Back to Journals » Journal of Inflammation Research » Volume 14
Necroptosis Underlies Neutrophilic Inflammation Associated with the Chronic Rhinosinusitis with Nasal Polyps (CRSwNP)
Authors Xie Y, Li M, Chen K, Zhu H, Tang M, Zhou C ![]() , Zheng Y, Wen J, Han M
, Zheng Y, Wen J, Han M ![]() , Zhang J
, Zhang J ![]() , Zhao K, Xiao H, Li H
, Zhao K, Xiao H, Li H
Received 1 June 2021
Accepted for publication 6 August 2021
Published 16 August 2021 Volume 2021:14 Pages 3969—3983
DOI https://doi.org/10.2147/JIR.S322875
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yadong Xie,1,2,* Min Li,1,3,* Kun Chen,4,* Haoxiang Zhu,5 Mengyao Tang,6 Chun Zhou,1 Yaoming Zheng,1 Jing Wen,2 Miaomiao Han,1 Jia Zhang,1 Keqing Zhao,1 Hui Xiao,2 Huabin Li1
1ENT Institute and Department of Otorhinolaryngology, Eye & ENT Hospital, Fudan University, Shanghai, 200031, People’s Republic of China; 2The Center for Microbes, Development and Health, Institut Pasteur of Shanghai; University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, 200031, People’s Republic of China; 3Department of Otolaryngology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, 310003, People’s Republic of China; 4Department of Otolaryngology-Head and Neck Surgery, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200092, People’s Republic of China; 5Department of Infectious Diseases, Huashan Hospital, Fudan University, Shanghai, 200040, People’s Republic of China; 6Department of Plastic and Reconstructive Surgery, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, 200072, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huabin Li
ENT Institute and Department of Otorhinolaryngology, Eye & ENT Hospital, Fudan University, 83 Fenyang Road, Shanghai, 200031, People’s Republic of China
Tel + 86-21-6437-7134
Email [email protected]
Hui Xiao
The Center for Microbes, Development and Health, Institut Pasteur of Shanghai; University of Chinese Academy of Sciences, Chinese Academy of Sciences, 320 Yueyang Road, Shanghai, 200031, People’s Republic of China
Tel + 86-21-5492-3127
Email [email protected]
Background: Necroptosis is an inflammatory cell death associated with a variety of chronic diseases. Chronic rhinosinusitis with nasal polyps (CRSwNP) is a chronic inflammatory disease accompanied by eosinophil and neutrophil infiltration. The role of necroptosis in the pathogenesis of CRSwNP remains elusive.
Methods: Cell death, including apoptosis, pyroptosis and necroptosis in control sinonasal mucosa and CRSwNP, were analyzed by immunoblotting, immunohistochemistry (IHC) and immunofluorescence (IF) staining for cleaved caspase 3, cleaved gasdermin D and p-MLKL, respectively. Correlations between necroptosis, inflammatory cytokines and neutrophil infiltration were assessed and a possible role of necroptosis in CRSwNP was evaluated. Primary nasal polyp cells (DNPCs) were stimulated with damage-associated molecular patterns (DAMPs) including ATP or IL-1α and their expression of inflammatory cytokines was analyzed using RT-PCR. The expression of TNF-α and IFNs in nasal polyps was measured by ELISA; human monocyte THP-1 cells were treated with TNF-α or IFN-γ and cell death was measured by LDH release.
Results: Necroptosis, rather than apoptosis or pyroptosis, was overtly activated in both eosinophilic and non-eosinophilic CRSwNP as evidenced by the presence of prominent phosphorylation of MLKL compared to controls. The abundance of DAMPs (IL-1α, HMGB1), inflammatory cytokines (IL-6) and chemokines (IL-8, CXCL-1) were all increased especially in non-eosinophilic CRSwNP. The extent of necroptosis was positively correlated with the abundance of DAMPs and cytokines, and neutrophil infiltration in CRSwNP. In DNPCs, ATP and IL-1α induced the expression of IL-8 and CXCL-1. Macrophage was found to be the predominant cell type positive for p-MLKL in CRSwNP. Concomitant treatment with TNF-α and IFN-γ, which were abundantly present in CRSwNP, triggered marked necroptosis in THP-1 cells.
Conclusion: Necroptosis induced by TNF-α and IFN-γ may facilitate the production and release of a myriad of proinflammatory cytokines and entailed neutrophil infiltration to exacerbate inflammation in CRSwNP.
Keywords: chronic rhinosinusitis with nasal polyps, CRSwNP, necroptosis, macrophage, IL-1α, neutrophil
Graphical Abstract:

Introduction
Chronic rhinosinusitis (CRS) is a chronic and relapsing inflammatory disorder characterized by persistent inflammation in the sinonasal mucosa. While CRS affects 4–12% population worldwide, the clinical treatments for CRS are still limited and often give rise to unsatisfactory outcomes when nasal polyps are concurrent.1 CRS with nasal polyps (CRSwNP) is heterogeneous and can be generally classified into eosinophilic and non-eosinophilic endotypes based on the immune signatures. Eosinophilic CRSwNP characterized by extensive eosinophils infiltration and type 2 inflammation accounts for 80% of CRSwNP cases in white patients and has been extensively studied.2,3 However, the non-eosinophilic CRSwNP associated with Th1/Th17 activation is more prevalent in Asian countries including China and Korea, but has been less studied.4 Recently, enhanced neutrophil infiltration was reported in both non-eosinophilic CRSwNP and eosinophilic CRSwNP with severe type 2 inflammation.5,6 Importantly, neutrophils may contribute to disease severity by disrupting the epithelial barrier or inducing tissue remodeling through oncostatin M or TGF-β2, respectively.7 Indeed, patients with higher neutrophil infiltration were reported to be more resistant to oral corticosteroid therapy.8 However, the precise mechanisms underlying the regulation of neutrophilic inflammation in CRSwNP remain largely unknown.
Programmed cell death, which comprises apoptosis, necroptosis, pyroptosis and NETosis, plays important roles in tissue homeostasis and inflammation.9 Apoptosis is the best-known programmed cell death executed by caspase-3,7 upon the activation of upstream initiator caspases caspase-8/9/10.10 Necroptosis is executed by RIPK3 kinase-mediated MLKL phosphorylation and oligomerization.11 Pyroptosis is generally induced by inflammasome-dependent- and caspase-1/11-mediated cleavage of gasdermin D and contributes to the release of cytokines IL-1β and IL-18.12 NETosis is a specialized lytic cell death of neutrophils and is accompanied by the release of neutrophil extracellular traps (NETs) to trap and kill microbes.13 While apoptosis is historically considered as a homeostatic non-inflammatory cell death, necroptosis, pyroptosis and NETosis are highly inflammatory lytic cell death.9,14 Unlike pyroptosis and NETosis, necroptosis can take place in a broad range of tissues and cell types, and thus has been implicated in a broad range of physiological and pathological settings.15 A number of inflammatory cytokines like TNF and IFNs can initiate the activation of RIPK3 especially when Caspase-8 or FADD is inactivated. Besides, pathogen-associated molecular patterns (PAMPs) and live pathogens such as influenza virus, Candida albicans and Staphylococcus aureus can also cause necroptosis through different mechanisms.16–19 Not surprisingly, necroptosis is implicated in the pathogenesis of multiple inflammatory diseases, such as inflammatory bowel diseases, dermatitis and hepatitis.15,20,21 In these settings, necroptosis can promote inflammation through at least two distinct mechanisms. First, the necroptosis of epithelial cells may impair the barrier function and cause microflora to penetrate the intestine or skin to activate immune cells in these tissues.20–22 Second, necroptotic cells often release proinflammatory cytokines and DAMPs including IL-1α, ATP and HMGB1, which act as second messengers to alert or activate the immune system.23,24 Therefore, necroptosis and inflammation can form a vicious loop to drive disease progression.
Recent studies also imply an important role of cell death in the pathogenesis of CRSwNP. For instance, the proinflammatory cytokine IFN-γ was reported to be involved in epithelial apoptosis in CRSwNP.25,26 In neutrophilic CRSwNP, our group identified that the anti-microbial peptide LL-37 was able to elicit NETosis, which may in turn exacerbate neutrophilic inflammation.27 However, whether other types of cell death like necroptosis and pyroptosis are involved in CRSwNP pathogenesis requires further investigation. In this study, we uncovered that necroptosis was mainly responsible for the overt cell death in CRSwNP. Moreover, our data demonstrated that TNFα/IFN-γ were the culprits for necroptosis. Overall, this study links necroptosis to Th17 inflammation and neutrophil recruitment, thus providing new insight into the pathogenesis of CRSwNP.
Materials and Methods
Patients and Tissue Samples
This study complies with the Declaration of Helsinki and was approved by the Ethics Committee of Eye & ENT Hospital, Fudan University (approval No. [2019][2019097–1]). Written informed consent was obtained from each patient. Nasal polyps were collected from patients diagnosed with CRSwNP according to the European Position Paper on Rhinosinusitis and Nasal Polyps (EPOS) guidelines. Patients with allergic fungal nasal polyps, antrochoanal polyps, or had taken oral corticosteroids or other immunosuppressive agents in the month before surgery were excluded from the study. Control nasal mucosae were obtained from patients who were treated for cysts and deviations from the nasal septum. Eos CRSwNP was defined as >10 eosinophils/high-power field (HPF). The characteristics of the subjects are listed in Table 1.
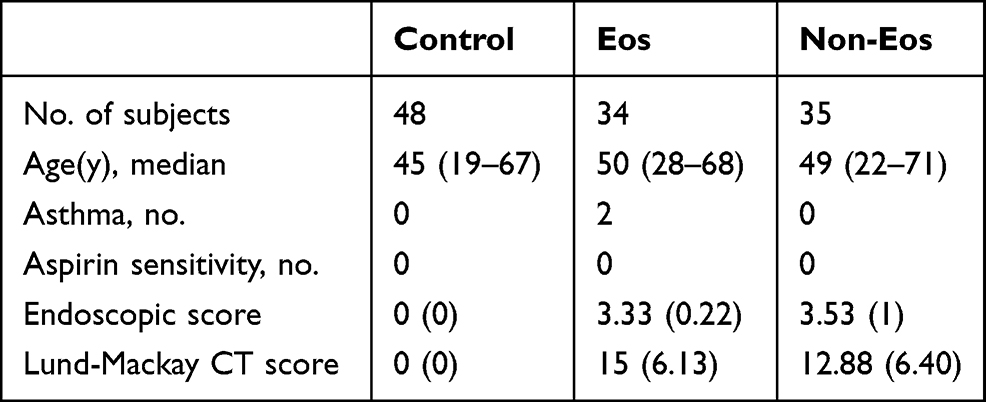 |
Table 1 Characteristics of Subjects |
Cell Culture
THP-1 cells were purchased from ATCC and cultured in RPMI-1640 medium supplemented with 10% FBS, 1% penicillin (100 IU/mL)/streptomycin (100 μg/mL) and 50 μM 2-mercaptoethanol. Primary DNPCs were isolated from patients with CRSwNP as previously described.28 Briefly, fresh nasal polyp tissues were washed with PBS 3 times, cut into small pieces of approximately 1 mm and digested in RPMI medium with 1 mg/mL collagenase IV and 5 U/mL DNase I at 37°C for 1 h. The cell suspensions were vortexed rigorously and passed through a 70 μm cell strainer. Then, cells were collected by spin at 500 g for 5 mins and further washed 3 times with RPMI medium. DNPCs were seeded at a density of 2×106 cells in 12-well plates and cultured in RPMI-1640 medium supplemented with 10% FBS, 1% penicillin (100 IU/mL)/streptomycin (100 μg/mL). DNPCs were rested overnight before treatment with DAMPs.
Immunohistochemical Staining (IHC) and Immunofluorescence (IF)
IHC and IF were conducted as previously described.29 Briefly, fresh tissues were fixed by 4% paraformaldehyde overnight. Following ethyl alcohol dehydration, tissues were embedded in paraffin and sectioned (3 μm) sequentially. Heat-induced antigen retrieval was performed in a 0.01 m citrate buffer (pH 6.0) or EDTA buffer (pH 9.0) by microwave for 15 min. The sections were incubated overnight at 4°C with primary antibodies against Cleaved Caspase-3 (1:200, #9664, Cell Signaling Technology), cleaved GSDM-D (1:100, #36425, Cell Signaling Technology), p-MLKL (1:100, ab187091, Abcam), CD68 (1:500, ab955, Abcam), MPO (1:500, ab208670, Abcam), NE (1:500, ab68672, Abcam), ECP (1:800, ab207429, Abcam). Next, sections were incubated with a biotinylated secondary antibody (Jackson immunoresearch) for 1 h and Streptavidin-HRP (Vector Laboratories) for 30 min at RT. Staining was visualized with DAB substrate (Vector Laboratories) and counterstained with hematoxylin.
For IF experiments, double staining was performed according to manufacturer’s instructions (PerkinElmer, Opal kit). Sections were incubated with primary antibodies overnight at 4°C and then with HRP conjugated secondary antibodies (Vector Laboratories). Visualization of each target was achieved by fluorescein TSA plus (1:200). The slides were then subjected to antigen retrieval in citrate buffer (pH 6.0) using microwave to remove redundant antibodies and followed by staining with another primary antibody. The sections were counterstained with DAPI (Sigma). TUNEL assay was conducted with In-Situ Cell Death Kit (Roche) according to the manufacturer’s recommendations. Sections were examined under an Olympus IX73 Microscope (Olympus Optical, Hamburg, Germany).
RNA Extraction and Quantitative RT-PCR Analysis
RNA was isolated by TRIzol reagent in accordance with the manufacturer’s protocol. cDNAs were synthesized with the PrimeScript RT-PCR Kit (TaKaRa). The real-time polymerase chain reaction (RT-PCR) was carried out with SYBR Premix Taq (Takara) and performed on an ABI PRISM 7500 Detection System (Applied Biosystems, Foster City, CA, USA). Gene expression analysis was normalized to β-actin.
Western Blotting
Tissues and THP-1 cells were lysed in RIPA lysis buffer supplemented with protease inhibitor cocktail (Roche). Proteins were then separated by 8–10% SDS-PAGE and transferred to a PVDF membrane. The membrane was first blocked in 5% milk, and then incubated overnight at 4°C with primary antibodies against p-MLKL (1:1000, ab187091, Abcam), MLKL (1:1000, ab184718, Abcam), p-RIPK3 (1:1000, ab209384, Abcam), RIPK3 (1:1000, #2238, Prosci), FADD (1:1000, ab108601, Abcam), Cleaved Caspase-3 (1:1000, #9664, Cell Signaling Technology), Cleaved GSDM-D (1:1000, #36425, Cell Signaling Technology), GSDM-D (1:1000; #39754, Cell Signaling Technology), p-S6 (1:1000, #5364, Cell Signaling Technology) and ACTIN (1:5000; #3700, Cell Signaling Technology). Subsequently, the membrane was incubated with a secondary antibody for another 1 h. Images were visualized by Tanon image system (Tanon Technology Co., Shanghai, China).
ELISA
Freshly obtained samples were weighed and homogenized in ice-cold PBS supplemented with protease inhibitor cocktail (Roche). The protein concentration was measured by BCA kits. The concentrations of cytokines were determined by Milliplex Multiplex Assays Using Luminex system (Millipore) or commercially available ELISA kits (R&D Systems) in accordance with the manufacturer’s protocols. The detection limits of individual cytokines are as follows: IL-8 (0.34–3425 pg/mL), IFN-β (0.84–13,254 pg/mL), IL-1β (1.09–23,091 pg/mL), IFN-γ (2.79–56,496 pg/mL), TRAIL (1.86–24,073 pg/mL), IL-1α (0.49–4349 pg/mL), FasL (0.57–9219 pg/mL), CXCL1 (23.01–22,008 pg/mL), TNF-α (0.50–10,285 pg/mL), IL-6 (0.30–6113 pg/mL), HMGB1 (31.25–2000 pg/mL).
Ligands and Inhibitors
Pam3CSK4 (tlrl-pms), Poly(I:C) (tlrl-piclv), LPS (tlrl-peklps) and Flagellin (tlrl-pafla) were purchased from Invivogen. TNF-α (300–01A), IFN-γ (300–02) and IL-1α (200–01A) were purchased from Peprotech. HMGB1 (1690-HMB) was from R&D system. Rapamycin (S1039), Torin-1 (S2827), z-DEVD-FMK (S7312), z-VAD-FMK (S7023), GSK’872 (S8465) and ATP (S5260) were purchased from Selleck. Ac-YVAD-CMK (SML0429) and necrosulfonamide (480,073) were purchased from Sigma.
Cell Viability and Cytotoxicity Assay
The viability of THP-1 cells was measured by a CellTiter-Glo Luminescent Cell Viability Assay kit (Promega). The cell cytotoxicity was measured using a CytoTox 96® Non-Radioactive Cytotoxicity Assay kit (Promega).
Statistical Analysis
All statistical analyses were carried out with GraphPad Prism Software 8.0 (GraphPad Software, La Jolla, Calif). The Mann–Whitney U 2-tailed test was used for between-group comparisons of clinical samples. Spearman’s rank correlation test was performed when appropriate. The paired Student’s t-test was used to compare differences before and after DAMPs stimulation in DNPCs. Cell viability and cytotoxicity data were analyzed by one-way analysis of variance (ANOVA). P values less than 0.05 were considered statistically significant.
Results
Prominent Necroptosis is Associated with CRSwNP
As cell death is closely related to infections and inflammatory conditions, we sought to test whether aberrant cell death is linked to CRSwNP. By TUNEL staining, only scarcely scattered TUNEL-positive cells were observed in the non-CRSwNP control nasal mucosa, whereas pronounced TUNEL-positive cells were detected in both eosinophilic and non-eosinophilic CRSwNP group (Figure 1A). In line with previous report,26 epithelial cell death was evident in the CRSwNP group, likely contributing to impaired barrier function. Of note, cell death was more pronounced in the subepithelial area of nasal polyps, suggesting the presence of immune cell death (Figure 1A). As TUNEL-positive cells might comprise both apoptotic and necrotic cells, we further used specific markers to define what type of cell death is involved. Immunoblotting revealed no significant difference in the apoptosis marker cleaved caspase-3 (C-Casp3) or pyroptosis marker cleaved gasdermin D (C-GSDMD) between the control and CRSwNP groups (Figure 1B and C). However, the phosphorylation of RIPK3 and MLKL, both of which are specific markers for necroptosis, was significantly increased in the CRSwNP group (Figure 1B and C). IHC staining also revealed unaltered apoptosis between the control and CRSwNP groups, albeit pyroptosis was slightly increased in the non-eosinophilic group compared to the control group (Figure 1D). Consistent with the immunoblotting data, p-MLKL staining was markedly enhanced in nasal polyps compared to the control tissues. Of note, MLKL and p-MLKL positive cells were mainly located in the sub-epithelium area (Figures 1D and S1A), and the non-eosinophilic group exhibited more necroptosis than the eosinophilic group (Figure 1D). Thus, necroptosis rather than apoptosis or pyroptosis was specifically activated in the CRSwNP tissue.
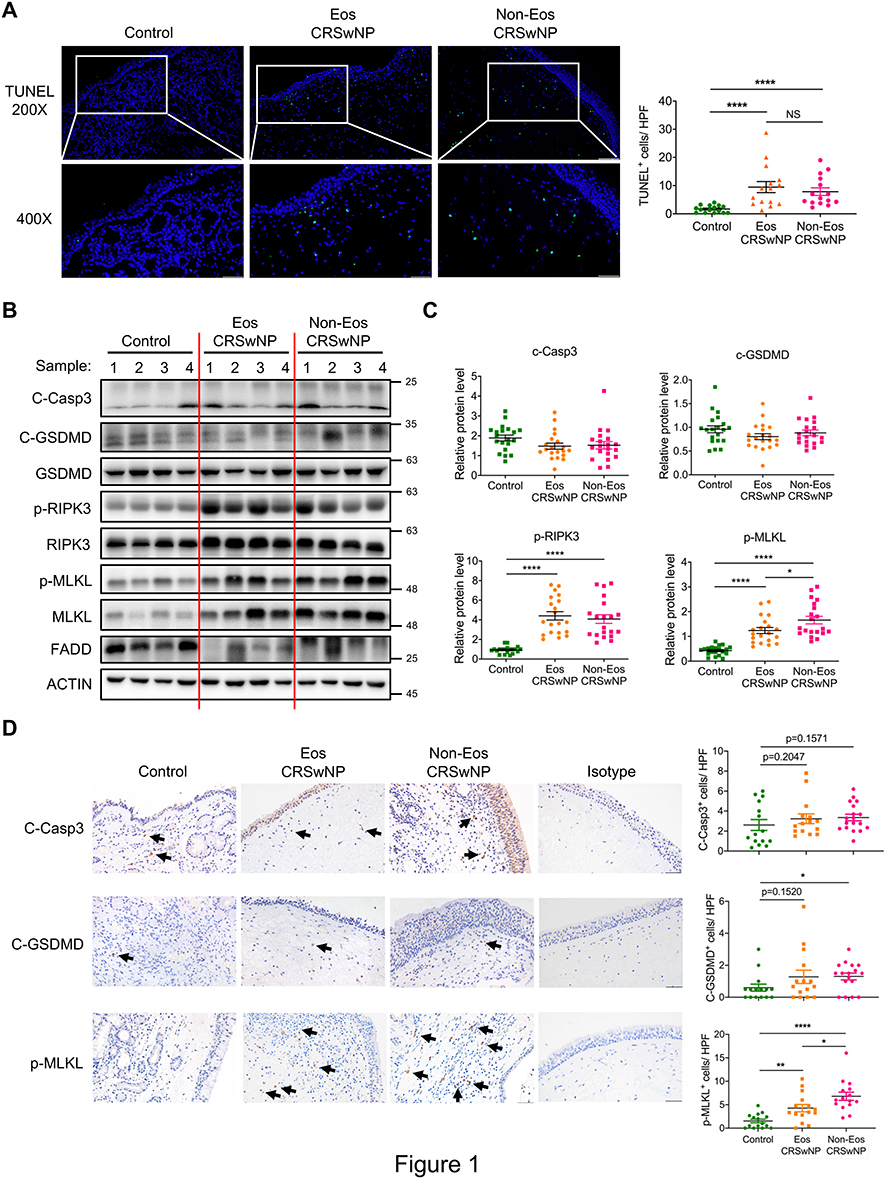 |
Figure 1 Prominent necroptosis is associated with CRSwNP. (A) TUNEL staining of control tissues and nasal polyps (Eos, Eosinophilic; Non-Eos, Non-eosinophilic). TUNEL positive cells were counted over high power field (400×) (n=15 per group). Scale bars: 200×, 100 μm; 400×, 50 μm. (B and C) Immunoblots and quantification of the protein levels over actin in control tissues and nasal polyps (n=20 per group). (D) IHC staining of cleaved caspase-3 (C-Casp3), cleaved gasdermin D (C-GSDMD) and p-MLKL in control tissues and nasal polyps. The positive cells were counted over high power field (400×) (n=15 per group). Scale bars: 50 μm. The data were shown as mean±SEM. NS, not significant; *p<0.05, **p<0.01, ****p<0.0001; by Mann–Whitney U 2-tailed test. |
Necroptosis is Correlated with Neutrophilic Inflammation in CRSwNP
Necroptosis can amplify inflammation by releasing DAMPs and inflammatory cytokines when the plasma membrane is disrupted. To test whether the increased necroptosis in CRSwNP may lead to DAMPs release, we measured IL-1α and HMGB1 in tissue homogenates by ELISA. The IL-1α and HMGB1 protein levels were remarkably increased in CRSwNP tissues compared to the controls, especially in the non-eosinophilic nasal polyps (Figure 2A). Proinflammatory cytokines and chemokines including IL-8, CXCL1, IL-1β and IL-6 were also highly expressed in the non-eosinophilic nasal polyps (Figures 2B and S1B). Importantly, the extent of necroptosis was positively correlated with the protein levels of IL-1α, HMGB1, IL-8 and CXCL1 (Figure 2C), implying a possible role of necroptosis in DAMPs release and tissue inflammation. As IL-8 and CXCL1 are important chemokines for neutrophil recruitment, we reasoned that necroptosis might affect neutrophil infiltration in CRSwNP. So, we detected eosinophil and neutrophil cell numbers in the nasal mucosa by IHC staining. Similar to a previous report,6 we found more extensive eosinophil infiltration in the eosinophilic CRSwNP than non-eosinophilic CRSwNP as evidenced by Eosinophil Cationic Protein (ECP) staining (Figure 2D). However, neutrophil infiltration was more pronounced in the non-eosinophilic group than in the eosinophilic group as shown by Myeloperoxidase (MPO) and Neutrophil Elastase (NE) staining (Figure 2D). Correspondingly, neutrophil but not eosinophil infiltration was also positively correlated with necroptosis (Figure 2E). Collectively, these results suggest necroptosis may promote the expression and release of inflammatory cytokines and chemokines and entailed neutrophil infiltration in CRSwNP.
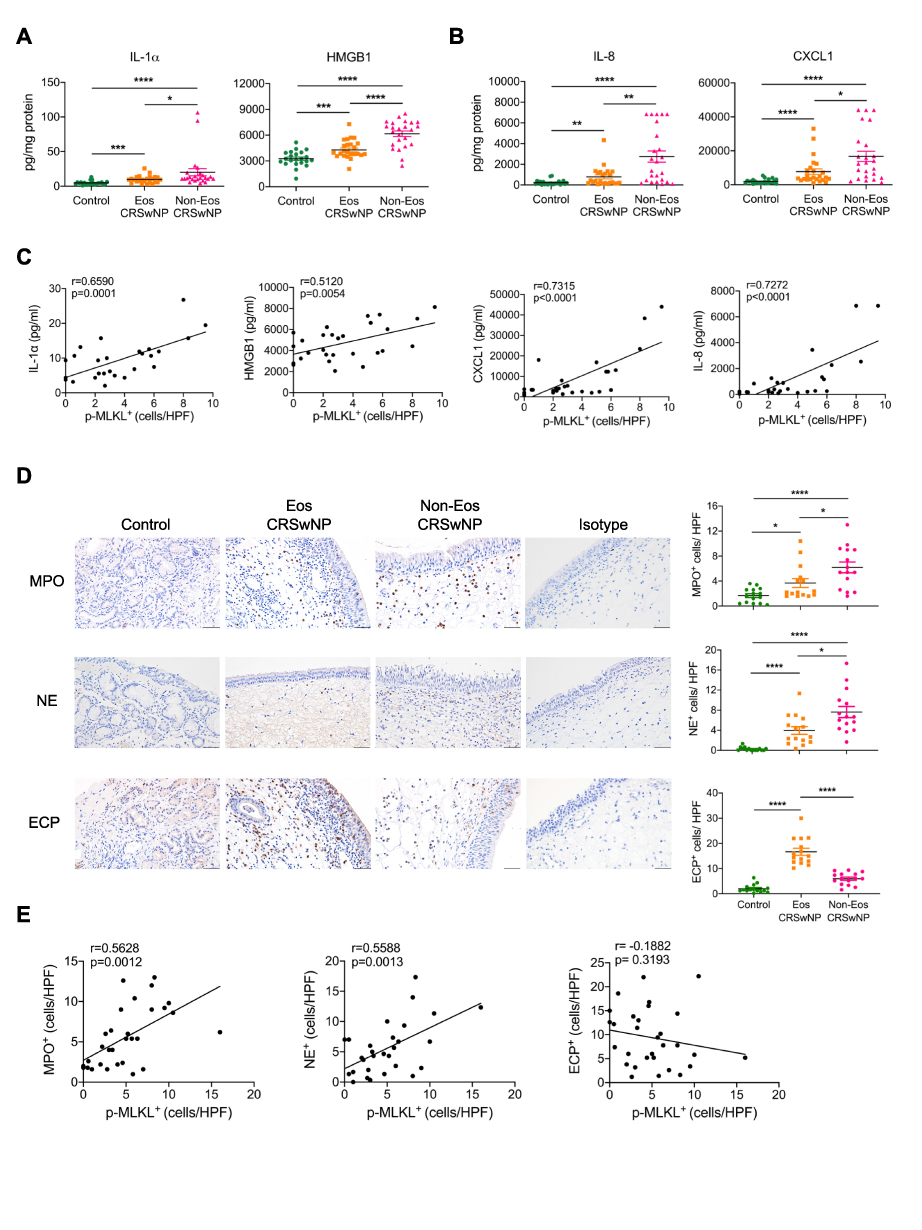 |
Figure 2 Necroptosis is correlated with neutrophilic inflammation in CRSwNP. (A and B) The protein levels of IL-8 and CXCL1 in tissue homogenates were detected by ELISA (n=22–24 per group). (C) Spearman correlations between IL-1α, HMGB1, IL-8, CXCL1 protein levels with p-MLKL positive cell numbers in control tissues and nasal polyps (n=28) (D) IHC staining of Myeloperoxidase (MPO), Neutrophil Elastase (NE) and Eosinophil Cationic Protein (ECP) in control tissues and nasal polyps. The positive cells were counted over high power field (400×) (n=15 per group). Scale bars: 50 μm. (E) Spearman correlations between MPO, NE or ECP positive cell numbers with p-MLKL positive cell numbers and in control tissues and nasal polyps (n=30). The data were shown as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; by Mann–Whitney U 2-tailed test. |
DAMPs Facilitate Inflammation in CRSwNP
The data presented above imply a possible link between necroptosis and neutrophilic inflammation in CRSwNP, we then explored the underlying mechanisms. It’s generally believed DAMPs released by necrotic cells are potent triggers for inflammatory response through engaging pattern recognition receptors (PRRs). For example, IL-1α and HMGB1 can initiate NF-κB signaling through IL-1R and TLR-4 signaling respectively, while ATP can trigger NLRP3 inflammasome activation.23 As IL-1α and HMGB1 expressions were increased in CRSwNP, we hypothesized that necroptosis may promote inflammation through DAMPs. To test this notion, we treated primary nasal polyp cells with DAMPs including IL-1α, ATP, HMGB1 and checked their effects on inflammatory cytokines expression. As shown in Figure 3A, chemokines like IL-8, CXCL1were pronouncedly induced by IL-1α and ATP. In addition, cytokines that mediate Th17 development and activation like IL-1β, IL-6 and IL-23 also showed a significant elevation after IL-1α and ATP treatment (Figure 3B). Similar to IL-1α and ATP, HMGB1 also induced the expression of these inflammatory genes but to a lesser extent (Figure 3A and B). In contrast, cytokines produced by T helper cells like IFN-γ, IL-4, IL-13 and IL-17A were not affected by DAMPs stimulation (Figure S1C). Both the DNPCs from eosinophilic and non-eosinophilic CRSwNP showed a similar response to stimulation with DAMPs (Figure 3A and B), thus DAMPs may promote inflammation in both groups. In line with the effects of DAMPs on inflammatory genes expression, we found IL-1α protein levels were positively correlated with IL-8 and CXCL1 protein levels in CRSwNP tissues (Figure 3C). Therefore, we established an interesting link between DAMPs release and inflammatory cytokines and chemokines expression in CRSwNP.
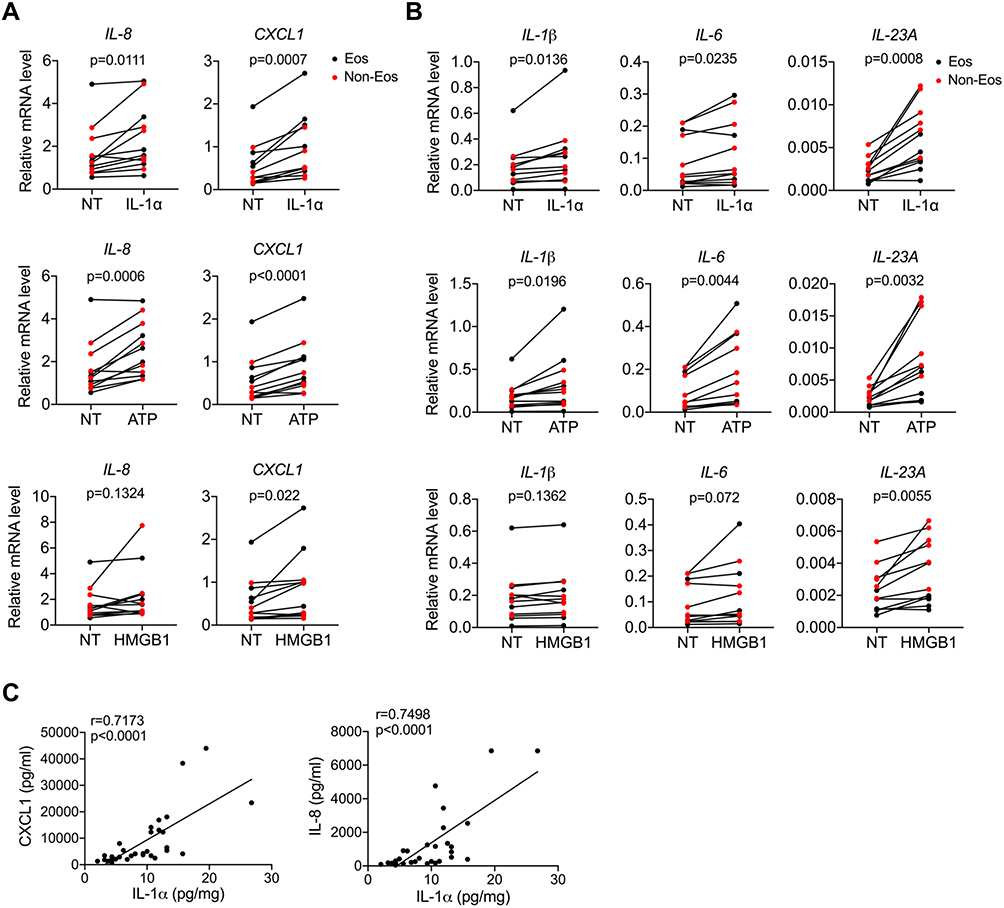 |
Figure 3 DAMPs facilitate inflammation in CRSwNP. (A and B) qPCR analysis of inflammatory cytokines and chemokines mRNAs (normalized to beta-actin) in primary nasal polyp cells before and after IL-1α (50 ng/mL), ATP (1 mM) or HMGB1 (1 μg/mL) stimulation for 8 h (n=12). (C) Spearman correlations between IL-1α and IL-8, CXCL1 protein levels in control tissues and nasal polyps (n=33). The data were analyzed by paired Student’s t-test (A and B). |
High Expression of p-MLKL in Macrophages in CRSwNP
The data presented in Figure 1D demonstrated that necroptosis signaling was activated in the subepithelial immune cells within the nasal polyps. We further dissected which immune cell type underwent necroptosis in CRSwNP tissues by immunofluorescence (IF) staining of p-MLKL along with specific immune cell markers. As presented in Figure 4A and B, macrophages (CD68+) accounted for about 70% of the total numbers of p-MLKL positive cells detected in the nasal submucosal area, whereas only a low percentage (about 10%) of p-MLKL positive cells were neutrophils (MPO+) or dendritic cells (CD11c+). On the contrary, other immune cells, such as mast cells (Tryptase+), B cells (CD20+) and T cells (CD3+) which were reported to be highly enriched in CRSwNP’s nasal mucosae,30 did not show a notable p-MLKL staining (Figure 4A). The percentage of p-MLKL positive cells among CD68 positive macrophages was about 25%, with no significant difference between eosinophilic and non-eosinophilic CRSwNPs (Figure 4C). Similar to the previous reports,31 we found markedly increased macrophage infiltration in CRSwNP tissues compared to control tissues (Figure 4D). Together, these results demonstrated massive macrophage infiltration and necroptosis were associated with CRSwNP, and suggested a possible role of macrophage necroptosis in CRSwNP.
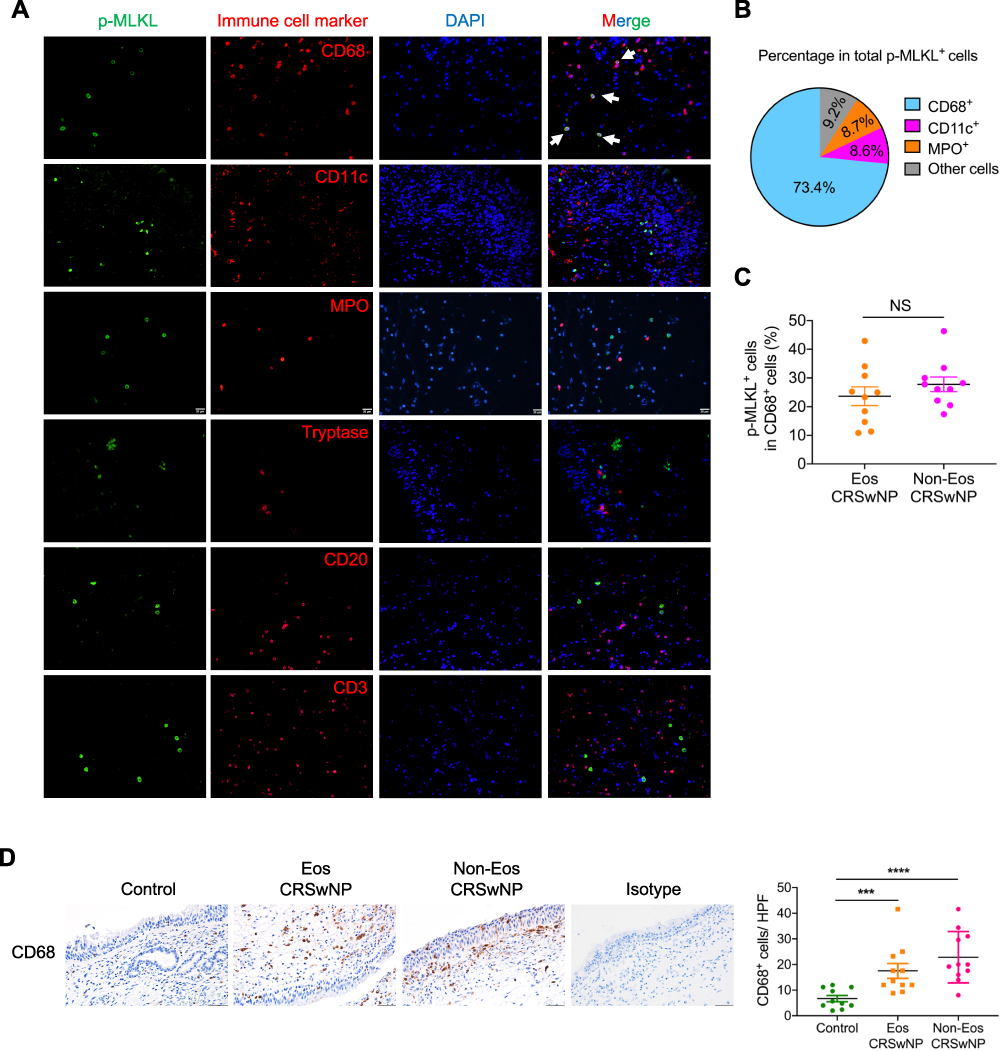 |
Figure 4 Prominent p-MLKL expression in macrophages in CRSwNP. (A) IF staining of p-MLKL (green) with specific immune cell markers (red) in nasal polyp sections. Arrows indicate double positive cells. The sections were counterstained with DAPI (blue). Scale bars: 20 μm. (B) The mean percentages of different cell types expressing p-MLKL in total p-MLKL+ cells in nasal polyps were shown (n=11). (C) The mean percentages of p-MLKL+ cells in total CD68+ cells were calculated in nasal polyps (n=10). (D) IHC staining of CD68 in control tissues and nasal polyps. The positive cells were counted over high power field (400×) (n=10–11 per group). Scale bars: 50 μm. The data were shown as mean±SEM. ***p<0.001, ****p<0.0001; by Mann–Whitney U 2-tailed test. Abbreviation: NS, not significant. |
TNF-α and IFN-γ Promote mTOR Activation and Necroptosis in Macrophages
Next, we sought to explore the possible triggers for macrophage necroptosis in CRSwNPs. We first checked the presence of legitimate necroptosis inducers of TNF and IFN family such as TNF-α, FasL, TRAIL, IFN-β and IFN-γ in control tissues and CRSwNPs. We found TNF-α and FasL protein levels were significantly higher in the non-eosinophilic nasal polyps compared to the eosinophilic and control tissues, whereas TRAIL expression was similar among the three groups (Figure 5A). IFN-γ protein levels were notably higher in both eosinophilic and non-eosinophilic CRSwNPs than in control tissues, while IFN-β expression was elevated only in the eosinophilic nasal polyps (Figure 5A). Therefore, we focused on TNF-α FasL and IFN-γ next since their expression was all increased in the non-eosinophilic CRSwNPs, which also had the highest necroptotic cell numbers (Figure 1D). Recently, a study reported TNF-α and IFN-γ can induce macrophage cell death accompanied by MLKL activation.32 We then set out to test whether they could also induce cell death in the human monocyte/macrophage cell-line THP-1 cells. Though TNF-α or IFN-γ alone failed to induce cell death, they acted together to induce pronounced cell necrosis in a time-dependent manner, as indicated by the drastic decrease in intracellular ATP and potent release of extracellular LDH (Figure 5B). Notably, the TNF-α- and IFN-γ-induced cell necrosis could not be rescued by caspase-1 inhibitor YVAD, caspase-3 inhibitor DEVD or pan-caspase inhibitor z-VAD, excluding a possible role for apoptosis or pyroptosis. However, the RIPK3 inhibitor GSK’872 and MLKL inhibitor necrosulfonamide (NSA) significantly attenuated THP-1 cell death (Figure 5C), suggesting an important role of RIPK3 and MLKL in TNF-α/IFN-γ induced cell necrosis. Besides, TNF-α and IFN-γ induced potent phosphorylation of RIPK3 and MLKL, hallmarks of necroptosis signaling activation (Figure 5D).
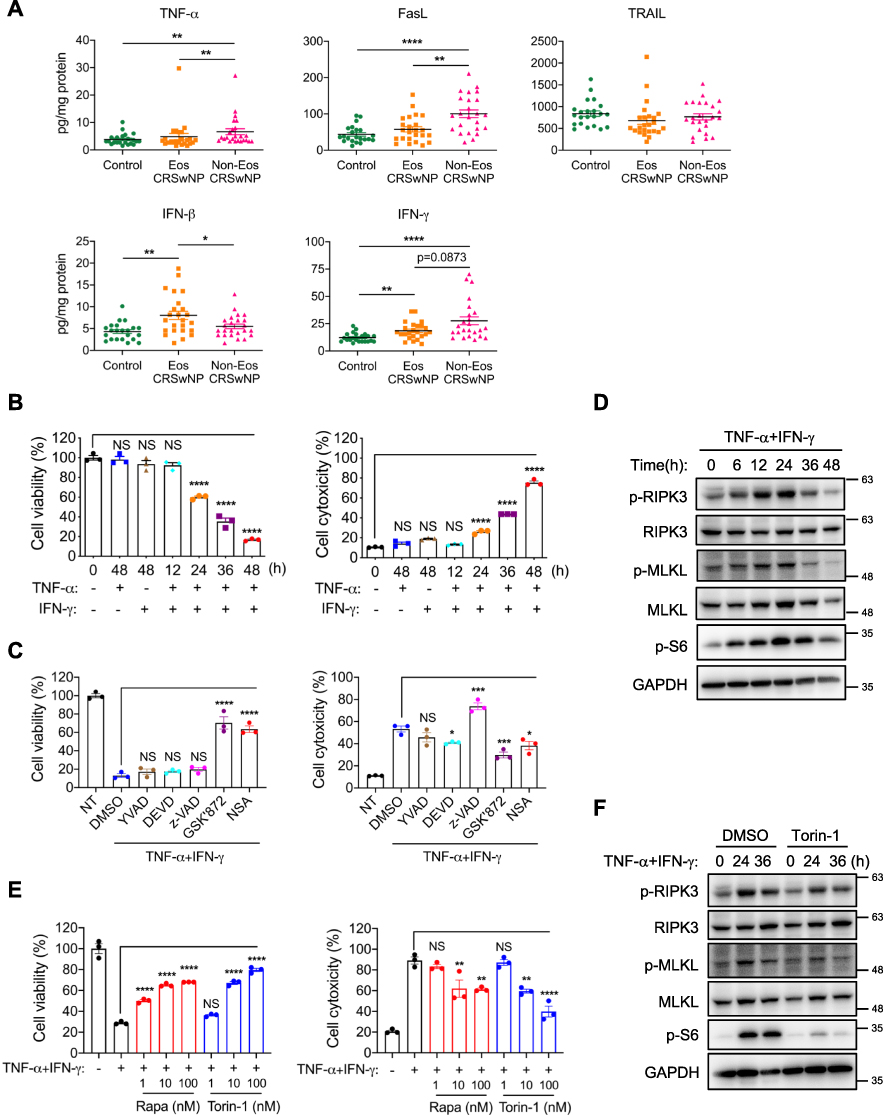 |
Figure 5 TNF-α and IFN-γ promote mTOR activation and necroptosis in macrophages. (A) The protein levels of TNF-α FasL, TRAIL, IFN-β and IFN-γ in tissue homogenates were detected by ELISA (n=22–24 per group). (B) THP-1 cells were treated with TNF-α (50 ng/mL) and IFN-γ (50 ng/mL) alone or together for indicated times. Cell viability and cytotoxicity were measured by intracellular ATP content or lactate dehydrogenase (LDH) release respectively. (C) THP-1 cells were treated with TNF-α (50 ng/mL) plus IFN-γ (50 ng/mL) in the presence or absence of Ac-YVAD-CMK (YVAD, 40 μM), z-DEVD-FMK (DEVD, 40 μM), z-VAD (40 μM), GSK’872 (5 μM) or NSA (1 μM) for 48 h. Cell viability and cytotoxicity were measured as in (B). (D) Immunoblots of cell lysates from THP-1 cells treated with TNF-α (50 ng/mL) plus IFN-γ (50 ng/mL) for indicated times. (E) THP-1 cells were treated with TNF-α (50 ng/mL) plus IFN-γ (50 ng/mL) with or without various doses of mTOR inhibitors rapamycin (rapa) or torin-1 for 48 h. Cell viability and cytotoxicity were measured as in (B). (F) Immunoblots of cell lysates from THP-1 cells treated with TNF-α (50 ng/mL) plus IFN-γ (50 ng/mL) with or without torin-1 (100 nM) for indicated times. The data were representative of 3 independent experiments (B–F) and shown as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; by Mann–Whitney U 2-tailed test (A) or one-way ANOVA (B, C and E). Abbreviation: NS, not significant. |
Our previous studies reported mTOR signaling can promote necroptosis in intestinal epithelial cells and murine macrophages upon induction by PAMPs or fungal pathogen.18,33 We wondered whether mTOR also played a role in TNF-α and IFN-γ mediated necroptosis. Intriguingly, mTOR was greatly activated upon TNF-α plus IFN-γ treatment as evidenced by increased phosphorylation of the mTOR substrate S6 (Figure 5D). Of note, the mTOR-specific inhibitors rapamycin and torin-1 remarkably suppressed THP-1 cell death in a dose-dependent manner (Figure 5E). Torin-1 also decreased RIPK3 and MLKL phosphorylation (Figure 5F), further supporting the crucial role of mTOR in promoting necroptosis.
The impaired barrier in CRSwNP often causes subepithelial translocation of nasal commensals, which can be recognized by toll-like receptors (TLRs), and previous studies have suggested that TLRs can promote macrophage necroptosis when caspases are inhibited.17,34 We then tested whether TLRs can also induce necroptosis in human macrophages. We found that TLR ligands alone could not induce THP-1 cell death (data not shown). However, the TLR2 ligand pam3CSK4 and the TLR5 ligand flagellin can induce robust cell death in combination with IFN-γ but not TNF-α (Figure S2A and B). Additionally, in the presence of IFN-γ pam3CSK4 and flagellin also triggered mTOR activation, and the phosphorylation of RIPK3 and MLKL (Figure S2C). Taken together, these results demonstrated TNF-α and IFN-γ were potent triggers for macrophages necroptosis either alone or in combination with PAMPs.
Discussion
Neutrophil infiltration is critically involved in the pathogenesis of both eosinophilic and non-eosinophilic CRSwNP,5,6 but the underlying mechanisms are poorly understood. In this study, we found that necroptosis was selectively activated in CRSwNP, a phenomenon possibly triggered by the presence of proinflammatory cytokines like TNF-α, IFN-γ and nasal commensal. In addition, the mTOR pathway positively regulated TNF-α and IFN-γ-induced necroptosis. Our data further revealed that necroptosis-associated DAMPs such as ATP and IL-1α can boost inflammatory cytokines and chemokines expression to mediate neutrophil recruitment to the CRSwNP tissues (Figure 6). Collectively, we established an interesting link between necroptosis, DAMPs release and neutrophilic inflammation in CRSwNP.
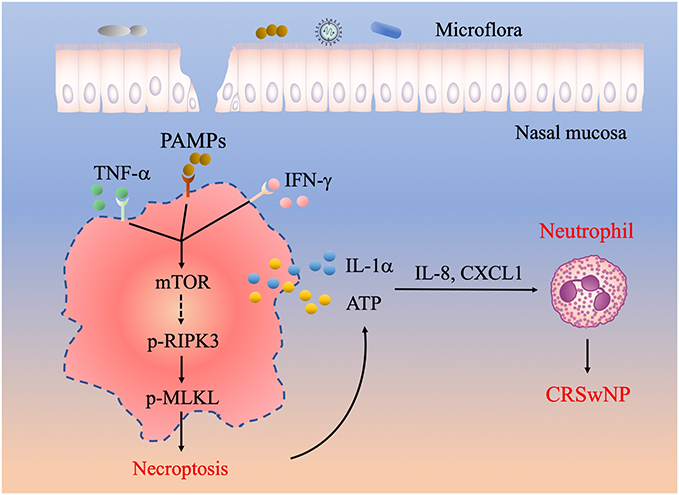 |
Figure 6 A working model illustrating the potential role of necroptosis in the pathogenesis of CRSwNP. The presence of TNF-α, IFN-γ and PAMPs in the nasal mucosa can cause RIPK3/MLKL dependent necroptosis and cell membrane rupture, which leads to the release of DAMPs such as IL-1α and ATP. DAMPs further stimulate the expression of proinflammatory cytokines and chemokines including IL-8 and CXCL1 to recruit neutrophils and exacerbate inflammation in CRSwNP. |
Necroptosis participates in multiple human inflammatory diseases such as inflammatory bowel diseases (IBD), systemic inflammatory response syndrome (SIRS) and atherosclerosis.14,15 It is not surprising that necroptosis is also activated in chronic rhinosinusitis since the nasal mucosa closely resembles that of the intestine. Nevertheless, we did not observe obvious necroptosis of nasal epithelial cells in CRSwNP, which frequently takes place in IBD to cause epithelial barrier dysfunction.22,33 This is likely due to low expression of MLKL in nasal epithelial cells as evidenced by our IHC data. On the contrary, macrophages appear to be the major cells with prominent MLKL phosphorylation. Given the fact macrophages express a broad range of PRRs, it is conceivable that macrophage might be prone to cell death under the circumstances like inflammatory and infectious conditions. However, it is also possible that macrophages may actively take up necroptotic cells and thus exhibit strong MLKL phosphorylation. In addition to necroptosis, other types of cell death like apoptosis or NETosis were also reported in CRSwNP. While our data revealed no significant increase in apoptosis, pyroptosis was elevated in the non-eosinophilic CRSwNP. In line with this phenomenon, a recent study found IL-1β protein level was upregulated in neutrophilic CRSwNP.35 Since pyroptosis can be mediated by other gasdermin members,36 the role of pyroptosis in CRSwNP warrants further investigation.
While necroptosis can be initiated by inflammatory cytokines and pathogens in vitro, the physiological or pathological triggers for necroptosis in vivo remain undefined, especially when Caspase-8 activity is intact. Our data showed RIPK3 and MLKL protein levels were increased notably in CRSwNP, while the expression of FADD was significantly decreased, which may explain why necroptosis was actively induced in this scenario. A recent study reported TNF-α and IFN-γ can trigger a mixed type of cell death termed PANoptosis in macrophages, in which case the caspase, gasdermin and MLKL were all activated.32 Here, we found TNF-α plus IFN-γ can promote RIPK3 and MLKL phosphorylation in THP-1 cells. Importantly, both RIPK3 and MLKL inhibitors reduced TNF-α plus IFN-γ-induced cell death, suggesting an important role of necroptosis in this case. Of note, PAMPs also induced remarkable cell death in combination with TNF-α or IFN-γ. It is possible that both the inflammatory cytokines and the nasal commensals work together to induce macrophage necroptosis in CRSwNP. Our recent studies revealed mTOR hyperactivation sensitized cells to necroptosis by promoting RIPK3 and MLKL expression in both intestinal epithelial cells and murine macrophages,18,33 here we uncovered a positive role of mTOR in facilitating TNF-α and IFN-γ-induced cell death without affecting RIPK3 and MLKL protein abundance. However, the detailed mechanisms of how mTOR regulates this process need to be further elucidated.
Although CRSwNP is traditionally regarded as a Th2-skewed and eosinophilic disease, recent studies5,6 and our data showed that neutrophils were also abundantly present in both eosinophilic and non-eosinophilic CRSwNP. Neutrophils are recruited by the chemokines including CXCL1, CXCL2 and IL-8, which can be induced by PAMPs or IL-17 family members. Consistent with the previous studies,5,6 we found IL-8 and CXCL1 protein levels were elevated in CRSwNP. Previous studies have found ATP or IL-1α can boost Th17 differentiation through inducing inflammasome activation or IL-6 and IL-23 expression.37,38 In addition, extracellular ATP and IL-1R signaling can mediate neutrophils recruitment to the lung or brain by inducing neutrophil chemoattractants.39,40 However, it remains unclear whether necroptosis and the released DAMPs are involved in neutrophil recruitment in CRSwNP. Here, we found that ATP and IL-1α could directly stimulate IL-8 and CXCL1 transcription in primary nasal polyp cells. Moreover, ATP and IL-1α upregulated IL-1β, IL-6 and IL-23 expression, which may further promote Th17 differentiation and function in CRSwNP. As Th17 cells play an important role in neutrophil recruitment via producing IL-17, DAMPs may regulate neutrophil infiltration through both mechanisms. Indeed, necroptosis and IL-1α were positively correlated with IL-8 and CXCL-1, as well as neutrophil infiltration in CRSwNP. However, the major cell types that respond to DAMPs and the relative contributions of DAMPs to proinflammatory cytokines and chemokines expression in CRSwNP still remain unclear.
There are several limitations to this study. First, we only used immunoblotting and IHC staining to check for necroptosis in nasal polyps, and electron microscopy will be decisive to verify necrotic cell death in CRSwNP. Second, we tested the effect of TNF-α/IFN-γ on necroptosis primarily in cell culture system; however, necroptosis in nasal polyps may be affected by other cytokines in the tissue microenvironment as well. Third, while we observed a positive correlation between necroptosis and neutrophilic inflammation in CRSwNP, the causative role of necroptosis in neutrophil infiltration needs to be validated by animal models or more specific settings.
In summary, we identified the presence of TNF-α/IFN-γ-induced necroptosis and the release of DAMPs in CRSwNP, which may further promote inflammatory cytokines and chemokines production and neutrophil recruitment. We propose the necroptosis-inflammation as a vicious loop feed-off of each other in the pathogenesis of CRSwNP. Thus, our findings also offer new targets for the treatment of CRSwNP.
Abbreviations
CRSwNP, chronic rhinosinusitis with nasal polyps; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; DNPCs, dispersed nasal polyp cells; TLRs, toll-like receptors; PRRs, pathogen recognition receptors; EETs, eosinophilic extracellular traps; NETs, neutrophil extracellular traps; IBD, inflammatory bowel diseases; SIRS, systemic inflammatory response syndrome; S. aureus, Staphylococcus aureus.
Acknowledgments
We are very grateful to all the clinicians and researchers involved in the study of CRSwNP.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval for the version to be published; and agreed to be accountable for all aspects of the work.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant numbers 81725004, 81720108019), and the Shanghai Municipal Science and Technology Major Project (2019SHZDZX02).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol. 2017;12:331–357. doi:10.1146/annurev-pathol-052016-100401
2. Tomassen P, Vandeplas G, Van Zele T, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. 2016;137(5):1449–1456. doi:10.1016/j.jaci.2015.12.1324
3. Bachert C, Marple B, Hosemann W, Cavaliere C, Wen W, Zhang N. Endotypes of chronic rhinosinusitis with nasal polyps: pathology and possible therapeutic implications. J Allergy Clin Immunol Pract. 2020;8(5):1514–1519. doi:10.1016/j.jaip.2020.03.007
4. Zhang Y, Gevaert E, Lou H, et al. Chronic rhinosinusitis in Asia. J Allergy Clin Immunol. 2017;140(5):1230–1239. doi:10.1016/j.jaci.2017.09.009
5. Delemarre T, Holtappels G, De Ruyck N, et al. A substantial neutrophilic inflammation as regular part of severe type 2 chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2021;147(1):179–188. doi:10.1016/j.jaci.2020.08.036
6. Wang H, Li ZY, Jiang WX, et al. The activation and function of IL-36 gamma in neutrophilic inflammation in chronic rhinosinusitis. J Allergy Clin Immunol. 2018;141(5):1646–1658. doi:10.1016/j.jaci.2017.12.972
7. Pothoven KL, Norton JE, Suh LA, et al. Neutrophils are a major source of the epithelial barrier disrupting cytokine oncostatin M in patients with mucosal airways disease. J Allergy Clin Immunol. 2017;139(6):1966–1978. doi:10.1016/j.jaci.2016.10.039
8. Wen W, Liu W, Zhang L, et al. Increased neutrophilia in nasal polyps reduces the response to oral corticosteroid therapy. J Allergy Clin Immunol. 2012;129(6):1522–1528. doi:10.1016/j.jaci.2012.01.079
9. Kolb JP, Oguin TH, Oberst A, Martinez J. Programmed cell death and inflammation: winter is coming. Trends Immunol. 2017;38(10):705–718. doi:10.1016/j.it.2017.06.009
10. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50(6):1352–1364. doi:10.1016/j.immuni.2019.05.020
11. Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem. 2016;85:743–763. doi:10.1146/annurev-biochem-060815-014830
12. He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
13. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–147. doi:10.1038/nri.2017.105
14. Kim EH, Wong SW, Martinez J. Programmed necrosis and disease: we interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 2019;26(1):25–40. doi:10.1038/s41418-018-0179-3
15. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4(15):e128834. doi:10.1172/jci.insight.128834
16. Zhang T, Yin C, Boyd DF, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180(6):1115–1129. doi:10.1016/j.cell.2020.02.050
17. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–20059. doi:10.1073/pnas.1116302108
18. Li T, Xie Y, Shi L, et al. TSC1 suppresses macrophage necroptosis for the control of infection by fungal pathogen Candida albicans. Immunohorizons. 2021;5(2):90–101. doi:10.4049/immunohorizons.2000093
19. Kitur K, Parker D, Nieto P, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015;11(4):e1004820. doi:10.1371/journal.ppat.1004820
20. Welz PS, Wullaert A, Vlantis K, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477(7364):330–334. doi:10.1038/nature10273
21. Bonnet MC, Preukschat D, Welz PS, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35(4):572–582. doi:10.1016/j.immuni.2011.08.014
22. Gunther C, Martini E, Wittkopf N, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477(7364):335–339. doi:10.1038/nature10400
23. Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and consequences of necroinflammation. Physiol Rev. 2018;98(2):727–780. doi:10.1152/physrev.00041.2016
24. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–223. doi:10.1016/j.immuni.2013.02.003
25. Wang BF, Cao PP, Wang ZC, et al. Interferon-gamma-induced insufficient autophagy contributes to p62-dependent apoptosis of epithelial cells in chronic rhinosinusitis with nasal polyps. Allergy. 2017;72(9):1384–1397. doi:10.1111/all.13153
26. Soyka MB, Wawrzyniak P, Eiwegger T, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-gamma and IL-4. J Allergy Clin Immunol. 2012;130(5):1087–1096. doi:10.1016/j.jaci.2012.05.052
27. Cao Y, Chen F, Sun Y, et al. LL-37 promotes neutrophil extracellular trap formation in chronic rhinosinusitis with nasal polyps. Clin Exp Allergy. 2019;49(7):990–999. doi:10.1111/cea.13408
28. Chen K, Han M, Tang M, et al. Differential Hrd1 expression and B-cell accumulation in eosinophilic and non-eosinophilic chronic rhinosinusitis with nasal polyps. Allergy Asthma Immunol Res. 2018;10(6):698–715. doi:10.4168/aair.2018.10.6.698
29. Li M, Xie Y, Zhao K, et al. Endoplasmic reticulum stress exacerbates inflammation in chronic rhinosinusitis with nasal polyps via the transcription factor XBP1. Clin Immunol. 2021;223:108659. doi:10.1016/j.clim.2020.108659
30. Bachert C, Han JK, Wagenmann M, et al. EUFOREA expert board meeting on uncontrolled severe chronic rhinosinusitis with nasal polyps (CRSwNP) and biologics: definitions and management. J Allergy Clin Immunol. 2021;147(1):29–36. doi:10.1016/j.jaci.2020.11.013
31. Wang ZC, Yao Y, Wang N, et al. Deficiency in interleukin-10 production by M2 macrophages in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2018;8(11):1323–1333. doi:10.1002/alr.22218
32. Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184(1):149–168. doi:10.1016/j.cell.2020.11.025
33. Xie Y, Zhao Y, Shi L, et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J Clin Invest. 2020;130(4):2111–2128. doi:10.1172/JCI133264
34. Kaiser WJ, Sridharan H, Huang C, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–31279. doi:10.1074/jbc.M113.462341
35. Wei Y, Zhang J, Wu X, et al. Activated pyrin domain containing 3 (NLRP3) inflammasome in neutrophilic chronic rhinosinusitis with nasal polyps (CRSwNP). J Allergy Clin Immunol. 2020;145(3):1002–1005. doi:10.1016/j.jaci.2020.01.009
36. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–157. doi:10.1038/s41577-019-0228-2
37. Atarashi K, Nishimura J, Shima T, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–812. doi:10.1038/nature07240
38. Ketelut-Carneiro N, Souza COS, Benevides L, et al. Caspase-11-dependent IL-1alpha release boosts Th17 immunity against Paracoccidioides brasiliensis. PLoS Pathog. 2019;15(8):e1007990. doi:10.1371/journal.ppat.1007990
39. Shah D, Romero F, Stafstrom W, Duong M, Summer R. Extracellular ATP mediates the late phase of neutrophil recruitment to the lung in murine models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306(2):L152–61. doi:10.1152/ajplung.00229.2013
40. Liu X, Nemeth DP, McKim DB, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity. 2019;50(2):317–333. doi:10.1016/j.immuni.2018.12.012
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.