Back to Journals » International Journal of Nanomedicine » Volume 21
Nanozyme-Driven Ferroptosis–Cuproptosis Interplay in Lung Cancer
Received 22 April 2026
Accepted for publication 25 June 2026
Published 15 July 2026 Volume 2026:21 618979
DOI https://doi.org/10.2147/IJN.S618979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Dong Wang
Man Sun, Dan Zang, Jun Chen
Department of Oncology, The Second Hospital of Dalian Medical University, Dalian, Liaoning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Chen, Email [email protected]
Abstract: Lung cancer is uniquely amenable to local administration and is characterized by sustained exposure to an oxygen-rich exchange environment and marked metabolic heterogeneity. These features make it a distinctive therapeutic setting for nanozyme-mediated induction of ferroptosis and cuproptosis, while simultaneously imposing more stringent requirements on delivery precision and safety control for therapies based on intense oxidative and metal-induced stress. From the organ-specific context of lung cancer, this review focuses on the catalytic design of nanozymes, strategies for localized pulmonary delivery, and the interplay between ferroptosis and cuproptosis, while evaluating their potential applications in metabolic reprogramming, tumor suppression, and immune sensitization. Current evidence suggests that iron- and copper-associated catalysis, multiple enzyme-mimetic activities, and cascade reactions can amplify lipid peroxidation, deplete reductive capacity, and exacerbate mitochondrial proteotoxic stress, thereby enhancing tumor cell killing. In certain systems, these effects are further accompanied by increased immunogenic cell death and activation of antitumor immunity. Nevertheless, the field is constrained less by a shortage of additional nanoplatforms than by the absence of clear and comparable criteria for defining cuproptosis, insufficient evidence supporting lung cancer-directed local delivery, and the lack of biomarker-guided patient stratification and rational combination strategies.
Keywords: lung cancer, nanozymes, ferroptosis, cuproptosis, immune modulation, local delivery
Background
Lung cancer remains one of the leading causes of cancer-related mortality worldwide.1 Although surgery, chemotherapy, molecularly targeted therapy, and immune checkpoint blockade have improved outcomes for a subset of patients, durable clinical benefit continues to be constrained by primary or acquired resistance, an immunosuppressive tumor microenvironment, heterogeneity across metastatic lesions, and systemic toxicity.2 Conventional nanodrug delivery systems can enhance drug solubility, prolong circulation time, and improve tumor accumulation; however, the function of most platforms remains largely confined to cargo loading and delivery, with limited capacity to actively modulate tumor metabolic networks or cell death programs.3,4 This limitation is particularly relevant to lung cancer, where therapeutic resistance often reflects not a single pathway defect but a coordinated state involving redox adaptation, metabolic rewiring, metal-ion homeostasis, and immune escape.5 The lung provides a distinctive but demanding setting for nanozyme-based therapy. On the one hand, pulmonary tumors are located in an oxygen-rich gas-exchange organ and may be accessed through local administration routes such as inhalation or intratracheal delivery. On the other hand, pulmonary surfactants, mucociliary clearance, and phagocytic uptake by alveolar macrophages collectively impose stringent constraints on deposition, retention, catalytic activation, and safety.6 Accordingly, the central challenge in lung cancer therapy is not merely to increase drug deposition at the target site, but to confine intense oxidative or metal-induced stress as selectively as possible to tumor lesions while preserving the tolerance of normal lung tissue. This requirement makes local pulmonary delivery particularly important for ferroptosis and cuproptosis based nanozyme therapy, because both strategies require sufficient intralesional redox or metal ion stress while avoiding diffuse lipid peroxidation, systemic metal homeostasis disruption, and off target injury in normal tissues.
Against this backdrop, nanozymes, which combine structural tunability with enzyme-mimetic catalytic activity, have attracted increasing attention. Unlike conventional nanoplatforms that primarily serve as delivery vehicles, nanozymes can, through the regulation of active sites, electronic structure, metal valence cycling, surface interfaces, and tumor-responsive activation, sustain oxidative stress amplification, weaken antioxidant defenses, and disrupt metal ion homeostasis within the tumor microenvironment, thereby directly influencing metabolic processes associated with regulated cell death.7,8 Their significance, therefore, lies not merely in functioning as drug carriers, but in serving as catalytic therapeutic platforms capable of exploiting the intrinsic metabolic vulnerabilities of lung cancer cells.
In lung cancer, ferroptosis and cuproptosis represent two forms of non-apoptotic programmed cell death that are closely linked to disruption of metabolic homeostasis. Unlike canonical apoptosis, which is primarily mediated by caspase cascades, these two death programs are more directly driven by redox disequilibrium, disturbances in metal ion homeostasis, and aberrant mitochondrial metabolism.9,10 Ferroptosis is defined by the uncontrolled accumulation of iron-dependent lipid peroxidation and is closely associated with dysfunction of the cystine/glutamate antiporter system (System Xc−)–glutathione (GSH)–glutathione peroxidase 4 (GPX4) axis.11,12 By contrast, cuproptosis is triggered by excessive intracellular copper accumulation and is characterized by aggregation of lipoylated tricarboxylic acid (TCA) cycle proteins, loss of Fe-S cluster proteins, and proteotoxic stress.13 These two processes are not biologically isolated. Copper can intensify ferroptosis-related stress by increasing reactive oxygen species (ROS), whereas GSH levels, mitochondrial respiratory activity, and maintenance of metal-ion homeostasis simultaneously influence cellular susceptibility to both forms of death. Thus, ferroptosis and cuproptosis are mechanistically distinct but converge on several shared constraints, including GSH buffering, redox balance, metal-ion regulation, mitochondrial respiration, and proteostatic capacity.14,15
This convergence provides the biological basis for considering whether ferroptosis and cuproptosis can be jointly exploited in lung cancer.16 However, in this review, ferroptosis–cuproptosis synergy is not defined as the simple coexistence of ferroptosis-associated and cuproptosis-associated markers within the same experimental system. Instead, we reserve this term for conditions in which three levels of evidence are considered together. First, ferroptosis should be supported by lipid peroxidation-specific evidence, such as lipid ROS accumulation, C11-BODIPY oxidation, malondialdehyde or 4-hydroxynonenal elevation, suppression of GPX4, SLC7A11, FSP1, or DHODH, and rescue by ferroptosis inhibitors or iron chelation. Second, cuproptosis should be supported by canonical mitochondrial proteotoxic evidence, including aggregation of lipoylated TCA cycle proteins, loss of Fe-S cluster proteins, FDX1-, LIAS-, or DLAT-related changes, and reversibility by copper chelation. Third, the relationship between the two pathways should be supported by functional or causal evidence, such as temporal coordination, mitochondrial dependence, pathway-specific rescue patterns, or enhanced tumor suppression beyond parallel nonspecific oxidative or metal-associated stress responses.17,18 When formal quantitative combination analyses, such as combination index- or independence model-based assessments, are unavailable, we interpret synergy conservatively. Platforms that only increase ROS, deplete GSH, disturb mitochondrial function, or alter metal-ion distribution are described as inducing oxidative stress, copper-associated stress, concurrent pathway activation, or putative synergy unless the above criteria are fulfilled. This distinction is particularly important for nanozyme-based lung cancer therapy, because intense catalytic reactions may generate overlapping stress phenotypes that do not necessarily prove mechanistic cooperation between ferroptosis and cuproptosis.
For lung cancer, the significance of nanozyme-mediated dual-death induction lies not only in showing that ferroptosis and cuproptosis can occur concurrently, but also in defining the molecular, metabolic, and delivery conditions under which these death programs interact in a mechanistically meaningful and therapeutically useful manner.19,20 On this basis, the present review discusses the design principles of nanozymes, the mechanistic basis of ferroptosis–cuproptosis interplay, and the major translational constraints currently limiting their application in lung cancer, with particular emphasis on mechanistic definition, pulmonary delivery, safety boundaries, immune consequences, and patient stratification.
Nanozyme Design for Ferroptosis–Cuproptosis Regulation
Nanozymes are nanomaterials endowed with enzyme-mimetic catalytic activity that are capable of catalyzing substrate conversion under physiologically relevant conditions and eliciting defined biological effects.21 In the regulation of ferroptosis and cuproptosis, their functional impact is not determined solely by material composition, but more importantly by the configuration of active sites, the mode of component integration, interfacial modification, and the conditions governing intratumoral activation. These factors are particularly critical in lung cancer, where the local oxygen-exchange environment, complex clearance barriers, and the tolerance limits of normal lung tissue collectively shape both catalytic output and biosafety. This section therefore discusses the design principles underlying nanozyme-mediated regulation of ferroptosis and cuproptosis from five perspectives, active sites, multimetal combinations, interfacial engineering, lesion-responsive activation, and pulmonary delivery (Figure 1). To avoid overlap with later mechanistic sections, this section focuses on design variables that determine catalytic output and delivery behavior, whereas the biological interaction between ferroptosis and cuproptosis and the resulting immune consequences are discussed in Nanozyme-Mediated Synergistic Intervention Through Ferroptosis and Cuproptosis. Together, these design variables determine whether nanozyme activity can reshape redox balance, metal ion homeostasis, catalytic reaction pathways, and pulmonary lesion selectivity in a manner that supports safe and effective regulation of ferroptosis and cuproptosis.
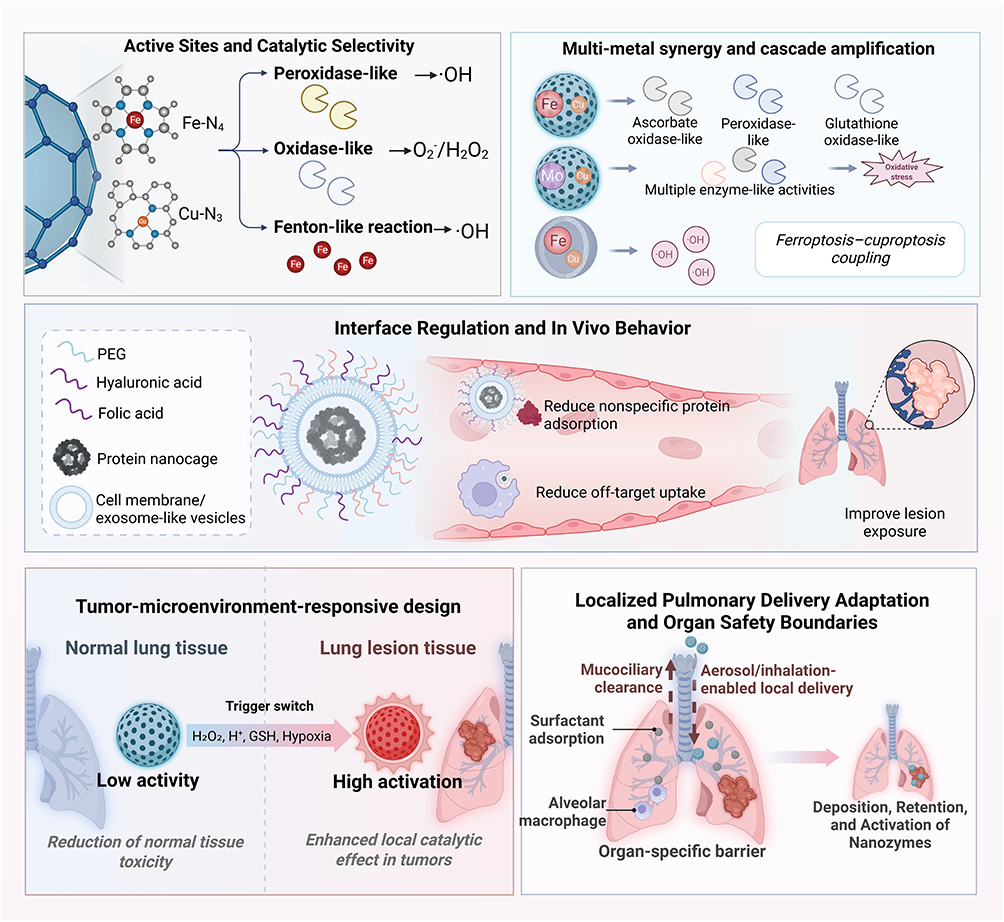 |
Figure 1 Design principles for nanozyme-mediated regulation of ferroptosis and cuproptosis in lung cancer. Active-site engineering defines catalytic selectivity by tuning POD-, OXD-, and Fenton-like activities, thereby shaping ROS output. Multi-metal integration and cascade catalysis further amplify oxidative stress and promote ferroptosis-cuproptosis coupling. Interfacial engineering, including surface functionalization and biomimetic encapsulation, improves in vivo performance by reducing nonspecific adsorption and off-target uptake while enhancing lesion exposure. Tumor microenvironment-responsive design enables preferential activation under acidic, H2O2-rich, glutathione-rich, and hypoxic conditions. Pulmonary delivery must also account for mucociliary clearance, surfactant adsorption, and alveolar macrophage uptake to optimize local deposition and retention. |
Active Sites and Catalytic Selectivity
The configuration of active sites is a critical determinant of the catalytic selectivity of nanozymes. Distinct coordination environments can alter local electronic structures at the atomic scale, thereby influencing substrate adsorption, electron transfer, and reaction pathways, and ultimately dictating the relative outputs of peroxidase (POD)-, oxidase (OXD)-, and Fenton-like catalytic activities.22–24 Single-atom nanozymes provide a representative example. These systems are centered on atomically dispersed metal sites and can enhance site utilization and reaction selectivity through optimization of the coordination environment.25 Previous studies have shown that structures such as M-N-C and M-N_x can markedly modulate POD-, OXD-, and Fenton-like activities, thereby establishing a foundation for amplifying oxidative stress within the tumor microenvironment (TME).26,27
Beyond the identity of the metal center itself, the catalytic behavior of nanozymes is jointly determined by active site distribution, electronic structure, coordination environment, defects, vacancies, exposed facets, surface functional groups, particle size, morphology, and surface charge.28 These parameters influence substrate binding, reactant accessibility, electron transfer efficiency, local intermediate stabilization, and the direction of reaction pathways.29 For example, the type and valence state of the metal center can determine whether a nanozyme preferentially supports POD-like, OXD-like, CAT-like, GSHOx-like, or Fenton-like activity, whereas defects and vacancies may generate unsaturated coordination sites that enhance substrate adsorption and accelerate redox cycling. Surface functional groups and charge can further regulate interactions with H2O2, GSH, oxygen, copper ions, lipid substrates, and cellular membranes, thereby affecting both catalytic activity and biological selectivity.30,31
For ferroptosis and cuproptosis, these structural variables are important because they determine not only catalytic intensity, but also pathway directionality.29 Active sites favoring Fenton-like reactions or POD-like activity are more directly linked to ferroptosis through lipid peroxidation amplification, whereas copper-containing sites or sites capable of regulating copper release and redox cycling may be more relevant to cuproptosis when they increase effective intracellular or mitochondrial copper availability and promote lipoylated protein aggregation.32 Particle size, morphology, and surface charge also affect cellular uptake, endosomal escape, mitochondrial accessibility, pulmonary retention, and macrophage clearance, which may alter whether catalytic output is converted into tumor-selective ferroptosis, cuproptosis, or nonspecific oxidative injury.33 Thus, the same nanozyme composition may produce different biological outcomes depending on how its active sites and interfacial properties control substrate access, redox flux, metal availability, and subcellular localization.
A major unresolved challenge is that the true active sites operating in biological environments may differ from those identified in simplified catalytic assays. Protein corona formation, pulmonary surfactant adsorption, ligand exchange, partial degradation, aggregation, oxidation-state transition, and biomolecule-mediated surface remodeling can all reshape the catalytic interface and alter enzyme-like selectivity.30 Therefore, future studies should move beyond reporting overall POD-like, OXD-like, CAT-like, GSHOx-like, or Fenton-like activity and should establish clearer structure–activity–selectivity relationships.34,35 This will require integrated evaluation of active site structure, catalytic kinetics, substrate specificity, biological microenvironmental remodeling, pathway-specific rescue evidence, and in vivo catalytic behavior, especially in the pulmonary setting where surfactant interaction and macrophage uptake may substantially modify nanozyme function.36
Multimetal Catalysis and Cascade Reaction Design
If active sites determine the direction of a given catalytic reaction, multimetal combinations determine whether catalysis can progress from a single reaction event to a coordinated cascade.37 Compared with single metal nanozymes, bimetallic or polymetallic nanozymes can provide multiple neighboring catalytic centers, redistribute electronic density, accelerate interfacial electron transfer, stabilize reaction intermediates, and promote redox cycling among different metal valence states.38 These features may lower reaction energy barriers and improve catalytic activity, stability, and selectivity, especially when sequential reactions such as H2O2 activation, oxygen conversion, GSH oxidation, and metal ion release need to occur within the same platform.39
The catalytic behavior of multimetal nanozymes is strongly influenced by metal composition, oxidation state, coordination environment, spatial distribution, and interfacial coupling.37,38 Different metals may assume complementary catalytic functions, one metal center may facilitate H2O2 decomposition or Fenton like hydroxyl radical generation, whereas another may promote oxygen activation, GSH depletion, copper ion cycling, or mitochondrial metal stress.40,41 In this way, multimetal systems can coordinate ROS amplification, reductive capacity depletion, lipid oxidation, and mitochondrial proteotoxic stress more effectively than single metal systems.42 For ferroptosis and cuproptosis regulation, iron associated sites are mainly linked to lipid peroxidation through Fenton or Fenton like reactions, whereas copper associated sites become relevant to cuproptosis only when they increase effective intracellular or mitochondrial copper availability and promote lipoylated protein aggregation.5 In addition, multimetal design does not necessarily enhance cuproptosis. For example, zinc-containing combinations may induce metallothionein expression, thereby increasing copper buffering capacity and potentially attenuating cuproptosis. Therefore, Cu-based multimetal nanozymes should be evaluated not only for copper release but also for compensatory metal-sequestration responses.
Interfacial interactions are also critical for cascade catalysis. Core shell structures, dual atom sites, heterojunctions, and metal support interfaces can create electron transfer channels and spatially couple intermediate production with downstream reactions.39,40 For example, one catalytic site may generate H2O2 or hydroxyl radicals, while another consumes GSH or regulates copper availability, thereby converting independent catalytic events into a sequential or mutually reinforced reaction network.41 However, such coupling should not be interpreted automatically as biological synergy. The mechanistic interpretation of these outputs as ferroptosis, cuproptosis, or concurrent dual death activation requires pathway specific evidence and is discussed in Section 5.17
Several challenges remain. First, the precise distribution and oxidation states of multiple metals may change during synthesis, storage, degradation, or exposure to biological media, making it difficult to identify the true catalytic centers. Second, metal leaching, nonspecific ROS generation, excessive GSH consumption, and uncontrolled Fenton like reactions may cause unwanted side reactions and off target toxicity.38 Third, in biological or environmental applications, proteins, thiols, surfactants, salts, and other competing substrates can remodel the catalytic interface and alter selectivity.36 Therefore, future studies should establish clearer composition structure activity selectivity relationships for multimetal nanozymes by combining atomic level characterization, catalytic kinetic analysis, substrate specificity testing, in situ or operando monitoring, metal bioavailability assessment, and pathway specific biological validation.43
Interfacial Engineering and in vivo Behavior
Beyond the catalytic core, the in vivo performance of nanozymes is profoundly influenced by interfacial engineering.44 Surface coatings, functional groups, targeting ligands, polymer shells, biomimetic membranes, exosome mimetic vesicles, and protein nanocages can regulate how nanozymes interact with proteins, cells, extracellular matrix components, and biological barriers under physiological conditions.45,46 These modifications may improve colloidal stability, reduce aggregation, protect catalytic centers from premature inactivation, prolong circulation time, decrease nonspecific uptake, and enhance lesion accumulation.47 At the same time, surface chemistry can alter substrate accessibility and electron transfer at the catalytic interface, thereby changing enzyme like activity and catalytic selectivity.30
Interfacial design also affects biodistribution, cellular uptake, immune recognition, toxicity, clearance, and therapeutic efficiency. Polyethylene glycol, hyaluronic acid, folic acid, antibodies, peptides, or cell membrane coatings can reduce nonspecific protein adsorption, improve biocompatibility, and enhance tumor targeting through passive or active mechanisms.44 Biomimetic membranes may help evade macrophage clearance or provide homologous targeting, whereas ligand modification may increase receptor mediated uptake by tumor cells.48 However, these benefits are not automatic. Surface modification may also mask active sites, reduce substrate diffusion, alter ROS generation, trigger complement activation, change immune-cell recognition, or induce unexpected accumulation in the liver, spleen, or lung.49 Therefore, interfacial engineering should be evaluated not only by tumor accumulation, but also by catalytic retention, immune compatibility, organ distribution, degradation behavior, and clearance pathway.50
For lung cancer, interfacial behavior should be evaluated in the pulmonary microenvironment rather than only in systemic circulation. Surface designs optimized for intravenous administration may not directly translate to inhaled nanozyme systems, because pulmonary surfactants, mucus components, epithelial lining fluid, and alveolar macrophages can rapidly remodel the nanozyme interface after airway or alveolar deposition.51 Surfactant corona formation, mucus interaction, and macrophage uptake may alter surface charge, aggregation state, catalytic accessibility, pulmonary deposition, retention, clearance, and safety.30 These effects are particularly relevant for ferroptosis and cuproptosis based strategies, because small changes in catalytic accessibility or cellular localization may determine whether oxidative or metal associated stress is confined to tumor lesions or extended to normal lung tissue.52
A key challenge is that the interface observed at the time of synthesis may differ substantially from the interface operating in vivo. Protein corona formation, ligand exchange, enzymatic degradation, biomolecule adsorption, and pulmonary surfactant coating can dynamically reshape surface identity and catalytic output.30 Long term biosafety therefore requires evaluation of persistent metal retention, chronic inflammation, epithelial injury, macrophage activation, surfactant disturbance, fibrosis risk, and respiratory function.53 Future studies should integrate physicochemical characterization, catalytic activity after biological exposure, biodistribution, pharmacokinetics, immune response, degradation, clearance, and repeated dose safety assessment to establish more predictable in vivo performance for interfacially engineered nanozymes.54
Tumor-Responsive Design and Local Activation
The central objective of tumor-responsive nanozyme design is to ensure that catalytic activity is preferentially activated within tumor lesions while remaining relatively restrained in normal tissues.55 Common tumor microenvironmental cues include acidic pH, excess H2O2, high GSH levels, hypoxia, and abnormal redox balance.56,57 These cues can be used to trigger structural disassembly, active site exposure, metal ion release, valence state conversion, substrate access, or cascade reaction initiation.58 In this way, tumor-responsive design links material activation with local catalytic output rather than relying only on passive accumulation.59
Different tumor-specific conditions activate nanozyme catalysis through distinct mechanisms.60 Acidic pH can promote the degradation of acid-labile bonds, protonation of surface groups, framework disassembly, or release of Fe- or Cu-containing catalytic centers, thereby increasing local Fenton-like or copper-associated reactions.61 Excess H2O2 provides a substrate for POD-like and Fenton-like catalysis, leading to hydroxyl radical generation and lipid peroxidation amplification.62 High GSH levels can trigger GSH-responsive degradation or metal reduction while simultaneously weakening antioxidant defense, thereby promoting ferroptosis through GPX4-related vulnerability and facilitating copper-associated stress by altering copper redox cycling and buffering capacity. Hypoxia can activate hypoxia-responsive linkers or be modulated by CAT-like oxygen generation, thereby influencing ROS production, mitochondrial metabolism, and susceptibility to cuproptosis.56 Abnormal redox balance may further amplify these processes by promoting repeated metal valence cycling and sustained cascade catalysis.
For ferroptosis and cuproptosis based intervention, tumor-responsive activation should therefore be designed as a controlled catalytic switch rather than as unrestricted stress amplification.37 In ferroptosis-oriented systems, local activation should favor lipid peroxidation, GSH depletion, and impairment of GPX4/SLC7A11-related defenses while minimizing nonspecific oxidative injury in adjacent lung tissue.20 In cuproptosis-oriented systems, activation should increase effective intracellular or mitochondrial copper availability and mitochondrial proteotoxic stress without causing diffuse systemic copper toxicity.63 For dual-death platforms, multi-stimulus responsive or logic-gated designs may be particularly useful, because simultaneous requirements such as acidic pH plus high GSH or H2O2 plus hypoxia can improve spatial selectivity and reduce premature activation.64
Deep tumor penetration remains an additional challenge. Dense extracellular matrix, irregular vascularization, heterogeneous hypoxia, variable H2O2 or GSH distribution, and limited diffusion of catalytic substrates may cause nanozyme activation to occur mainly at the tumor periphery rather than throughout the lesion.65,66 Size-transformable systems, charge-switchable surfaces, degradable matrices, enzyme-responsive coatings, and staged activation strategies may help improve penetration and intratumoral distribution.67 However, stronger penetration should not be pursued at the expense of uncontrolled activity, because excessive diffusion of active catalytic species may increase off-target oxidative or metal-induced injury.68
Therefore, lesion-specific activation should not be defined solely by responsiveness to acidic pH, elevated H2O2, high GSH levels, hypoxia, or redox imbalance in simplified in vitro systems.69 More rigorous validation should demonstrate preferential catalytic activation within tumor tissue, limited activation in adjacent normal lung tissue, controlled intratumoral distribution, consistency between activation sites and biodistribution patterns, and pathway-specific rescue evidence.17,70 For lung cancer, tumor-responsive nanozyme systems should be evaluated by intratumoral activation, deep lesion penetration, pulmonary distribution, normal lung toxicity, degradation and clearance behavior, and respiratory safety rather than by stimulus responsiveness alone.71,72
Pulmonary Delivery and Safety Boundaries
In lung cancer, pulmonary delivery of nanozymes should be designed not only to increase local exposure, but also to control where, when, and how catalytic activity is activated within the respiratory tract.73 Effective inhaled or intratracheal nanozyme systems require appropriate particle size, aerosol stability, lung deposition efficiency, mucus penetration, tumor retention, and controlled degradation.74 Particle size and aerodynamic behavior determine whether nanozymes deposit mainly in the upper airway, bronchial region, or deep alveolar compartment, whereas morphology, surface charge, hydrophilicity, and aggregation state influence aerosol dispersion, mucus interaction, epithelial uptake, and macrophage clearance.75,76 Therefore, pulmonary delivery should be optimized as an integrated process involving formulation stability, device compatibility, reproducible dose delivery, and lesion specific activation.
The need for local delivery is particularly evident for ferroptosis and cuproptosis based nanozyme strategies, because both require sufficient intralesional catalytic activity while carrying a risk of off target redox or metal associated toxicity.73 Systemic administration may dilute nanozyme exposure at pulmonary tumors, increase liver and spleen sequestration, and expose normal tissues to iron or copper associated catalytic stress.77 For ferroptosis, uncontrolled systemic activation may amplify lipid peroxidation in normal tissues; for cuproptosis, systemic copper exposure may disturb physiological copper homeostasis before an effective mitochondrial copper burden is achieved in tumor cells. Therefore, pulmonary local delivery should be viewed not only as a deposition enhancing route, but also as a therapeutic window strategy that concentrates catalytic activity within lung lesions while reducing systemic toxicity.78
Surface chemistry and degradation behavior are central to pulmonary biocompatibility and therapeutic efficiency. PEGylation, zwitterionic coatings, biomimetic membranes, ligand modification, or charge switchable surfaces may improve aerosol stability, reduce nonspecific adsorption, enhance tumor cell uptake, or regulate macrophage recognition.79 However, excessive surface shielding may also reduce substrate access and weaken catalytic activity, whereas unstable coatings may cause premature activation or uncontrolled metal release.44 Similarly, prolonged retention may improve therapeutic exposure but also increase the risk of persistent metal accumulation, chronic oxidative stress, epithelial injury, immune activation, and fibrosis. Thus, inhaled nanozymes should be designed to balance retention with biodegradation and clearance.80
Pulmonary safety evaluation should therefore extend beyond acute cytotoxicity.33 Because the lung is continuously exposed to oxygen and contains surfactants, mucus, epithelial barriers, resident immune cells, and abundant capillary interfaces, nanozyme induced oxidative or metal associated reactions may provoke inflammation, surfactant disturbance, macrophage activation, immune cell recruitment, vascular leakage, or fibrotic remodeling if not spatially controlled.81,82 Dose, dosing frequency, degradation kinetics, pulmonary residence time, systemic leakage, and organ biodistribution should be evaluated together. For tumor responsive applications, it is also necessary to demonstrate that catalytic activation occurs preferentially within tumor lesions rather than in adjacent normal lung tissue.
Overall, the therapeutic performance of nanozymes in lung cancer depends on the coordinated interaction among active site characteristics, multimetal catalytic design, interfacial engineering, tumor responsive activation, and pulmonary delivery safety. Active sites and coordination structures determine catalytic activity, reaction pathway preference, and enzyme like selectivity, whereas multimetal composition and interfacial coupling regulate electron transfer, redox cycling, cascade reactions, and ROS activation. Surface modification influences stability, substrate accessibility, immune recognition, cellular uptake, biodistribution, and clearance, while tumor responsive design and pulmonary delivery determine local activation, lung deposition, intralesional penetration, retention, degradation, systemic leakage, and respiratory safety. These elements should be optimized as an integrated framework because catalytic potency, tumor selectivity, biocompatibility, predictable in vivo behavior, and therapeutic safety are jointly determined by their coordination.
Ferroptosis in Lung Cancer and Nanozyme-Based Intervention
Ferroptosis is a form of regulated cell death characterized by the uncontrolled accumulation of iron-dependent lipid peroxidation, offering an alternative metabolic intervention strategy in lung cancer that is distinct from the induction of classical apoptosis. Nanozymes can amplify oxidative reactions within tumor lesions, deplete reducing capacity, and weaken GPX4-related defense systems, thereby increasing the susceptibility of lung cancer cells to ferroptosis. In the context of lung cancer, the significance of this strategy extends beyond direct tumor cell killing, as it is also closely associated with radiotherapeutic responsiveness, metabolic stress, and the feasibility of local administration. This section focuses on how nanozymes promote ferroptosis, what evidence is sufficient to support such a conclusion, and the translational implications of this strategy in lung cancer (Figure 2). Ferroptosis is closely related to lung cancer progression and treatment resistance because lung cancer cells frequently develop enhanced antioxidant capacity, altered iron metabolism, increased lipid remodeling, and adaptive redox buffering during tumor evolution and therapeutic pressure.17 Upregulation of SLC7A11, GPX4, FSP1, DHODH, or NRF2-related antioxidant pathways can suppress lipid peroxide accumulation and allow tumor cells to survive chemotherapy, radiotherapy, targeted therapy, or immune stress.83,84 Conversely, weakening these defenses can lower the threshold for lipid peroxidation-driven cell death and may resensitize resistant lung cancer cells.85 Therefore, ferroptosis should be viewed not only as a terminal death phenotype, but also as a metabolic vulnerability linked to tumor progression, stress adaptation, and therapeutic resistance.
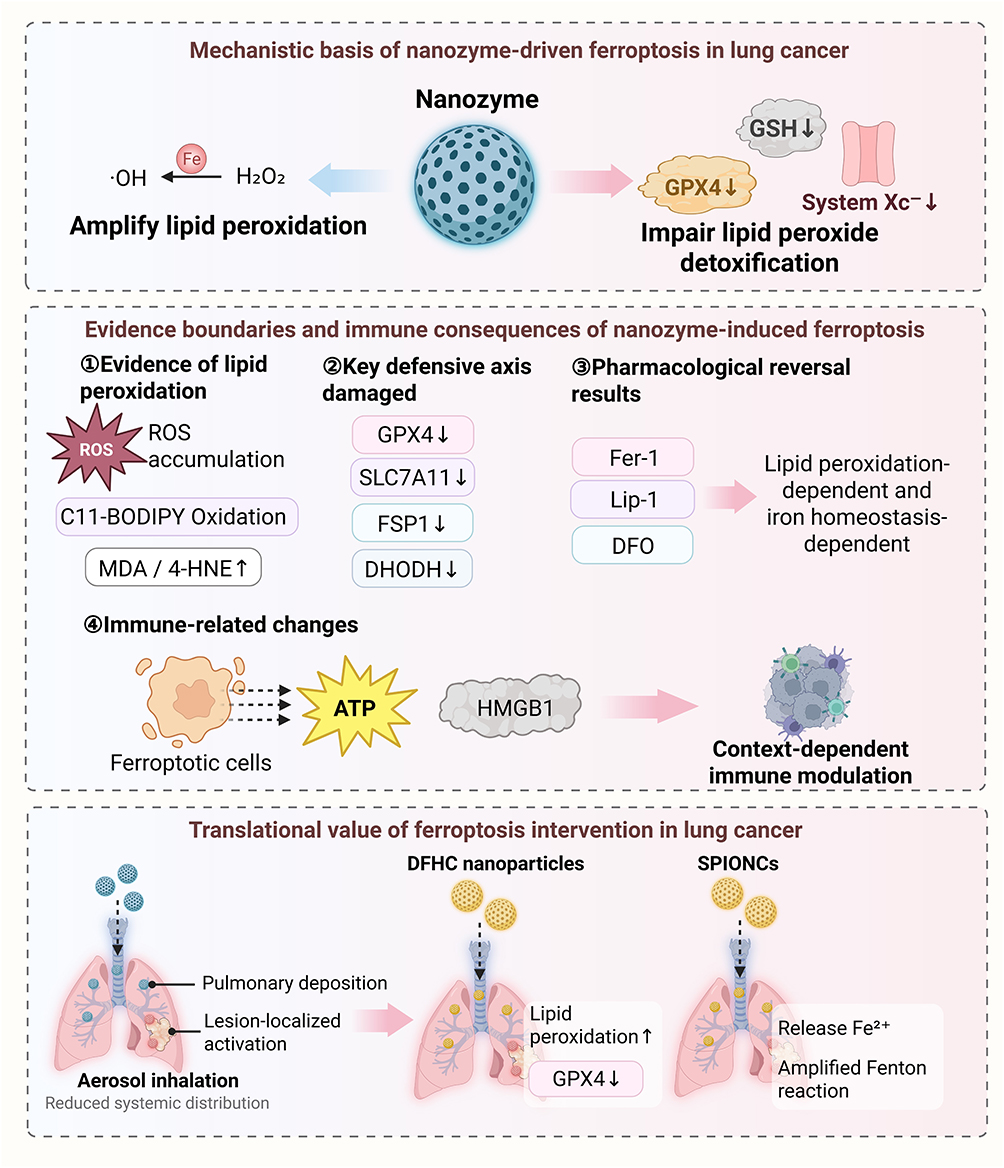 |
Figure 2 Mechanistic basis, evidentiary framework, and translational relevance of nanozyme-driven ferroptosis in lung cancer. Nanozymes induce ferroptosis by amplifying iron-dependent lipid peroxidation and impairing lipid peroxide detoxification through disruption of the System Xc−-GSH-GPX4 axis. Mechanistic assignment requires convergent evidence, including lipid peroxidation readouts, disruption of key defense pathways, and rescue by ferroptosis inhibitors or iron chelation. Nanozyme-induced ferroptosis may also be accompanied by ATP and HMGB1 release, indicating context-dependent immune modulation. In lung cancer, translational relevance is strengthened by pulmonary delivery strategies, including aerosol inhalation, that enhance lesion-confined activation while limiting systemic exposure. |
How Nanozymes Potentiate Ferroptosis
The ability of nanozymes to promote ferroptosis depends fundamentally on their capacity to sustain lipid peroxidation while impairing its detoxification. Iron-dependent catalytic reactions can convert H2O2 into hydroxyl radicals, thereby driving the chain amplification of membrane lipid peroxidation. Concurrently, GSH depletion, inhibition of GPX4, and impairment of System Xc− function further compromise the cellular capacity to buffer lipid peroxides.86–88 Thus, in lung cancer, the core feature of ferroptosis is not a generalized increase in ROS, but the sustained destabilization of the cellular defense system against lipid peroxidation. Nanozyme-based systems can enhance ferroptosis through several coordinated catalytic mechanisms. Iron-containing nanozymes or iron-releasing platforms can increase the labile iron pool and drive Fenton or Fenton-like reactions, thereby converting endogenous H2O2 into hydroxyl radicals and promoting lipid peroxide propagation. Nanozymes with POD-like or OXD-like activities can further amplify ROS generation, whereas GSHOx-like activity, GSH-responsive degradation, or catalytic GSH consumption can weaken cellular reductive capacity.89 In parallel, inhibition or downregulation of GPX4 and SLC7A11 prevents detoxification of lipid hydroperoxides and cystine-dependent GSH synthesis, thereby converting oxidative pressure into ferroptosis rather than nonspecific ROS injury.90 Selective ferroptosis induction therefore depends on the coordinated control of iron availability, lipid peroxidation, ROS production, GSH depletion, and GPX4/SLC7A11 pathway suppression within tumor cells.91
This process has been directly supported in lung cancer models. HSA@Pt(IV) nanoparticles have been shown to induce GSH depletion, prostaglandin-endoperoxide synthase 2 (PTGS2) upregulation, and lipid ROS accumulation in non-small cell lung cancer (NSCLC) cells, while their cytotoxic effects can be markedly reversed by ferrostatin-1 (Fer-1), liproxstatin-1 (Lip-1), and deferoxamine (DFO), indicating that ferroptosis is the predominant mode of cell death.92 Likewise, Cu2O@Au cascade nanozymes can enhance the sensitivity of lung cancer cells to ferroptosis by increasing H2O2 supply, depleting GSH, and downregulating solute carrier family 7 member 11 (SLC7A11) and GPX4.93 These findings indicate that nanozymes do not merely provoke isolated oxidative injury in lung cancer, but instead induce a coordinated state characterized by increased lipid peroxidation and concurrent weakening of antioxidant defense.
Evidence for Ferroptosis and Associated Immune Changes
In lung cancer, ferroptosis is not governed exclusively by the System Xc−–GSH–GPX4 axis. Ferroptosis suppressor protein 1–coenzyme Q (FSP1-CoQ) and dihydroorotate dehydrogenase (DHODH) also contribute to the restriction of lipid peroxidation, indicating that ferroptosis often arises from the simultaneous disruption of multiple layers of antioxidant defense.94,95 Accordingly, the conclusion that nanozymes induce ferroptosis should not rest on changes in a single molecular marker, but instead should be based on an integrated assessment incorporating evidence of lipid peroxidation, impairment of key defensive pathways, and pharmacological rescue results.
From the perspective of evidentiary standards, an increase in ROS or a reduction in GSH alone is insufficient to substantiate ferroptosis. More convincing evidence should include indicators of membrane lipid peroxidation, such as lipid ROS accumulation, C11-BODIPY oxidation, and elevated malondialdehyde (MDA) or 4-hydroxynonenal (4-HNE), interpreted together with alterations in critical defense regulators including GPX4, SLC7A11, FSP1, and DHODH.96 At the same time, reversal by Fer-1, Lip-1, or DFO remains highly informative, as such findings directly support dependence on lipid peroxidation and iron homeostasis.97 This point warrants particular emphasis in lung cancer research, because the lung is chronically exposed to an oxygen-exchange environment, making nonspecific oxidative stress more liable to be misinterpreted as ferroptosis. Compared with cuproptosis, the evidentiary standard for ferroptosis should place greater emphasis on lipid peroxidation specific injury and iron dependence.17,98 Therefore, nanozyme induced ferroptosis should be supported by a combination of lipid ROS accumulation, C11 BODIPY oxidation, MDA or 4 HNE elevation, suppression of key defensive regulators such as GPX4, SLC7A11, FSP1, or DHODH, and rescue by Fer 1, Lip 1, or iron chelation.99,100 In contrast, total ROS elevation, GSH depletion, or mitochondrial dysfunction alone should be interpreted as oxidative stress rather than definitive evidence of ferroptosis.91,101 Recent lung cancer evidence further supports the need to distinguish lipid peroxidation specific ferroptosis from generalized oxidative stress. Zhang et al reported that diethyldithiocarbamate copper (CuET) promoted both cuproptosis and ferroptosis in lung cancer cells by directly interacting with polyunsaturated phospholipids, increasing lipid peroxidation, and inducing copper mediated cell death. This finding suggests that copper associated stress can intersect with lipid peroxidation dependent cell death, but it also reinforces the need to validate ferroptosis through lipid specific evidence rather than through total ROS elevation alone.102
The significance of nanozyme-induced ferroptosis is not confined to direct tumor cell killing. Previous studies have shown that ferroptotic cells may be accompanied by the release of adenosine triphosphate (ATP), high-mobility group box 1 (HMGB1), and alterations in oxidized lipid-associated phagocytic signaling, thereby influencing subsequent antigen processing and immune recognition.103,104 However, these changes should not be equated directly with stable immune sensitization. Under different conditions, dysregulated lipid metabolism may also provoke inflammatory tolerance or immunosuppressive signaling, indicating that the immune consequences of ferroptosis are strongly context dependent.105,106 In lung cancer, both the direction and magnitude of these effects may further be shaped by molecular background, local inflammatory status, and treatment stage. Therefore, ferroptosis-associated immune alterations are more appropriately interpreted as accompanying phenomena than as stable and reproducible indicators of immune sensitization.
Translational Value of Ferroptosis-Based Intervention in Lung Cancer
In lung cancer, the true translational value of ferroptosis-based intervention depends on confining the relevant reactions as much as possible to pulmonary lesions, thereby maximizing local cytotoxic efficacy while minimizing systemic toxicity. Because lung cancer is characterized by both sustained oxygen exposure and accessibility to local administration, the design of nanozyme platforms should prioritize organ-directed targeting and intralesional activation rather than merely pursuing greater catalytic potency. Compared with intravenous administration, aerosol inhalation can enhance pulmonary deposition, reduce systemic distribution, and provide a more favorable local setting for the amplification of ferroptosis.107–109
This strategy has already been supported in lung cancer models. DFHC nanoparticles, a deep-lung-deliverable ferroptosis-inducing nanoplatform, achieved deep lung deposition and prolonged retention, accompanied by enhanced lipid peroxidation and downregulation of GPX4, thereby suppressing pulmonary tumor growth.110 Similarly, pH-responsive superparamagnetic iron oxide nanoclusters (SPIONCs), administered intratracheally, were delivered to the bronchial epithelium and deeply situated lung tumor tissues, where they released Fe2⁺ in the acidic microenvironment and amplified the Fenton reaction, resulting in a clearly ferroptosis-dependent radiosensitizing effect.78 These findings indicate that the significance of local delivery extends beyond improving drug deposition; more importantly, it enables ferroptosis-related catalytic reactions to be confined within a more controllable range of pulmonary lesions.
From a therapeutic perspective, nanozyme induced ferroptosis should not be viewed as a replacement for cisplatin, radiotherapy, targeted therapy, or immune checkpoint blockade, but rather as a local catalytic sensitization strategy.111 By amplifying lipid peroxidation, weakening antioxidant defenses, and increasing immunogenic stress within pulmonary lesions, ferroptosis inducing nanozymes may complement established treatments.112 Their translational value should therefore be judged by whether they enhance treatment responsiveness, overcome redox adaptation related resistance, and preserve normal lung safety.113,114
From a clinical translation perspective, the value of ferroptosis-based intervention lies in targeting residual, stress-adapted, and treatment-resistant tumor cell populations rather than replacing established anticancer therapies. Lung cancer recurrence and poor response to conventional therapy are often associated with enhanced antioxidant defense, redox adaptation, altered iron handling, lipid metabolic remodeling, and persistence of therapy-tolerant cell states.115 Ferroptosis-inducing strategies may counter these adaptive programs by lowering the threshold for lipid peroxidation-driven death, thereby improving sensitivity to chemotherapy, radiotherapy, targeted therapy, and immune checkpoint blockade.116 Nanozyme-based platforms are particularly relevant in this setting because they can integrate local ROS generation, iron-dependent Fenton or Fenton-like reactions, GSH depletion, GPX4/SLC7A11 pathway disruption, and tumor-responsive activation within one system.89,117 However, clinical applicability requires more than short-term tumor inhibition. Future studies should evaluate durable response, suppression of resistant clones, recurrence prevention, compatibility with standard regimens, biomarker-guided patient selection, immune consequences, long-term lung toxicity, degradation and clearance behavior, and preservation of respiratory function.
From a translational perspective, nanozyme-based delivery systems in lung cancer still face several organ-specific constraints, including mucociliary clearance, phagocytic uptake by alveolar macrophages, and the tolerance threshold of normal pulmonary epithelium to oxidative stress. Unless these factors are incorporated systematically into platform design, so-called lung cancer-oriented strategies may remain little more than a straightforward extension of general solid tumor platforms to the pulmonary setting.118 Accordingly, nanozyme platforms with genuine translational relevance are not those with the strongest catalytic activity, but those capable of establishing a stable balance among local delivery, lesion confinement, and safety boundaries. In lung cancer, local delivery is not a secondary optimization, but a prerequisite for rendering ferroptosis-based therapy feasible. Minimizing toxicity to normal lung tissue requires that ferroptosis-inducing nanozymes combine tumor-preferential deposition, lesion-responsive activation, controlled iron release, limited systemic leakage, and degradable or clearable material design.111 Because normal lung tissue is continuously exposed to oxygen and contains delicate epithelial, endothelial, surfactant, and immune interfaces, uncontrolled ROS generation or lipid peroxidation may cause epithelial injury, inflammation, vascular leakage, or fibrotic remodeling.119 Therefore, ferroptosis-oriented nanozyme systems should be evaluated by tumor-to-normal lung distribution, lipid peroxidation in adjacent lung tissue, inflammatory response, respiratory function, degradation kinetics, and pathway-specific rescue evidence rather than by tumor growth inhibition alone.
From a design perspective, improving ferroptosis-based nanozyme therapy requires the integration of tumor selectivity, catalytic efficiency, and biosafety. Tumor selectivity can be enhanced through pulmonary local delivery, ligand-mediated targeting, charge-switchable surfaces, and activation by tumor microenvironmental cues such as acidic pH, excess H2O2, high GSH levels, or hypoxia.120 Catalytic efficiency can be optimized by regulating metal valence states, coordination structures, active-site exposure, electron transfer, and cascade reactions that couple ROS generation with GSH depletion and lipid peroxidation.121 Biosafety requires controlled iron release, spatial confinement of catalytic activity, limited systemic leakage, biodegradable or clearable material design, and minimal lipid peroxidation in adjacent normal lung tissue.122 Therefore, the optimal ferroptosis-oriented nanozyme platform should not simply maximize ROS production, but should achieve tumor-confined lipid peroxidation with preserved pulmonary tolerance.
Cuproptosis in Lung Cancer with Evidence and Translational Constraints
Cuproptosis is a form of regulated cell death triggered by aberrant intracellular copper accumulation and characterized by the aggregation of lipoylated TCA cycle proteins and the loss of iron-sulfur (Fe-S) cluster proteins.13 Unlike generalized metal toxicity, cuproptosis depends on a specific mitochondrial metabolic state and is associated with relatively well-defined molecular hallmarks. For nanozyme-based platforms, the central issue is not merely how to increase local copper burden, but whether copper-related stress can be preferentially confined to tumor tissue through intralesional activation, metabolic reprogramming, and localized delivery. Compared with ferroptosis, research on cuproptosis in lung cancer remains at an early stage. At present, the most prominent challenges lie not in the conceptual framework itself, but in the absence of established evidentiary standards, the lack of clearly defined susceptible populations, and the underdevelopment of effective delivery strategies (Figure 3).
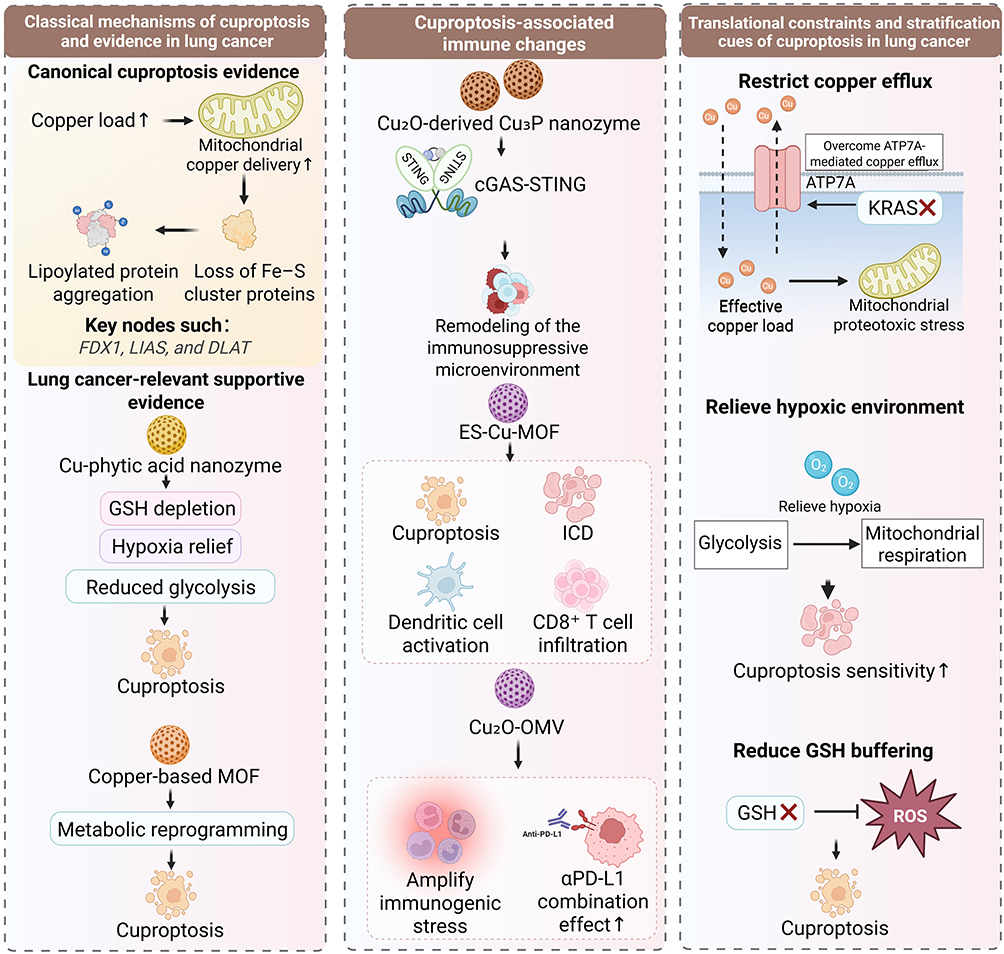 |
Figure 3 Canonical mechanisms, immune associations, and translational constraints of cuproptosis in lung cancer. Canonical cuproptosis is defined by increased effective copper burden, enhanced mitochondrial copper delivery, aggregation of lipoylated proteins, and loss of Fe-S cluster proteins, with FDX1, LIAS, and DLAT as key nodes. Emerging evidence in lung cancer suggests that copper-based nanozymes may promote cuproptosis through glutathione depletion, hypoxia relief, and metabolic reprogramming. Reported immune associations include cGAS-STING activation, dendritic-cell activation, increased CD8-positive T-cell infiltration, and remodeling of the immunosuppressive microenvironment, although these effects cannot yet be assigned specifically to cuproptosis. Effective translation will likely require restricting copper efflux, relieving hypoxia, and weakening glutathione buffering to increase intralesional copper stress and cuproptosis sensitivity. |
Canonical Mechanisms of Cuproptosis and Evidence in Lung Cancer
Nanoplatforms with the potential to induce cuproptosis generally need to satisfy at least one of the following conditions, increasing the effective intracellular copper burden, enhancing mitochondrial copper delivery, or increasing the exposure of lipoylated proteins. Certain copper-based nanozymes can serve as local copper sources to elevate intracellular copper concentrations, while also exacerbating copper homeostatic disruption and accelerating reductive depletion through GSH-mediated Cu2⁺/Cu⁺ conversion.123 Other platforms increase tumor cell susceptibility to cuproptosis by alleviating hypoxia, suppressing glycolysis, or enhancing dependence on mitochondrial respiration.124 However, these changes alone cannot be regarded as direct evidence of cuproptosis.
More convincing evidence should include the aggregation of lipoylated TCA cycle proteins, loss of Fe-S cluster proteins, and abnormalities in key regulators such as ferredoxin 1 (FDX1), lipoic acid synthetase (LIAS), and dihydrolipoamide S-acetyltransferase (DLAT); reversibility by copper chelation should also be considered an essential validation step.13 By contrast, elevated ROS, GSH depletion, mitochondrial injury, or generalized metabolic collapse only indicate the presence of copper-related stress and cannot substitute for canonical evidence of cuproptosis. This evidentiary threshold differs from that of ferroptosis.16 Whereas ferroptosis should be validated primarily by lipid peroxidation specific damage and iron dependent rescue, cuproptosis requires evidence of mitochondrial copper dependent proteotoxic stress. Accordingly, aggregation of lipoylated TCA cycle proteins, loss of Fe S cluster proteins, FDX1, LIAS, and DLAT related changes, and reversibility by copper chelation should be regarded as the core evidence for cuproptosis.32 Without these criteria, copper loading, ROS accumulation, GSH consumption, or mitochondrial injury should be described more cautiously as copper associated stress rather than cuproptosis.125,126
Direct evidence in lung cancer has begun to emerge. A pH/GSH dual-responsive Cu-phytic acid nanozyme has been shown to enhance cuproptosis in elesclomol-resistant A549 cells through GSH depletion, relief of hypoxia, and attenuation of glycolysis, while exhibiting antitumor effects both in vitro and in vivo.127 In NSCLC models, copper-based metal-organic frameworks (MOFs) have also demonstrated the potential to increase susceptibility to cuproptosis through metabolic reprogramming.128 These findings indicate that research on cuproptosis in lung cancer has progressed beyond conceptual proof-of-principle, although substantial distance remains before stable and comparable evidentiary standards can be established.
Cuproptosis-Associated Immune Changes
Emerging evidence suggests that copper-related nanoplatforms often induce a degree of immune modulation while eliciting cuproptosis-associated stress. In lung cancer models, a Cu2O-derived Cu3P nanozyme was shown to enhance cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) signaling and ameliorate the immunosuppressive microenvironment, suggesting that copper-related platforms may hold promise for combination with immunotherapy.129 In other tumor models, elesclomol-loaded copper-based metal-organic framework (ES-Cu-MOF) has been reported to trigger immunogenic cell death (ICD) while inducing cuproptosis, accompanied by dendritic cell activation and increased infiltration of CD8-positive T cells.130 Similarly, Cu2O-OMV has been shown to potentiate immunogenic stress through cooperation with other cell death programs and to enhance the therapeutic efficacy of anti-programmed death-ligand 1 (αPD-L1) treatment.131
However, these observations remain insufficient to be directly attributed to cuproptosis itself. In most systems within this field, cuproptosis occurs alongside ferroptosis, pyroptosis, ROS-induced ICD, or activation of the cGAS-STING pathway. Consequently, the observed immune effects are more likely to reflect the integrated output of broader immunogenic stress rather than the isolated consequence of cuproptosis alone.129 Thus, although cuproptosis-associated immune consequences merit close attention, they remain inadequate at present for defining a clear mechanistic boundary. For this field, the more critical question is not whether copper-related platforms can provoke immune alterations, but whether these changes exhibit a stable, separable, and reproducibly verifiable relationship with cuproptosis.
In addition, copper related proteotoxic stress does not necessarily lead to immune activation in all contexts.32 Excessive copper burden, mitochondrial injury, or tissue damage may also contribute to tolerogenic or immunosuppressive responses, including IL 10, TGF β, or lipid mediator associated signaling.132,133 Therefore, immune modulation induced by copper related platforms should be interpreted cautiously and validated by changes in antigen presentation, dendritic cell activation, T cell infiltration, cytokine profile, and therapeutic response rather than inferred solely from the presence of cuproptosis associated stress.19,64
Translational Constraints and Stratification Clues in Lung Cancer
Compared with ferroptosis, the translational investigation of cuproptosis in lung cancer remains substantially less developed. The principal limitation lies not simply in whether copper exposure can be increased, but in whether an effective intralesional copper burden sufficient to trigger mitochondrial proteotoxic stress can be achieved without disrupting copper homeostasis in normal tissues. Copper is not a tumor-specific element; normal tissues likewise depend on tightly regulated systems of transport, chelation, and efflux to maintain homeostasis.10 Even when tumor cells exhibit increased copper demand, intracellular copper accumulation may still be constrained by transport systems such as ATPase copper transporting alpha (ATP7A), thereby diminishing the practical efficacy of exogenous copper delivery.134,135
More importantly, cuproptosis directly targets lipoylated TCA cycle proteins, and its induction is highly dependent on mitochondrial respiratory status and metabolic context.136 Consequently, not all lung cancer subtypes are likely to exhibit equivalent susceptibility. Previous studies have shown that alleviating hypoxia and shifting cellular metabolism from glycolysis toward mitochondrial respiration can significantly enhance cuproptosis.124 In addition, KRAS-driven lung adenocarcinoma appears to depend on ATP7A-mediated copper homeostasis, and elesclomol can induce copper accumulation and cuproptosis by disrupting this axis, suggesting that the regulation of copper transport may itself represent a therapeutically actionable vulnerability.137 Furthermore, although the Phase III trial of elesclomol did not demonstrate an overall survival benefit, post hoc analyses suggested that patients with low plasma lactate dehydrogenase (LDH) levels might derive greater benefit, implying that LDH may serve as a stratification marker reflecting dependence on mitochondrial metabolism.138 Collectively, these findings indicate that cuproptosis in lung cancer is fundamentally an inducible process contingent on metabolic background, rather than a universal phenomenon that can be triggered with equal efficiency across all NSCLCs.
Accordingly, cuproptosis-based intervention in lung cancer should not remain limited to the simplistic intensification of copper delivery, but should instead advance toward more precise platform design, metabolic stratification, and localized delivery strategies. The aggregation of lipoylated proteins, loss of Fe-S cluster proteins, and reversibility by copper chelation should remain the fundamental criteria for establishing cuproptosis in lung cancer models, and should not be replaced by nonspecific findings such as increased ROS, GSH depletion, or mitochondrial injury.139 From the perspective of platform design, the more meaningful objective is not the indiscriminate elevation of copper burden, but the simultaneous resolution of key barriers, including copper efflux, GSH buffering, and hypoxic limitation, so as to increase effective intralesional copper exposure and intensify mitochondrial proteotoxic stress.140 This requirement also explains why localized delivery is particularly relevant to cuproptosis based intervention. Because copper is an essential but tightly regulated element in normal tissues, systemic copper loading may increase off target toxicity without ensuring sufficient mitochondrial copper accumulation in tumor cells. Local pulmonary delivery may help increase the lesion to normal tissue exposure ratio and make copper associated proteotoxic stress more spatially controllable. In lung cancer, the translational value of cuproptosis lies not in further amplifying nonspecific copper toxicity, but in developing intervention strategies that can be integrated with local delivery, metabolic stratification, and rational combination therapy. This point is also supported by recent small molecule evidence in lung cancer. Bai et al identified melampomagnolide B derivatives that triggered cuproptosis, ferroptosis, and apoptosis in lung cancer models, accompanied by mitochondrial dysfunction and ROS generation. Although this study was not based on a nanozyme platform, it provides direct lung cancer evidence that therapeutic benefit may arise from coordinated induction of multiple death programs. At the same time, it highlights the need to distinguish true pathway specific death from broader cytotoxic stress when evaluating ferroptosis and cuproptosis based strategies.141
Nanozyme-Mediated Synergistic Intervention Through Ferroptosis and Cuproptosis
The significance of ferroptosis and cuproptosis in lung cancer lies not merely in the possibility that these two forms of cell death can be induced concurrently, but more importantly in the capacity of nanozymes to simultaneously modulate redox balance, metal ion homeostasis, and mitochondrial proteostasis within the same tumor lesion. This is particularly relevant in lung cancer, where therapeutic resistance is frequently accompanied by enhanced antioxidant defenses, altered mitochondrial metabolism, and persistent immune suppression. This section focuses on the biological basis of dual-death synergy, representative nanozyme-based strategies, and their potential immunological implications (Figure 4). Building on the design principles summarized in Nanozyme Design for Ferroptosis–Cuproptosis Regulation, this section emphasizes the biological logic of ferroptosis–cuproptosis interaction, including shared limiting factors, pathway specific validation, potential antagonism, and immune consequences, rather than repeating detailed material design features.
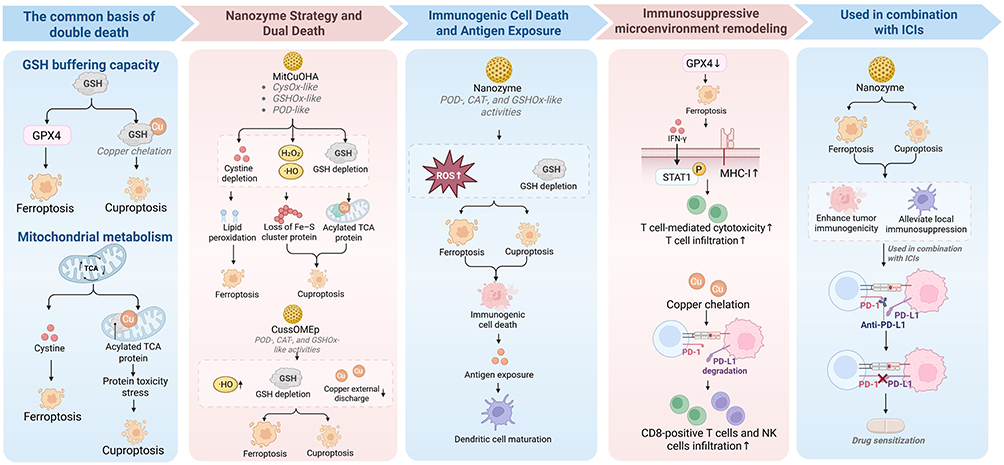 |
Figure 4 Biological basis, immunological consequences, and combination potential of nanozyme-mediated ferroptosis-cuproptosis synergy in lung cancer. Shared susceptibility to dual cell death arises from glutathione buffering, mitochondrial metabolism, and metal homeostasis. Representative nanozyme platforms can simultaneously promote ferroptosis and cuproptosis by amplifying oxidative stress, depleting glutathione, restricting copper efflux, and intensifying mitochondrial proteotoxic stress. These changes may enhance ICD, increase antigen exposure, and promote dendritic-cell maturation. Dual-death signaling may also remodel the immunosuppressive microenvironment by strengthening T-cell-mediated cytotoxicity, increasing CD8-positive T-cell and natural killer cell infiltration, and facilitating PD-L1 degradation. Together, these effects provide a mechanistic rationale for combining dual-death nanozyme strategies with immune checkpoint blockade. |
Shared Biological Basis of Dual Cell Death
The simultaneous enhancement of ferroptosis and cuproptosis in lung cancer is rooted in the fact that both processes are influenced by mitochondrial metabolism, metal homeostasis, and GSH buffering capacity. Ferroptosis is primarily characterized by the accumulation of lipid peroxidation and the collapse of membrane lipid antioxidant defenses, whereas cuproptosis is defined by the aggregation of lipoylated TCA cycle proteins, loss of Fe–S cluster proteins, and proteotoxic stress. Although their direct molecular targets differ, these two death programs are not biologically independent. Previous studies have shown that sorafenib and erastin, while intensifying ferroptosis-associated stress, can also inhibit FDX1 protein degradation and reduce intracellular GSH synthesis, thereby enhancing elesclomol-induced lipoylated protein aggregation and cuproptosis.142 This suggests that the concurrent emergence of these two forms of cell death is often not a phenotypic coincidence, but rather the consequence of simultaneously overcoming multiple shared constraints.
This common foundation is first reflected at the level of reductive metabolism. GSH not only restricts ferroptosis through the GSH–GPX4 axis, but also buffers cuproptosis through copper chelation; accordingly, GSH depletion often increases susceptibility to both forms of cell death simultaneously.143,144 Mitochondrial status is likewise critical. Cuproptosis depends on copper binding to lipoylated TCA cycle proteins and the subsequent induction of proteotoxic stress, whereas cystine deprivation-induced ferroptosis is also regulated by TCA cycle activity and mitochondrial respiratory status.13,145 Therefore, in discussing dual-death synergy in lung cancer, the central issue is not merely whether ferroptosis and cuproptosis occur concurrently, but whether they share a set of limiting factors that can be modulated simultaneously by nanozyme-based platforms. Only when GSH buffering, mitochondrial metabolism, and metal homeostasis are disrupted in parallel does the concept of dual-death synergy acquire a coherent biological basis. However, ferroptosis and cuproptosis may not always be cooperative. Excessive lipid peroxidation may impair mitochondrial respiration and thereby reduce cuproptosis susceptibility. Thus, concurrent dual death activation should be regarded as context dependent and validated by mitochondrial function assays, temporal evidence, and pathway specific rescue studies.
Representative Nanozyme Strategies and the Initiation of Dual Cell Death
Whether ferroptosis and cuproptosis can be simultaneously intensified within a single nanoplatform depends not on the mere co-incorporation of iron (Fe) and copper (Cu), but on whether the system can, in a sustained manner, elevate ROS, deplete GSH, restrict copper efflux, and exacerbate mitochondrial stress within the same biological context.146 Iron-containing components can promote ferroptosis by driving lipid peroxidation through Fenton or Fenton-like reactions, whereas copper-containing components are more likely to facilitate cuproptosis through proteotoxic stress associated with lipoylated proteins. Truly representative dual-death platforms are therefore not simple elemental combinations, but integrated systems capable of simultaneously weakening reductive defenses, intensifying oxidative stress, and increasing the proteostatic burden on mitochondria within the same tumor lesion.
Representative dual-death platforms illustrate that ferroptosis–cuproptosis interaction depends on more than the co-delivery of iron and copper elements. Platforms such as MitCuOHA, CussOMEp, and mitochondria-targeted Fe/Cu bimetallic systems can combine ROS amplification, GSH depletion, copper retention, and mitochondrial stress within the same biological context.64,147,148 However, in Nanozyme-Mediated Synergistic Intervention Through Ferroptosis and Cuproptosis, the key issue is not the material architecture itself, but whether these outputs produce pathway-specific and functionally connected cell death. Therefore, evidence for lipid peroxidation, lipoylated protein aggregation, Fe-S cluster loss, mitochondrial dependence, and pathway-specific rescue should be considered together before defining these systems as true ferroptosis–cuproptosis synergy.
The timing and subcellular localization of nanozyme activity may determine whether ferroptosis and cuproptosis act cooperatively or antagonistically.148 Severe lipid peroxidation may impair mitochondrial respiration and reduce cuproptosis susceptibility.149 Thus, simultaneous iron and copper delivery is insufficient to prove synergy without evidence of temporal coordination, mitochondrial dependence, and pathway specific rescue.
It should be noted, however, that the current literature does not always provide a robust basis for concluding true dual-death synergy. Many platforms simultaneously induce ROS accumulation, metabolic collapse, and additional cell death programs, such that the so-called coexistence of ferroptosis and cuproptosis may, in some cases, more accurately reflect a consequence of convergent stress rather than genuine co-dominance of the two pathways. In particular, when parallel evidence for lipoylated protein aggregation, Fe-S cluster loss, reversibility by copper chelation, and lipid peroxidation specificity is lacking, defining such mixed stress responses as dual-death synergy still carries a substantial risk of overinterpretation.150 Therefore, concurrent dual-death activation should be distinguished from true synergistic interaction. Concurrent activation indicates the coexistence of ferroptosis- and cuproptosis-related changes, whereas synergy requires evidence that one pathway enhances the other or that shared limiting factors are jointly disrupted to produce greater biological effects. Without pathway-specific rescue, temporal coordination, mitochondrial dependence, or quantitative enhancement beyond parallel stress responses, such platforms should be described as concurrent activation or putative synergy rather than definitive synergy.
ICD and Antigen Visibility
The immunological significance of dual-death nanozyme-based intervention lies, first and foremost, in its potential to convert metabolic collapse into enhanced antigen visibility and more robust release of danger-associated molecular signals. ROS are important inducers of ICD, and nanozymes can sustain ROS generation and deplete GSH through POD-, CAT-, and GSHOx-like activities, thereby creating conditions conducive to the release of damage-associated molecular patterns (DAMPs) and the exposure of tumor antigens.150 Consistent with this concept, a recent Nature Communications study reported that a quasi Fe MIL-53 nanozyme induced ferroptosis and immunogenic cell death, promoted dendritic cell maturation and T lymphocyte infiltration, and enhanced the antitumor efficacy of programmed cell death protein 1 antibody. This evidence supports the immunological relevance of nanozyme induced ferroptosis, although it should be interpreted as ferroptosis associated immune sensitization rather than direct proof of ferroptosis cuproptosis synergy. When this process is further accompanied by ferroptosis or cuproptosis, the resulting cell death is more likely to assume a form that can be recognized by the immune system.
Relevant evidence has begun to emerge. CuNTD has been shown to simultaneously induce ferroptosis and cuproptosis through ROS amplification, thereby enhancing ICD and promoting dendritic cell maturation.151 In addition, NP@ESCu has demonstrated a degree of immune-activating potential while inducing cuproptosis.152 These findings suggest that dual-death nanozyme-based intervention may not only enhance tumor cell killing, but also increase the immunological visibility of the death process, thereby creating favorable conditions for dendritic cell activation and the subsequent initiation of adaptive immunity.
Nevertheless, enhanced antigen visibility should not be equated with durable immune activation.153 Ferroptosis associated lipid peroxidation and cuproptosis associated proteotoxic stress may promote DAMP release and antigen presentation in some models, but excessive oxidative injury may also generate immunosuppressive lipid mediators, impair immune cell function, or favor IL 10 and TGF β dominated tolerogenic responses.154,155 Thus, the immune consequences of dual death induction should be evaluated by functional immune readouts rather than by ICD markers alone.64,156
Nevertheless, the current evidence more strongly supports the existence of immunogenic outcomes than it does the clear delineation of the independent contribution of each specific cell death program. In lung cancer, future studies will need to determine whether ferroptosis and cuproptosis each provide distinct and nonredundant functions in antigen priming, and whether such effects can remain stable and reproducible across different molecular backgrounds and treatment stages.
Remodeling of the Immunosuppressive Microenvironment
In lung cancer, the significance of dual-death synergy extends beyond the induction of ICD, as it may also enhance T cell-mediated antitumor activity and influence programmed death-ligand 1 (PD-L1)-associated immune evasion. Previous studies have shown that ferroptosis is closely linked to sensitivity to immunotherapy. Ras-selective lethal 3 (RSL3), a commonly used ferroptosis inducer that targets GPX4, triggers ferroptosis by promoting lipid peroxidation, increasing the intracellular labile iron pool and ROS levels, and weakening GPX4-dependent antioxidant defenses.157,158 RSL3 can enhance tumor cell sensitivity to interferon-γ (IFN-γ), promote signal transducer and activator of transcription 1 (STAT1) phosphorylation and nuclear translocation, upregulate major histocompatibility complex class I (MHC-I) expression, and strengthen T cell-mediated cytotoxicity, thereby improving the therapeutic efficacy of αPD-L1 treatment. In vivo studies have further shown that RSL3 increases intratumoral T cell infiltration and improves T cell function, suggesting that the amplification of ferroptosis-related stress may not only intensify tumor killing, but also enhance tumor immunological visibility and reinforce antitumor immune responses.159 Copper metabolism likewise participates in the regulation of immune evasion. Elevated intratumoral copper levels can upregulate PD-L1, whereas copper chelation can promote PD-L1 degradation, increase the infiltration of CD8-positive T cells and natural killer (NK) cells, and delay tumor growth.160
On this basis, multimetal nanozyme platforms have demonstrated a further capacity to modulate the tumor microenvironment. PEG@AuCZ@CC can reduce lactate accumulation through metabolic reprogramming, enhance dual-death signaling, promote dendritic cell maturation, reprogram tumor-associated macrophages (TAMs), and restore the function of CD4-positive and CD8-positive T cells.161 These findings indicate that the value of dual-death synergy lies not only in enhanced cytotoxicity, but also in its potential to convert local metabolic collapse into broader microenvironmental remodeling. In lung cancer, this process may be particularly relevant to the regulation of TAMs, regulatory T cells (Tregs), PD-L1 maintenance, and stromal barriers, thereby creating a more favorable initial context for subsequent immune checkpoint blockade. Recent evidence further indicates that altered cholesterol metabolism in aged regulatory T cells, including increased SOAT2 expression, may weaken antitumor immunity.162 This finding suggests that nanozyme-induced ferroptosis–cuproptosis intervention should not be evaluated only by tumor-cell death markers, but also by its capacity to remodel lipid-metabolic and Treg-associated immunosuppressive states within the tumor microenvironment.
However, current findings primarily suggest that dual-death signaling and microenvironmental remodeling may occur concurrently, while the causal sequence and relative contribution of each remain insufficiently dissected. On the basis of the available evidence, it is still premature to attribute all observed microenvironmental improvements directly to dual cell death itself.
Preclinical Evidence for Combination with Immune Checkpoint Inhibitors (ICIs) and Stratification Clues
Current preclinical studies suggest that platforms capable of coordinately inducing ferroptosis and cuproptosis may improve the biological basis for responsiveness to programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) blockade. However, such sensitization generally reflects the cumulative outcome of multiple immunoregulatory processes rather than a linear extension of any single death program. The theoretical rationale for combining nanozymes with ICIs lies in the ability of nanozymes to enhance tumor immunogenicity and alleviate local immune suppression, thereby creating more favorable conditions for response to PD-1 and PD-L1 blockade.
Available preclinical evidence has begun to support this combinatorial strategy. While inducing both ferroptosis and cuproptosis, dis-SAzyme-Dox@M was shown to increase tumor immunogenicity, promote infiltration of CD8-positive T cells, reduce the proportion of Tregs, and achieve a more pronounced in vivo antitumor effect when combined with αPD-L1 therapy.42 Fe/Cu-HPC@GOx/PEG, through the induction of dual death programs, amplification of ICD, and enhancement of CD8-positive T cell infiltration, has likewise demonstrated potential to overcome resistance to immunotherapy.163 In addition, CuP/Er nanoparticles have exhibited a relatively complete immune checkpoint-sensitizing cascade in non-pulmonary solid tumor models. This platform synergistically induced tumor cell death through copper-mediated cuproptosis and erastin-mediated ferroptosis, further amplified calreticulin (CRT) exposure, HMGB1 release, and ATP-associated immunogenic signals, promoted dendritic cell maturation and antigen presentation, and upregulated PD-L1 expression in tumor cells. When combined with αPD-L1 treatment, it further enhanced T cell proliferation, suppressed tumor growth, and reduced pulmonary metastasis.164 More importantly, in lung tumor models, CACuPDA synchronously enhanced ferroptosis and cuproptosis through dual depletion of GSH and GPX4 at the tumor site, accompanied by an increase in CD8-positive T cells and a reduction in Tregs, thereby significantly potentiating the therapeutic efficacy of αPD-L1 treatment.20 Representative preclinical nanozyme platforms relevant to ferroptosis–cuproptosis induction and ICI sensitization are summarized in Table 1.
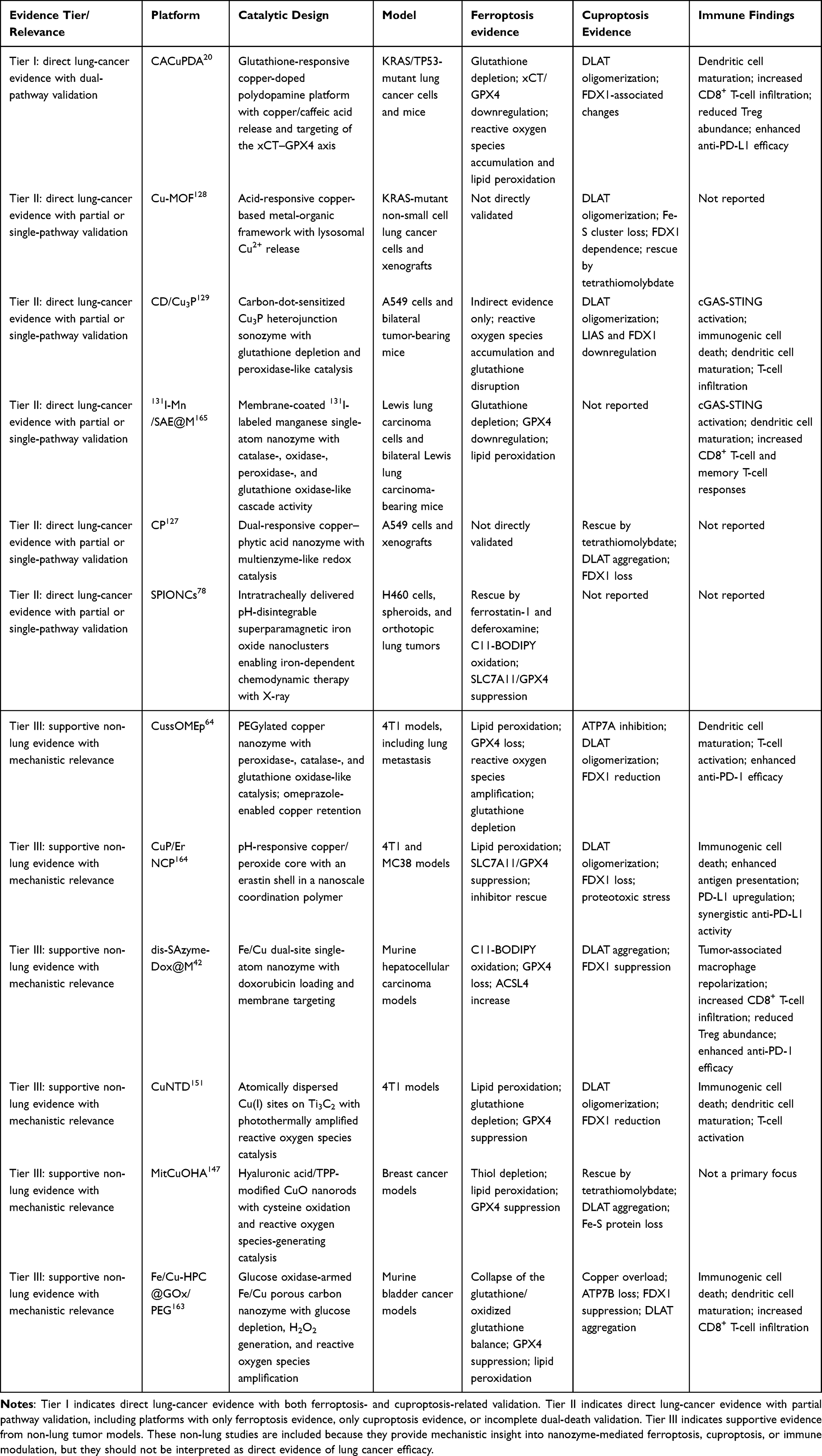 |
Table 1 Evidence-Tiered Summary of Representative Nanozyme Platforms Related to Ferroptosis, Cuproptosis, and Immune Modulation in Lung Cancer and Supportive Non-Lung Models |
At the same time, the available evidence also indicates that immune checkpoint sensitization is rarely explained by a single death program in isolation. Most combination systems are accompanied by ICD, activation of stimulator of interferon genes (STING), reprogramming of TAMs, or broader metabolic reprogramming. Accordingly, such sensitization is more likely to arise from the joint action of multiple mechanisms and should not be simplistically attributed to ferroptosis or cuproptosis alone.164 In lung cancer, the next priority should not be the continued accumulation of mechanistic layers within combination strategies, but rather the clarification of the relative contribution of cell death programs and immune remodeling across different nanozyme platforms, together with the establishment of stratification biomarkers applicable to patient selection.166,167
Current evidence further suggests that the potential benefit of combining dual-death synergy with ICIs is unlikely to be uniform, and is more plausibly shaped by molecular background and treatment stage. KRAS-mutant lung adenocarcinoma depends on ATP7A to maintain copper homeostasis, and elesclomol can induce copper accumulation and trigger cuproptosis by disrupting this axis, suggesting that a KRAS background may provide a metabolic entry point for cuproptosis-based intervention.137 By contrast, KEAP1 inactivation can upregulate FSP1 through nuclear factor erythroid 2-related factor 2 (NRF2) and activate the CoQ–FSP1 defense axis, thereby increasing ferroptosis tolerance and contributing to radioresistance, indicating that KEAP1 and NRF2 abnormalities may not only influence ferroptosis sensitivity but also constrain the therapeutic magnitude of dual-death synergy.95 In addition, epidermal growth factor receptor (EGFR)-driven lung cancer frequently exhibits enhanced oxidative phosphorylation, maintenance of Fe-S clusters, and metabolic adaptation under the selective pressure of targeted therapy, suggesting that susceptibility to ferroptosis and cuproptosis is stage-dependent and plastic.168 Anaplastic lymphoma kinase (ALK)-positive lung cancer and its resistant evolution likewise imply that dual-death sensitivity may shift during treatment, although the current evidence remains derived mainly from indirect extrapolation based on studies of lung cancer resistance and ferroptosis.114
Accordingly, the optimal integration of dual-death nanozyme-based intervention with ICIs will likely depend on stratified designs aligned with molecular background, resistance status, and treatment stage, rather than on a universal strategy applicable across all patients. Based on the currently available evidence, priority may reasonably be given to subgroups characterized by KRAS with concurrent serine/threonine kinase 11 (STK11) or KEAP1 abnormalities, resistant subtypes arising after EGFR- or ALK-targeted therapy and accompanied by enhanced mitochondrial metabolism, and tumors exhibiting substantial redox stress while still retaining local immunoreactive potential.137,169 By contrast, subgroups marked by strong NRF2- and FSP1-mediated protection, markedly enhanced copper efflux, or highly fixed immunosuppressive states may display relative resistance.170
Conclusions
Nanozyme mediated regulation of ferroptosis and cuproptosis in lung cancer is moving from the induction of individual death pathways toward integrated strategies that coordinate redox imbalance, metal homeostasis, mitochondrial metabolism, and antitumor immunity. The next stage of this field should focus less on adding increasingly complex material components and more on establishing experimentally reproducible links among nanozyme design, pathway specific cell death, pulmonary delivery, and therapeutic response.
First, clearer evidentiary standards are required. Nanozyme induced ferroptosis should be supported by lipid peroxidation specific evidence, including lipid ROS accumulation, C11 BODIPY oxidation, MDA or 4 HNE elevation, suppression of GPX4, SLC7A11, FSP1, or DHODH, and rescue by ferroptosis inhibitors or iron chelation. By contrast, cuproptosis should be validated by aggregation of lipoylated TCA cycle proteins, loss of Fe S cluster proteins, FDX1, LIAS, or DLAT related changes, and reversibility by copper chelation. For concurrent dual death activation, concurrent detection of ferroptosis associated and cuproptosis associated markers should be distinguished from true synergistic interaction. The former indicates that both pathways may be activated within the same system, whereas the latter requires evidence that the two processes are mechanistically or functionally connected. Future studies should therefore define temporal sequence, mitochondrial dependence, pathway specific rescue patterns, and, where possible, quantitative evidence showing that combined pathway activation produces effects beyond parallel nonspecific stress responses.
Second, lung cancer specific delivery and safety evaluation should be central to nanozyme development. Given continuous oxygen exposure, surfactant adsorption, mucociliary clearance, and alveolar macrophage uptake, pulmonary nanozyme systems should be assessed by aerosol stability, pulmonary deposition, intralesional retention, degradation behavior, surfactant interaction, macrophage clearance, metal accumulation, inflammation, fibrosis risk, and respiratory function. Translationally meaningful platforms should maintain controlled catalytic activation within pulmonary lesions while limiting oxidative or metal induced injury in normal lung tissue.
Third, patient stratification and rational combination therapy should be developed in parallel. Susceptibility to ferroptosis or cuproptosis is unlikely to be uniform across lung cancer subtypes. Potential stratification variables include copper transport status, ATP7A, GSH metabolism, GPX4, SLC7A11, FSP1, DHODH, FDX1, LIAS, DLAT, LDH level, mitochondrial respiratory dependence, KRAS, KEAP1, STK11, EGFR or ALK resistance status, and the immune microenvironment. Future studies should determine whether nanozyme induced ferroptosis or cuproptosis can sensitize lung cancer to cisplatin, radiotherapy, targeted therapy, or immune checkpoint blockade, and identify the molecular contexts most likely to benefit.
Overall, the clinical relevance of nanozyme based ferroptosis and cuproptosis will depend on whether this strategy can be converted from a mechanistic concept into a reproducible therapeutic framework. This will require pathway specific validation, lung focused delivery assessment, normal tissue safety evaluation, molecular stratification, and rational integration with established anticancer therapies. Only by connecting these elements can nanozyme mediated cell death regulation become a clinically actionable strategy for lung cancer.
Abbreviations
4-HNE, 4-hydroxynonenal; αPD-L1, anti-programmed death-ligand 1; ALK, anaplastic lymphoma kinase; ATP, adenosine triphosphate; ATP7A, ATPase copper transporting alpha; CAT, catalase; CoQ, coenzyme Q; CRT, calreticulin; Cys, cysteine; CysOx, cysteine oxidase; DAMPs, damage-associated molecular patterns; DFO, deferoxamine; DHODH, dihydroorotate dehydrogenase; DLAT, dihydrolipoamide S-acetyltransferase; ES-Cu-MOF, elesclomol-loaded copper-based metal-organic framework; FDX1, ferredoxin 1; Fer-1, ferrostatin-1; Fe-S, iron-sulfur; FSP1, ferroptosis suppressor protein 1; GPX4, glutathione peroxidase 4; GSH, glutathione; GSHOx, glutathione oxidase; H2O2, hydrogen peroxide; HMGB1, high-mobility group box 1; ICD, immunogenic cell death; ICIs, immune checkpoint inhibitors; IFN-γ, interferon-gamma; KEAP1, Kelch-like ECH-associated protein 1; KRAS, Kirsten rat sarcoma viral oncogene homolog; LDH, lactate dehydrogenase; LIAS, lipoic acid synthetase; Lip-1, liproxstatin-1; MDA, malondialdehyde; MHC-I, major histocompatibility complex class I; MOF, metal-organic framework; NK, natural killer; NRF2, nuclear factor erythroid 2-related factor 2; NSCLC, non-small cell lung cancer; OXD, oxidase; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PEG, polyethylene glycol; POD, peroxidase; PTGS2, prostaglandin-endoperoxide synthase 2; ROS, reactive oxygen species; RSL3, Ras-selective lethal 3; SLC7A11, solute carrier family 7 member 11; SPIONCs, superparamagnetic iron oxide nanoclusters; STAT1, signal transducer and activator of transcription 1; STING, stimulator of interferon genes; STK11, serine/threonine kinase 11; System Xc−, cystine/glutamate antiporter system; TAMs, tumor-associated macrophages; TCA, tricarboxylic acid; TME, tumor microenvironment; Tregs, regulatory T cells.
Data Sharing Statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Liaoning Provincial Science and Technology Plan Joint Program Project (2025JH2/101800202), the Chronic Disease Management Research Program of the Capacity Building and Continuing Education Center of the National Health Commission (GWJJMB202510022010), the United Foundation for the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, and the Second Hospital of Dalian Medical University (DMU-2&DICP UN202505), and the Chinese Young Medical Innovation Research Project (P250921129238).
Disclosure
The authors declare that they have no competing interests.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020, GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–28. doi:10.3322/caac.21660
2. Suresh K, Naidoo J, Lin CT, Danoff S. Immune checkpoint immunotherapy for non-small cell lung cancer, benefits and pulmonary toxicities. Chest. 2018;154(6):1416–1423. doi:10.1016/j.chest.2018.08.1048
3. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
4. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y
5. Guo Z, Chen D, Yao L, et al. The molecular mechanism and therapeutic landscape of copper and cuproptosis in cancer. Signal Transduction Targeted Ther. 2025;10(1):149. doi:10.1038/s41392-025-02192-0
6. Fernández-García R, Fraguas-Sánchez AI. Nanomedicines for pulmonary drug delivery, overcoming barriers in the treatment of respiratory infections and lung cancer. Pharmaceutics. 2024;16(12):1584. doi:10.3390/pharmaceutics16121584
7. Ren X, Chen D, Wang Y, et al. Nanozymes-recent development and biomedical applications. J Nanobiotechnol. 2022;20(1):92. doi:10.1186/s12951-022-01295-y
8. Zhang Y, Yu W, Chen M, Zhang B, Zhang L, Li P. The applications of nanozymes in cancer therapy, based on regulating pyroptosis, ferroptosis and autophagy of tumor cells. Nanoscale. 2023;15(29):12137–12156. doi:10.1039/d3nr01722b
9. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. 2018;36:489–517. doi:10.1146/annurev-immunol-042617-053010
10. Tian Z, Jiang S, Zhou J, Zhang W. Copper homeostasis and cuproptosis in mitochondria. Life Sci. 2023;334:122223. doi:10.1016/j.lfs.2023.122223
11. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–396. doi:10.1038/s41568-022-00459-0
12. Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG, Gao LC. System xc -/GSH/GPX4 axis, an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13:910292. doi:10.3389/fphar.2022.910292
13. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
14. Zhong G, Li Y, Ma F, et al. Copper exposure induced chicken hepatotoxicity, involvement of ferroptosis mediated by lipid peroxidation, ferritinophagy, and inhibition of FSP1-CoQ10 and Nrf2/SLC7A11/GPX4 axis. Biol Trace Elem Res. 2024;202(4):1711–1721. doi:10.1007/s12011-023-03773-2
15. Chu M, An X, Fu C, et al. Disulfiram/copper induce ferroptosis in triple-negative breast cancer cell line MDA-MB-231. Front Biosci. 2023;28(8):186. doi:10.31083/j.fbl2808186
16. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424–442. doi:10.1038/s41580-024-00703-5
17. Mishima E, Nakamura T, Doll S, et al. Recommendations for robust and reproducible research on ferroptosis. Nat Rev Mol Cell Biol. 2025;26(8):615–630. doi:10.1038/s41580-025-00843-2
18. Sorokin O, Hause F, Wedler A, et al. Comprehensive analysis of regulated cell death pathways, intrinsic disorder, protein-protein interactions, and cross-pathway communication. Apoptosis Int J Program Cell Death. 2025;30(9–10):2111–2162. doi:10.1007/s10495-025-02161-6
19. Liao Y, Wang D, Gu C, et al. A cuproptosis nanocapsule for cancer radiotherapy. Nat Nanotechnol. 2024;19(12):1892–1902. doi:10.1038/s41565-024-01784-1
20. Jiang C, Li X, Wan S, et al. Copper-doped polydopamine nanoparticles-mediated GSH/GPX4-depleted ferroptosis and cuproptosis sensitizes lung tumor to checkpoint blockade immunotherapy. Small. 2025;21(23):e2503208. doi:10.1002/smll.202503208
21. Wu W, Huang L, Wang E, Dong S. Atomic engineering of single-atom nanozymes for enzyme-like catalysis. Chem Sci. 2020;11(36):9741–9756. doi:10.1039/d0sc03522j
22. Xiang H, Feng W, Chen Y. Single-atom catalysts in catalytic biomedicine. Adv Mater. 2020;32(8):e1905994. doi:10.1002/adma.201905994
23. Zhang H, Lu XF, Wu ZP, Lou XWD. Emerging multifunctional single-atom catalysts/nanozymes. ACS Cent Sci. 2020;6(8):1288–1301. doi:10.1021/acscentsci.0c00512
24. Chang M, Hou Z, Wang M, et al. Cu single atom nanozyme based high-efficiency mild photothermal therapy through cellular metabolic regulation. Angew Chem. 2022;61(50):e202209245. doi:10.1002/anie.202209245
25. Wang Z, Wu FG. Emerging single-atom catalysts/nanozymes for catalytic biomedical applications. Adv Healthcare Mater. 2022;11(6):e2101682. doi:10.1002/adhm.202101682
26. Pei J, Zhao R, Mu X, Wang J, Liu C, Zhang XD. Single-atom nanozymes for biological applications. Biomater Sci. 2020;8(23):6428–6441. doi:10.1039/d0bm01447h
27. Liu Y, Zhao H, Zhao Y. Designing efficient single metal atom biocatalysts at the atomic structure level. Angew Chem. 2024;63(13):e202315933. doi:10.1002/anie.202315933
28. Wei S, Ma W, Sun M, et al. Atom-pair engineering of single-atom nanozyme for boosting peroxidase-like activity. Nat Commun. 2024;15(1):6888. doi:10.1038/s41467-024-51022-4
29. Zhang R, Yan X, Gao L, Fan K. Nanozymes expanding the boundaries of biocatalysis. Nat Commun. 2025;16(1):6817. doi:10.1038/s41467-025-62063-8
30. Cong Y, Qiao R, Wang X, et al. Protein Corona-mediated inhibition of nanozyme activity, impact of protein shape. J Am Chem Soc. 2024;146(15):10478–10488. doi:10.1021/jacs.3c14046
31. R A, Han Z, Wang T, Zhu M, Zhou M, Sun X. Pulmonary delivery of nano-particles for lung cancer diagnosis and therapy, recent advances and future prospects. Wiley Interdiscip Rev Nanomed Nanobiotechnology. 2024;16(1):e1933. doi:10.1002/wnan.1933
32. Hao Q, Gan Y, Zhou X. Tackling cuproptosis, from metabolic rewiring to therapeutic exploitation in cancer. Cell Mol Immunol. 2026;23(3):239–260. doi:10.1038/s41423-026-01387-x
33. Trautmann-Rodriguez M, Fromen CA. Nanoparticle-based pulmonary immune engineering. Annu Rev Chem Biomol. 2025;16(1):249–270. doi:10.1146/annurev-chembioeng-082223-105117
34. Li L, Liu X, Liu G, Xu S, Hu G, Wang L. Valence-engineered catalysis-selectivity regulation of molybdenum oxide nanozyme for acute kidney injury therapy and post-cure assessment. Nat Commun. 2024;15(1):8720. doi:10.1038/s41467-024-53047-1
35. Shen X, Wang Z, Gao XJ, Gao X. Reaction mechanisms and kinetics of nanozymes, insights from theory and computation. Adv Mater. 2024;36(10):e2211151. doi:10.1002/adma.202211151
36. Fu F, Crespy D, Landfester K, Jiang S. In situ characterization techniques of protein Corona around nanomaterials. Chem Soc Rev. 2024;53(22):10827–10851. doi:10.1039/d4cs00507d
37. Jia X, Wang E, Wang J. Rational design of nanozymes for engineered cascade catalytic cancer therapy. Chem Rev. 2025;125(5):2908–2952. doi:10.1021/acs.chemrev.4c00882
38. He Q, Zhang L. Bimetallic and multimetallic nanozymes, synergistic catalysis for advanced biomedical and health applications. RSC Adv. 2025;15(55):47648–47665. doi:10.1039/d5ra08106h
39. Cheng H, Chen Y, Liu M, et al. Theory-guided design of S-doped Fe/Co dual-atom nanozymes for highly efficient oxidase mimics. Chem Sci. 2024;15(36):14816–14828. doi:10.1039/d4sc03101f
40. Wang Y, Paidi VK, Wang W, et al. Spatial engineering of single-atom Fe adjacent to Cu-assisted nanozymes for biomimetic O2 activation. Nat Commun. 2024;15(1):2239. doi:10.1038/s41467-024-46528-w
41. Li Z, Ding B, Li J, et al. Multi-enzyme mimetic MoCu dual-atom nanozyme triggering oxidative stress cascade amplification for high-efficiency synergistic cancer therapy. Angew Chem. 2025;64(1):e202413661. doi:10.1002/anie.202413661
42. Yang LJ, Pan MM, Hu H, et al. Bioinspired dual-ionic-site single-atom nanozymes for synergistic ferroptosis/cuproptosis and enhanced immune checkpoint blockade therapy. Small. 2025;21(35):e2501076. doi:10.1002/smll.202501076
43. Zheng JJ, Zhu F, Song N, et al. Optimizing the standardized assays for determining the catalytic activity and kinetics of peroxidase-like nanozymes. Nat Protoc. 2024;19(12):3471–3488. doi:10.1038/s41596-024-01034-7
44. Chen X, Willner I. Surface-modified nanozymes for enhanced and selective catalysis. ACS Appl Mater Interfaces. 2025;17(31):44011–44029. doi:10.1021/acsami.5c07647
45. Zhong Z, Deng W, Wu J, et al. Cell membrane coated nanoparticles as a biomimetic drug delivery platform for enhancing cancer immunotherapy. Nanoscale. 2024;16(18):8708–8738. doi:10.1039/d4nr00284a
46. Liu Q, Gao Z, Zhang X, et al. Assembly of genetically engineered ionizable protein nanocage-based nanozymes for intracellular superoxide scavenging. Nat Commun. 2025;16(1):1123. doi:10.1038/s41467-025-56414-8
47. Xu K, Cui Y, Guan B, et al. Nanozymes with biomimetically designed properties for cancer treatment. Nanoscale. 2024;16(16):7786–7824. doi:10.1039/d4nr00155a
48. Lin J, Li W, Aboushanab AR, Sun J. Cancer cell membrane-coated NPs as a biomimetic strategy for precision tumor therapy. Pharmaceutics. 2025;17(10):1322. doi:10.3390/pharmaceutics17101322
49. He Y, Wang Y, Wang L, Jiang W, Wilhelm S. Understanding nanoparticle-liver interactions in nanomedicine. Expert Opin Drug Delivery. 2024;21(6):829–843. doi:10.1080/17425247.2024.2375400
50. Moghimi SM, Simberg D. Complement activation turnover on surfaces of nanoparticles. Nano Today. 2017;15:8–10. doi:10.1016/j.nantod.2017.03.001
51. Fan Y, Zhou Y, Zhao J, Zhao Y. Advances in inhaled nanoparticle drug delivery for pulmonary disease management. FASEB J Off Publ Fed Am Soc Exp Biol. 2025;39(21):e71191. doi:10.1096/fj.202502624R
52. Tagaras N, Song H, Sahar S, Tong W, Mao Z, Buerki-Thurnherr T. Safety landscape of therapeutic nanozymes and future research directions. Adv Sci. 2024;11(46):e2407816. doi:10.1002/advs.202407816
53. Fu Q, Liu Y, Peng C, Muluh TA, Anayyat U, Liang L. Recent advancement in inhaled nano-drug delivery for pulmonary, nasal, and nose-to-brain diseases. Curr Drug Deliv. 2025;22(1):3–14. doi:10.2174/0115672018268047231207105652
54. Li X, Zhang J, Wang M, et al. Pulmonary surfactant homeostasis dysfunction mediates multiwalled carbon nanotubes induced lung fibrosis via elevating surface tension. ACS Nano. 2024;18(4):2828–2840. doi:10.1021/acsnano.3c05956
55. Liu C, Xing J, Akakuru OU, et al. Nanozymes-engineered metal-organic frameworks for catalytic cascades-enhanced synergistic cancer therapy. Nano Lett. 2019;19(8):5674–5682. doi:10.1021/acs.nanolett.9b02253
56. Zhang H, Lv J, Wu H, et al. Endogenous/exogenous dual-responsive nanozyme for photothermally enhanced ferroptosis-immune reciprocal synergistic tumor therapy. Sci Adv. 2025;11(20):eadq3870. doi:10.1126/sciadv.adq3870
57. Han D, Ding B, Zheng P, et al. NADPH oxidase-like nanozyme for high-efficiency tumor therapy through increasing glutathione consumption and blocking glutathione regeneration. Adv Healthcare Mater. 2024;13(11):e2303309. doi:10.1002/adhm.202303309
58. Wang W, Zhu Y, Feng L, et al. Anchoring Ru single-atoms on MXene achieves dual-enzyme activities for mild photothermal augmented nanocatalytic therapy. Nanoscale. 2025;17(9):5191–5203. doi:10.1039/d4nr04609a
59. Xiong R, Zhu X, Zhao J, Ling G, Zhang P. Nanozymes-mediated cascade reaction system for tumor-specific diagnosis and targeted therapy. Small Methods. 2024;8(10):e2301676. doi:10.1002/smtd.202301676
60. Wang S, Wang L, Yan X, Jiang B. Cascade catalytic nanozymes, design, classification, and biomedical applications. ACS Appl Mater Interfaces. 2025;17(32):45354–45381. doi:10.1021/acsami.5c09544
61. Qin S, Zhao HY, Luo XY, et al. Photothermally reinforced nanozyme remodeling tumor microenvironment of redox and metabolic homeostasis to enhance ferroptosis in tumor therapy. ACS Nano. 2024;18(46):32235–32254. doi:10.1021/acsnano.4c13087
62. Zhang S, Gao XJ, Ma Y, et al. A bioinspired sulfur-Fe-heme nanozyme with selective peroxidase-like activity for enhanced tumor chemotherapy. Nat Commun. 2024;15(1):10605. doi:10.1038/s41467-024-54868-w
63. Tang D, Kroemer G, Kang R. Targeting cuproplasia and cuproptosis in cancer. Nat Rev Clin Oncol. 2024;21(5):371–388. doi:10.1038/s41571-024-00876-0
64. Gu L, Sun Y, Bai T, et al. Functional nanozyme system for synergistic tumor immunotherapy via cuproptosis and ferroptosis activation. J Nanobiotechnol. 2025;23(1):212. doi:10.1186/s12951-025-03284-3
65. Sheng J, Yuan W, Zhang M, et al. Overcoming tumor microenvironment barriers, transformable and bioinspired nanomedicine strategies for deep tumor penetration. J Nanobiotechnol. 2026;24(1):189. doi:10.1186/s12951-026-04028-7
66. Shin D, Choi J, Park B, Kim SH, Shim MK. Factors governing tumor penetration of nanomedicines, intrinsic and extrinsic determinants. Nanoscale. 2026;18(5):2363–2381. doi:10.1039/d5nr04462f
67. Ma T, Tran TB, Lin E, et al. Size-transformable nanotherapeutics for cancer therapy. Acta Pharm Sin B. 2025;15(2):834–851. doi:10.1016/j.apsb.2024.11.012
68. Gong P, Wang F, Hua Y, Ying J, Chen J, Qiao Y. Collagenase-mediated extracellular matrix targeting for enhanced drug penetration and therapeutic efficacy in nanoscale delivery systems for cancer therapy. J Nanobiotechnol. 2025;23(1):733. doi:10.1186/s12951-025-03815-y
69. Joyce P, Allen CJ, Alonso MJ, et al. A translational framework to DELIVER nanomedicines to the clinic. Nat Nanotechnol. 2024;19(11):1597–1611. doi:10.1038/s41565-024-01754-7
70. May JN, Moss JI, Mueller F, et al. Histopathological biomarkers for predicting the tumour accumulation of nanomedicines. Nat Biomed Eng. 2024;8(11):1366–1378. doi:10.1038/s41551-024-01197-4
71. Wang Y, Zhao H, Sun K, et al. Conductive coordination nanozyme prodrugs precisely trigger pyroptosis, cuproptosis and ferroptosis for in situ cancer vaccination. Signal Transduction Targeted Ther. 2026;11(1):96. doi:10.1038/s41392-026-02607-6
72. Lim YX, Choo YN, Looi YT, et al. Emerging therapeutic strategies in asthma, advances in treatment, drug delivery, drug adherence, and disease management. Curr Allergy Asthma Rep. 2026;26(1):17. doi:10.1007/s11882-026-01265-6
73. Woodward IR, Fromen CA. Recent developments in aerosol pulmonary drug delivery, new technologies, new cargos, and new targets. Annu Rev Biomed Eng. 2024;26(1):307–330. doi:10.1146/annurev-bioeng-110122-010848
74. Berkenfeld K, Carneiro S, Corzo C, et al. Formulation strategies, preparation methods, and devices for pulmonary delivery of biologics. Eur J Pharm Biopharm. 2024;204:114530. doi:10.1016/j.ejpb.2024.114530
75. Anderson S, Atkins P, Bäckman P, et al. Inhaled medicines, past, present, and future. Pharmacol Rev. 2022;74(1):48–118. doi:10.1124/pharmrev.120.000108
76. Darquenne C, Corcoran TE, Lavorini F, Sorano A, Usmani OS. The effects of airway disease on the deposition of inhaled drugs. Expert Opin Drug Delivery. 2024;21(8):1175–1190. doi:10.1080/17425247.2024.2392790
77. Zelepukin IV, Shevchenko KG, Deyev SM. Rediscovery of mononuclear phagocyte system blockade for nanoparticle drug delivery. Nat Commun. 2024;15(1):4366. doi:10.1038/s41467-024-48838-5
78. Li Y, Yang J, Gu G, et al. Pulmonary delivery of theranostic nanoclusters for lung cancer ferroptosis with enhanced chemodynamic/radiation synergistic therapy. Nano Lett. 2022;22(3):963–972. doi:10.1021/acs.nanolett.1c03786
79. Guo Y, Ma Y, Chen X, et al. Mucus penetration of surface-engineered nanoparticles in various pH microenvironments. ACS Nano. 2023;17(3):2813–2828. doi:10.1021/acsnano.2c11147
80. Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. doi:10.1016/j.jconrel.2013.09.013
81. Miller RS, Goodnough R, Durrani TS. The potential adverse human health effects of metal-containing nanoparticles, a scoping review. J Occup Med Toxicol. 2025;20(1):39. doi:10.1186/s12995-025-00491-4
82. Zhou X, Jin W, Ma J. Lung inflammation perturbation by engineered nanoparticles. Front Bioeng Biotechnol. 2023;11:1199230. doi:10.3389/fbioe.2023.1199230
83. Kim JW, Kim MJ, Han TH, et al. FSP1 confers ferroptosis resistance in KEAP1 mutant non-small cell lung carcinoma in NRF2-dependent and -independent manner. Cell Death Dis. 2023;14(8):567. doi:10.1038/s41419-023-06070-x
84. Chen Y, Jiang Z, Li X. New insights into crosstalk between Nrf2 pathway and ferroptosis in lung disease. Cell Death Dis. 2024;15(11):841. doi:10.1038/s41419-024-07224-1
85. Yan R, Liu D, Guo H, et al. LAPTM4B counteracts ferroptosis via suppressing the ubiquitin-proteasome degradation of SLC7A11 in non-small cell lung cancer. Cell Death Dis. 2024;15(6):436. doi:10.1038/s41419-024-06836-x
86. Winterbourn CC. Toxicity of iron and hydrogen peroxide, the fenton reaction. Toxicol Lett. 1995;82-83:969–974. doi:10.1016/0378-4274(95)03532-x
87. Wiktorowska-Owczarek A, Berezińska M, Nowak JZ. PUFAs, structures, metabolism and functions. Adv Clin Exp Med. 2015;24(6):931–941. doi:10.17219/acem/31243
88. Jiang X, Stockwell BR, Conrad M. Ferroptosis, mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
89. Wang M, Xu W, Ding Y, Liu X, Fu J, Zhang P. Nanozymes for regulation of reactive oxygen species in biomedical applications, design, enzyme-like activity and therapeutic mechanisms. Colloids Surf B. 2025;254:114874. doi:10.1016/j.colsurfb.2025.114874
90. Zhang M, Zheng H, Jin H, et al. Regulating SLC7A11/GSH/GPX4 axis by glucose dyshomeostasis to simultaneously promote disulfidptosis, cuproptosis and ferroptosis. Bioact Mater. 2025;54:744–758. doi:10.1016/j.bioactmat.2025.09.003
91. Zhou Q, Meng Y, Li D, et al. Ferroptosis in cancer, from molecular mechanisms to therapeutic strategies. Signal Transduction Targeted Ther. 2024;9(1):55. doi:10.1038/s41392-024-01769-5
92. Tian HX, Mei J, Cao L, et al. Disruption of iron homeostasis to induce ferroptosis with albumin-encapsulated Pt(IV) nanodrug for the treatment of non-small cell lung cancer. Small. 2023;19(49):e2206688. doi:10.1002/smll.202206688
93. Li S, Wang B, Tao J, et al. Chemodynamic therapy combined with endogenous ferroptosis based on “sea urchin-like” copper sulfide hydrogel for enhancing anti-tumor efficacy. Int J Pharm. 2024;660:124330. doi:10.1016/j.ijpharm.2024.124330
94. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
95. Koppula P, Lei G, Zhang Y, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13(1):2206. doi:10.1038/s41467-022-29905-1
96. Wang X, Shen T, Lian J, et al. Resveratrol reduces ROS-induced ferroptosis by activating SIRT3 and compensating the GSH/GPX4 pathway. Mol Med. 2023;29(1):137. doi:10.1186/s10020-023-00730-6
97. Shen Z, Zhao L, Yoo SA, et al. Emodin induces ferroptosis in colorectal cancer through NCOA4-mediated ferritinophagy and NF-κb pathway inactivation. Apoptosis Int J Program Cell Death. 2024;29(9–10):1811–1823. doi:10.1007/s10495-024-01973-2
98. Nakamura T, Conrad M. Exploiting ferroptosis vulnerabilities in cancer. Nat Cell Biol. 2024;26(9):1407–1419. doi:10.1038/s41556-024-01425-8
99. Wu K, Vaughan AJ, Bossowski JP, et al. Targeting FSP1 triggers ferroptosis in lung cancer. Nature. 2026;649(8096):487–495. doi:10.1038/s41586-025-09710-8
100. Yang C, Zhao Y, Wang L, et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat Cell Biol. 2023;25(6):836–847. doi:10.1038/s41556-023-01146-4
101. Yan Z, Bai Y, Zhang S, et al. Quasi Fe MIL-53 nanozyme inducing ferroptosis and immunogenic cell death for cancer immunotherapy. Nat Commun. 2025;16(1):2290. doi:10.1038/s41467-025-57542-x
102. Zhang J, Lei C, Zheng G, et al. CuET promotes cuproptosis and ferroptosis of lung cancer cells by interacting directly with polyunsaturated phospholipids. iScience. 2026;29(4):115438. doi:10.1016/j.isci.2026.115438
103. Efimova I, Catanzaro E, Van der Meeren L, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J ImmunoTher Cancer. 2020;8(2):e001369. doi:10.1136/jitc-2020-001369
104. Luo X, Gong HB, Gao HY, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021;28(6):1971–1989. doi:10.1038/s41418-020-00719-2
105. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19(7):405–414. doi:10.1038/s41568-019-0149-1
106. Ma X, Xiao L, Liu L, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33(5):1001–1012.e5. doi:10.1016/j.cmet.2021.02.015
107. Yu XY, Jin X, Shou ZX. Surface-engineered smart nanocarrier-based inhalation formulations for targeted lung cancer chemotherapy, a review of current practices. Drug Delivery. 2021;28(1):1995–2010. doi:10.1080/10717544.2021.1981492
108. Labiris NR, Dolovich MB. Pulmonary drug delivery. Part I, physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol. 2003;56(6):588–599. doi:10.1046/j.1365-2125.2003.01892.x
109. Rotolo L, Vanover D, Bruno NC, et al. Species-agnostic polymeric formulations for inhalable messenger RNA delivery to the lung. Nat Mater. 2023;22(3):369–379. doi:10.1038/s41563-022-01404-0
110. Feng Q, Fang W, Guo Y, Hu P, Shi J. Nebulized therapy of early orthotopic lung cancer by iron-based nanoparticles, macrophage-regulated ferroptosis of cancer stem cells. J Am Chem Soc. 2023;145(44):24153–24165. doi:10.1021/jacs.3c08032
111. Khan RU, Qian L, Min J, Peng H, Wang F. Nanozymes for ferroptosis-based cancer theranostics. Bioact Mater. 2026;62:862–882. doi:10.1016/j.bioactmat.2026.03.043
112. Mao C, Wu S, Rajacharya GH, et al. DHODH-mediated suppression of ferroptosis supports radioresistance and represents a therapeutic vulnerability in lung cancer. Cancer Res. 2026. doi:10.1158/0008-5472.CAN-25-3728
113. Zhang L, Xu Y, Cheng Z, et al. The EGR1/miR-139/NRF2 axis orchestrates radiosensitivity of non-small-cell lung cancer via ferroptosis. Cancer Lett. 2024;595:217000. doi:10.1016/j.canlet.2024.217000
114. Sheng P, Jin J, Liu H, et al. Targeting ferroptosis to overcome drug resistance in lung cancer. Transl Lung Cancer Res. 2025;14(12):5509–5526. doi:10.21037/tlcr-2025-915
115. Jiang C, Xie S, Jia K, Feng G, Ren X, Wang Y. Exploring cellular plasticity and resistance mechanisms in lung cancer, innovations and emerging therapies. J Pharm Anal. 2025;15(5):101179. doi:10.1016/j.jpha.2024.101179
116. Dai Z, Liu J, Zeng L, et al. Targeting ferroptosis in cancer therapy, mechanisms, strategies, and clinical applications. Cell Investig. 2025;1(4):100049. doi:10.1016/j.clnves.2025.100049
117. Yuan D, Luo T, Su Q, et al. Nanozyme-based catalytic therapeutics, applications in infectious diseases, cancer therapy, and bone regeneration. Chin Chem Lett. 2026;37(3):111842. doi:10.1016/j.cclet.2025.111842
118. Hussain Z, Abdulelah AA, Tahseen SH, Mashkoor AA, Thu HE. Stimuli-responsive nanomedicines for lung cancer therapy, design strategies, advances, challenges, and future directions. J Drug Delivery Sci Technol. 2026;116:107900. doi:10.1016/j.jddst.2025.107900
119. Yoshida M, Arzili R, Nikolić MZ. Immune-epithelial cell interactions in lung development, homeostasis and disease. Int J Biochem Cell Biol. 2025;178:106703. doi:10.1016/j.biocel.2024.106703
120. Ge D, Ma S, Sun T, et al. Pulmonary delivery of dual-targeted nanoparticles improves tumor accumulation and cancer cell targeting by restricting macrophage interception in orthotopic lung tumors. Biomaterials. 2025;315:122955. doi:10.1016/j.biomaterials.2024.122955
121. Duan Y, Chen D, Zhang R, Du Y, Wang L. Optimizing the coordination environment of active sites to enhance heterogeneous electrocatalytic performances. Coord Chem Rev. 2025;522:216243. doi:10.1016/j.ccr.2024.216243
122. Ta YN, Wang SN, Chen Y. Enzyme-based nanomedicine for tumor microenvironment modulation in cancer therapy. J Control Release. 2026;115101. doi:10.1016/j.jconrel.2026.115101
123. Wang S, Liu X, Wei D, et al. Polyvalent aptamer nanodrug conjugates enable efficient tumor cuproptosis therapy through copper overload and glutathione depletion. J Am Chem Soc. 2024;146(44):30033–30045. doi:10.1021/jacs.4c06338
124. Kim K, Lee J, Park OK, et al. Enhanced cuproptosis via metabolic reprogramming using copper-delivering Co-N-C single-atom nanozyme. ACS Nano. 2025;19(24):21969–21982. doi:10.1021/acsnano.5c00012
125. Xie J, Yang Y, Gao Y, He J. Cuproptosis, mechanisms and links with cancers. Mol Cancer. 2023;22(1):46. doi:10.1186/s12943-023-01732-y
126. Mao L, Lu J, Wen X, et al. Cuproptosis, mechanisms and nanotherapeutic strategies in cancer and beyond. Chem Soc Rev. 2025;54(13):6282–6334. doi:10.1039/d5cs00083a
127. Han XW, Chen X, Yang TL, et al. Cu-phytic acid nanozyme-induced cuproptosis therapy for the inhibition of tumor growth. Nanoscale Horiz. 2025;10(9):2111–2122. doi:10.1039/d5nh00183h
128. L K, W L, W H, et al. Apoptosis and cuproptosis Co-activated copper-based metal-organic frameworks for cancer therapy. J Nanobiotechnol. 2024;22(1). doi:10.1186/s12951-024-02828-3
129. Xu S, Wu Y, Cai J, et al. Carbon dot sensitized Cu3P sonozymes for cuproptosis-enhanced and heterojunction-amplified sono-immunotherapy through activating cGAS-STING pathway. J Nanobiotechnol. 2025;23(1):802. doi:10.1186/s12951-025-03877-y
130. Luo Y, Luo X, Ru Y, et al. Copper(II)-based nano-regulator correlates cuproptosis burst and sequential immunogenic cell death for synergistic cancer immunotherapy. Biomater Res. 2024;28:39. doi:10.34133/bmr.0039
131. Wu M, Zhao K, Tao X, et al. Biomimetic copper nanozyme reprograms cold tumor via cuproptosis-pyroptosis crosstalk for potent renal carcinoma immunotherapy. ACS Appl Mater Interfaces. 2025;17(20):29291–29304. doi:10.1021/acsami.5c03559
132. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6(3):1671–1690. doi:10.3390/cancers6031670
133. Zhang S, Lv K, Liu Z, Zhao R, Li F. Fatty acid metabolism of immune cells, a new target of tumour immunotherapy. Cell Death Discovery. 2024;10(1):39. doi:10.1038/s41420-024-01807-9
134. La Fontaine S, Jf M. Trafficking of the copper-ATPases, ATP7A and ATP7B, role in copper homeostasis. Arch Biochem Biophys. 2007;463(2). doi:10.1016/j.abb.2007.04.021
135. Lukanović D, Herzog M, Kobal B, Černe K. The contribution of copper efflux transporters ATP7A and ATP7B to chemoresistance and personalized medicine in ovarian cancer. Biomed Pharmacother. 2020;129:110401. doi:10.1016/j.biopha.2020.110401
136. Tang D, Chen X, Kroemer G. Cuproptosis, a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–418. doi:10.1038/s41422-022-00653-7
137. Zhao J, Zhang W, Zeng Y, et al. Targeting ATP7A-dependent copper metabolic homeostasis induces cuproptosis and suppresses the progression of mutant KRAS-driven lung cancer. Cancer Res. 2025;85(20):3999–4017. doi:10.1158/0008-5472.CAN-24-2558
138. O’Day SJ, Eggermont AMM, Chiarion-Sileni V, et al. Final results of phase III SYMMETRY study, randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J Clin Oncol. 2013;31(9):1211–1218. doi:10.1200/JCO.2012.44.5585
139. Li SR, Bu LL, Cai L. Cuproptosis, lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduction Targeted Ther. 2022;7(1):158. doi:10.1038/s41392-022-01014-x
140. Chen W, Lin C, Gao Z, et al. A tumor microenvironment-responsive nanocomposite for enhanced copper retention and hypoxia reversal to promote cuproptosis in tumor treatment. Acta Biomater. 2025;202:463–475. doi:10.1016/j.actbio.2025.07.013
141. Bai Y, Chen L, Liu H, et al. Identification of melampomagnolide B derivatives as triggers of cuproptosis, ferroptosis, and apoptosis for treatment of lung cancer. J Med Chem. 2026;69(7):8255–8284. doi:10.1021/acs.jmedchem.5c03682
142. Wang W, Lu K, Jiang X, et al. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J Exp Clin Cancer Res CR. 2023;42(1):142. doi:10.1186/s13046-023-02720-2
143. Cobine PA, Brady DC. Cuproptosis, cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell. 2022;82(10):1786–1787. doi:10.1016/j.molcel.2022.05.001
144. Badgley MA, Kremer DM, Maurer HC, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–89. doi:10.1126/science.aaw9872
145. Liu Y, Lu S, Wu LL, Yang L, Yang L, Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023;14(8):519. doi:10.1038/s41419-023-06045-y
146. Zhang W, Chen Z, Xiong C, et al. A peptide-copper self-assembled nanoparticle for enhanced cuproptosis by metabolic reprogramming in tumor cells. ACS Nano. 2024;18(50):34244–34256. doi:10.1021/acsnano.4c12123
147. Bai J, Zhang X, Zhao Z, et al. CuO nanozymes catalyze cysteine and glutathione depletion induced ferroptosis and cuproptosis for synergistic tumor therapy. Small. 2024;20(40):e2400326. doi:10.1002/smll.202400326
148. Liu C, Guo L, Cheng Y, et al. A mitochondria-targeted nanozyme platform for multi-pathway tumor therapy via ferroptosis and cuproptosis regulation. Adv Sci. 2025;12(36):e17616. doi:10.1002/advs.202417616
149. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. 2020;99(1):151058. doi:10.1016/j.ejcb.2019.151058
150. Li X, Gao J, Wu C, et al. Precise modulation and use of reactive oxygen species for immunotherapy. Sci Adv. 2024;10(20):eadl0479. doi:10.1126/sciadv.adl0479
151. Zhang Y, Ya S, Huang J, et al. Spatial isolation of single copper(I) sites for cascade enzyme-like catalysis and simultaneous ferroptosis/cuproptosis boosted immunotherapy. Exploration. 2025;5(3):20240275. doi:10.1002/EXP.20240275
152. Guo B, Yang F, Zhang L, et al. Cuproptosis induced by ROS responsive nanoparticles with elesclomol and copper combined with αPD-L1 for enhanced cancer immunotherapy. Adv Mater. 2023;35(22):e2212267. doi:10.1002/adma.202212267
153. Galluzzi L, Vitale I, Warren S, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J ImmunoTher Cancer. 2020;8(1):e000337. doi:10.1136/jitc-2019-000337
154. Lei G, Zhuang L, Gan B. The roles of ferroptosis in cancer, tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell. 2024;42(4):513–534. doi:10.1016/j.ccell.2024.03.011
155. Chen R, Zou J, Liu J, Kang R, Tang D. DAMPs in the immunogenicity of cell death. Mol Cell. 2025;85(20):3874–3889. doi:10.1016/j.molcel.2025.09.007
156. Janssens S, Rennen S, Agostinis P. Decoding immunogenic cell death from a dendritic cell perspective. Immunol Rev. 2024;321(1):351–370. doi:10.1111/imr.13301
157. Kim JW, Min DW, Kim D, et al. GPX4 overexpressed non-small cell lung cancer cells are sensitive to RSL3-induced ferroptosis. Sci Rep. 2023;13(1):8872. doi:10.1038/s41598-023-35978-9
158. Cheff DM, Huang C, Scholzen KC, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023;62:102703. doi:10.1016/j.redox.2023.102703
159. Zhu J, Li X, Chen W, et al. Ferroptosis triggers anti-tumor immunity via promoting chaperone-mediated autophagic degradation of SHP2. Redox Biol. 2025;86:103796. doi:10.1016/j.redox.2025.103796
160. Voli F, Valli E, Lerra L, et al. Intratumoral copper modulates PD-L1 expression and influences tumor immune evasion. Cancer Res. 2020;80(19):4129–4144. doi:10.1158/0008-5472.CAN-20-0471
161. Luo J, Huang K, Yi X, et al. Bimetallic nanozyme amplifier for synergistic ferroptosis-cuproptosis and metabolic reprogramming to reshape immunosuppressive tumor microenvironment. Adv Sci. 2026;13(1):e12764. doi:10.1002/advs.202512764
162. Zhang M, Cui J, Chen H, et al. Increased SOAT2 expression in aged regulatory T cells is associated with altered cholesterol metabolism and reduced anti-tumor immunity. Nat Commun. 2025;16(1):630. doi:10.1038/s41467-025-56002-w
163. Chen T, Huang C, Liu Y, Nie D, Chen J. Bimetallic nanozyme-mediated dual ferroptosis/cuproptosis synergy potentiates immunotherapy in bladder cancer. Colloids Surf B Biointerfaces. 2025;255:114954. doi:10.1016/j.colsurfb.2025.114954
164. Li Y, Liu J, Chen Y, Weichselbaum RR, Lin W. Nanoparticles synergize ferroptosis and cuproptosis to potentiate cancer immunotherapy. Adv Sci. 2024;11(23):e2310309. doi:10.1002/advs.202310309
165. Yang MD, Zhu CY, Yang G, et al. Camouflaged membrane-bridged radionuclide/Mn single-atom enzymes target lipid metabolism disruption to evoke antitumor immunity. Mil Med Res. 2025;12(1):59. doi:10.1186/s40779-025-00647-7
166. Zhang R, Li Y, Fu H, et al. Nanomedicine strategies for cuproptosis, metabolic reprogramming and tumor immunotherapy. Acta Pharm Sin B. 2025;15(9):4582–4613. doi:10.1016/j.apsb.2025.07.007
167. Cui K, Wang K, Huang Z. Ferroptosis and the tumor microenvironment. J Exp Clin Cancer Res CR. 2024;43(1):315. doi:10.1186/s13046-024-03235-0
168. Wang H, Hu Q, Chen Y, et al. Ferritinophagy mediates adaptive resistance to EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Nat Commun. 2024;15(1):4195. doi:10.1038/s41467-024-48433-8
169. Sun L, Handorf EA, Zhou Y, Borghaei H, Aggarwal C, Bauman J. Outcomes in patients treated with frontline immune checkpoint inhibition (ICI) for advanced NSCLC with KRAS mutations and STK11/KEAP1 comutations across PD-L1 levels. Lung Cancer. 2024;190:107510. doi:10.1016/j.lungcan.2024.107510
170. Behinaein P, Gadgeel SM, Rattan R, et al. Resistance mechanisms in anaplastic lymphoma kinase-positive lung cancer. Crit Rev Oncol Hematol. 2025;214:104821. doi:10.1016/j.critrevonc.2025.104821
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
New Targets for Immune Inflammatory Response in Rheumatoid Arthritis: Focus on the Potential Significance of N6-Methyladenosine, Ferroptosis and Cuproptosis
Wang S, Wan L, Zhang M, Yan D, Li F
Journal of Inflammation Research 2025, 18:8085-8106
Published Date: 19 June 2025
Machine Learning-Based Identification and Experimental Validation of Hub Ferroptosis-Related Cuproptosis Genes in Lupus Nephritis
Zhang S, Hu W, Zhang Y, Huang C, He Z, Xu J, Lin S, Yang B, Chen X
Journal of Inflammation Research 2025, 18:11335-11353
Published Date: 18 August 2025
Targeting Lactylation Offers Therapy to Reverse Cell Death Resistance
Wang Y, Chen J, Wang Y, Cao Y, Li Y, Zhu Y, Zhang Z, Wu S, Wang H
Drug Design, Development and Therapy 2025, 19:11371-11394
Published Date: 20 December 2025
Biomimetic Nanoplatform Based on Macrophage Membrane-Coated Fe3O4 Nanoparticles for Synergistic Ferroptosis and Sonodynamic Therapy of Lung Cancer
Zhang H, Zhuang Y, Song J, Shao F, Shi W
International Journal of Nanomedicine 2026, 21:573349
Published Date: 13 January 2026