Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 8
Nedd4-2 regulation of voltage-gated ion channels: an update on structure–function relationships and the pathophysiological consequences of dysfunction
Authors Arévalo JC
Received 23 May 2015
Accepted for publication 14 July 2015
Published 18 August 2015 Volume 2015:8 Pages 53—63
DOI https://doi.org/10.2147/JRLCR.S52534
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Trevor W. Stone
Juan Carlos Arévalo1,2
1Department of Cell Biology and Pathology, Instituto de Neurociencias de Castilla y León, Universidad de Salamanca, 2Institute of Biomedical Research of Salamanca, Salamanca, Spain
Abstract: Neuronal excitability is mediated mainly by voltage-gated ion channels (VGICs), which include voltage-gated Na+, K+, and Cl– channels located along the axon and at neuronal synapses. Voltage-gated channels play pivotal roles in the proper functioning of the nervous system because they set the resting membrane potential, initiate and propagate action potentials, and regulate neurotransmitter release. The abnormal activity or misregulation of VGICs caused by mutations has been directly linked to neurological and cardiac diseases. Among other posttranslational modifications, the ubiquitination of VGICs is a key to the regulation of the number of channels in the cell surface, and hence, neuronal excitability. Nedd4-2 is an E3 ubiquitin ligase that ubiquitinates several proteins, including different VGICs. Accordingly, understanding the molecular mechanisms underlying channel regulation will provide insights to design drugs to treat illnesses. The focus of the present review is to provide an update about the regulation of VGICs upon ubiquitination by Nedd4-2 and the relevance of such regulation in the pathophysiological consequences of dysfunction.
Keywords: Nedd4-2, voltage-gated ion channels, disease
Introduction
The degradation of a protein is as crucial as its synthesis since it ensures the elimination of proteins that participate in different cellular functions. If this does not occur, an unwanted protein will be accumulated, and this will consequently lead to functional disorders. In certain cancers, oncogenic targets are mutated such that they are no longer ubiquitinated; thus, they escape degradation and accumulate in the cell. In the nervous system, correct degradation ensures cell-surface protein turnover and substrate delivery to the proteolytic machineries that are required for both synaptic plasticity and self-renewal,1 while abnormal protein accumulation could lead to chronic neurodegenerative processes, such as Alzheimer’s disease, Parkinson’s disease, Lewy body dementia, and amyotrophic lateral sclerosis.2 Voltage-gated ion channels (VGICs) are subject to different posttranslational modifications that regulate their function and stability. Among them, ubiquitination is a key in regulating VGIC protein levels and insertion into the plasma membrane, and alterations in VGICs are therefore associated with different pathologies. Understanding the mechanisms regulating VGICs will be crucial to develop treatments for several pathophysiological dysfunctions in which they have been implicated directly. The present review aims to provide an update of the regulation of different VGICs that are substrates of Nedd4-2, an E3 ubiquitin ligase belonging to the Homologous to E6AP Carboxy Terminus (HECT) family, and the diseases linked to these channels.
Voltage-gated ion channels
VGICs are proteins expressed in electrically responsive cells that allow the movement of ions across the cell membrane and that are regulated by the voltage difference across the membrane they span. Among their functions are the initiation and propagation of action potentials, setting the resting membrane potential, and responding to the electrical signals that regulate neurotransmitter release and muscle contraction. The first indication of the existence of VGICs was proposed in the late 1940s by Alan Lloyd Hodgkin and Andrew Fielding Huxley using voltage clamp techniques. They demonstrated that changes in the permeability of Na+ and K+ were both necessary and sufficient to produce action potentials and that membrane potential-evoked ionic currents flow across the membrane.3 In response to a change to a level more positive than the resting membrane potential in neurons, a redistribution of charge across the axonal membrane occurs; this is caused by two effects: an early, fast and transient inward current of Na+, and a delayed, slow, and sustained outward current of K+.
There are several classes of VGICs that include voltage-gated Na+, K+, and Cl− channels. VGICs are composed of several transmembrane-spanning domains, assembled to form a central ion pore, and a C-terminal tail, which includes different domains and motifs involved in the interaction with different proteins that regulate their insertion in the cell membrane.4–6 The number of VGICs present in the membrane is critical for maintaining normal patterns of excitability and is regulated by the rate of insertion, retention, and endocytosis, which are modulated by different conditions and proteins (Figure 1). Mutations in VGICs affecting any of the above processes have been directly implicated in different neurological diseases, such as epilepsy and neuropathic pain, and also in several cardiac diseases.
 | Figure 1 Nedd4-2 ubiquitinates voltage-gated ion channels. |
Voltage-gated sodium channels
One family of VGICs is the voltage-gated sodium (Nav) channel. Nav channels are present in many electrically active tissues, including neurons and both cardiac and skeletal muscle cells,7 allowing sodium to flow from the extracellular solution into the cytosol, and consequently causing a depolarization of the cell. Nav channels comprise a family consisting of Nav1.1 through Nav1.9 channels.7–9 Nav channels initiate action potentials in neurons, whereas they trigger contraction in skeletal and cardiac muscle cells. In response to an action potential in neurons, Nav channels open, allowing a large inward-flowing sodium current, rendering the membrane potential more positive, after which the action potential can propagate to the next segment and trigger another action potential. The depolarization due to sodium influx activates a calcium-signaling cascade, causing contraction in skeletal and cardiac muscle cells that respectively result in body movement or blood pumping.
All the Nav channel subtypes share a common structure.7 The largest subunit of the sodium channel is the α-subunit, which alone is sufficient to conduct sodium and consists of 24 alpha-helical transmembrane segments arranged in four domains surrounding a central pore.8 The first four transmembrane segments of the Nav channel form a voltage-sensing domain, and transmembrane segments five and six form a pore domain. The transmembrane segments of the voltage-sensing domain and of the pore domain are connected by the extracellular linker (the p-loop), which constitutes the ion selectivity filter for the channel.10 In addition, there are smaller beta subunits associated with the channel modulated by them.11
Different tissues within the human body predominantly express different Nav channel subtypes, whereas Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are mainly expressed in the central nervous system (CNS), Nav1.7, Nav1.8, and Nav1.9 are found in the peripheral nervous system (PNS). Similarly, Nav1.4 is found mostly in skeletal muscle, and Nav1.5 is found in cardiac muscle.7,8 The expression pattern of different Nav channels is highly relevant, since these channels show different voltage dependencies and drug-binding affinities.8,12 Accordingly, the differences in the distribution of these channels may have large impacts with regard to the effects of mutations and pharmacological intervention.
Voltage-gated potassium channels
Voltage-gated K+ (Kv) channels are membrane proteins that allow the rapid and selective flow of K+ ions out of the cell membrane in order to generate electrical signals in cells. These channels, which are present in all animal cells, open and close with the changes in the transmembrane potential. Together with Nav channels, Kv channels are key components in the generation and propagation of electrical impulses in the nervous system. Following changes in transmembrane potential, these channels open and allow a passive flow of K+ ions from the cell, which restores the membrane potential.
Kv channels are formed of four subunits arranged symmetrically around the central pore that is formed of transmembrane segments S5–S6 of each subunit. The voltage-sensor domains consist of four transmembrane segments (S1–S4) and are located at the periphery of the channel.13 A sequence of five amino acids that is highly conserved among potassium channels, voltage-gated or not, is responsible for the high selectivity of the channel.13 A functional Kv channel can be formed by either two or four subunits co-assembled as monomers or heteromers, with dimer or tetramer combination displaying a unique set of gating properties.6,14
The Kv channel is the largest family of potassium channels; it includes 40 members and can be classified into 12 subfamilies.6,15 In this review, we shall mainly focus on Kv7 channels since some of them have PY motifs and/or are regulated by Nedd4-2 protein. The Kv7 channel subfamily is composed of Kv7.1 (KCNQ1), Kv7.2 (KCNQ2), Kv7.3 (KCNQ3), Kv7.4 (KCNQ4), and Kv7.5 (KCNQ5). The expression pattern of these channels varies from the heart (KCNQ1) to the nervous system (KCNQ2, KCNQ3, and KCNQ5) and the ear (KCNQ4).16,17
Voltage-gated chloride channels
Chloride channels (ClCs) constitute an evolutionarily well-conserved family of voltage-gated channels that are structurally unrelated to the other known voltage-gated channels, since they function as dimers. They can be classified in bona fide Cl− channels, including ClC-1, ClC-2, ClC-Ka, and ClC-Kb, and Cl−/H+ antiporters, including ClC-3, ClC-4, ClC-5, ClC-6, and ClC-7.18 ClCs have been highly conserved along evolution and – among other functions – they regulate cell volume and control electrical excitability and transepithelial transport. They are expressed in several different tissues, some of them having a broad distribution (ClC-2, ClC-3, ClC-4, and ClC-7), while others are more restricted to certain tissues such as skeletal muscle (ClC-1), kidney (ClC-Ka/b, ClC-5), and neuronal tissue (ClC-6).18
Ubiquitination and E3 ubiquitin ligases
Ubiquitination is a posttranslational modification carried out by a set of enzymes to tag ubiquitin, a 76-amino acid polypeptide, to a substrate protein. In this process, ubiquitin is covalently conjugated to the ubiquitin-activating enzyme (E1) in an ATP-dependent manner, after which it is transferred to the ubiquitin-conjugating enzyme (E2), and finally to the substrate protein through the ubiquitin-protein ligase (E3) bridge.19 Ubiquitin is bound to a substrate protein through the formation of isopeptide bonds between the C-terminus of ubiquitin and lysine (K) present on substrates. Depending on the number of ubiquitin moieties added to the substrate, there are 1) monoubiquitination, which involves the attachment of a single ubiquitin to a protein; 2) multimonoubiquitination, in which more than one ubiquitin is added to the target protein; and 3) polyubiquitination, in which several moieties of ubiquitin are added in tandem. Since ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, and K63), the conjugated ubiquitin can serve as a substrate for the binding of the next ubiquitin to form a chain.20
Protein ubiquitination was first identified as a tag of protein degradation through the proteasome, the substrate being labeled with K48 polyubiquitination.19 Numerous lines of evidence have also shown that ubiquitin labeling is not always fatal for a protein, since it has been found that monoubiquitination is involved in histone regulation, endocytosis, virus budding, Golgi reassembly, gene transcription, DNA repair, and tumor suppression,21 and K63 polyubiquitination contributes to intracellular trafficking.19,22,23 Thus, different forms of ubiquitination are related to different fates of the ubiquitinated substrate proteins, and hence, different cell biological functions. This means that protein ubiquitination is a complex process that is still not fully understood.
Regarding the ubiquitination process, much attention has focused on ubiquitin-related enzymes. Interestingly, it has been found that mammalian genomes encode just two E1s, dozens of E2s, but >500 E3s.24 Moreover, whereas E2s share many well-conserved catalytic domains, E3 ubiquitin ligases share only a few conserved motifs. Therefore, E3 ubiquitin ligases are believed to carry out the important task of recognizing substrate proteins. E3 ubiquitin ligases can be divided into several families based on their structural characteristics, which include 1) the Really Interesting New Gene (RING) family, such as MDM2 and c-Cbl; 2) the HECT family, such as the Neural precursor cell-Expressed Developmentally Down-regulated 4 (Nedd4) family; and 3) the UFD2 homology (U-box) family.25 The functional differences between the RING-family and the HECT-family of E3 ubiquitin ligases lie in the fact that RING E3 ubiquitin ligases add ubiquitin carried by E2s directly to the substrates, whereas HECT E3 ubiquitin ligases form a thiol ester intermediate, with ubiquitin transferred first from the E2s and then to the substrate. In addition, HECT E3 ubiquitin ligases play a direct role in catalysis during ubiquitination, whereas RING and U-box E3 ubiquitin ligases facilitate protein ubiquitination.26 These latter two E3 ubiquitin ligase types act as adaptor-like molecules by bringing an E2 and a substrate into sufficiently close proximity to promote the ubiquitination of the substrate. While RING-type E3 ubiquitin ligases, such as MDM2 and c-Cbl, can apparently act alone, other E3 ubiquitin ligases are found as components of much larger multi-protein complexes.27
Nedd4 E3 ubiquitin ligases
Nedd4-2 belongs to the Nedd4 family, which in humans includes nine members: Nedd4, Nedd4-2 (Nedd4L), Itch, WWP1, WWP2, SMURF1, SMURF2, Bul1, and NEDL2 (Figure 2). All Nedd4 family members contain a C2 domain in the N-terminus, several WW domains, and a catalytic HECT domain at the C-terminus28 (Figure 2). The C2 domain translocates the protein to the membrane upon calcium binding, and the WW domains are known to bind substrate proteins containing PY (PPXY) motifs, with lower affinity for LPXY motifs and phosphoserine and phosphothreonine modules.29 The catalytic cysteine within the HECT domain is responsible for ubiquitin transfer.30 Although there is some redundancy in their functions, it has been demonstrated that different Nedd4 proteins are involved in different biological processes.29
 | Figure 2 The human Nedd4 family of E3 ubiquitin ligases. |
Nedd4-2 is the closest homolog of Nedd4, the first member of the HECT family identified.31 Nedd4-2 is present in several tissues, but is most highly expressed in heart, kidney, brain, lung, and liver.1,32 The first physiological role for Nedd4-2 was its involvement in Liddle’s syndrome (an inherited form of hypertension). Mutations that delete or disrupt a C-terminal PY motif in the Epithelial Na Channel (ENaC), a nonvoltage-gated channel, prevent the interaction of Nedd4-2 with the ENaC, increasing renal Na+ absorption and causing Liddle’s syndrome.33 The ENaC is responsible for salt and fluid reabsorption in the distal nephron, distal colon, and lung epithelia, and abnormal elevations of ENaC activity lead to hypertension.34 Nedd4-2 regulates ENaC degradation and activity. It has been found that Nedd4-2 catalyzes both the monoubiquitination and polyubiquitination of ENaC.35 The roles of Nedd4-2 in ENaC activity have also been detected in the clearance of lung fluid in newborn animals. Mice knocked out for Nedd4-2 die perinatally, with increased ENaC expression and activity in the lung, which leads to the failure to inflate the lungs, in turn resulting in the inability of pups to breathe.36,37 In addition, Nedd4-2 has been reported to play a protective regulatory role against the development of cystic fibrosis in lung through the regulation of ENaC function.37 Other substrates of Nedd4-2 include serum and glucocorticoid kinase (SGK),38 14-3-3,38 TrkA,39–43 ACK1,44 EAAT1/2,45,46 Dvl2,47,48 etc. For a complete and updated list of Nedd4-2-interacting proteins, readers can consult a recent review.49 Since the subject of the present review is the regulation of VGIC by Nedd4-2, we shall focus on these substrates.
Regulation of VGICs by Nedd4-2
Voltage-gated sodium channels
Seven out of the Nav channels (Nav1.1, Nav1.2, Nav1.3, Nav1.5, Nav1.6, Nav1.7, and Nav1.8) include in their α-subunit the highly conserved PY motif (L/PPXY) in their C-terminus, recognized by the WW domains of Nedd4-2 (Table 1). Nedd4-2 can bind directly to these channels in vitro and regulate Nav currents in different cells.50–52 The cardiac Nav1.5 was originally reported to be ubiquitinated by Nedd4,53 and it has later been extensively investigated in the context of Nedd4-2.52,54 Nedd4-2 expression causes a decrease in Nav1.5 current density that depends on the PY motif of the channel and on the catalytic activity of Nedd4-2, leading to the ubiquitination of the channel.52 Nav1.2, Nav1.3, Nav1.6, Nav1.7, and Nav1.8 are also substrates of Nedd4-2 in vitro,28,50,51 but the biological significance of the regulation of Navs by Nedd4-2 has been lacking for many years. Cachemaille et al reported that Nedd4-2 expression was downregulated upon induction of neuropathic pain by the spared nerve injury model, and the authors postulated that this might contribute to the dysregulation of Nav1.7 and Nav1.8 associated with peripheral nerve injury.55 Recently, a seminal work performed by the same group demonstrated that Nedd4-2 indeed regulates the surface expression of Nav1.7 and Nav1.8 in adult dorsal root ganglion (DRG) neurons in response to neuropathic pain.54 Moreover, the levels of Nav1.7 and Nav1.8 were enhanced in a knockout of Nedd4-2 in nociceptive neurons. These knockout mice showed an altered pain phenotype, suggesting that the regulation of Navs by Nedd4-2 is critical for the neuronal excitability of the sensory system and that Nedd4-2 downregulation may be required for the central sensitization.54 Therefore, Nedd4-2 regulation may be an alternative to Nav blockers to tackle chronic pain. Regarding the regulation of Nav channels in the CNS, it has been shown that Nedd4-2 is essential for the downregulation of the channels in response to elevated intracellular Na+ levels in cortical neurons,56 although additional work will be necessary to fully understand the role of Nedd4-2 in the CNS.
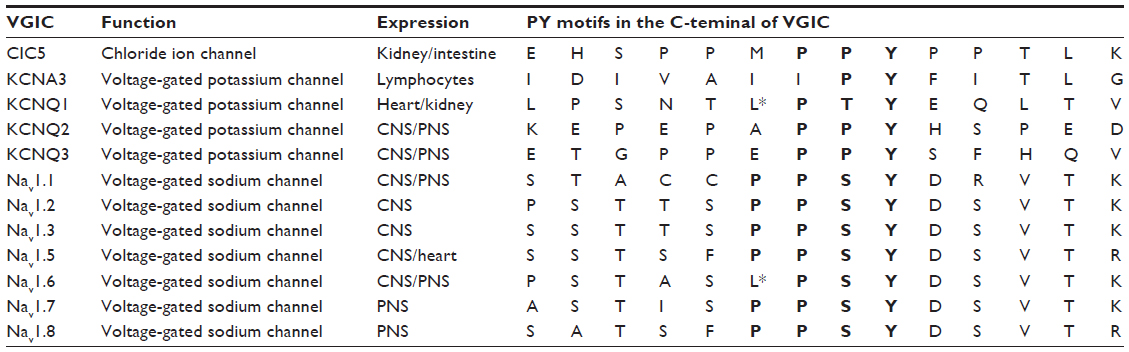 | Table 1 Voltage-gated ion channels regulated by Nedd4-2 |
Voltage-gated potassium channels
Similar to the effects described for Navs, Nedd4-2 regulates some Kvs. KCNQ1 has an atypical PY motif (LPXY) in its C-terminus (Table 1) that is well conserved across species and constitutes a binding site for Nedd4-2 and other E3 ubiquitin ligases of the Nedd4 family. The expression of Nedd4-2 in HEK293 cells and cardiomyocytes reduced the amount of KCNQ1 and the current mediated by KCNQ1, respectively.57 These effects were dependent on the catalytic activity of Nedd4-2 and on the presence of the PY motif in KCNQ1.57 The effect of Nedd4-2 can be counteracted by direct binding of the deubiquitinating enzyme USP2 to KCNQ158 and by PI3K and SGK1 inhibition of Nedd4-2.59 However, Nedd4-2 activity on KCNQ1 can be induced by adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK)60 and 17β-oestradiol.61
It is now clear that Nedd4-2 regulates heteromeric KCNQ2/3 y KCNQ3/5 channels, although these channels lack the classical PY motif in the C-terminus (Table 1). Nedd4-2 expression decreases the amplitude of the K+ currents mediated by KCNQ2/3 and KCNQ3/5 through a direct interaction with the C-terminus of KCNQ3 and its ubiquitination.62 However, the binding motif of KCNQ2 and KCNQ3 recognized by Nedd4-2 remains unknown. Likewise, the peak K+ current amplitude of Kv1.3, which does not contain a PY motif, is reduced by 50% by Nedd4-2 in Xenopus oocytes.63 The effects on KCNQ2, KCNQ3, and Kv1.3 can be counteracted by the inhibition of Nedd4-2 by SGK1.63,64 Accordingly, together these in vitro studies suggest that Nedd4-2 regulates Kv channel surface expression, controlling cell excitability. Future work should validate these results in vivo using genetically modified animals.
Voltage-gated chloride channels
Among the Cl− channel family, ClC-2, ClC-Ka, and ClC-Kb have been shown to be regulated by Nedd4-2,65–67 although none of these channels contains a PY motif. Upon phosphorylation of Nedd4-2, ClC-2 expression in the plasma membrane is regulated by SGK1-3.67 However, it is unknown whether Nedd4-2 binds directly to ClC-2. To be functional, ClC-Ka and ClC-Kb channels need an accessory protein, Barttin, containing a PY motif, whose mutation enhances the activity of the ClC-Kb/Barttin channel.66 In addition, it has been demonstrated that the expression of ClC-Ka/Barttin in Xenopus oocytes induces an inwardly rectifying current that is significantly decreased when Nedd4-2, but not the catalytically inactive Nedd4-2C938S, is coexpressed.65 The effect of Nedd4-2 can be inhibited by the coexpression of SGK1 or SGK3, due to the phosphorylation of Nedd4-2, which prevents its binding to its target.65 A mutation of the proline-rich stretch amino acids (PPMPPY) of ClC-5 that resembles the PY motif (Table 1), increases surface expression,68 but in vivo disruption of this motif has no effect.69 So far, all the reports indicating that Nedd4-2 regulates ClCs are based on in vitro studies and, therefore, in vivo studies are required to validate previous results.
Regulation of Nedd4-2 function
Intramolecular interactions
Nedd4-2 itself contains a LPPY motif within the HECT that is involved in the interaction with its own WW domains70 (Figure 3A). The consequences of this intramolecular interaction are 1) Nedd4-2 autoubiquitination is reduced; 2) the stability of Nedd4-2 is increased; and 3) the binding of Nedd4-2 to ENaC is decreased, and this results in increased ENaC activity.70 Thus, in basal conditions, the self-interaction of Nedd4-2 seems to protect it from degradation, and it regulates Nedd4-2 interaction with its targets. At present, it is unknown whether the Nedd4-2 intramolecular interaction modulates VGIC levels and functions.
 | Figure 3 Nedd4-2 activity and binding to substrates can be regulated in several ways. |
SGKs, Akt, PKA, and 14-3-3 proteins
Another way by which Nedd4-2 is regulated in response to different stimuli is through phosphorylation by SGK, which decreases the binding of Nedd4-2 and the ubiquitination of ENaC,71 Nav1.5,72 ClC-2,67 and different membrane transporters45,73,74 (Figure 3B). However, SGK1 modulation of ENaC, Kv4.3, EAAT4, or hERG independently of Nedd4-2 phosphorylation has also been reported.75–78 The effect of SGK1 in regulating ENaC channels has been suggested previously in SGK1-knockout mice, which had problems in reducing renal Na+ output and in maintaining blood pressure under dietary salt restriction.79 SGK levels are regulated by Nedd4-2-mediated ubiquitination,80 supporting a regulatory loop of both proteins.
However, SGKs are not the only kinases that are able to phosphorylate and inactivate Nedd4-2. Akt and PKA can also phosphorylate and inactivate it81,82 (Figure 3B). The residues of Nedd4-2 phosphorylated by the above kinases are binding sites for 14-3-3 proteins,38,83–85 and it is known that this interaction prevents the recruitment of Nedd4-2 to ENaC84–86 (Figure 3B). However, it remains to be addressed whether other substrates of Nedd4-2, such as the VGICs, are regulated by the above kinases and 14-3-3 proteins.
DUBs
Another protein that has been demonstrated to regulate Nedd4-2 substrates is the deubiquitinase enzyme USP2, which binds directly to Nedd4-2 and counteracts the effects of Nedd4-2 on ENaC87 and on KCNQ1.58 Nedd4-2 seems to locate USP2 in close proximity to the ubiquitinated substrates in order to deubiquitinate them (Figure 3C). However, it remains to be seen whether this DUB, or others, may regulate Nedd4-2 activity and/or stability and the effects on other substrates of Nedd4-2.
Ndfip
As previously indicated, there are substrates of Nedd4-2 that do not contain PY or other motifs that allow the direct binding of the E3 ubiquitin ligase to the substrate. Among Nedd4-2 targets, ClC-Ka/b uses Barttin as a protein adaptor to bring Nedd4-2 close enough to allow ubiquitination.66 Other proteins that have been identified as interacting with Nedd4-2, allowing or competing with the binding to different substrates, are the Ndfips (Ndfip1 and Ndfip2).88–90 These adaptors can prevent the effect of Nedd4-2 on different substrates such as ENaC89 and EGFR,91 whereas they can promote the Nedd4-2-mediated degradation of the divalent metal transporter 1 (DMT1)92 and water-channel aquaporin 2 (AQP2)93 (Figure 3D). It has been described that Ndfip1 is upregulated in ischemic conditions, and it has been suggested that Ndfip1, together with Nedd4-2 and Itch, may provide neuroprotection.94,95 Moreover, it has been reported that Ndfips recruit Nedd4-2 to exosomes to terminate the actions of Nedd4-2.96 Whether Ndfips modulate VGICs protein expression and/or function in nonpathological conditions or in response to injury remains to be addressed.
AMPK
AMPK is activated by increases in the cytoplasmic AMP/ATP concentration ratio, which corresponds to the cellular energy status,97 but can also be further activated by the increase in cytoplasmic Ca2+ even in the absence of metabolic stress.98,99 Among many different proteins, AMPK regulates some VGICs, including KCNQ160,100,101 and Kv1.5,102 not directly but rather by acting through Nedd4-2. In some cases, it has been reported that AMPK phosphorylates Nedd4-2, enhancing the binding to some of its substrates,103 and therefore, regulating the ubiquitination of these substrates. However, it is unknown which Nedd4-2 residues are phosphorylated by AMPK, although they must be different from those recognized by 14-3-3 proteins. Identification of the Nedd4-2 amino acids that are phosphorylated by AMPK will provide mechanistic insight regarding the binding and/or activation of Nedd4-2. In addition, it remains to be addressed whether other substrates of Nedd4-2 can be downregulated by AMPK.
Voltage-gated ion channels and disease
Numerous studies have provided convincing evidence that VGICs play an important role in the functioning of electrical cells and in different pathologies. Mutations in the Nav channels expressed in the CNS, such as Nav1.1 and Nav1.2, cause neurological disorders, including different forms of epilepsy, such as Dravet syndrome (DS), generalized epilepsy with febrile seizure plus, and benign familial neonatal–infantile seizures.104,105 These mutations lead to loss of function of Nav channels mainly in inhibitory interneurons, thereby impairing their inhibitory function on excitatory neurons and leading to hyperexcitability.106,107 Antiepileptic drugs used to control both partial and generalized seizures arising from many different causes may be contraindicated for the treatment of inherited seizure syndromes caused by loss-of-function mutations in Nav channels. An alternative approach to rebalance excitation and inhibition is to enhance inhibitory neurotransmission using benzodiazepines that enhance GABA receptors in combination with other drugs.108 This has been proved to be a successful strategy in preventing seizures in DS, although most patients are not seizure free.108
Mutations of Nav expressed in the PNS, mainly Nav1.7, cause different kind of pain syndromes.109 Gain-of-function mutations are associated with erythromelalgia and paroxysmal extreme pain disorder as a result of sensory neuron hyperexcitability, whereas loss-of-function mutations result in congenital indifference to pain because of an attenuation in action potential firing that causes complete loss of pain sensation.109–111 Moreover, mutations in Nav cardiac channels, mainly in Nav1.5, cause cardiac arrhythmias such as Brugada syndrome, Long QT (LQT) syndrome, ventricular fibrillation, and sudden infant death syndrome.112–114 Treatment of LQT syndrome using sodium current blockers, such as mexiletine, flecainide, or ranolazine, may represent a therapeutic option,115,116 although the response to the drug is mutation-specific showing benefits in some LQT patients, but not in others.
In addition to the altered functionality of Nav due to mutations, regulation of its localization and expression are highly relevant for the development of peripheral neuropathic pain. In a spared nerve injury model, Nedd4-2 expression was decreased in DRG neurons,54,55 leading to increased Nav1.7 and Nav1.8 levels that enhanced the currents meditated by these channels.54 These results were further confirmed in Nedd4-2 knockout mice, which exhibited increased thermal and inflammatory pain.54 Restoration of Nedd4-2 levels in the SNI model eased the mechanical allodynia experienced by these mice. Together, these results point to Nedd4-2 as a key regulator of Nav in chronic pain that may be of use as a target to control different pain pathologies. Specific blockers of Nav1.7, Nav1.8, and Nav1.9 that are currently in development may represent an appropriate drug for the treatment of patients with gain-of-function mutations in these channels.
Kv channels have been implicated in different pathologies, including LQT syndrome (KCNQ1), deafness (KCNQ1, KCNQ4), and benign familial neonatal seizures (KCNQ2, KCNQ3).117,118 The mutations described for these channels can lead to impaired protein stability, misfolding, trafficking, and alterations in channel gating. Some drugs acting on KCNQ channels to modulate their activity are currently being developed as treatments for epilepsy. One of them is retigabine, which stabilizes neuronal KCNQ channels in the open position without affecting cardiac KCNQ1 channels, and therefore, decreasing the potential for cardiac side effects. This drug leads to hyperpolarization of the subthreshold membrane potential by increasing the outward K+ current.119–121
A number of human disease-causing mutations have been identified in the genes encoding ClCs. Mutations in ClC-1 lead to both recessively- and dominantly inherited forms of muscle stiffness or myotonia.122 Mutations in ClC-2 in humans result in leukodystrophy,123 whereas mutations in ClC-Ka and ClC-Kb lead to Bartter syndrome, which combines deafness and a severe renal phenotype.124,125 Similarly, mutations in ClC-5 lead to several forms of inherited kidney stone disease named Dent’s disease,126,127 while other mutations lead only to proteinuria.128 These mutations have been shown to reduce or abolish ClC function.
Conclusion
VGICs are crucial components for the proper functioning of electrically stimulated cells, and hence, their numbers and presence at the plasma membrane must be tightly controlled. Ubiquitination is a posttranslational modification that regulates protein stability, as well as protein trafficking and localization, and Nedd4-2 ubiquitinates several VGICs and regulates their functions. Misregulation or mutations that change the location or number of the VGICs are the basis of several different neurological and cardiac diseases.
Several questions remain to be answered, and further studies should address the following issues, among others: 1) the identification of other VGICs that are substrates of Nedd4-2; 2) a better understanding of the molecular mechanisms involved in the regulation of VGICs, with the identification of accessory proteins that participate in the ubiquitination of VGICs by Nedd4-2; 3) the role of Nedd4-2 in the development of cardiac diseases with the use of genetically modified mice. The seminal in vivo studies indicating that Nedd4-2 protein levels are regulated in response to chronic pain and that Nedd4-2 modulates Nav1.7 and Nav1.8 channels in neuropathic pain should also be the basis for future studies. Gaining further knowledge about interactions between VGICs and Nedd4-2 from recent advances in solving structures of transmembrane proteins may also allow the development of drugs that potentiate or block these complexes in a specific manner. Such drugs could be of potential therapeutic use in diseases in which VGICs are involved directly.
Acknowledgments
The work in my laboratory has been supported by Ministerio de Ciencia e Innovación Grant BFU2008-00162, by Ministerio de Economía y Competitividad Grants BFU2011-22898 and BFU2014-51846-R, and within the VII European Community Framework Programme by a Marie Curie International Reintegration Grant and by the PAINCAGE integrative project.
Disclosure
The author reports no conflicts of interest in this work.
References
Araki N, Umemura M, Miyagi Y, et al. Expression, transcription, and possible antagonistic interaction of the human Nedd4L gene variant: implications for essential hypertension. Hypertension. 2008;51(3):773–777. | |
Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10(1):29–46. | |
Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952; 116(4):449–472. | |
Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57(4):397–409. | |
Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72:95–121. | |
Gutman GA, Chandy KG, Grissmer S, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508. | |
Goldin AL. Diversity of mammalian voltage-gated sodium channels. Ann N Y Acad Sci. 1999;868:38–50. | |
Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57(4):411–425. | |
Watanabe E, Fujikawa A, Matsunaga H, et al. Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J Neurosci. 2000;20(20):7743–7751. | |
Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. | |
Egri C, Ruben PC. A hot topic: temperature sensitive sodium channelopathies. Channels. 2012;6(2):75–85. | |
Vilin YY, Peters CH, Ruben PC. Acidosis differentially modulates inactivation in na(v)1.2, na(v)1.4, and na(v)1.5 channels. Front Pharmacol. 2012;3:109. | |
Doyle DA, Morais Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. | |
Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. | |
Sharman JL, Mpamhanga CP, Spedding M, et al. IUPHAR-DB: new receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res. 2011;39(Database issue):D534–D538. | |
Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1(1):21–30. | |
Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch. 2010;460(2):277–288. | |
Jentsch TJ. Discovery of CLC transport proteins: cloning, structure, function and pathophysiology. J Physiol. Epub 2015 Feb 19. | |
Hershko A, Ciechanover A, Varshavsky A. Basic medical research award. The ubiquitin system. Nat Med. 2000;6(10):1073–1081. | |
Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. | |
Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9(6):536–542. | |
Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315(5809):201–205. | |
Geetha T, Jiang J, Wooten MW. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell. 2005;20(2):301–312. | |
Kawabe H, Brose N. The role of ubiquitylation in nerve cell development. Nat Rev Neurosci. 2011;12(5):251–268. | |
Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol. 2011;12(9):605–620. | |
Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta. 2014;1843(1):61–74. | |
Ardley HC, Robinson PA. E3 ubiquitin ligases. Essays Biochem. 2005;41:15–30. | |
Gasser A, Cheng X, Gilmore ES, Tyrrell L, Waxman SG, Dib-Hajj SD. Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J Biol Chem. 2010;285(34):26149–26161. | |
Yang B, Kumar S. Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010;17(1):68–77. | |
Chen C, Sun X, Guo P, et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene. 2007;26(16):2386–2394. | |
Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185(3):1155–1161. | |
Kumar S, Harvey KF, Kinoshita M, Copeland NG, Noda M, Jenkins NA. cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics. 1997;40(3):435–443. | |
Knight KK, Olson DR, Zhou R, Snyder PM. Liddle’s syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci U S A. 2006;103(8):2805–2808. | |
Hummler E. Implication of ENaC in salt-sensitive hypertension. J Steroid Biochem Mol Biol. 1999;69(1–6):385–390. | |
Zhou R, Patel SV, Snyder PM. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J Biol Chem. 2007;282(28):20207–20212. | |
Boase NA, Rychkov GY, Townley SL, et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat Commun. 2011;2:287. | |
Kimura T, Kawabe H, Jiang C, et al. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc Natl Acad Sci U S A. 2011;108(8):3216–3221. | |
Bhalla V, Daidié D, Li H, et al. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol Endocrinol. 2005;19(12):3073–3084. | |
Arévalo JC, Waite J, Rajagopal R, et al. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50(4):549–559. | |
Georgieva MV, de Pablo Y, Sanchis D, Comella JX, Llovera M. Ubiquitination of TrkA by Nedd4-2 regulates receptor lysosomal targeting and mediates receptor signaling. J Neurochem. 2011;117(3):479–493. | |
Persaud A, Alberts P, Amsen EM, et al. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol Syst Biol. 2009;5:333. | |
Yu T, Calvo L, Anta B, et al. In vivo regulation of NGF-mediated functions by Nedd4-2 ubiquitination of TrkA. J Neurosci. 2014; 34(17):6098–6106. | |
Yu T, Calvo L, Anta B, et al. Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic. 2011;12(4):521–534. | |
Chan W, Tian R, Lee YF, Sit ST, Lim L, Manser E. Down-regulation of active ACK1 is mediated by association with the E3 ubiquitin ligase Nedd4-2. J Biol Chem. 2009;284(12):8185–8194. | |
Boehmer C, Henke G, Schniepp R, et al. Regulation of the glutamate transporter EAAT1 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid-inducible kinase isoforms SGK1/3 and protein kinase B. J Neurochem. 2003;86(5):1181–1188. | |
Boehmer C, Palmada M, Rajamanickam J, Schniepp R, Amara S, Lang F. Post-translational regulation of EAAT2 function by co-expressed ubiquitin ligase Nedd4-2 is impacted by SGK kinases. J Neurochem. 2006;97(4):911–921. | |
Ding Y, Zhang Y, Xu C, Tao QH, Chen YG. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J Biol Chem. 2013; 288(12):8289–8298. | |
Zhang Y, Ding Y, Chen YG, Tao Q. NEDD4L regulates convergent extension movements in Xenopus embryos via disheveled-mediated non-canonical Wnt signaling. Dev Biol. 2014;392(1):15–25. | |
Goel P, Manning JA, Kumar S. NEDD4-2 (NEDD4L): the ubiquitin ligase for multiple membrane proteins. Gene. 2015;557(1):1–10. | |
Fotia AB, Ekberg J, Adams DJ, Cook DI, Poronnik P, Kumar S. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem. 2004;279(28):28930–28935. | |
Rougier JS, van Bemmelen MX, Bruce MC, et al. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am J Physiol Cell Physiol. 2005;288(3):C692–C701. | |
van Bemmelen MX, Rougier JS, Gavillet B, et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004;95(3):284–291. | |
Abriel H, Kamynina E, Horisberger JD, Staub O. Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase Nedd4. FEBS Lett. 2000;466(2–3):377–380. | |
Laedermann CJ, Cachemaille M, Kirschmann G, et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J Clin Invest. 2013;123(7):3002–3013. | |
Cachemaille M, Laedermann CJ, Pertin M, Abriel H, Gosselin RD, Decosterd I. Neuronal expression of the ubiquitin ligase Nedd4-2 in rat dorsal root ganglia: modulation in the spared nerve injury model of neuropathic pain. Neuroscience. 2012;227:370–380. | |
Ekberg JA, Boase NA, Rychkov G, Manning J, Poronnik P, Kumar S. Nedd4-2 (NEDD4L) controls intracellular Na(+)-mediated activity of voltage-gated sodium channels in primary cortical neurons. Biochem J. 2014;457(1):27–31. | |
Jespersen T, Membrez M, Nicolas CS, et al. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc Res. 2007;74(1):64–74. | |
Krzystanek K, Rasmussen HB, Grunnet M, et al. Deubiquitylating enzyme USP2 counteracts Nedd4-2-mediated downregulation of KCNQ1 potassium channels. Heart Rhythm. 2012;9(3):440–448. | |
Andersen MN, Krzystanek K, Petersen F, et al. A phosphoinositide 3-kinase (PI3K)-serum- and glucocorticoid-inducible kinase 1 (SGK1) pathway promotes Kv7.1 channel surface expression by inhibiting Nedd4-2 protein. J Biol Chem. 2013;288(52):36841–36854. | |
Alzamora R, Gong F, Rondanino C, et al. AMP-activated protein kinase inhibits KCNQ1 channels through regulation of the ubiquitin ligase Nedd4-2 in renal epithelial cells. Am J Physiol Renal Physiol. 2010;299(6):F1308–F1319. | |
Rapetti-Mauss R, O’Mahony F, Sepulveda FV, Urbach V, Harvey BJ. Oestrogen promotes KCNQ1 potassium channel endocytosis and postendocytic trafficking in colonic epithelium. J Physiol. 2013;591 (pt 11):2813–2831. | |
Ekberg J, Schuetz F, Boase NA, et al. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J Biol Chem. 2007;282(16):12135–12142. | |
Henke G, Maier G, Wallisch S, Boehmer C, Lang F. Regulation of the voltage gated K+ channel Kv1.3 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid inducible kinase SGK1. J Cell Physiol. 2004;199(2):194–199. | |
Schuetz F, Kumar S, Poronnik P, Adams DJ. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by serum- and glucocorticoid-regulated kinase-1. Am J Physiol Cell Physiol. 2008;295(1):C73–C80. | |
Embark HM, Böhmer C, Palmada M, et al. Regulation of CLC-Ka/barttin by the ubiquitin ligase Nedd4-2 and the serum- and glucocorticoid-dependent kinases. Kidney Int. 2004;66(5):1918–1925. | |
Estévez R, Boettger T, Stein V, et al. Barttin is a Cl-channel beta-subunit crucial for renal Cl-reabsorption and inner ear K+ secretion. Nature. 2001;414(6863):558–561. | |
Palmada M, Dieter M, Boehmer C, Waldegger S, Lang F. Serum and glucocorticoid inducible kinases functionally regulate ClC-2 channels. Biochem Biophys Res Commun. 2004;321(4):1001–1006. | |
Schwake M, Friedrich T, Jentsch TJ. An internalization signal in ClC-5, an endosomal Cl-channel mutated in Dent’s disease. J Biol Chem. 2001; 276(15):12049–12054. | |
Rickheit G, Wartosch L, Schaffer S, et al. Role of ClC-5 in renal endocytosis is unique among ClC exchangers and does not require PY-motif-dependent ubiquitylation. J Biol Chem. 2010;285(23):17595–17603. | |
Bruce MC, Kanelis V, Fouladkou F, Debonneville A, Staub O, Rotin D. Regulation of Nedd4-2 self-ubiquitination and stability by a PY motif located within its HECT-domain. Biochem J. 2008;415(1):155–163. | |
Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277(1):5–8. | |
Boehmer C, Wilhelm V, Palmada M, et al. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc Res. 2003;57(4):1079–1084. | |
Boehmer C, Embark HM, Bauer A, et al. Stimulation of renal Na+ dicarboxylate cotransporter 1 by Na+/H+ exchanger regulating factor 2, serum and glucocorticoid inducible kinase isoforms, and protein kinase B. Biochem Biophys Res Commun. 2004;313(4):998–1003. | |
Dieter M, Palmada M, Rajamanickam J, et al. Regulation of glucose transporter SGLT1 by ubiquitin ligase Nedd4-2 and kinases SGK1, SGK3, and PKB. Obes Res. 2004;12(5):862–870. | |
Baltaev R, Strutz-Seebohm N, Korniychuk G, Myssina S, Lang F, Seebohm G. Regulation of cardiac shal-related potassium channel Kv 4.3 by serum- and glucocorticoid-inducible kinase isoforms in Xenopus oocytes. Pflugers Arch. 2005;450(1):26–33. | |
Diakov A, Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s alpha-subunit. J Biol Chem. 2004;279(37):38134–38142. | |
Lamothe SM, Zhang S. The serum- and glucocorticoid-inducible kinases SGK1 and SGK3 regulate hERG channel expression via ubiquitin ligase Nedd4-2 and GTPase Rab11. J Biol Chem. 2013;288(21):15075–15084. | |
Rajamanickam J, Palmada M, Lang F, Boehmer C. EAAT4 phosphorylation at the SGK1 consensus site is required for transport modulation by the kinase. J Neurochem. 2007;102(3):858–866. | |
Lang F, Henke G, Embark HM, et al. Regulation of channels by the serum and glucocorticoid-inducible kinase – implications for transport, excitability and cell proliferation. Cell Physiol Biochem. 2003;13(1):41–50. | |
Zhou R, Snyder PM. Nedd4-2 phosphorylation induces serum and glucocorticoid-regulated kinase (SGK) ubiquitination and degradation. J Biol Chem. 2005;280(6):4518–4523. | |
Ismail NA, Baines DL, Wilson SM. The phosphorylation of endogenous Nedd4-2 In Na(+)-absorbing human airway epithelial cells. Eur J Pharmacol. 2014;732:32–42. | |
Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J Biol Chem. 2007;282(41):29866–29873. | |
Chandran S, Li H, Dong W, et al. Neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2) regulation by 14-3-3 protein binding at canonical serum and glucocorticoid kinase 1 (SGK1) phosphorylation sites. J Biol Chem. 2011;286(43):37830–37840. | |
Ichimura T, Yamamura H, Sasamoto K, et al. 14-3-3 Proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J Biol Chem. 2005;280(13):13187–13194. | |
Liang X, Butterworth MB, Peters KW, Walker WH, Frizzell RA. An obligatory heterodimer of 14-3-3beta and 14-3-3epsilon is required for aldosterone regulation of the epithelial sodium channel. J Biol Chem. 2008;283(41):27418–27425. | |
Nagaki K, Yamamura H, Shimada S, et al. 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry. 2006;45(21):6733–6740. | |
Oberfeld B, Ruffieux-Daidie D, Vitagliano JJ, Pos KM, Verrey F, Staub O. Ubiquitin-specific protease 2-45 (Usp2-45) binds to epithelial Na+ channel (ENaC)-ubiquitylating enzyme Nedd4-2. Am J Physiol Renal Physiol. 2011;301(1):F189–F196. | |
Harvey KF, Shearwin-Whyatt LM, Fotia A, Parton RG, Kumar S. N4WBP5, a potential target for ubiquitination by the Nedd4 family of proteins, is a novel Golgi-associated protein. J Biol Chem. 2002; 277(11):9307–9317. | |
Konstas AA, Shearwin-Whyatt LM, Fotia AB, et al. Regulation of the epithelial sodium channel by N4WBP5A, a novel Nedd4/Nedd4-2-interacting protein. J Biol Chem. 2002;277(33):29406–29416. | |
Mund T, Pelham HR. Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 2009;10(5):501–507. | |
Shearwin-Whyatt LM, Brown DL, Wylie FG, Stow JL, Kumar S. N4WBP5A (Ndfip2), a Nedd4-interacting protein, localizes to multivesicular bodies and the Golgi, and has a potential role in protein trafficking. J Cell Sci. 2004;117(pt 16):3679–3689. | |
Howitt J, Putz U, Lackovic J, et al. Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons. Proc Natl Acad Sci U S A. 2009;106(36):15489–15494. | |
Velic A, Gabriëls G, Hirsch JR, et al. Acute rejection after rat renal transplantation leads to downregulation of NA+ and water channels in the collecting duct. Am J Transplant. 2005;5(6):1276–1285. | |
Lackovic J, Howitt J, Callaway JK, Silke J, Bartlett P, Tan SS. Differential regulation of Nedd4 ubiquitin ligases and their adaptor protein Ndfip1 in a rat model of ischemic stroke. Exp Neurol. 2012; 235(1):326–335. | |
Sang Q, Kim MH, Kumar S, et al. Nedd4-WW domain-binding protein 5 (Ndfip1) is associated with neuronal survival after acute cortical brain injury. J Neurosci. 2006;26(27):7234–7244. | |
Putz U, Howitt J, Lackovic J, et al. Nedd4 family-interacting protein 1 (Ndfip1) is required for the exosomal secretion of Nedd4 family proteins. J Biol Chem. 2008;283(47):32621–32627. | |
Steinberg GR, O’Neill HM, Dzamko NL, et al. Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity. J Biol Chem. 2010;285(48):37198–37209. | |
Bair AM, Thippegowda PB, Freichel M, et al. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J Biol Chem. 2009;284(1):563–574. | |
Wu WN, Wu PF, Zhou J, et al. Orexin-A activates hypothalamic AMP-activated protein kinase signaling through a Ca(2)(+)-dependent mechanism involving voltage-gated L-type calcium channel. Mol Pharmacol. 2013;84(6):876–887. | |
Alesutan I, Föller M, Sopjani M, et al. Inhibition of the heterotetrameric K+ channel KCNQ1/KCNE1 by the AMP-activated protein kinase. Mol Membr Biol. 2011;28(2):79–89. | |
Andersen MN, Krzystanek K, Jespersen T, Olesen SP, Rasmussen HB. AMP-activated protein kinase downregulates Kv7.1 cell surface expression. Traffic. 2012;13(1):143–156. | |
Mia S, Munoz C, Pakladok T, et al. Downregulation of Kv1.5 K channels by the AMP-activated protein kinase. Cell Physiol Biochem. 2012;30(4):1039–1050. | |
Bhalla V, Oyster NM, Fitch AC, et al. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem. 2006;281(36):26159–26169. | |
Catterall WA. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol. 2014;54:317–338. | |
Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360(9336):851–852. | |
Ogiwara I, Miyamoto H, Morita N, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27(22):5903–5914. | |
Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9(9):1142–1149. | |
Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol. 2011;53(Suppl 2):16–18. | |
Waxman SG, Merkies IS, Gerrits MM, et al. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 2014;13(11):1152–1160. | |
Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci. 2008;28(46):11768–11777. | |
Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. | |
Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115(8):2010–2017. | |
Wang J, Ou SW, Wang YJ, Kameyama M, Kameyama A, Zong ZH. Analysis of four novel variants of Nav1.5/SCN5A cloned from the brain. Neurosci Res. 2009;64(4):339–347. | |
Tfelt-Hansen J, Winkel BG, Grunnet M, Jespersen T. Inherited cardiac diseases caused by mutations in the Nav1.5 sodium channel. J Cardiovasc Electrophysiol. 2010;21(1):107–115. | |
Moss AJ, Goldenberg I. Importance of knowing the genotype and the specific mutation when managing patients with long QT syndrome. Circ Arrhythm Electrophysiol. 2008;1(3):213–226; discussion 226. | |
Moss AJ, Windle JR, Hall WJ, et al. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol. 2005;10(4 Suppl):59–66. | |
Maljevic S, Wuttke TV, Lerche H. Nervous system KV7 disorders: breakdown of a subthreshold brake. J Physiol. 2008;586(7):1791–1801. | |
Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71(1):15–25. | |
Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58(2):253–262. | |
Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61(4):921–927. | |
Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24(19):4614–4624. | |
Lehmann-Horn F, Mailander V, Heine R, George AL. Myotonia levior is a chloride channel disorder. Hum Mol Genet. 1995;4(8):1397–1402. | |
Depienne C, Bugiani M, Dupuits C, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12(7):659–668. | |
Nozu K, Inagaki T, Fu XJ, et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet. 2008;45(3):182–186. | |
Schlingmann KP, Konrad M, Jeck N, et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350(13):1314–1319. | |
Fisher SE, Black GC, Lloyd SE, et al. Isolation and partial characterization of a chloride channel gene which is expressed in kidney and is a candidate for Dent’s disease (an X-linked hereditary nephrolithiasis). Hum Mol Genet. 1994;3(11):2053–2059. | |
Wrong OM, Norden AG, Feest TG. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87(8):473–493. | |
Sekine T, Komoda F, Miura K, et al. Japanese Dent disease has a wider clinical spectrum than dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant. 2014;29(2):376–384. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.