106,108–113 Loss of PTPRD enhances the tumor-forming capacity of immortalized human astrocytes in mouse-xenograft models.105 Heterologous loss of PTPRD leads to a significant increase in STAT3 phosphorylation and expression of the STAT3 target genes within glioblastoma multiforme.106 Loss-of-function mutations in PTPRD also promote cell growth and phosphorylation of STAT3 (Y705) in head-and-neck SCC cells.112 Interestingly, head-and-neck SCC cells with a PTPRD mutation are more sensitive to a STAT3 inhibitor, providing an important clue to treatment of head-and-neck SCC patients.112 Additionally, exogenous PTPRD significantly decreases tumor-cell proliferation and induces increased apoptosis in PTPRD-deficient primary melanoma cells.111
Protein tyrosine phosphatase–receptor type T and tumors
PTPRT is also a member of the PTP-receptor family. Like PTPRD, PTPRT can act as a tumor suppressor, mainly through inhibition of STAT3 signaling via directly dephosphorylating STAT3 at Y705.114,115 PTPRT is inactivated by mutations in many cancers, including colorectal cancer, lung cancer, gastric cancer, and head-and-neck SCC.114–117 It has been reported that PTPRT mutations enhance STAT3 activation and promote cell survival in head-and-neck SCC.117 Many human tumors exhibit aberrant hypermethylation of the PTPRT promoter, which is associated with decreased expression of PTPRT.118 Reduction in PTPRT correlates with an increase in phosphorylated STAT3 and sensitivity to STAT3 inhibition in head-and-neck SCC.118 Therefore, it is possible that PTPRT hypermethylation can function as a biomarker for evaluating the efficacy of STAT3 inhibitors against cancer.
Protein tyrosine phosphatase–receptor type K and tumors
PTPRK is another member of the PTP-receptor family. In 2015, Chen et al firstly reported that PTPRK can interact directly with STAT3 by dephosphorylating phosphorylated STAT3 at Y705.119 In addition, they found that expression of PTPRK decreased in NKTCL cells. Furthermore, restored PTPRK dephosphorylates phospho-STAT3 at Y705, which can inhibit the proliferation, migration, and invasion of NKTCL cells. Low expression of PTPRK is related to its promoter's hypermethylation.119 Finally, PTPRK is able to inhibit EGFR signaling to suppress tumor growth.120–122
Src homology region 2 domain–containing phosphatase 1 and tumors
SHP1, also known as PTPN6, is a member of the nonreceptor PTP family and encoded by the PTPN6 gene. The tumor-suppressive activity of SHP1 is mediated by its negative regulation of JAK and STAT. Downregulation or loss of SHP1 protein is often detected in human lymphoma and leukemia, and inhibition of SHP1 expression is proven to correlate with methylation of the PTPN6 promoter in anaplastic large-cell lymphoma, multiple myeloma, T-cell lymphoma, and B-cell lymphoma.123–129 Another study demonstrated that overexpression of SHP1 is able to decrease STAT3 activity in non-Hodgkin's lymphoma cells with loss of SHP1.123 It has also been reported that guggulsterone can induce apoptosis and suppress the proliferation of multiple cancer types, such as head-and-neck SCC, leukemia, and melanoma, by induction of SHP1 expression, which significantly decreases activities of JAK2 and STAT3.130 In sorafenib-resistant HCC, dovitinib, a receptor-kinase inhibitor, can induce apoptosis and overcome sorafenib resistance through SHP1-mediated inhibition of STAT3 signaling.131 Sorafenib derivatives have been reported to be able to block STAT3 activity and suppress sorafenib-resistant HCC-cell growth through promoting SHP1 activity.132 Plumbagin, a vitamin K3 analogue, has also been found to induce SHP1 expression in human myeloma cells, leading to inhibition of STAT3 phosphorylation by inactivation of cSrc, JAK1, and JAK2.133 These findings further suggest that activity of STAT3 is vital to tumorigenesis and drug resistance, and SHP1-activating agents may be valuable in cancer therapy.
Src homology region 2 domain–containing phosphatase 2 and tumors
SHP2 is another member of the non-receptor PTP family, and is encoded by PTPN11. Studies have revealed that the tumor-suppressive capabilities of SHP2 occur mainly through its inactivation of STAT3. For example, SHP2-deficient mice present increased STAT3 activity, increased spontaneous hepatocellular adenomas, and chemically induced HCCs.134 Accordingly, Bard-Chapeau et al reported that SHP2 is downregulated in a small number of human HCCs.134 Moreover, SHP2 expression is depressed in human ESCC, and SHP2 knockdown enhances ESCC-cell proliferation in vitro and in vivo, along with a significant increase in phosphorylated STAT3.135 More importantly, low SHP2 expression and high phosphorylated STAT3 correlate with poor prognosis and vice versa in colorectal cancer.136 However, SHP2 was initially established as an oncogenic protein, and PTPN11 the first identified proto-oncogene to encode a tyrosine phosphatase.137 Germline gain-of-function mutations of PTPN11 are associated with increased risk of solid tumors and leukemia.138–141 Promotion of cancer by SHP2 is associated with activation of Ras GTPase–ERK signaling and the PI3K–AKT pathway mediated by SHP2.138–140
MEG2/protein tyrosine phosphatase–nonreceptor type 9 and tumors
PTPN9 (PTP-MEG2 or MEG2) is a cytosolic nonreceptor PTP encoded by PTPN9. In 2012, PTP-MEG2 was first reported to dephosphorylate STAT3 and suppress tumor growth in breast cancer.142 This study also demonstrated that MEG2 can interact directly with STAT3 in vitro and in vivo by dephosphorylating STAT3 at Y705 in a time- and dose-dependent manner.142 Other research has revealed that upregulated expression of MEG2 can effectively inhibit tumor-cell growth and induce cell apoptosis by reducing STAT3 activity in prostate carcinoma cells.143 In addition, MEG2 is able to dephosphorylate the NGF receptor TrkA, the insulin receptor, and VEGFR2,144–146 while the detailed mechanisms need to be further explored.
T-cell protein tyrosine phosphatase and tumors
TC-PTP, encoded by PTPN2, also belongs to the nonreceptor PTP family. STAT3 is one of several substrates of TC-PTP.147 TC-PTP has been reported to be tumor-suppressive through its modulation of STAT3 signaling. For instance, deletion of the PTPN2 gene is present in some T-cell acute lymphoblastic leukemia cells and results in increased JAK–STAT signaling.148 Biallelic inactivation of PTPN2 correlates with activation of the JAK–STAT pathway in the Hodgkin’s lymphoma cell line SUPHD1 and T-cell non-Hodgkin’s lymphoma tissue.149 In addition, TC-PTP deficiency in triple-negative primary breast cancer leads to increased cell proliferation via elevated STAT3 signaling and SFK.150 Restoration of TC-PTP expression in human breast cancer cell lines apparently suppresses cell proliferation in vitro and xenograft growth in vivo.150 These findings suggest that TC-PTP can act as a suppressor of human tumors.
Direct inhibitors of STAT3
Given the critical role of constitutively active STAT3 in human tumors, STAT3 has become an attractive target for small-molecule therapeutics. Since the first peptide inhibitor of STAT3, PY*LKTK (where Y* represents phosphotyrosine) was reported,151 a number of small molecules (Table 1) have been developed to inhibit directly the function of STAT3 for cancer therapies.152–171 The mechanisms of direct STAT3 inhibition include disruption of phosphorylation, dimerization, nuclear translocation, and/or DNA binding of STAT3. Both peptides/peptidomimetics and small nonpeptidic molecules are able to target the STAT3 SH2 domain/STAT3 DNA-binding domain (details shown in Table 1). Because peptides/peptidemimics have poor membrane permeability and stability,172 small nonpeptidic molecules have become attractive candidates of STAT3 inhibitors for tumor treatment. In this regard, STA21 has completed a phase I/II trial for psoriasis, and OPB51602 has completed a phase I trial for refractory hematologic and solid malignancies.158,167,168 However, it will take a long time to transfer these molecules to the clinic. One of the major reasons is that STAT3:STAT3 dimerization is a protein–protein interaction involving a large surface area and difficult to be influenced by small molecules.163 Additionally, high concentrations of small-molecule inhibitors are required to counteract STAT3 activity, and in this case off-target toxicity is very likely increased. Up to now, although significant progress has been made in preclinical trials, few small-molecule inhibitors have been used in clinical cancer therapies.
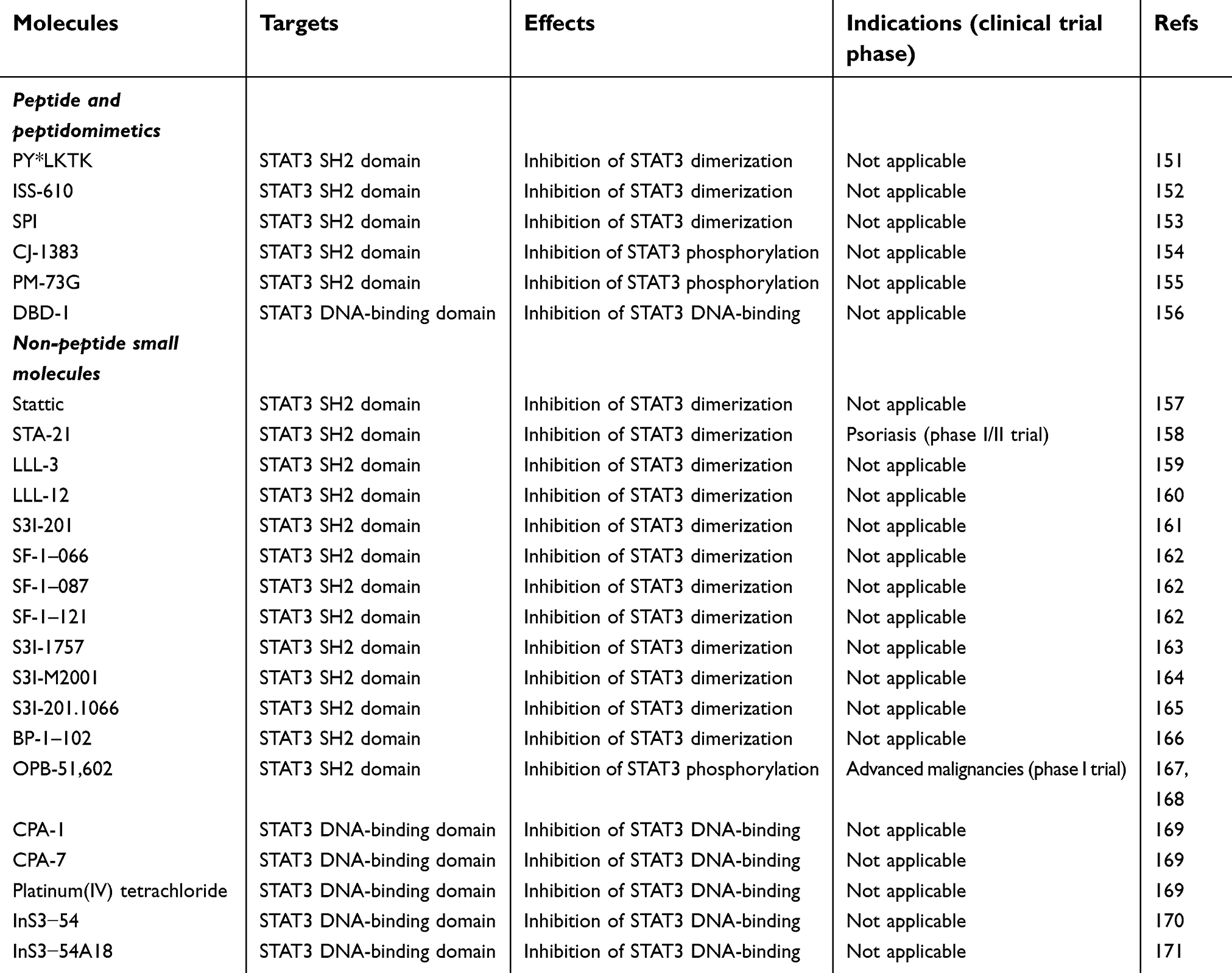 | Table 1 Direct inhibitors of STAT3 |
Conclusion
STAT3 is an ideal target for tumor therapy, because of its pivotal biological functions in tumors. So far, various STAT3 inhibitors have been developed for tumor therapy, including peptides, small nonpeptidic molecules and natural-product inhibitors.173 Some STAT3 inhibitors are currently in clinical trials; however, few are suitable for clinical application,172,173 so a new strategy for STAT3 inhibitors needs to be further explored. PIAS, SOCS, and PTP can effectively prevent tumor progression; therefore, negative regulation of STAT3 signaling would be a valuable strategy. These studies showed that PIAS proteins can inhibit STAT3-transcription activity by binding to active STAT dimers and blocking the DNA-binding activity of STAT. SOCSs are known as a negative-feedback loop of STAT3 signaling, and interact with JAK domains or intracellular portions of cytokine receptors to reduce STAT3 activation. STAT3 is also inactivated through dephosphorylation of Tyr705 by such PTPs as PTPRD, SHP1, SHP2, and TC-PTP. The data presented in this review prove the important role of negative regulators of STAT3 signaling in tumor suppression, which will pave a new avenue for cancer treatment.
Abbreviation list
ESCC, esophageal squamous-cell cancer; HCC, hepatocellular carcinoma; NKTCL, NK/T-cell lymphoma; SCC, squamous-cell carcinoma.
Acknowledgments
We thank Dr Xinlin Yang for helping us improve our English writing. This work was supported by grants from the National Natural Science Foundation of China (81672945 and 81072063), and the Science Project of Liaoning Province (201602234).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(Pt 8):1281–1283. doi:10.1242/jcs.00963
2. Kim M, Morales LD, Jang IS, Cho YY, Kim DJ. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int J Mol Sci. 2018;19(9):
3. D‘Amico S, Shi J, Martin BL, Crawford HC, Petrenko O, Reich NC. STAT3 is a master regulator of epithelial identity and KRAS-driven tumorigenesis. Genes Dev. 2018;32(17–18):1175–1187. doi:10.1101/gad.311852.118
4. Chen X, Wei J, Li C, Pierson CR, Finlay JL, Lin J. Blocking interleukin-6 signaling inhibits cell viability/proliferation, glycolysis, and colony forming activity of human medulloblastoma cells. Int J Oncol. 2018;52(2):571–578. doi:10.3892/ijo.2017.4211
5. Yu LJ, Wu ML, Li H, et al. Inhibition of STAT3 expression and signaling in resveratrol-differentiated medulloblastoma cells. Neoplasia. 2008;10(7):736–744.
6. Li H, Chen L, Li JJ, et al. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J Hematol Oncol. 2018;11(1):70. doi:10.1186/s13045-018-0618-0
7. Wen W, Lowe G, Roberts CM, et al. Pterostilbene suppresses ovarian cancer growth via induction of apoptosis and blockade of cell cycle progression involving inhibition of the STAT3 Pathway. Int J Mol Sci. 2018;19(7):
8. Li WM, Huang CN, Lee YC, et al. Over-expression of activated signal transducer and activator of transcription 3 predicts poor prognosis in upper tract urothelial carcinoma. Int J Med Sci. 2017;14(13):1360–1367. doi:10.7150/ijms.17367
9. Yao Y, Ye H, Qi Z, et al. B7-H4(B7x)-mediated cross-talk between glioma-initiating cells and macrophages via the il6/jak/stat3 pathway lead to poor prognosis in glioma patients. Clin Cancer Res. 2016;22(11):2778–2790. doi:10.1158/1078-0432.CCR-15-0858
10. Won C, Kim BH, Yi EH, et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology. 2015;62(4):1160–1173. doi:10.1002/hep.27968
11. Laudisi F, Cherubini F, Monteleone G, Stolfi C. STAT3 interactors as potential therapeutic targets for cancer treatment. Int J Mol Sci. 2018;19(6):
12. Kluge A, Dabir S, Vlassenbroeck I, Eisenberg R, Dowlati A. Protein inhibitor of activated STAT3 expression in lung cancer. Mol Oncol. 2011;5(3):256–264. doi:10.1016/j.molonc.2011.03.004
13. Saydmohammed M, Joseph D, Syed V. Curcumin suppresses constitutive activation of STAT-3 by up-regulating protein inhibitor of activated STAT-3 (PIAS-3) in ovarian and endometrial cancer cells. J Cell Biochem. 2010;110(2):447–456.
14. Baek SH, Ko JH, Lee H, et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine. 2016;23(5):566–577. doi:10.1016/j.phymed.2016.02.011
15. Yu H, Liu Y, McFarland BC, et al. SOCS3 deficiency in myeloid cells promotes tumor development: involvement of STAT3 activation and myeloid-derived suppressor cells. Cancer Immunol Res. 2015;3(7):727–740. doi:10.1158/2326-6066.CIR-15-0004
16. Koh JS, Joo MK, Park JJ, et al. Inhibition of STAT3 in gastric cancer: role of pantoprazole as SHP-1 inducer. Cell Biosci. 2018;8:50. doi:10.1186/s13578-018-0248-9
17. Wen LZ, Ding K, Wang ZR, et al. SHP-1 acts as a tumor suppressor in hepatocarcinogenesis and HCC progression. Cancer Res. 2018;78(16):4680–4691. doi:10.1158/0008-5472.CAN-17-3896
18. Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006;16(2):196–202. doi:10.1038/sj.cr.7310027
19. Chung CD, Liao J, Liu B, et al. Specific inhibition of STAT3 signal transduction by PIAS3. Science. 1997;278(5344):1803–1805.
20. Liu B, Liao J, Rao X, et al. Inhibition of STAT1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A. 1998;95(18):10626–10631. doi:10.1073/pnas.95.18.10626
21. Rabellino A, Andreani C, Scaglioni PP. The role of PIAS SUMO E3-Ligases in cancer. Cancer Res. 2017;77(7):1542–1547. doi:10.1158/0008-5472.CAN-16-2958
22. Heppler LN, Frank DA. Targeting oncogenic transcription factors: therapeutic implications of endogenous STAT inhibitors. Trends Cancer. 2017;3(12):816–827. doi:10.1016/j.trecan.2017.10.004
23. Wang L, Banerjee S. Differential PIAS3 expression in human malignancy. Oncol Rep. 2004;11(6):1319–1324.
24. Zhang Q, Raghunath PN, Xue L, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-celllymphoma. J Immunol. 2002;168(1):466–474.
25. Zhang C, Mukherjee S, Tucker-Burden C, et al. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol Oncol. 2017;11(3):280–294. doi:10.1002/1878-0261.12034
26. Brantley EC, Nabors LB, Gillespie GY, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 2008;14(15):4694–4704. doi:10.1158/1078-0432.CCR-08-0618
27. Dabir S, Kluge A, Kresak A, et al. Low PIAS3 expression in malignant mesothelioma is associated with increased STAT3 activation and poor patient survival. Clin Cancer Res. 2014;20(19):5124–5132. doi:10.1158/1078-0432.CCR-14-1233
28. Ogata Y, Osaki T, Naka T, et al. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia. 2006;8(10):817–825. doi:10.1593/neo.06409
29. Abbas R, McColl KS, Kresak A, et al. PIAS3 expression in squamous cell lung cancer is low and predicts overall survival. Cancer Med. 2015;4(3):325–332. doi:10.1002/cam4.372
30. Lee JH, Kim C, Sethi G, Ahn KS. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget. 2015;6(8):6386–6405. doi:10.18632/oncotarget.3443
31. Wible BA, Wang L, Kuryshev YA, Basu A, Haldar S, Brown AM. Increased K+ efflux and apoptosis induced by the potassium channel modulatory protein KChAP/PIAS3beta in prostate cancer cells. J Biol Chem. 2002;277(20):17852–17862. doi:10.1074/jbc.M201689200
32. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancergenomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
33. Hoefer J, Schäfer G, Klocker H, et al. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am J Pathol. 2012;180(5):2097–2107. doi:10.1016/j.ajpath.2012.01.026
34. Puhr M, Hoefer J, Eigentler A, et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene. 2016;35(18):2322–2332. doi:10.1038/onc.2015.292
35. Driscoll JJ, Pelluru D, Lefkimmiatis K, et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010;115(14):2827–2834. doi:10.1182/blood-2009-03-211045
36. Li H, Gao H, Bijukchhe SM, Wang Y, Li T. PIAS3 may represent a potential biomarker for diagnosis and therapeutic of human colorectal cancer. Med Hypotheses. 2013;81(6):1151–1154. doi:10.1016/j.mehy.2013.09.022
37. McCormick SM, Heller NM. Regulation of macrophage, dendritic cell, and microglial phenotype and function by the SOCS PRoteins. Front Immunol. 2015;6:549. doi:10.3389/fimmu.2015.00549
38. Hilton DJ, Richardson RT, Alexander WS, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95(1):114–119.
39. Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387(6636):917–921. doi:10.1038/43206
40. Endo TA, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387(6636):921–924. doi:10.1038/43213
41. Jiang M, Zhang WW, Liu P, Yu W, Liu T, Yu J. Dysregulation of SOCS-mediated negative feedback of cytokine signaling in carcinogenesisand its significance in cancer treatment. Front Immunol. 2017;8:70. doi:10.3389/fimmu.2017.00070
42. Sasi W, Sharma AK, Mokbel K. The role of suppressors of cytokine signalling in human neoplasms. Mol Biol Int. 2014;2014:630797. doi:10.1155/2014/630797
43. Kim MH, Kim MS, Kim W, et al. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PLoS One. 2015;10(4):e0123133. doi:10.1371/journal.pone.0123133
44. Sugase T, Takahashi T, Serada S, et al. Suppressor of cytokine signaling-1 gene therapy induces potent antitumor effect in patient-derived esophageal squamous cell carcinoma xenograft mice. Int J Cancer. 2017;140(11):2608–2621. doi:10.1002/ijc.30666
45. Jueliger S, Lyons J, Cannito S, et al. Efficacy and epigenetic interactions of novel DNA hypomethylating agent guadecitabine (SGI-110) in preclinical models of hepatocellular carcinoma. Epigenetics. 2016;11:1–12. doi:10.1080/15592294.2015.1107695
46. Sutherland KD, Lindeman GJ, Choong DY, et al. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 2004;23(46):7726–7733. doi:10.1038/sj.onc.1207787
47. Zhou H, Miki R, Eeva M, et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res. 2007;13(8):2344–2353. doi:10.1158/1078-0432.CCR-06-2303
48. Lee MC, Kuo YY, Chou WC, Hou HA, Hsiao M, Tien HF. Gfi-1 is the transcriptional repressor of SOCS1 in acute myeloid leukemia cells. J Leukoc Biol. 2014;95(1):105–115. doi:10.1189/jlb.0912475
49. Liu TC, Lin SF, Chang JG, Yang MY, Hung SY, Chang CS. Epigenetic alteration of the SOCS1 gene in chronic myeloid leukaemia. Br J Haematol. 2003;123(4):654–661. doi:10.1046/j.1365-2141.2003.04660.x
50. Mafanda EK, Kandhi R, Bobbala D, et al. Essential role of suppressor of cytokine signaling 1 (SOCS1) in hepatocytes and macrophages in the regulation of liver fibrosis. Cytokine. 2018. pii:S1043-4666(18)30330-2. doi:10.1016/j.cyto.2018.07.032
51. Juskevicius D, Jucker D, Dietsche T, et al. Novel cell enrichment technique for robust genetic analysis of archival classical Hodgkin lymphoma tissues. Lab Invest. 2018;98(11):1487–1499. doi:10.1038/s41374-018-0096-6
52. Mottok A, Renné C, Seifert M, et al. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood. 2009;114(20):4503–4506. doi:10.1182/blood-2009-06-225839
53. Watanabe D, Ezoe S, Fujimoto M, et al. Suppressor of cytokine signalling-1 gene silencing in acute myeloid leukaemia and human haematopoietic cell lines. Br J Haematol. 2004;126(5):726–735. doi:10.1111/j.1365-2141.2004.05107.x
54. Park Y, Shon SK, Kim A, et al. SOCS1 induced by NDRG2 expression negatively regulates STAT3 activation in breast cancer cells. Biochem Biophys Res Commun. 2007;363(2):361–367. doi:10.1016/j.bbrc.2007.08.195
55. Bagnyukova TV, Tryndyak VP, Muskhelishvili L, Ross SA, Beland FA, Pogribny IP. Epigenetic downregulation of the suppressor of cytokine signaling 1 (SOCs1) gene is associated with the STAT3 activation and development of hepatocellular carcinoma induced by methyl-deficiency in rats. Cell Cycle. 2008;7(20):3202–3210. doi:10.4161/cc.7.20.6816
56. Oshimo Y, Kuraoka K, Nakayama H, et al. Epigenetic inactivation of SOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int J Cancer. 2004;112(6):1003–1009. doi:10.1002/ijc.20521
57. Hanada T, Kobayashi T, Chinen T, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203(6):1391–1397. doi:10.1084/jem.20060436
58. Kang XC, Chen ML, Yang F, et al. Promoter methylation and expression of SOCS-1 affect clinical outcome and epithelial-mesenchymal transition in colorectal cancer. Biomed Pharmacother. 2016;80:23–29. doi:10.1016/j.biopha.2016.02.011
59. Sasi W, Jiang WG, Sharma A, Mokbel K. Higher expression levels of SOCS 1,3,4,7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer. 2010;10:178. doi:10.1186/1471-2407-10-663
60. Smolkova B, Mego M, Horvathova Kajabova V, et al. Expression of SOCS1 and CXCL12 proteins in primary breast cancer are associated with presence of circulating tumor cells in peripheral blood. Transl Oncol. 2016;9(3):184–190. doi:10.1016/j.tranon.2016.03.004
61. Shimada K, Serada S, Fujimoto M, et al. Molecular mechanism underlying the antiproliferative effect of suppressor of cytokine signaling-1 in non-small-cell lung cancer cells. Cancer Sci. 2013;104(11):1483–1491. doi:10.1111/cas.12266
62. Neuwirt H, Puhr M, Santer FR, et al. Suppressor of cytokine signaling (SOCS)-1 is expressed in human prostate cancer and exerts growth-inhibitory function through down-regulation of cyclins and cyclin-dependent kinases. Am J Pathol. 2009;174(5):1921–1930. doi:10.2353/ajpath.2009.080751
63. David M, Naudin C, Letourneur M, et al. Suppressor of cytokine signaling 1 modulates invasion and metastatic potential of colorectal cancer cells. Mol Oncol. 2014;8(5):942–955. doi:10.1016/j.molonc.2014.03.014
64. Haffner MC, Petridou B, Peyrat JP, et al. Favorable prognostic value of SOCS2 and IGF-I in breast cancer. BMC Cancer. 2007;7:136. doi:10.1186/1471-2407-7-136
65. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. doi:10.1038/nrc3818
66. Weber A, Hengge UR, Bardenheuer W, et al. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005;24(44):6699–6708. doi:10.1038/sj.onc.1208818
67. He B, You L, Uematsu K, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100(24):14133–14138. doi:10.1073/pnas.2232790100
68. Martini M, Pallini R, Luongo G, Cenci T, Lucantoni C, Larocca LM. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int J Cancer. 2008;123(12):2955–2960. doi:10.1002/ijc.23805
69. Isomoto H. Epigenetic alterations in cholangiocarcinoma-sustained IL-6/STAT3 signaling in cholangiocarcinoma due to SOCS3 epigenetic silencing. Digestion. 2009;79(Suppl 1):2–8. doi:10.1159/000167859
70. Pierconti F, Martini M, Pinto F, et al. Epigenetic silencing of SOCS3 identifies a subset of prostate cancer with an aggressive behavior. Prostate. 2011;71(3):318–325. doi:10.1002/pros.21245
71. Tischoff I, Hengge UR, Vieth M, et al. Methylation of SOCS-3 and SOCS-1 in the carcinogenesis of Barrett‘s adenocarcinoma. Gut. 2007;56(8):1047–1053. doi:10.1136/gut.2006.111633
72. Li Y, Deuring J, Peppelenbosch MP, Kuipers EJ, de Haar C. van der Woude CJ. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis. 2012;33(10):1889–1896. doi:10.1093/carcin/bgs214
73. Tokita T, Maesawa C, Kimura T, et al. Methylation status of the SOCS3 gene in human malignant melanomas. Int J Oncol. 2007;30(3):689–694.
74. Yuan K, Lei Y, Chen HN, et al. HBV-induced ROS accumulation promotes hepatocarcinogenesis through Snail-mediated epigenetic silencing of SOCS3. Cell Death Differ. 2016;23(4):616–627. doi:10.1038/cdd.2015.129
75. Ogata H, Kobayashi T, Chinen T, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131(1):179–193. doi:10.1053/j.gastro.2006.04.025
76. Tang Q, Jiang J, Liu J. CCR5 blockade suppresses melanoma development through inhibition of il-6-STAT3 pathway via upregulation of SOCS3. Inflammation. 2015;38(6):2049–2056. doi:10.1007/s10753-015-0186-1
77. Yu ZB, Bai L, Qian P, et al. Restoration of SOCS3 suppresses human lung adenocarcinoma cell growth by downregulating activation of Erk1/2, Akt apart from STAT3. Cell Biol Int. 2009;33(9):995–1001. doi:10.1016/j.cellbi.2009.06.002
78. Xiong H, Zhang Y, Chen S, et al. Induction of SOCS3 by liver X receptor suppresses the proliferation of hepatocellular carcinoma cells. Oncotarget. 2017;8(38):64083–64094. doi:10.18632/oncotarget.19321
79. Lin YC, Lin CK, Tsai YH, et al. Adenovirus-mediated SOCS3 gene transfer inhibits the growth and enhances the radiosensitivity of human non-small cell lung cancer cells. Oncol Rep. 2010;24(6):1605–1612.
80. Puhr M, Santer FR, Neuwirt H, Marcias G, Hobisch A, Culig Z. SOCS-3 antagonises the proliferative and migratory effects of fibroblast growth factor-2 in prostate cancer by inhibition of p44/p42 MAPK signalling. Endocr Relat Cancer. 2010;17(2):525–538. doi:10.1677/ERC-10-0007
81. Iwahori K, Serada S, Fujimoto M, et al. Overexpression of SOCS3 exhibits preclinical antitumor activity against malignant pleural mesothelioma. Int J Cancer. 2011;129(4):1005–1017. doi:10.1002/ijc.25716
82. Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26(33):4833–4841. doi:10.1038/sj.onc.1210286
83. Calvisi DF, Ladu S, Gorden A, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Invest. 2007;117(9):2713–2722. doi:10.1172/JCI31457
84. Kobayashi D, Nomoto S, Kodera Y, et al. Suppressor of cytokine signaling 4 detected as a novel gastric cancer suppressor gene using double combination array analysis. World J Surg. 2012;36(2):362–372. doi:10.1007/s00268-011-1358-2
85. Scheitz CJ, Lee TS, McDermitt DJ, Tumbar T. Defining a tissue stem cell-driven Runx1/Stat3 signalling axis in epithelial cancer. Embo J. 2012;31(21):4124–4139. doi:10.1038/emboj.2012.270
86. Yoon S, Yi YS, Kim SS, Kim JH, Park WS, Nam SW. SOCS5 and SOCS6 have similar expression patterns in normal and cancer tissues. Tumour Biol. 2012;33(1):215–221. doi:10.1007/s13277-011-0264-4
87. Francipane MG, Eterno V, Spina V, et al. Suppressor of cytokine signaling 3 sensitizes anaplastic thyroid cancer to standard chemotherapy. Cancer Res. 2009;69(15):6141–6148. doi:10.1158/0008-5472.CAN-09-0994
88. Lai RH, Hsiao YW, Wang MJ, et al. SOCS6, down-regulated in gastric cancer, inhibits cell proliferation and colony formation. Cancer Lett. 2010;288(1):75–85. doi:10.1016/j.canlet.2009.06.025
89. Sriram KB, Larsen JE, Savarimuthu Francis SM, et al. Array-comparative genomic hybridization reveals loss of SOCS6 is associated with poor prognosis in primary lung squamous cell carcinoma. PLoS One. 2012;7(2):e30398. doi:10.1371/journal.pone.0030398
90. Yuan D, Wang W, Su J, et al. SOCS6 functions as a tumor suppressor by inducing apoptosis and inhibiting angiogenesis in human prostate cancer. Curr Cancer Drug Targets. 2018;18(9):894–904. doi:10.2174/1568009618666180102101442
91. Letellier E, Schmitz M, Baig K, et al. Identification of SOCS2 and SOCS6 as biomarkers in human colorectal cancer. Br J Cancer. 2014;111(4):726–735. doi:10.1038/bjc.2014.377
92. Qiu X, Zheng J, Guo X, et al. Reduced expression of SOCS2 and SOCS6 in hepatocellular carcinoma correlates with aggressive tumor progression and poor prognosis. Mol Cell Biochem. 2013;378(1–2):99–106. doi:10.1007/s11010-013-1599-5
93. Tanaka T, Arai M, Jiang X, et al. Downregulation of microRNA-431 by human interferon-β inhibits viability of medulloblastoma and glioblastoma cells via upregulation of SOCS6. Int J Oncol. 2014;44(5):1685–1690. doi:10.3892/ijo.2014.2317
94. Cheng L, Kong B, Zhao Y, Jiang J. miR-494 inhibits cervical cancer cell proliferation through upregulation of SOCS6 expression. Oncol Lett. 2018;15(3):3075–3080. doi:10.3892/ol.2017.7651
95. Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9(5):435–443. doi:10.7150/ijbs.6087
96. Zadjali F, Pike AC, Vesterlund M, et al. Structural basis for c-KIT inhibition by the suppressor of cytokine signaling 6 (SOCS6) ubiquitin ligase. J Biol Chem. 2011;286(1):480–490. doi:10.1074/jbc.M110.173526
97. Bayle J, Letard S, Frank R, Dubreuil P, De Sepulveda P. Suppressor of cytokine signaling 6 associates with KIT and regulates KIT receptor signaling. J Biol Chem. 2004;279(13):12249–12259. doi:10.1074/jbc.M313381200
98. Liu XH, Xu SB, Yuan J, et al. Defective interleukin-4/Stat6 activity correlates with increased constitutive expression of negative regulators SOCS-3, SOCS-7, and CISH in colon cancer cells. J Interferon Cytokine Res. 2009;29(12):809–816. doi:10.1089/jir.2009.0004
99. Ge D, Gao AC, Zhang Q, Liu S, Xue Y, You Z. LNCaP prostate cancer cells with autocrine interleukin-6 expression are resistant to IL-6-induced neuroendocrine differentiation due to increased expression of suppressors of cytokine signaling. Prostate. 2012;72(12):1306–1316. doi:10.1002/pros.22479
100. Roman-Gomez J, Jimenez-Velasco A, Castillejo JA, et al. The suppressor of cytokine signaling-1 is constitutively expressed in chronic myeloid leukemia and correlates with poor cytogenetic response to interferon-alpha. Haematologica. 2004;89(1):42–48.
101. Kobawala TP, Trivedi TI, Gajjar KK, Patel GH, Ghosh NR. Significance of expression of suppressor of cytokine signaling proteins: suppressor of cytokine signaling-1, suppressor of cytokine signaling-2, and suppressor of cytokine signaling-3 in papillary thyroid cancer. J Cancer Res Ther. 2017;13(2):337–345. doi:10.4103/0973-1482.174172
102. Raccurt M, Tam SP, Lau P, et al. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br J Cancer. 2003;89(3):524–532. doi:10.1038/sj.bjc.6601115
103. Komyod W, Böhm M, Metze D, Heinrich PC, Behrmann I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol Cancer Res. 2007;5(3):271–281. doi:10.1158/1541-7786.MCR-06-0274
104. Bogazzi F, Ultimieri F, Raggi F, et al. Changes in the expression of suppressor of cytokine signalling (SOCS) 2 in the colonic mucosa of acromegalic patients are associated with hyperplastic polyps. Clin Endocrinol (Oxf). 2009;70(6):898–906. doi:10.1111/j.1365-2265.2008.03431.x
105. Veeriah S, Brennan C, Meng S, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009;106(23):9435–9440. doi:10.1073/pnas.0900571106
106. Ortiz B, Fabius AW, Wu WH, et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc Natl Acad Sci U S A. 2014;111(22):8149–8154. doi:10.1073/pnas.1401952111
107. Chan TA, Heguy A. The protein tyrosine phosphatase receptor D, a broadly inactivated tumor suppressor regulating STAT function. Cell Cycle. 2009;8(19):3063–3064. doi:10.4161/cc.8.19.9455
108. Funato K, Yamazumi Y, Oda T, Akiyama T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp Ther Med. 2011;2(3):457–463. doi:10.3892/etm.2011.231
109. Stallings RL, Nair P, Maris JM, et al. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006;66(7):3673–3680. doi:10.1158/0008-5472.CAN-05-4154
110. Purdie KJ, Lambert SR, Teh MT, et al. Allelic imbalances and microdeletions affecting the PTPRD gene in cutaneous squamous cell carcinomas detected using single nucleotide polymorphism microarray analysis. Genes Chromosomes Cancer. 2007;46(7):661–669. doi:10.1002/gcc.20447
111. Solomon DA, Kim JS, Cronin JC, et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008;68(24):10300–10306. doi:10.1158/0008-5472.CAN-08-3272
112. Peyser ND, Du Y, Li H, et al. Loss-of-function PTPRD mutations lead to increased STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. PLoS One. 2015;10(8):e0135750. doi:10.1371/journal.pone.0135750
113. Walia V, Prickett TD, Kim JS, et al. Mutational and functional analysis of the tumor-suppressor PTPRD in human melanoma. Hum Mutat. 2014;35(11):1301–1310. doi:10.1002/humu.22630
114. Zhao Y, Zhang X, Guda K, et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc Natl Acad Sci U S A. 2010;107(6):2592–2597. doi:10.1073/pnas.0914884107
115. Zhang X, Guo A, Yu J, et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A. 2007;104(10):4060–4064. doi:10.1073/pnas.0611665104
116. Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304(5674):1164–1166. doi:10.1126/science.1096096
117. Lui VW, Peyser ND, Ng PK, et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc Natl Acad Sci U S A. 2014;111(3):1114–1119. doi:10.1073/pnas.1319551111
118. Peyser ND, Freilino M, Wang L, et al. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene. 2016;35(9):1163–1169. doi:10.1038/onc.2015.171
119. Chen YW, Guo T, Shen L, et al. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood. 2015;125(10):1589–1600. doi:10.1182/blood-2014-07-588970
120. Xu Y, Tan LJ, Grachtchouk V, Voorhees JJ, Fisher GJ. Receptor-type protein-tyrosine phosphatase-kappa regulates epidermal growth factor receptor function. J Biol Chem. 2005;280(52):42694–42700. doi:10.1074/jbc.M507722200
121. Wang C, Yang Y, Yang Z, et al. EGF-mediated migration signaling activated by N-acetylglucosaminyltransferase-V via receptor protein tyrosine phosphatase kappa. Arch Biochem Biophys. 2009;486(1):64–72. doi:10.1016/j.abb.2009.02.005
122. Xu Y, Xue S, Zhou J, Voorhees JJ, Fisher GJ. Notch and TGF-β pathways cooperatively regulate receptor protein tyrosine phosphatase-κ (PTPRK) gene expression in human primary keratinocytes. Mol Biol Cell. 2015;26(6):1199–1206. doi:10.1091/mbc.E14-12-1591
123. Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108(8):2796–2803. doi:10.1182/blood-2006-04-017434
124. Khoury JD, Rassidakis GZ, Medeiros LJ, Amin HM, Lai R. Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood. 2004;104(5):1580–1581. doi:10.1182/blood-2004-03-1151
125. Chim CS, Fung TK, Cheung WC, Liang R, Kwong YL. SOCS1 and SHP1 hypermethylation in multiple myeloma: implications for epigenetic activation of the Jak/STAT pathway. Blood. 2004;103(12):4630–4635. doi:10.1182/blood-2003-06-2007
126. Oka T, Ouchida M, Koyama M, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62(22):6390–6394.
127. Koyama M, Oka T, Ouchida M, et al. Activated proliferation of B-cell lymphomas/leukemias with the SHP1 gene silencing by aberrant CpG methylation. Lab Invest. 2003;83(12):1849–1858.
128. Witkiewicz A, Raghunath P, Wasik A, et al. Loss of SHP-1 tyrosine phosphatase expression correlates with the advanced stages of cutaneous T-cell lymphoma. Hum Pathol. 2007;38(3):462–467. doi:10.1016/j.humpath.2006.09.012
129. Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102(19):6948–6953. doi:10.1073/pnas.0501959102
130. Ahn KS, Sethi G, Sung B, Goel A, Ralhan R, Aggarwal BB. Guggulsterone, a farnesoid X receptor antagonist, inhibits constitutive and inducible STAT3 activation through induction of a protein tyrosine phosphatase SHP-1. Cancer Res. 2008;68(11):4406–4415. doi:10.1158/0008-5472.CAN-07-6696
131. Tai WT, Cheng AL, Shiau CW, et al. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol Cancer Ther. 2012;11(2):452–463. doi:10.1158/1535-7163.MCT-11-0412
132. Chen KF, Tai WT, Hsu CY, et al. Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP-1 phosphatase activity. Eur J Med Chem. 2012;55:220–227. doi:10.1016/j.ejmech.2012.07.023
133. Sandur SK, Pandey MK, Sung B, Aggarwal BB. 5-hydroxy-2-methyl-1,4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: potential role in chemosensitization. Mol Cancer Res. 2010;8(1):107–118. doi:10.1158/1541-7786.MCR-09-0257
134. Bard-Chapeau EA, Li S, Ding J, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011;19(5):629–639. doi:10.1016/j.ccr.2011.03.023
135. Qi C, Han T, Tang H, et al. Shp2 inhibits proliferation of esophageal squamous cell cancer via dephosphorylation of STAT3. Int J Mol Sci. 2017;18(1):
136. Huang Y, Wang J, Cao F, et al. SHP2 associates with nuclear localization of STAT3: significance in progression and prognosis of colorectal cancer. Sci Rep. 2017;7(1):17597. doi:10.1038/s41598-017-17604-7
137. Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. 2007;109(3):862–867. doi:10.1182/blood-2006-07-028829
138. Bentires-Alj M, Paez JG, David FS, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64(24):8816–8820. doi:10.1158/0008-5472.CAN-04-1923
139. Mohi MG, Neel BG. The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev. 2007;17(1):23–30. doi:10.1016/j.gde.2006.12.011
140. Ostman A, Hellberg C, Böhmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6(4):307–320. doi:10.1038/nrc1837
141. Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem. 2006;281(10):6785–6792. doi:10.1074/jbc.M513068200
142. Su F, Ren F, Rong Y, et al. Protein tyrosine phosphatase MEG2 dephosphorylates signal transducer and activator of transcription 3 and suppresses tumor growth in breast cancer. Breast Cancer Res. 2012;14(2):R38. doi:10.1186/bcr3169
143. Jin Y, Kim YH, Park JY, et al. Methyllucidone inhibits STAT3 activity by regulating the expression of the protein tyrosine phosphatase MEG2 in DU145 prostate carcinoma cells. Bioorg Med Chem Lett. 2018;28(5):853–857. doi:10.1016/j.bmcl.2018.02.012
144. Hao Q, Samten B, Ji HL, Zhao ZJ, Tang H. Tyrosine phosphatase PTP-MEG2 negatively regulates vascular endothelial growth factor receptor signaling and function in endothelial cells. Am J Physiol Cell Physiol. 2012;303(5):C548–C553. doi:10.1152/ajpcell.00415.2011
145. Zhang D, Marlin MC, Liang Z, et al. The protein tyrosine phosphatase MEG2 regulates the transport and signal transduction of tropomyosin receptor kinase A. J Biol Chem. 2016;291(46):23895–23905. doi:10.1074/jbc.M116.728550
146. Cho CY, Koo SH, Wang Y, et al. Identification of the tyrosine phosphatase PTP-MEG2 as an antagonist of hepatic insulin signaling. Cell Metab. 2006;3(5):367–378. doi:10.1016/j.cmet.2006.03.006
147. Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr Biol. 2002;12(6):446–453.
148. Kleppe M, Lahortiga I, El Chaar T, et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42(6):530–535. doi:10.1038/ng.587
149. Kleppe M, Tousseyn T, Geissinger E, et al. Mutation analysis of the tyrosine phosphatase PTPN2 in Hodgkin‘s lymphoma and T-cell non-Hodgkin‘s lymphoma. Haematologica. 2011;96(11):1723–1727. doi:10.3324/haematol.2011.041921
150. Shields BJ, Wiede F, Gurzov EN, et al. TCPTP regulates SFK and STAT3 signaling and is lost in triple-negative breast cancers. Mol Cell Biol. 2013;33(3):557–570. doi:10.1128/MCB.01016-12
151. Turkson J, Ryan D, Kim JS, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276(48):45443–45455. doi:10.1074/jbc.M107527200
152. Turkson J, Kim JS, Zhang S, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261–269.
153. Zhao W, Jaganathan S, Turkson J. A cell-permeable STAT3 SH2 domain mimetic inhibits STAT3 activation and induces antitumor cell effects in vitro. J Biol Chem. 2010;285(46):35855–35865. doi:10.1074/jbc.M110.154088
154. Chen J, Bai L, Bernard D, et al. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem Lett. 2010;1(2):85–89. doi:10.1021/ml100010j
155. Auzenne EJ, Klostergaard J, Mandal PK, et al. A phosphopeptide mimetic prodrug targeting the SH2 domain of STAT3 inhibits tumor growth and angiogenesis. J Exp Ther Oncol. 2012;10(2):155–162.
156. Nagel-Wolfrum K, Buerger C, Wittig I, Butz K, Hoppe-Seyler F, Groner B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerizationdomain of the transcription factor STAT3 inhibits transactivation and induces apoptosis in tumor cells. Mol Cancer Res. 2004;2(3):170–182.
157. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–1242. doi:10.1016/j.chembiol.2006.09.018
158. Miyoshi K, Takaishi M, Nakajima K, et al. Stat3 as a therapeutic target for the treatment of psoriasis: a clinical feasibility study with STA-21, a STAT3 inhibitor. J Invest Dermatol. 2011;131(1):108–117. doi:10.1038/jid.2010.255
159. Mencalha AL, Du Rocher B, Salles D, Binato R, Abdelhay E. LLL-3, a STAT3 inhibitor, represses BCR-ABL-positive cell proliferation, activates apoptosis and improves the effects of Imatinib mesylate. Cancer Chemother Pharmacol. 2010;65(6):1039–1046. doi:10.1007/s00280-009-1109-3
160. Lin L, Hutzen B, Li PK, et al. A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia. 2010;12(1):39–50.
161. Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104(18):7391–7396. doi:10.1073/pnas.0609757104
162. Fletcher S, Singh J, Zhang X, et al. Disruption of transcriptionally active STAT3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. Chembiochem. 2009;10(12):1959–1964. doi:10.1002/cbic.200900172
163. Zhang X, Sun Y, Pireddu R, et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73(6):1922–1933. doi:10.1158/0008-5472.CAN-12-3175
164. Siddiquee KA, Gunning PT, Glenn M, et al. An oxazole-based small-molecule STAT3 inhibitor modulates STAT3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2(12):787–798. doi:10.1021/cb7001973
165. Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts STAT3 SH2 domain-phosphotyrosine interactions and STAT3-dependent tumor processes. Biochem Pharmacol. 2010;79(10):1398–1409. doi:10.1016/j.bcp.2010.01.001
166. Zhang X, Yue P, Page BD, et al. Orally bioavailable small-molecule inhibitor of transcription factor STAT3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109(24):9623–9628. doi:10.1073/pnas.1121606109
167. Wong AL, Soo RA, Tan DS, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015;26(5):998–1005. doi:10.1093/annonc/mdv026
168. Ogura M, Uchida T, Terui Y, et al. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015;106(7):896–901. doi:10.1111/cas.12683
169. Turkson J, Zhang S, Palmer J, et al. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol Cancer Ther. 2004;3(12):1533–1542.
170. Huang W, Dong Z, Wang F, Peng H, Liu JY, Zhang JT. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem Biol. 2014;9(5):1188–1196. doi:10.1021/cb500071v
171. Huang W, Dong Z, Chen Y, et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2016;35(6):783–792. doi:10.1038/onc.2015.215
172. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11(2):308–318. doi:10.1021/acschembio.5b00945
173. Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12(8):611–629. doi:10.1038/nrd4088
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.