Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
NCOA4-Mediated Ferroptosis in Bronchial Epithelial Cells Promotes Macrophage M2 Polarization in COPD Emphysema
Authors Liu J ![]() , Zhang Z, Yang Y, Di T, Wu Y, Bian T
, Zhang Z, Yang Y, Di T, Wu Y, Bian T
Received 20 December 2021
Accepted for publication 7 March 2022
Published 30 March 2022 Volume 2022:17 Pages 667—681
DOI https://doi.org/10.2147/COPD.S354896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Zhang
Jiaxin Liu,1 Zixiao Zhang,1 Yue Yang,1 Tingting Di,2 Yan Wu,1 Tao Bian1
1Department of Respiratory Medicine, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, 214023, People’s Republic of China; 2Department of Respiratory Medicine, First People’s Hospital of Nantong, Nantong, Jiangsu, 226006, People’s Republic of China
Correspondence: Tao Bian; Yan Wu, Department of Respiratory Medicine, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu, 214023, People’s Republic of China, Email [email protected]; [email protected]
Background: Macrophage polarization plays an important role in the pathogenesis of COPD emphysema. Changes in macrophage polarization in COPD remain unclear, while polarization and ferroptosis are essential factors in its pathogenesis. Therefore, this study investigated the relationship between macrophage polarization and ferroptosis in COPD emphysema.
Methods: We measured macrophage polarization and the levels of matrix metalloproteinases (MMPs) in the lung tissues of COPD patients and cigarette smoke (CS)-exposed mice. Flow cytometry was used to determine macrophage (THP-M cell) polarization changes. Ferroptosis was examined by FerroOrange, Perls’ DAB, C11-BODIPY and 4-HNE staining. Nuclear receptor coactivator 4 (NCOA4) was measured in the lung tissues of COPD patients and CS-exposed mice by western blotting. A cell study was performed to confirm the regulatory effect of NCOA4 on macrophage polarization.
Results: Increased M2 macrophages and MMP9 and MMP12 levels were observed in COPD patients, CS-exposed mice and THP-M cells cocultured with CS extract (CSE)-treated human bronchial epithelial (HBE) cells. Increased NCOA4 levels and ferroptosis were confirmed in COPD. Treatment with NCOA4 siRNA and the ferroptosis inhibitor ferrostatin-1 revealed an association between ferroptosis and M2 macrophages. These findings support a role for NCOA4, which induces an increase in M2 macrophages, in the pathogenesis of COPD emphysema.
Conclusion: In our study, CS led to the dominance of the M2 phenotype in COPD. We identified NCOA4 as a regulator of M2 macrophages and emphysema by mediating ferroptosis, which offers a new direction for research into COPD diagnostics and treatment.
Keywords: NCOA4, ferroptosis, macrophage polarization, MMP9, MMP12, COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory respiratory disease. It is the third leading cause of death worldwide. COPD is characterized by the progressive limitation of airflow associated with an intense inflammatory reaction in the lung in response to harmful particles or gases.1 Cigarette smoke (CS) is the main risk factor for the development of COPD. Emphysema, which constitutes the major pathological change in COPD, can lead to lung parenchyma destruction and lung elasticity loss.2 Matrix metalloproteinases (MMPs) participate in the destruction of the alveolar extracellular matrix and promote the pathogenesis of emphysema, and the underlying mechanisms warrant further investigation.
Macrophages are innate immune cells that play a critical role in resolving inflammation and defending against exogenous pathogens, contributing to lung homeostasis.3,4 Macrophages are classified as classically activated (M1) or alternatively activated (M2).5–7 M1 macrophages are induced by interferon-γ (IFN-γ) and lipopolysaccharide (LPS), produce inducible nitric oxide synthase (iNOS) and tumor necrosis factor α (TNF-α), and exert proinflammatory and cytotoxic effects.8,9 M2 macrophages are stimulated by interleukin 4 (IL-4), express CD163 and CD206, secrete Arg1 and transforming growth factor-β (TGF-β) and are involved in immunoregulatory and tissue remodeling processes.5,10,11 Studies have shown that macrophage activation is closely related to COPD development. Total macrophage numbers increase significantly with smoking and COPD severity.12 Currently, there are different views on macrophage polarization in COPD. It has been reported that M1 macrophages dominate the sputum of patients with COPD.13 However, multiple research groups have reported alterations in the immunophenotypes of macrophages toward the M2 phenotype in blood and bronchoalveolar lavage (BAL) samples of COPD subjects.14 The study also confirmed an increased deposition of alveolar M2 macrophages in the mouse COPD model.15 Although macrophage polarization is involved in COPD emphysema, the direction and mechanisms of macrophage polarization are undefined. A better understanding of the mechanisms that regulate macrophage polarization is critical for improving COPD emphysema treatment.
Cell death is critical for normal development and homeostasis.16 Ferroptosis is a recently defined iron-dependent cell death that occurs as a consequence of lethal levels of lipid peroxidation.17,18 Enyong et al found that autophagy-dependent ferroptosis drives tumor-associated macrophage polarization.19,20 They also found that Gpx4 depletion or a high-iron diet increased ferroptosis, which promoted subsequent macrophage accumulation and activation.21 In addition, the ferroptosis driver SOCS1 and suppressor FTH1 served as independent prognostic factors that independently correlated with M1 and M2 macrophage infiltration in HNSCC, suggesting that inducing ferroptosis directly influences the infiltration of macrophages.22 Targeting ferroptosis immunomodulation may serve as a strategy to enhance the efficacy of immunotherapy. Yoshida’s research group has shown that ferroptosis is involved in the development of COPD. Meanwhile, nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy is a critical regulator of ferroptosis.23 Although NCOA4-mediated ferroptosis has been documented in COPD, reports regarding macrophage polarization regulation remain limited, and further investigation into these mechanisms is required.
Here, we hypothesized that NCOA4-mediated ferroptosis in human bronchial epithelial (HBE) cells would lead to M2 macrophage polarization, which is involved in the MMPs production and tissue destruction observed in COPD. Therefore, the aim of this study was to determine the changes in macrophages in COPD and the effect of NCOA4-mediated ferroptosis on macrophage polarization and MMPs production. It is critical to seek new therapeutic targets for COPD.
Materials and Methods
Cell Culture and Treatment
HBE cells, an SV40-transformed, normal HBE cell line, were obtained from Chi Scientific (Jiangsu, China). HBE cells were cultured in a humidified incubator containing 95% air and 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Biological Industries, Italy), 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific, USA).
THP-1 cells were purchased from the National Collection of Authenticated Cell Cultures and cultured in a humidified incubator containing 95% air and 5% CO2 at 37°C in RPMI 1640 (Gibco, USA) supplemented with 10% FBS (Biological Industries, Italy) and β-Mer (Invitrogen, USA). THP-1 cells were treated with 10 µM PMA for 48 h to form THP-1 macrophages (THP-M cells). THP-M cells were seeded in a six-well plate at a density of 1×106 cells per well. HBE cells were cocultured on 24 mm diameter inserts with 0.4 μm pores (#3412, Corning, USA). HBE cells were treated with 5% CS extract (CSE) with or without 10 µM ferrostatin-1 (Fer-1) (SML0583, Sigma–Aldrich, USA) for 48 h before they were cocultured with THP-M cells. HBE cells were transfected with NCOA4 siRNA and treated with 5% CSE for 48 h before they were cocultured with THP-M cells for 48 h.
Preparation of CSE
CSE was prepared as previously reported with some modifications.24 Briefly, the smoke of Da Qian Men (10 mg of tar and 0.8 mg of nicotine/cigarette, Shanghai, China) was bubbled through 10 mL of serum-free DMEM. The resulting suspension was adjusted to pH 7.4 and then filtered through a 0.22 μm pore filter (Merck Millipore, USA) to remove bacteria and large particles. The CSE was standardized by measuring the absorbance at 320 nm and was defined as 100% CSE. The CSE was diluted to the desired concentration with medium and used in experiments within 30 min.
Establishment of the Murine COPD Model
Male C57BL6J mice (6–8 weeks old) were purchased from Changzhou Kawensi Experimental Animal Co., Ltd. (China) and housed in animal facilities at Wuxi People’s Hospital, Jiangsu Province. The weights and numbers of mice are detailed in our previous article.25 The animals used in this study were treated humanely according to a protocol approved by Wuxi People’s Hospital, Jiangsu Province Institutional Animal Care and Use Committee, in compliance with the laws of Jiangsu Province on the administration of experimental animals. The mice were exposed to smoke from Da Qian Men (10 mg of tar and 0.8 of mg nicotine/cigarette, Shanghai, China) as reported previously. During each cigarette smoke exposure, ten cigarettes per hour for 2 h each session were used. In summary, the mice underwent whole-body exposure to CS in a tempered glass box for 2 h twice per day, 6 h apart, 7 days a week for a total of 24 weeks. Age-matched mice were housed in a similar environment without exposure to CS and served as controls.
Lung Function Measurement
The lung function of the mice was measured at the Jiangsu Provincial Center for Disease Control and Prevention using whole-body plethysmography (Buxco Electronics, Ltd., USA), as previously reported.25 Briefly, mice were placed unrestrained in a chamber connecting a sensitive pressure transducer to measure pressure changes inside the chamber. Enhanced pause (Penh) was recorded using FinePointe software (Buxco Electronics, Ltd., USA) when the mice were quiet. Penh generally reflects pulmonary resistance in all recorded Respiratory parameters. Values were averaged and expressed as absolute Penh values.
Western Blotting
Total lysates were prepared according to the manufacturer’s recommendations (Beyotime Institute of Biotechnology, Shanghai, China). Protein concentrations were measured with the BCA protein assay according to the manufacturer’s instructions (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts (20 μg-40 µg) of protein were separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA). The membranes were incubated overnight at 4°C with mouse anti-GAPDH (ab8245, Abcam), mouse anti-β-actin (66009-1-Ig, Proteintech), rabbit anti-NCOA4 (ab86707, Abcam), rabbit anti-ferritin (ab65080, Abcam), rabbit anti-Ptgs2 (#12282, CST), rabbit anti-MMP9 (ab38898, Abcam) and rabbit anti-MMP12 (22989-1-AP, Proteintech) antibodies. After several washing steps, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for one hour. Detection was performed with the Immobilon ECL system (Millipore, S.p.A., Italy). Three independent experiments were carried out. Densitometric analyses of the bands were performed with ImageJ software.
Transmission Electron Microscopy (TEM)
HBE cells were exposed to 5% CSE for 48 h, treated with trypsin-EDTA (Gibco, USA) and centrifuged at 1500 rpm for 10 min. After the supernatant was removed, the cell pellets were fixed in 1 mL of 2.5% glutaraldehyde at 4°C. The fixed HBE cells were then embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate. Photos were taken with a transmission electron microscope.
Iron Assay
The iron concentration was assessed using FerroOrange (F374, Dojindo) and Perls/DAB staining. The working fluid was prepared in advance according to the instructions. The cells were seeded in 35 mm dishes and cultured overnight at 37°C in a 5% CO2 incubator. Serum-free culture medium was added, and the cells were incubated overnight. Subsequently, the cells were cultured with 5% CSE for 48 h. Then, the supernatant was removed, and the cells were washed three times with phosphate-buffered saline (PBS). After 1 µmol/l working solution was added, the cells were cultured for 30 min and observed with a microscope.
Lung tissue sections were deparaffinized at 60°C for 2 h and washed in distilled water. Equal volumes of potassium ferrocyanide and hydrochloric acid solutions were mixed to make a working iron stain solution. The lung tissues were then incubated in the working solution for 60 min. The samples were viewed using light microscopy.
Measurement of Lipid Peroxidation
HBE cells were inoculated in a 6-well microplate and cultured overnight at 37°C in a 5% CO2 incubator. Lipid peroxidation was measured using a C11-BODIPY 581/591 probe (C10445, Invitrogen). After the HBE cells were exposed to 5% CSE for 48 h, the cells were incubated with C11-BODIPY 581/591 (10 μM) for 30 min. If lipid peroxidation occurred, the fluorescence of C11-BODIPY shifted from red (581/610 nm) to green (484/510 nm).
Flow Cytometry
After 48-h coculture, THP-M cells were washed with ice-cold PBS and removed from the dishes with trypsin-EDTA. To obtain the cell pellet, the cell suspension was transferred to a 5 mL sterile conical tube and centrifuged at 1000 × g for 5 min. The supernatant was completely removed by rapid decanting, and the cell pellet was immediately resuspended in 200 µL of PBS containing 10% FBS. The cells were blocked with Fc Block (564219, BD) to block or significantly reduce potential nonspecific antibody staining. The cells were incubated with fluorescence-conjugated antibodies against APC CD86 (564544, BD) and BB515 CD206 (550889, BD) in PBS for 30 min at 4°C. CD86 was used as a marker of M1 macrophages, and CD206 was used as a marker of M2 macrophages. All samples were analyzed using a flow cytometer. The data collected for each sample were analyzed using FlowJo software.
Immunofluorescence Analysis
Cells were fixed with a 4% paraformaldehyde solution for 30 min and washed with PBS 3 times after being permeabilized for 20 min. The cells were then blocked with PBS containing 5% goat serum for 1 h and incubated with primary antibodies (ferritin and NCOA4) overnight at 4°C. The cells were incubated with fluorescent secondary antibodies at room temperature for 1 h, and 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) was added for 5 min. Images were obtained with a fluorescence microscope.
Human lung tissue sections were incubated overnight with antibodies against CD68 (13395-1-AP, Proteintech), iNOS (ab3523-200 µL, Abcam) or CD206 (ab64693-100 µg, Abcam). After being washed with PBS, the sections were incubated with Alexa 594-labeled goat anti-mouse and Alexa 488-labeled goat anti-rabbit secondary antibodies for 1 h at 37°C, and then DAPI (Beyotime, Shanghai, China) was added for nuclear staining. Colocalization of CD68 and iNOS or CD206 was evaluated with confocal microscopy. The analysis of CD68+ cells (total macrophages), iNOS+CD68+ cells (M1 macrophages) and CD206+CD68+ cells (M2 macrophages) was performed using ImageJ software on randomly chosen fields [the average of 5 to 10 fields (× 200) in the section].
Immunohistochemistry and Hematoxylin and Eosin (H&E) Staining
The right middle lobe of the lung was fixed with 4% neutral paraformaldehyde for 24 h. Tissues were embedded in paraffin and sectioned into 4 µm sections. The sample slides were stained with H&E (Solarbio G1120, China) according to the manufacturer’s instructions to evaluate emphysema. After staining, the slides were examined under a light microscope at a photograph documentation facility (Olympus, Tokyo, Japan).
The lung sections were deparaffinized and rinsed at room temperature. Then, the lung tissues were incubated overnight at 4°C with anti-4-HNE antibodies (1:200 dilution, Abcam, USA). The samples were then washed extensively and incubated with the appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. After being washed, the samples were incubated with a 3.39-diaminobenzidine-4HCl (DAB; Sigma, Missouri, USA)-H2O2 solution to visualize immunolabeling. Some sections were then counterstained with H&E and mounted with a coverslip. The lung sections were observed under a microscope. The size of the alveolar space was examined by measuring the mean interalveolar septal wall distance (MLI) using Image-Pro Plus 6.0 software, as previously described.26 Briefly, the MLI was measured by dividing the length of the diagonals of 10 random fields of each lung tissue by the sum of intercepts counted within the diagonals at 100× magnification (the bronchial regions were not included).
RNA Extraction and Real-Time PCR
Total RNA was isolated using RNAiso Plus (9108/9109, Takara, Japan). For mRNA detection, total RNA (1 μg) was transcribed into cDNA using the Prime Script™ RT reagent kit with gDNA eraser (RR047A, Takara, Japan) according to the manufacturer’s recommendations. All primers were synthesized by Sangon Biotech (Shanghai, China). The primer sequences are listed in Table 1. RT–PCR was performed with TB Green™ Premix Ex Taq™ II (RR820A, Takara, Japan) and an ABI 9600 real-time PCR detection system (Applied Biosystems). GAPDH served as an internal control for mRNA. Fold changes in the expression levels of each gene were calculated by a comparative threshold cycle (Ct) method using the formula 2—(ΔΔCt). Three independent experiments were carried out.
 |
Table 1 Primer Sequences Used |
Human Samples Collection
All lung tissues were donated voluntarily. This study was conducted in accordance with the tenets of the Declaration of Helsinki, and written informed consent was obtained from all participants. Lung tissue from normal individuals and smokers was collected at Wuxi People’s Hospital during pulmonary surgery, and tissue from COPD patients was collected during lung transplantation. All COPD patients met the GOLD 2019 diagnosis criteria. The lung function and Medical history of each subject in the study are reported in Table 2. All lung tissues were frozen quickly in liquid nitrogen and stored at −80°C for later use.
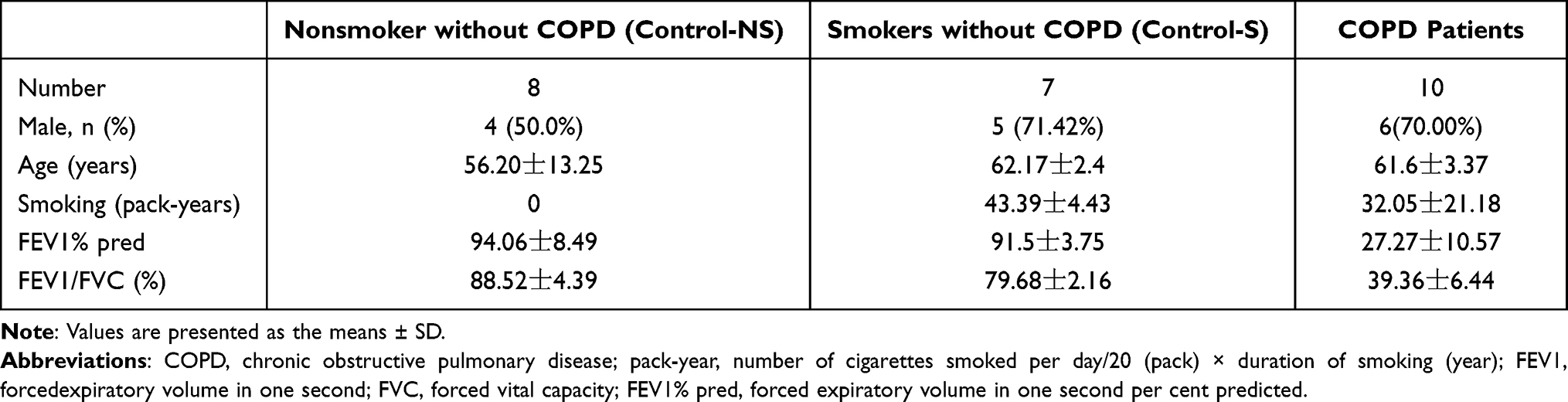 |
Table 2 Clinical Characteristics of the Subjects |
Cell Transfection
When HBE cells were grown to 70% confluency, NCOA4-siRNA or control-siRNA was transfected into cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, United States) according to the corresponding experimental groups. Then, the stable NCOA4 knockdown HBE cell line was established by using lentivirus transfection. Lentiviruses expressing si-NCOA4 or si-Con were purchased from Jikai Gene (Shanghai, China). After 48 h of transfection, the cells were collected and analyzed for NCOA4 expression using quantitative PCR (qPCR) and Western blotting. The siRNA sequences were as follows: NCOA4-siRNA 5’-CAGGAAGTATTACTTAATT-3’; scrambled control-siRNA 5’- TTCTCCGAACGTGTCACGT −3’.
Statistical Analysis
All relevant data are expressed as the mean ± SD of three independent experiments. Differences between the mean values of normally distributed data were analyzed using one-way ANOVA (Dunnett’s t test), two-tailed Student’s t test and the Kruskal–Wallis test. Statistical analyses were conducted using SPSS 20 software, and values of P < 0.05 were considered statistically significant (*P<0.05; **P < 0.01).
Results
M2 Macrophages Polarization is Increased in COPD
Table 2 shows the clinical characteristics of the subjects. The samples were divided into a nonsmoker group, a smoker group, and a COPD smoker group based on smoking history and diagnosis. According to the GOLD guidelines, COPD patients were diagnosed when dyspnea and a forced expiratory volume in one second/forced vital capacity (FEV1/FVC) ratio <70% was present. The number of total macrophages was increased in COPD patient samples (Figure 1A). M1 macrophages and M2 macrophages represent the extremes of macrophage polarization. Total macrophages are CD68+, M1 macrophages are iNOS+CD68+, and M2 macrophages are CD206+CD68+. Immunofluorescence analysis showed that the levels of M2 macrophages in COPD patients were higher than those in nonsmokers and smokers (Figure 1B and C). MMPs are one of the important causes of emphysema. MMP9 and MMP12 were upregulated in the peripheral lung tissue of COPD patients (Figure 1D and E).
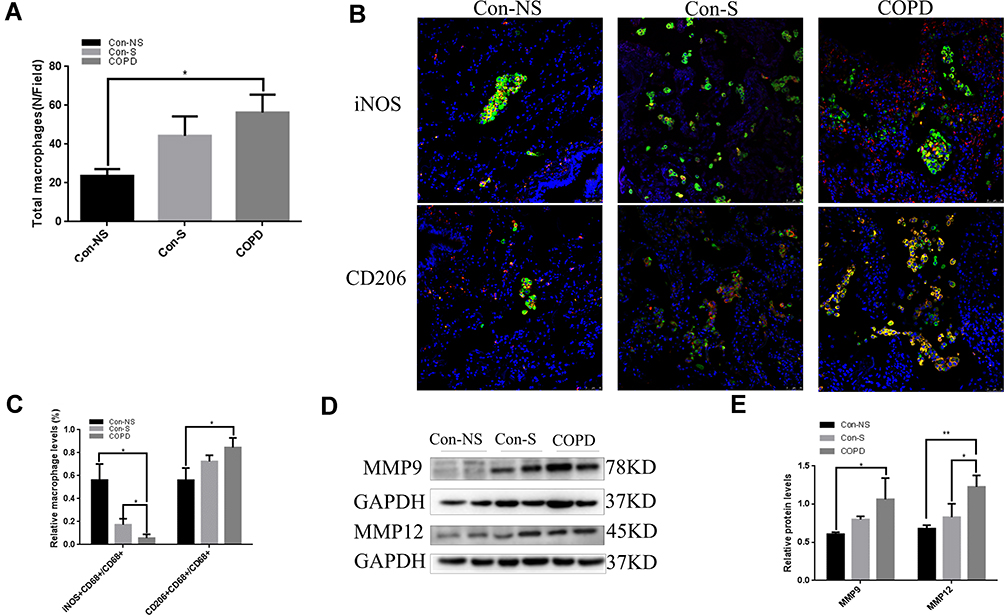 |
Figure 1 M2 macrophage numbers are increased in COPD patients. Con-NS, nonsmokers without COPD; Con-S, smokers without COPD; COPD, COPD patients. (A) The increased numbers of lung macrophages were assessed by immunofluorescence. (B) Immunofluorescence analysis of lung sections in the Con-NS (n=4), Con-S (n=5), and COPD (n=6) groups. Scale bars, 50 μm. CD68: green; iNOS: red; CD206: red. (C) Quantification of the ratio of M1 or M2 macrophages to the total number of macrophages. Total macrophages: CD68+. M1: CD68+ and iNOS+. M2: CD68+ and CD206+. (D) Western blots. (E) Relative protein levels of MMP9 and MMP12 in the lung were determined. The data are the mean ± SD (n=3). *P<0.05; **P < 0.01. |
CS Promotes M2 Macrophage Polarization in Mouse Models
After 24 weeks of exposure to CS, the mice exhibited airflow limitations and emphysema. Penh was increased in CS-exposed mice compared with that in air-exposed mice (Figure 2A). H&E staining showed that the mean linear intercept (MLI) increased after 24 weeks of exposure in the CS group (Figure 2B and C). Furthermore, Western blot analysis revealed that MMP9 and MMP12 levels were higher in CS-exposed mice than in the controls (Figure 2D and E). qRT–PCR showed that the levels of the M2 macrophage markers Arg1 and Tgf-β1 were increased, but the levels of M1 macrophage markers iNOS and Tnf-α were decreased after the mice were exposed to CS (Figure 2F).
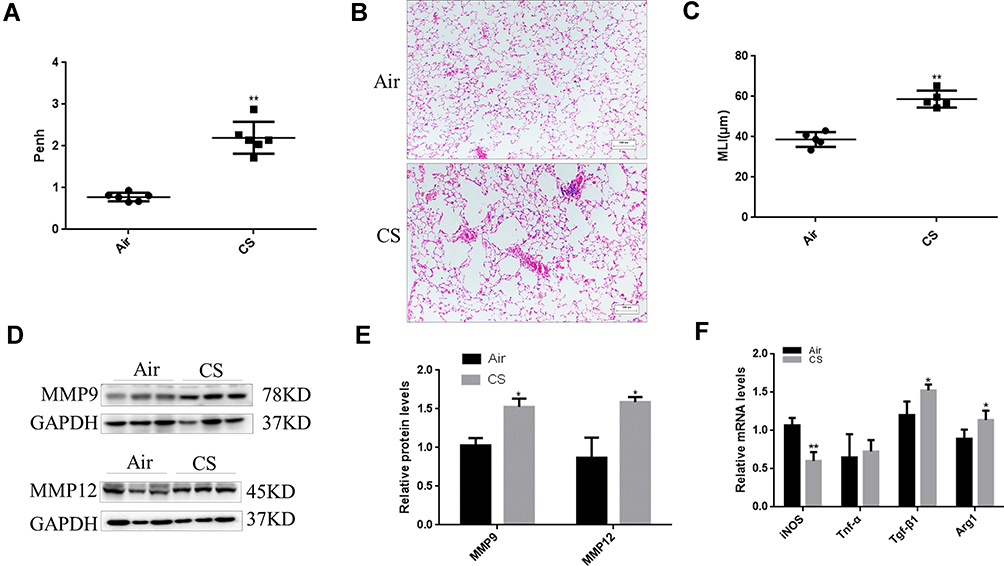 |
Figure 2 CS-exposed mice. Male C57BL/6J mice at 6–8 weeks of age were exposed to CS for 24 weeks. (A) Pulmonary function is shown as Penh in air-exposed mice and CS-exposed mice. (B) Emphysema was assessed by H&E staining of lung sections. Scale bars, 100 μm. (C) Quantification of the MLI of the mice. (D) Western blotting was performed, and (E) the relative protein levels of MMP9 and MMP12 in the lung were determined. (F) The mRNA levels of M2 macrophage markers (Arg1 and Tgf-β1) and M1 macrophage markers (iNOS and Tnf-α) in lung tissues of mice were measured by quantitative RT–PCR. The data are the mean ± SD (n=6). *P<0.05; **P < 0.01. |
The Ratio of M2/M1 Phenotype is Increased in THP-M Cells Cocultured with CHBE Cells
To determine whether macrophages are an important factor for COPD emphysema, THP-M cells were differentiated from THP-1 monocytes and then cocultured with control or 5% CSE-treated HBE cells for 48 h (Figure 3A and B). MMP9 and MMP12 were upregulated in THP-M cells cocultured with CSE-treated HBE cells (Figure 3C and D). The extracellular ferroptosis inhibitor Fer-1 could modulate the COPD phenotype in vitro. Fer-1 decreased the expression of MMP9 and MMP12 in THP-M cells (Figure 3C and D). Additionally, to examine macrophage polarization, we assessed the expression of the phenotypic markers CD86 and CD206 by flow cytometry. The results showed that in THP-M cells cocultured with CHBE cells, the polarization of M2/M1 macrophages was increased more than that in HBE cells (Figure 3E and F).
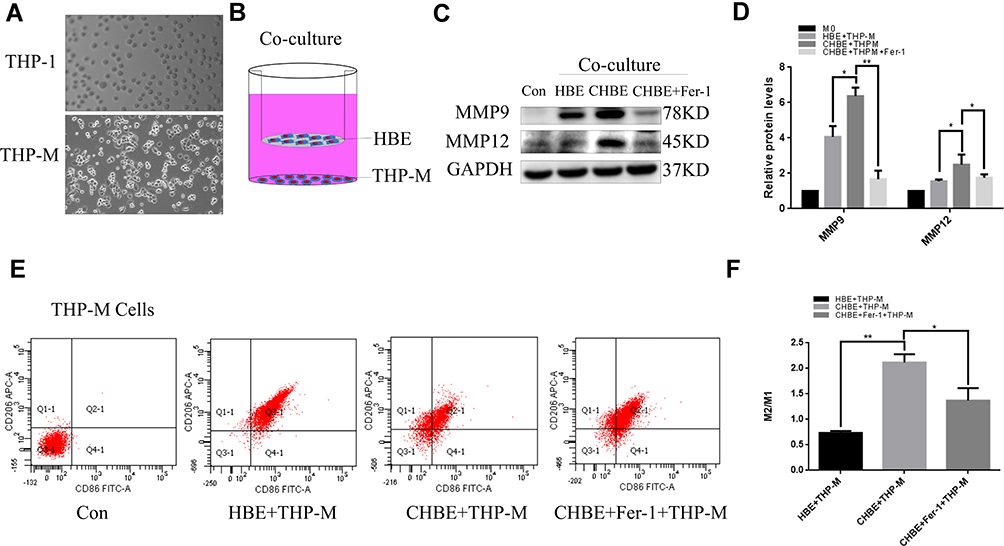 |
Figure 3 M2/M1 polarization was increased in THP-M cells cocultured with CHBE cells for 48 h. Con, normal THP-M cells; CHBE, HBE cells treated with 5% CSE for 48h; CHBE+Fer-1, HBE cells pretreated with Fer-1 for one hour and then treated with 5% CSE for 48h. THP-M cells were cocultured with HBE, CHBE and CHBE+Fer-1 cells for 48h. Band densities were quantified by ImageJ software. GAPDH levels were measured in parallel and served as controls. (A) Cell morphology shifted from THP-1 to THP-M. (B) Schematic graph illustrating the HBE cell and THP-M cell coculture model. Coculture assays were performed for 48 h with THP-M cells in the lower compartments of the Boyden chambers and HBE cells in the upper compartments. (C) and (D) THP-M cells were cocultured with HBE, CHBE, and CHBE+Fer-1 cells, and the levels of MMP9 and MMP12 were determined using Western blotting. (E) Representative histograms showing the flow cytometry results and (F) the ratios of M2/M1 macrophages. The data are the mean ± SD (n=3). *P<0.05; **P < 0.01. |
Ferroptosis is Involved in vivo and in vitro
As shown in Figure 4A and B, we showed that expression of Ptgs2, a marker of ferroptosis, was upregulated in human COPD lungs. Moreover, Perls’ DAB staining revealed that free iron was increased in human COPD lung sections compared with in those from nonsmokers or smokers (Supplementary Figure 1A). The accumulation of lipid peroxidation was shown by immunohistochemical (IHC) staining (Supplementary Figure 1B). After 24 weeks of exposure to CS, ferroptosis was also observed in mouse lungs, as indicated by increased Ptgs2 expression (Figure 4C and D). Perls’ DAB staining revealed that HBE cells from CS-exposed mice had higher levels of nonheme iron than those from air-exposed mice (Supplementary Figure 1C). IHC staining demonstrated that 4-HNE expression levels in the CS groups were higher than those in the control groups (Supplementary Figure 1D). Increased free iron and phospholipid peroxidation play important roles in ferroptosis.27 CCK-8 assays showed that the viability of HBE cells was decreased at 24 h and 48 h after 5% CSE exposure (Figure 4E). To elucidate the involvement of ferroptosis in CSE-mediated cell death, we measured the expression of Ptgs2, which increased in a time-dependent manner at 6 h, 12 h, 24 h, and 48 h after 5% CSE exposure (Figure 4F and G). In addition, free iron was examined by FerroOrange, and the results showed that iron increased in HBE cells after 48 h of 5% CSE treatment (Supplementary Figure 1E). CSE-induced lipid peroxidation was evaluated by C11-BODIPY staining. The red fluorescence partially shifted to green fluorescence (Supplementary Figure 1F). TEM revealed smaller mitochondria with increased membrane density and decreased membrane cristae (Figure 4H). These results confirmed the occurrence of ferroptosis in CHBE cells.
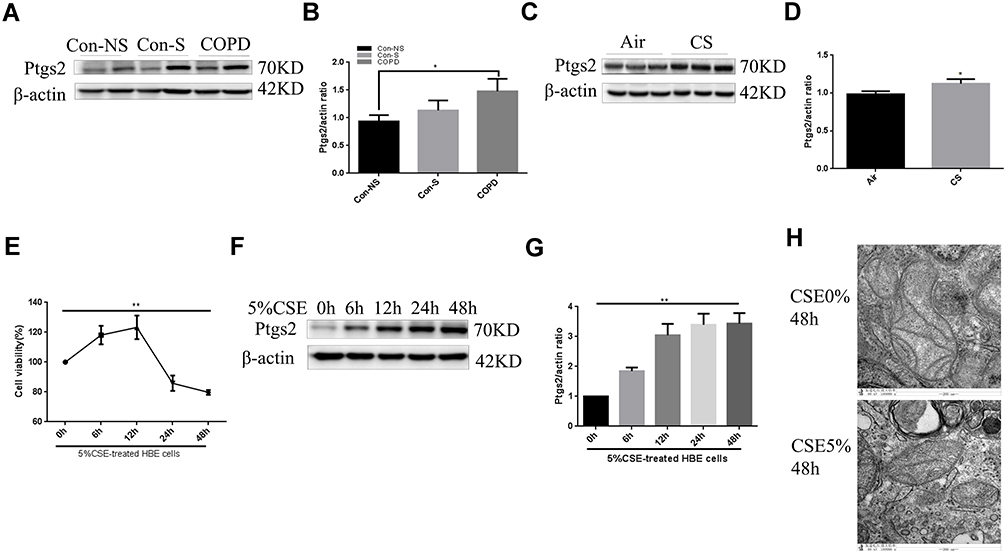 |
Figure 4 Ferroptosis occurs in COPD patients, CS-exposed mice and CHBE cells. (A) and (B) Relative expression of Ptgs2 in human lung tissues was measured by Western blotting. (C) and (D) Western blot showing the expression levels of Ptgs2 and β-actin in the lung homogenates of air-exposed and CS-exposed mice. (E) CCK-8 assays showed that the viability of CHBE cells decreased at 24 h and 48 h. The most severe cell death occurred after treatment with 5% CSE for 48 h. (F) and (G) The time course of Ptgs2 expression in response to 5% CSE was assessed by Western blotting. (H) Representative TEM images of HBE cells treated with 0% or 5% CSE for 48 h. Scale bar, 200 nm. The data are the mean ± SD. *P<0.05; **P < 0.01. |
NCOA4 is Increased in vivo and in vitro
To elucidate the potential involvement of NCOA4 in COPD pathogenesis, we evaluated NCOA4 expression levels in the lung tissue of nonsmokers, smokers and COPD patients. The levels of NCOA4 were higher in COPD lungs than in the lungs of nonsmokers and smokers (Figure 5A and B). In addition, Western blot analysis showed an increase in NCOA4 (Figure 5C and D) and a decrease in ferritin (Supplementary Figure 2A and 2B) in mouse lung homogenates. IHC staining also showed the same results (Figure 5E and Supplementary Figure 2C). Western blotting revealed that NCOA4 levels increased in a time-dependent manner, while ferritin levels decreased (Figure 5F and G). Immunofluorescence staining of CHBE cells showed that NCOA4 expression was enhanced by CSE exposure, and ferritin and NCOA4 colocalization was detected (Figure 5H).
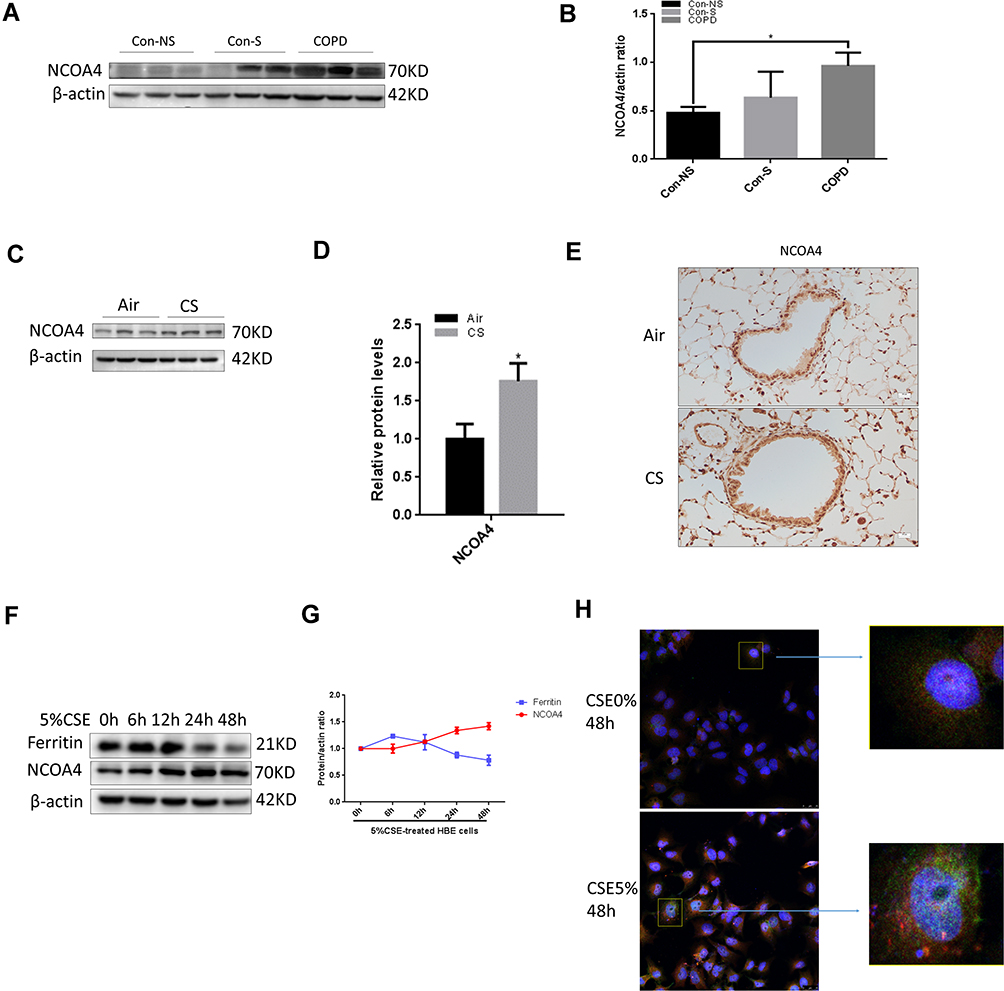 |
Figure 5 Expression of NCOA4 in vivo and in vitro. (A and B) Assessment of NCOA4 and β-actin expression levels by Western blotting. (C–E) Wild-type (WT) mice were exposed to air or CS for 24 weeks. (C) Western blotting was performed, and (D) the relative protein levels of NCOA4 in the lung were determined. (E) IHC staining of NCOA4 is shown. (F–H) HBE cells were treated with 5% CSE for 48 h. (F) The time course of NCOA4 and ferritin expression in response to 5% CSE was assessed by Western blotting. (G) The data shown represent the mean ± SD (n = 3). (H) Representative images of HBE cells showing the colocalization (yellow) of NCOA4 (red) with ferritin (green). Scale bars = 20 µm. *P<0.05. |
NCOA4 Modulates Ferroptosis in CHBE Cells
A cell study was performed to demonstrate the role of NCOA4 in ferroptosis. To explore whether NCOA4 could modulate ferroptosis, we successfully transfected NCOA4 siRNA into HBE cells (Figure 6A and B). Subsequently, Western blot analysis showed that ferritin levels were increased in NCOA4 siRNA-transfected CHBE cells (Figure 6C and D). The increase in ferritin indicates that the iron store was increased. Western blotting revealed that in CHBE cells transfected with NCOA4 siRNA, Ptgs2 expression levels were decreased more than those in CSE-treated HBE cells (Figure 6E and F).
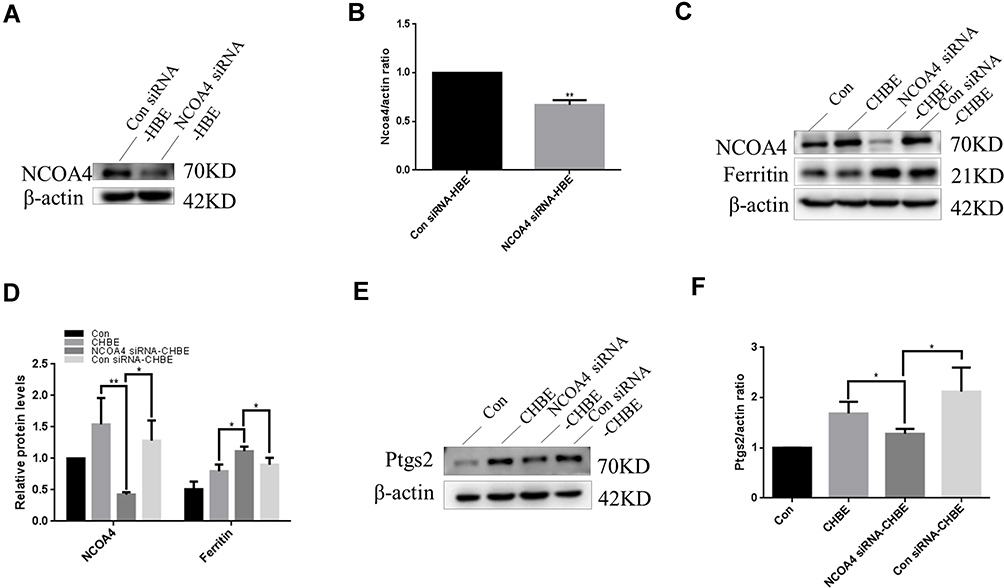 |
Figure 6 NCOA4 can regulate ferroptosis in HBE cells. Con siRNA-HBE, HBE cells transfected with control siRNA; NCOA4 siRNA-HBE, HBE cells transfected with NCOA4 siRNA; Con, normal HBE cells; CHBE, HBE cells treated with 5% CSE for 48 h; Con siRNA-CHBE, HBE cells transfected with control siRNA and then treated with 5% CSE for 48 h; NCOA4 siRNA-CHBE, HBE cells transfected with NCOA4 siRNA and then treated with 5% CSE for 48 h. Band densities were quantified by ImageJ software. β-actin levels were measured in parallel and served as controls. (A) and (B) HBE cells were transfected with control siRNA or NCOA4 siRNA before control or 5% CSE treatment. (C) and (D) Western blotting was performed to measure the expression of NCOA4 and ferritin in HBE cells before and after transfection. (E) and (F) The expression of Ptgs2 was determined. The data are the mean ± SD (n=3). *P<0.05; **P < 0.01. |
NCOA4-Mediated Ferroptosis Increases the Ratio of M2/M1 Phenotype in THP-M Cells Cocultured with CHBE Cells
Based on the results of a previous study, we hypothesized that NCOA4 could modulate macrophage polarization and the production of MMPs. Validation studies showed that the ratio of M2/M1 polarization was lower in THP-M cells cocultured with CHBE cells transfected with NCOA4 siRNA than in THP-M cells cocultured with CHBE cells (Figure 7A and B). Moreover, Western blotting revealed that MMP9 and MMP12 levels were decreased in THP-M cells cocultured with NCOA4-downregulated CHBE cells (Figure 7C and D). Therefore, we concluded that NCOA4-mediated ferroptosis in CHBE cells increased the ratio of M2/M1 polarization and the levels of MMPs in THP-M cells cocultured with CHBE cells.
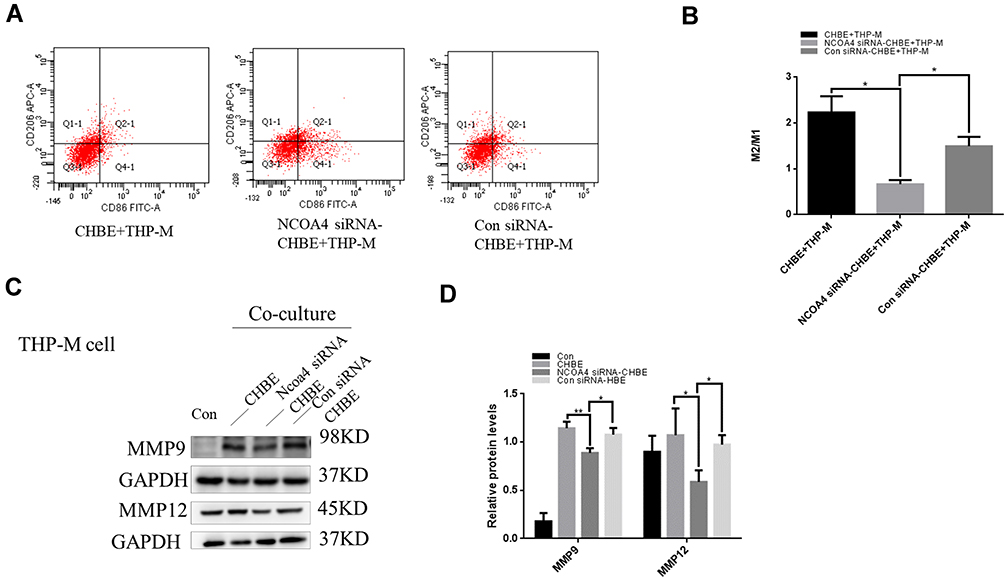 |
Figure 7 NCOA4 in HBE cells promotes the polarization of THP-M cells. Con, normal THP-M cells; CHBE, HBE cells treated with 5% CSE for 48 h; Con siRNA-CHBE, HBE cells transfected with control siRNA and then treated with 5% CSE for 48h; NCOA4 siRNA-CHBE, HBE cells transfected with NCOA4 siRNA and then treated with 5% CSE for 48h. THP-M cells were cocultured with CHBE, Con siRNA-CHBE and NCOA4 siRNA-CHBE cells for 48 h. (A) Representative histograms showing the flow cytometry results and (B) the percentages of M2/M1 macrophages. (C) and (D) THP-M cells were cocultured with CHBE, Con siRNA-CHBE and NCOA4 siRNA-CHBE cells, and the levels of MMP9 and MMP12 were determined using Western blotting. The data are the mean ± SD (n=3). *P<0.05; **P < 0.01. |
Discussion
COPD is a chronic respiratory disease, and its prevalence and mortality rates are increasing worldwide.28 Cigarette smoking is one of the most important causes of irreversible loss of lung function in COPD.29,30 CS-induced oxidative stress, airway inflammation, and macrophage polarization have been reported to be involved in the pathogenesis of COPD and lead to emphysema.2,31 Emphysema is a pathophysiological change in COPD. Our study showed that the polarization of M2 macrophages was dominant in severe COPD. We proposed that the upregulation of NCOA4 induced by CS plays an important role in this process and is an underlying mechanism.
Emphysema in COPD is associated with abnormal airspace enlargement distal to the terminal bronchioles accompanied by destructive changes in the alveolar walls and chronic inflammation.32 MMPs are involved in the pathogenesis of CS-induced COPD and emphysema, its serious sequelae.30,33 In mice, overexpression of MMP9 in macrophages results in emphysema.34 Moreover, MMP12 knockdown attenuated CS-related emphysema in mice.35 These observations are consistent with our findings that the levels of MMP9 and MMP12 were increased in the lung tissues of COPD patients and CS-exposed mice. These changes suggest that MMP9 and MMP12 may play important roles in the development and progression of emphysema related to CS exposure.
Macrophages are thought to produce MMP9 and MMP12, which are associated with the degradation of elastin in the alveoli walls, resulting in the functional destruction of lung tissue.36 In addition, M2 macrophages contribute to the breakage of alveolar walls.28,37 A growing body of evidence indicates that M2 macrophages can secrete MMP9 and MMP12.38,39 However, the relationship between macrophage polarization and MMPs production is complex and remains controversial. Macrophages can be polarized in one of two directions. M1 macrophages, which express iNOS, secrete cytokines such as TNF-α, thus promoting inflammation.6,40 Conversely, M2 macrophages resist inflammation, promote fibrosis and express Arg-1 and CD206.7,41 In the present study, we showed that M2-related cytokine levels were increased and that the M2 phenotype dominated in COPD. However, the results of some other studies were different from the results of this study. The Bazzan group showed that dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity.42 We believe that both M1 and M2 macrophages increased in severe COPD, but which M1 or M2 macrophages dominated was not determined in the study. However, Fang et al showed that cigarette smoke exposure could induce M1 macrophage polarization.43 In Fang’s study, macrophages were only exposed to cigarette smoke for 12 hours and mice for 12 weeks, while our lab exposed cells to smoke for 48 hours and mice for 24 weeks. The process of macrophage polarization is dynamic. At present, monitoring can only be performed at certain times and in specific environments. In the future, dynamic monitoring should be considered to better observe these changes.
We further aimed to uncover how cigarette smoke promoted alterations in macrophage phenotypes. Ferroptosis is a necrotic form of regulated cell death (RCD) mediated by phospholipid peroxidation in association with free iron-mediated Fenton reactions.44,45 Ferroptosis has been shown to play an important role in COPD pathogenesis.46 We confirmed this result. The expression of Ptgs2, a biomarker of ferroptosis, was increased in vivo and in vitro.47 In fact, we observed increased free iron with concomitant lipid peroxidation in response to CSE exposure, which was mainly attributable to ferritinophagy.
NCOA4 is a cargo receptor that mediates ferritinophagy and is important for the selective autophagy of ferritin.48–50 Western blotting showed that NCOA4 levels were increased in COPD.51,52 To further examine the role of NCOA4, we transfected NCOA4 siRNA into HBE cells to clarify the involvement of NCOA4 in ferroptosis. NCOA4-mediated ferritinophagy has been demonstrated to promote ferroptosis. It has been shown that ferroptosis-mediated damage-associated molecular patterns (DAMPs) release is tightly connected to inflammation and immunity.19,53 Although ferroptosis in CHBE cells results in the release of DAMPs that participate in COPD pathogenesis,23 the effect of ferroptosis on immune cells is largely unclear. DAMPs, including IL-33 and HMGB-1, in bronchoalveolar lavage fluid (BALF) were significantly increased by CS exposure in mice.23 IL-33 may promote M2 polarization, but HMGB-1 may promote M1 polarization. There are numerous DAMPs, and which DAMPs plays the most important role in COPD needs to be established in future studies. Therefore, we used a ferroptosis inhibitor to inhibit all cytokines released from CHBE cells. In addition, some DAMPs can be exported actively from live cells by exocytosis of secretory exosomes and ectosomes and the activation of cell membrane channel pores.54 HBE cells induce M2 macrophage polarization via the exosomes secreted by cells. We hypothesized that CS promoted ferroptosis, which was regulated by NCOA4, leading to M2 macrophage polarization. Then, we conducted experiments to verify this hypothesis.
To confirm the effects of NCOA4, we cocultured THP-M cells with HBE cells. After inhibiting ferroptosis in CHBE cells, we found that the ratio of M2/M1 polarization in THP-M cells was reduced, as shown by flow cytometry. Downregulation of NCOA4 reduced the smoking-induced M2/M1 polarization of macrophages. Moreover, MMP9 and MMP12 were reduced after ferroptosis was inhibited. Therefore, NCOA4 promoted M2 polarization of macrophages by targeting ferroptosis, which increased the levels of MMP9 and MMP12. CS increased the levels of NCOA4 in COPD. However, whether the overexpression of NCOA4 in CHBE cells can increase the level of ferroptosis and M2 polarization depends on the degree of NCOA4 expression and whether iron released from ferritin is saturated. Based on all the previous studies and what we found in our present study, we could make a hypothesis that the effects of NCOA4 overexpression can be rescued by a ferroptosis inhibitor. NCOA4 overexpression leads to the generation of excess free iron and ferroptosis through Fenton reactions, which can be rescued by ferrostatin-1.55
In summary, our results reveal that the M2 phenotype is dominant in severe COPD emphysema. NCOA4, as a mediator of ferroptosis, can promote M2 polarization and emphysema (Figure 8). Blocking NCOA4 may be a promising therapeutic strategy for COPD. However, our study had several limitations. To elucidate the changes in macrophages, we need to analyze additional clinical samples. Furthermore, ferroptosis may be part of a complex network regulating macrophage polarization. Additional studies are warranted to establish whether CS also acts through other mechanisms.
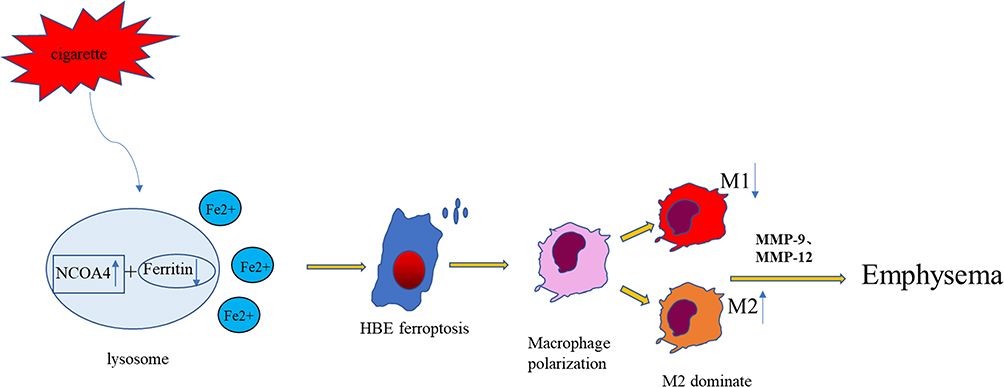 |
Figure 8 Schematic representation of the potential role of NCOA4 and macrophage polarization. CS increases intracellular NCOA4 and induces ferroptosis. Endogenous molecules released from ferroptotic cells promote M2 polarization. Macrophages can secrete MMP9 and MMP12, leading to emphysema in COPD. |
Data Sharing Statement
All data generated or analyzed during this study are included in this published article [and its Supplementary Information Files].
Ethics Approval and Consent to Participate
All experimental work was approved by the ethical review board of Wuxi People’s Hospital Affiliated to Nanjing Medical University. This study was conducted in accordance with the tenets of the Declaration of Helsinki. The procedures for the care and use of animals were approved by the Ethics Committee of Nanjing Medical University (KY21033). The animals used in this study were maintained in accordance with the ethical guidelines of the Guiding Principles in the Care and Use of Animals (China) and the Policy of Animal Care and Use Committee of Nanjing Medical University. Humane care was provided according to the 3R principles of animal experiments.
Author Contributions
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all of these areas. All authors took part in drafting, revising or critically reviewing the article, gave final approval of the version to be published, have agreed on the journal to which the article has been submitted and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundations of China (82173472), the project of the Translational Medicine Program of Wuxi City, Jiangsu Province (2020ZHYB13), the projects of the Health Department of Wuxi City, Jiangsu Province (M202032) and the Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (BJ2020006).
Disclosure
The authors declare that they have no competing interests.
References
1. Giordano L, Farnham A, Dhandapani PK, et al. Alternative oxidase attenuates cigarette smoke-induced lung dysfunction and tissue damage. Am J Respir Cell Mol Biol. 2019;60(5):515–522. doi:10.1165/rcmb.2018-0261OC
2. Kubo H, Asai K, Kojima K, et al. Astaxanthin suppresses cigarette smoke-induced emphysema through Nrf2 Activation in Mice. Mar Drugs. 2019;17(12):673. doi:10.3390/md17120673
3. Gimenes JA, Srivastava V, ReddyVari H, et al. Rhinovirus-induces progression of lung disease in a mouse model of COPD via IL-33/ST2 signaling axis. Clin Sci. 2019;133(8):983–996. doi:10.1042/CS20181088
4. Wang L, Zhang Y, Zhang N, Xia J, Zhan Q, Wang C. Potential role of M2 macrophage polarization in ventilator-induced lung fibrosis. Int Immunopharmacol. 2019;75:105795. doi:10.1016/j.intimp.2019.105795
5. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
6. Zizzo G, Cohen PL. Antibody Cross-Linking of CD14 Activates MerTK and Promotes Human Macrophage Clearance of Apoptotic Neutrophils: the Dual Role of CD14 at the Crossroads Between M1 and M2c Polarization. Inflammation. 2018;41(6):2206–2221. doi:10.1007/s10753-018-0864-x
7. Bosco MC. Macrophage polarization: reaching across the aisle? J Allergy Clin Immunol. 2019;143(4):1348–1350. doi:10.1016/j.jaci.2018.12.995
8. Huang T, Wu H, Chen S, Wang Y, Wu C. Thrombomodulin facilitates peripheral nerve regeneration through regulating M1/M2 switching. J Neuroinflamm. 2020;17(1). doi:10.1186/s12974-020-01897-z
9. Tong Y, Yu Z, Chen Z, et al. The HIV protease inhibitor Saquinavir attenuates sepsis-induced acute lung injury and promotes M2 macrophage polarization via targeting matrix metalloproteinase-9. Cell Death Dis. 2021;12(1). doi:10.1038/s41419-020-03320-0
10. Xiao T, Zou Z, Xue J, et al. LncRNA H19-mediated M2 polarization of macrophages promotes myofibroblast differentiation in pulmonary fibrosis induced by arsenic exposure. Environ Pollut. 2021;268:115810. doi:10.1016/j.envpol.2020.115810
11. Uchiyama R, Toyoda E, Maehara M, et al. Effect of Platelet-Rich Plasma on M1/M2 Macrophage Polarization. Int J Mol Sci. 2021;22(5):2336. doi:10.3390/ijms22052336
12. Feng H, Yin Y, Ren Y, et al. Effect of CSE on M1/M2 polarization in alveolar and peritoneal macrophages at different concentrations and exposure in vitro. In Vitro Cellular Dev Biol. 2020;56(2):154–164. doi:10.1007/s11626-019-00426-4
13. Lea SR, Reynolds SL, Kaur M, et al. The effects of repeated Toll-like receptors 2 and 4 stimulation in COPD alveolar macrophages. Int J Chron Obstruct Pulmon Dis. 2018;13:771–780. doi:10.2147/COPD.S97071
14. Han H, Peng G, Meister M, et al. Electronic Cigarette Exposure Enhances Lung Inflammatory and Fibrotic Responses in COPD Mice. Front Pharmacol. 2021:12. doi:10.3389/fphar.2021.726586
15. He S, Xie L, Lu J, Sun S. Characteristics and potential role of M2 macrophages in COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:3029–3039. doi:10.2147/COPD.S147144
16. Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234. doi:10.1038/s41556-020-0461-8
17. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J Diabetes Res. 2021;2021:1–10. doi:10.1155/2021/9999612
18. Zhang Y, Kong Y, Ma Y, et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene. 2021;40(8):1425–1439. doi:10.1038/s41388-020-01622-3
19. Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069–2083. doi:10.1080/15548627.2020.1714209
20. Hou W, Zhang Q, Yan Z, et al. Strange attractors: dAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4(12):e966. doi:10.1038/cddis.2013.493
21. Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. doi:10.1038/s41467-020-20154-8
22. Hu ZW, Wen YH, Ma RQ, et al. Ferroptosis Driver SOCS1 and Suppressor FTH1 Independently Correlate With M1 and M2 Macrophage Infiltration in Head and Neck Squamous Cell Carcinoma. Front Cell Dev Biol. 2021;9:727762. doi:10.3389/fcell.2021.727762
23. Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1). doi:10.1038/s41467-019-10991-7
24. Di T, Yang Y, Fu C, et al. Let‐7 mediated airway remodelling in chronic obstructive pulmonary disease via the regulation of IL‐6. Eur J Clin Invest. 2021;51(4). doi:10.1111/eci.13425
25. Yang Y, Di T, Zhang Z, et al. Dynamic evolution of emphysema and airway remodeling in two mouse models of COPD. BMC Pulm Med. 2021;21(1). doi:10.1186/s12890-021-01456-z
26. He S, Li L, Sun S, et al. Chronic Obstructive Pulmonary Disease Model and the Pathogenic Role of MicroRNA-21. Front Physiol. 2018;9:503. doi:10.3389/fphys.2018.00503
27. Bebber CM, Muller F, Prieto CL, Weber J, von Karstedt S. Ferroptosis in Cancer Cell Biology. Cancers. 2020;12(1):164. doi:10.3390/cancers12010164
28. Sun X, Liu Y, Feng X, Li C, Li S, Zhao Z. The key role of macrophage depolarization in the treatment of COPD with ergosterol both in vitro and in vivo. Int Immunopharmacol. 2020;79:106086. doi:10.1016/j.intimp.2019.106086
29. Lu Z, Van Eeckhoutte HP, Liu G, et al. Necroptosis Signalling Promotes Inflammation, Airway Remodelling and Emphysema in COPD. Am J Respir Crit Care Med. 2021;204:667–681. doi:10.1164/rccm.202009-3442OC
30. Cornwell WD, Kim V, Fan X, et al. Activation and polarization of circulating monocytes in severe chronic obstructive pulmonary disease. BMC Pulm Med. 2018;18(1):101. doi:10.1186/s12890-018-0664-y
31. Kawasaki T, Sugihara F, Fukushima K, et al. Loss of FCHSD1 leads to amelioration of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2021;118(26). doi:10.1073/pnas.2019167118
32. Kaku Y, Imaoka H, Morimatsu Y, et al. Overexpression of CD163, CD204 and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PLoS One. 2014;9(1):e87400–e87400. doi:10.1371/journal.pone.0087400
33. Gharib SA, Manicone AM, Parks WC. Matrix metalloproteinases in emphysema. Matrix Biol. 2018;73:34–51. doi:10.1016/j.matbio.2018.01.018
34. Chukowry PS, Spittle DA, Turner AM. Small Airways Disease, Biomarkers and COPD: where are We? Int J Chron Obstruct Pulmon Dis. 2021;16:351–365. doi:10.2147/COPD.S280157.
35. Xia H, Wu Y, Zhao J, et al. The aberrant cross-talk of epithelium–macrophages via METTL3-regulated extracellular vesicle miR-93 in smoking-induced emphysema. Cell Biol Toxicol. 2021. doi:10.1007/s10565-021-09585-1
36. Jiang Y, Zhao Y, Wang Q, Chen H, Zhou X. Fine particulate matter exposure promotes M2 macrophage polarization through inhibiting histone deacetylase 2 in the pathogenesis of chronic obstructive pulmonary disease. Ann Transl Med. 2020;8(20):1303. doi:10.21037/atm-20-6653
37. Liu C, Li B, Tang K, et al. Aquaporin 1 alleviates acute kidney injury via PI3K-mediated macrophage M2 polarization. Inflamm Res. 2020;69(5):509–521. doi:10.1007/s00011-020-01334-0
38. Huang WC, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-kappaB. PLoS One. 2012;7(8):e42507. doi:10.1371/journal.pone.0042507
39. Le Y, Cao W, Zhou L, et al. Infection of Mycobacterium tuberculosis Promotes Both M1/M2 Polarization and MMP Production in Cigarette Smoke-Exposed Macrophages. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.01902
40. Yang S, Chen Y, Hsu C, et al. Activation of M1 Macrophages in Response to Recombinant TB Vaccines With Enhanced Antimycobacterial Activity. Front Immunol. 2020:11. doi:10.3389/fimmu.2020.01298
41. He S, Chen D, Hu M, et al. Bronchial epithelial cell extracellular vesicles ameliorate epithelial–mesenchymal transition in COPD pathogenesis by alleviating M2 macrophage polarization. NanoMedicine. 2019;18:259–271. doi:10.1016/j.nano.2019.03.010
42. Bazzan E, Turato G, Tine M, et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir Res. 2017;18(1):40. doi:10.1186/s12931-017-0522-0
43. Feng H, Yin Y, Zheng R, Kang J. Rosiglitazone ameliorated airway inflammation induced by cigarette smoke via inhibiting the M1 macrophage polarization by activating PPARgamma and RXRalpha. Int Immunopharmacol. 2021;97:107809. doi:10.1016/j.intimp.2021.107809
44. Otasevic V, Vucetic M, Grigorov I, Martinovic V, Stancic A. Ferroptosis in Different Pathological Contexts Seen through the Eyes of Mitochondria. Oxid Med Cell Longev. 2021;2021:5537330. doi:10.1155/2021/5537330
45. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
46. Tang R, Xu J, Zhang B, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13(1):110. doi:10.1186/s13045-020-00946-7
47. Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting Ferroptosis through Disrupting the NCOA4–FTH1 Interaction: a New Mechanism of Action. Acs Central Sci. 2021;7(6):980–989. doi:10.1021/acscentsci.0c01592
48. Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: a New Mechanism of Action. ACS Cent Sci. 2021;7(6):980–989. doi:10.1021/acscentsci.0c01592
49. Gryzik M, Asperti M, Denardo A, Arosio P, Poli M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim Biophys Acta Mol Cell Res. 2021;1868(2):118913. doi:10.1016/j.bbamcr.2020.118913
50. Zhang Z, Yao Z, Wang L, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14(12):2083–2103. doi:10.1080/15548627.2018.1503146
51. Santana-Codina N, Gikandi A, Mancias JD. The Role of NCOA4-Mediated Ferritinophagy in Ferroptosis. Adv Exp Med Biol. 2021;1301:41–57. doi:10.1007/978-3-030-62026-4_4
52. Chen X, Yu C, Kang R, Tang D. Iron Metabolism in Ferroptosis. Front Cell Dev Biol. 2020;8:590226. doi:10.3389/fcell.2020.590226
53. Gu Z, Liu T, Liu C, et al. Ferroptosis-Strengthened Metabolic and Inflammatory Regulation of Tumor-Associated Macrophages Provokes Potent Tumoricidal Activities. Nano Lett. 2021;21(15):6471–6479. doi:10.1021/acs.nanolett.1c01401
54. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis. 2021;26(3–4):152–162. doi:10.1007/s10495-021-01663-3
55. Zhao L, Zhou X, Xie F, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022;42(2):88–116. doi:10.1002/cac2.12250
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.