Back to Journals » International Journal of Nanomedicine » Volume 18
Nanodepots Encapsulating a Latency Reversing Agent and Broadly Neutralizing Antibody Enhance Natural Killer Cell Cytotoxicity Against an in vitro Model of Latent HIV
Authors Ghofrani J ![]() , Bowen A, Chen J, Balakrishnan PB, Powell AB, Cherukula K, Cruz CRY, Jones RB, Lynch RM, Sweeney EE, Fernandes R
, Bowen A, Chen J, Balakrishnan PB, Powell AB, Cherukula K, Cruz CRY, Jones RB, Lynch RM, Sweeney EE, Fernandes R ![]()
Received 22 December 2022
Accepted for publication 10 July 2023
Published 25 July 2023 Volume 2023:18 Pages 4055—4066
DOI https://doi.org/10.2147/IJN.S401304
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Joshua Ghofrani,1,2 Allan Bowen,2 Jie Chen,2 Preethi Bala Balakrishnan,2 Allison B Powell,1,2 Kondareddy Cherukula,2 Conrad Russell Y Cruz,2,3 R Brad Jones,4 Rebecca M Lynch,5 Elizabeth E Sweeney,2,6 Rohan Fernandes1,2,7
1The Institute for Biomedical Sciences, School of Medicine and Health Sciences, George Washington University, Washington, DC, USA; 2The George Washington Cancer Center, The George Washington University, Washington, DC, USA; 3Center for Cancer and Immunology Research, Children’s National Hospital, Washington, DC, USA; 4Division of Infectious Diseases, Department of Medicine, Weill Cornell Medicine, New York, NY, USA; 5Department of Microbiology, Immunology, and Tropical Medicine, The George Washington University School of Medicine and Health Sciences, Washington, DC, USA; 6Department of Biochemistry and Molecular Medicine, School of Medicine and Health Sciences, George Washington University, Washington, DC, USA; 7Department of Medicine, School of Medicine and Health Sciences, George Washington University, Washington, DC, USA
Correspondence: Rohan Fernandes, 800 22nd St. NW, Science and Engineering Hall, Suite 8410, Washington, DC, 20052, USA, Tel +1 202 994 0899, Email [email protected]
Purpose: Current antiretroviral therapies (ART) for human immunodeficiency virus (HIV) are not curative, as the virus persists in latent reservoirs, requiring lifelong adherence to ART and increasing the risk of co-morbidities. “Shock and kill” approaches to reactivate HIV from latent reservoirs followed by administration of anti-HIV drugs represent a promising strategy for eradicating latent HIV. To achieve effective shock and kill, we describe a strategy to eradicate the HIV reservoir that combines latency reversing agents (LRAs), broadly neutralizing antibodies (bnAbs), and natural killer (NK) cells. This strategy utilizes a polymer nanodepot (ND) that co-encapsulates the LRA and bnAb to reactivate latent infection and elicit enhanced cytotoxicity from co-administered NK cells.
Methods: Poly(lactic-co-glycolic acid) (PLGA) NDs were synthesized using the nanoprecipitation method to co-encapsulate an LRA (TNF-α) and a bnAb (3BNC117) (TNF-α-3BNC117-NDs). ACH-2 cells were used as a cellular model of latent HIV infection. An NK92 subline, genetically modified to constitutively express the Fc receptor CD16, was administered to ACH-2 cells in combination with TNF-α-3BNC117-NDs. ACH-2 cell death and extracellular p24 were measured via flow cytometry and ELISA, respectively.
Results: Stable PLGA NDs co-encapsulated TNF-α and 3BNC117 with high efficiencies and released these agents in physiological conditions. NK92 phenotype remained similar in the presence of TNF-α-3BNC117-NDs. TNF-α released from NDs efficiently reactivated HIV in ACH-2 cells, as measured by a 3.0-fold increase in the frequency of intracellular p24 positive cells. Released 3BNC117 neutralized and bound reactivated virus, targeting 57.5% of total ACH-2 cells. Critically, TNF-α-3BNC117-NDs significantly enhanced NK92 cell-mediated killing of ACH-2 cells (1.9-fold) and reduced extracellular levels of p24 to baseline.
Conclusion: These findings suggest the therapeutic potential of our novel ND-based tripartite strategy to reactivate HIV from latently infected cells, generate an HIV-specific site for bnAb binding, and enhance the killing of reactivated HIV-infected target cells by NK92 cells.
Keywords: latent HIV reservoirs, latency reversing agent, broadly neutralizing antibody, shock and kill strategies, NK cell therapy, PLGA nanodepots, TNF-α, 3BNC117, ACH-2 cells
Introduction
Antiretroviral therapy (ART) effectively suppresses human immunodeficiency virus (HIV) replication but does not lead to a sterilizing cure, as the virus persists within latent reservoirs.1 Upon interruption or cessation of ART, viral rebound occurs rapidly, requiring patients to adhere to a lifelong treatment regimen.2 Chronic exposure to ART is associated with co-morbidities such as cardiovascular disease and neurocognitive disorders;3,4 thus, a novel therapy capable of completely eradicating HIV is urgently needed.
Natural killer (NK) cells are attractive immune cells for treating HIV, as they are insusceptible to CD4-mediated infection, allowing them to kill infected target cells without the risk of viral spread.5,6 NK cells also contribute to protection against viral infections, including HIV.7,8 NK cells can kill target cells by several mechanisms, but in particular, NK cell-mediated antibody-dependent cell-cytotoxicity (ADCC) is linked with slower progression of HIV infection,9–12 increased capacity of NK cells to lyse allogeneic HIV-infected T cells,13 and enhanced viral control in elite controllers,14 suggesting the critical role of ADCC in eliciting beneficial clinical responses. However, NK cell-mediated ADCC of HIV-infected cells requires the local availability of an HIV-specific antibody in the physical context of NK cells and an exposed HIV-specific target on an HIV-infected cell, which is often absent in latently infected cells.
Broadly neutralizing antibodies (bnAbs) recognize envelope regions on HIV typically expressed in reactivated cells, and have shown promise in neutralizing HIV.15 However, latently infected cells do not produce ample virus, so bnAb binding sites are inaccessible in latent reservoirs. In this context, we have developed a coordinated approach that combines the release of a latency reversing agent (LRA) and a bnAb using biodegradable polymeric nanodepots (NDs)16–19 in combination with NK cells for enhanced HIV-specific NK cell-mediated cytotoxicity. Encapsulation of these agents within NDs may allow their sustained release over time, thereby prolonging their function and enhancing HIV eradication by spatiotemporally coordinating their effects.20–22
To that end, we have generated a poly(lactic-co-glycolic acid) (PLGA) ND platform that co-encapsulates TNF-α, an LRA that reactivates HIV in our model of HIV latency,23 and 3BNC117, an HIV-specific bnAb.24 To our knowledge, the encapsulation of two protein biologics within the same PLGA ND has not been pursued in the context of HIV. PLGA is biodegradable, FDA-approved, and enables the sustained release of encapsulated agents.21,25 The PLGA NDs co-encapsulating both TNF-α and 3BNC117 (termed TNF-α-3BNC117-NDs) were synthesized using the nanoprecipitation technique.22,26,27 We seek to illustrate that the administration of TNF-α-3BNC117-NDs in combination with NK cells (here, NK92 cells genetically engineered to constitutively express CD16) as a therapeutic ensemble co-localizes the actions of the three components. TNF-α reactivates HIV from target cells, exposing an HIV-specific binding site for 3BNC117 to subsequently bind and neutralize reactivated virus. This, in turn, enables enhanced cytotoxicity mediated by the NK92 cell line.28 We hypothesize that the co-localization of an LRA, an HIV-specific bnAb, and NK92 cells will trigger a coordinated and enhanced eradication of latently infected T cells. To test this hypothesis, we present the physicochemical characterization of the TNF-α-3BNC117-NDs, evaluate the TNF-α-3BNC117-ND capacity to reactivate latent HIV, bind the surface of reactivated target cells, and diminish the presence of soluble viral antigen (p24), and establish the proof-of-concept efficacy of our combination ND and NK92 cell therapeutic approach in the ACH-2 cell model of latent HIV.29
Material and Methods
PLGA ND Synthesis
Poly(lactic-co-glycolic acid) (PLGA; lactide: glycolide (50:50), MW 30,000–60,000), polyvinyl alcohol (PVA), acetonitrile, and phosphate-buffered saline (PBS) were purchased from Millipore Sigma (USA). TNF-α was purchased from R&D Systems (USA). 3BNC117 was obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH, contributed by Dr. Michel Nussenzweig, and from the laboratory of Dr. Rebecca Lynch. Antibody protein was expressed from transient transfection of heavy- and light-chain expression vectors into 293F cells and purified by affinity chromatography using HiTrap Protein A HP Columns (GE Healthcare, USA). PLGA NDs were synthesized using the nanoprecipitation technique.22,26,27 Briefly, TNF-α (7.5 µg in 7.5 µL PBS) or the equivalent amount of PBS was added to 5 mg/mL PLGA in 1 mL acetonitrile and vortexed for 20s. Subsequently, 3BNC117 (75 µg in 17 µL PBS) or the equivalent amount of PBS was added to the solution, vortexed for 20s, and sonicated using a microtip probe (Q500; QSonica, USA) on ice at 40% amplitude for 30s. The resultant emulsion was then added to 10 mL of 5% PVA while stirring on a magnetic stirrer (initially 400 RPM, increased to 600 RPM upon addition of emulsion). After stirring for 30 min, resultant NDs were collected by centrifugation at 10,000 ×g for 30 minutes. The supernatant was then centrifuged at 10,000 ×g for 30 minutes to collect residual particles. Resultant pellets were combined and sonicated on ice at 30% amplitude for 30s. All NDs were stored at 4 °C in deionized water. The supernatants were saved for determining encapsulation efficiencies.
Cells
NK92 cells engineered to constitutively express CD16 were obtained from the American Type Culture Collection (USA) and were cultured in αMEM cell media (Millipore Sigma) supplemented with 12.5% fetal bovine serum (FBS; Gibco, USA), 12.5% horse serum (Gibco) and stimulated every four days with 500 IU/mL IL-2 (Proleukin; Clinigen Group, UK). ACH-2 cells were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH, contributed by Dr. Thomas Folks, and cultured in αMEM supplemented with 10% FBS. Tzm-bl reporter cells were acquired from the NIH AIDS Reagent Program and cultured in DMEM cell media (ThermoFisher, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% gentamicin (Millipore Sigma).
Physical Characterization of NDs
ND size (hydrodynamic diameter) and surface charge (zeta potential) were measured using dynamic light scattering (DLS) and electrophoretic light scattering, respectively, on a Zetasizer Nano ZS (Malvern Instruments, UK). Scanning electron microscopy (SEM) was performed on a FEI Teneo LV Field Emission Scanning Electron Microscope (USA).
Quantification of ND Encapsulation and Release of Cargo
Encapsulation efficiencies of TNF-α and 3BNC117 within PLGA NDs were determined by measuring their content in the supernatants after collection of the NDs by centrifugation at the end of the synthesis. ND encapsulation of TNF-α was determined via a TNF-α ELISA (Human TNF-α Quantikine ELISA, R&D Systems, USA) per the manufacturer’s specifications. ND encapsulation of 3BNC117 was determined by a resurfaced stabilized core 3 (RSC3) protein ELISA. Briefly, 2 µg/mL RSC3 was adsorbed on 96-well high binding microplates (Grenier Bio-One, USA) overnight at 4 °C. The following day, samples were added to the RSC3-coated plate and incubated at 37 °C for 2 h. Subsequently, the plates were washed, and incubated at 37 °C for 1 h with horseradish peroxidase (HRP) labeled goat anti-human IgG (H+L) cross-absorbed secondary antibody (Invitrogen, USA) diluted 1:3000 in blocking buffer (5% BSA in PBS). Finally, the plate was washed and 100 µL Turbo TMB (3,3’,5,5’-tetramethylbenzidine) substrate (ThermoFisher) was added to each well for 10 min, prior to quantifying absorbance at 450 nm on a Spectramax i3x multimodal plate reader (Molecular Devices, USA). RSC3 recombinant protein was expressed from HEK293 cells that were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH, contributed by Drs. Zhi-Yong Yang, Peter Kwong, Gary Nabel, and John Mascola. To determine the release kinetics of TNF-α and 3BNC117 from the NDs, the NDs were incubated in PBS at 37°C at dilutions of 10% or 2% (v/v). At predetermined time intervals, the NDs were centrifuged at 15,000 ×g for 10 minutes, and supernatants were analyzed for TNF-α content by the TNF-α ELISA and 3BNC117 content by the RSC3 ELISA, as described above. After supernatant collection at each timepoint, the ND pellets were re-suspended in fresh PBS.
Characterization of NK Cell Phenotype
To characterize the phenotype of NK cells upon treatment with the NDs or controls, NK92 cells were treated with vehicle (PBS), TNF-α, 3BNC117, TNF-α+3BNC117, NDs containing PBS (Blank NDs), NDs containing TNF-α (TNF-α-NDs), NDs containing 3BNC117 (3BNC117-NDs), or TNF-α-3BNC117-NDs for 24 hours (n≥2/group). The equivalent dose of 4 ng/mL TNF-α and/or 33 ng/mL 3BNC117 was maintained in the free agent and ND formulations. These doses were determined based on the kinetics of viral reactivation in ACH-2 cells treated with TNF-α as described below. After 24 h, the NK92 cells were incubated with Fc Block (BD Biosciences, USA), and stained with Fixable Viability Stain 660 (BD Biosciences), and antibodies against CD56 (clone 5.1H11), CD16 (3G8), NKp46 (9E2), CD25 (BC96), NKG2D (1D11), PD1 (EH12.2H7), TIM-3 (F38-2E2), and CD69 (FN50) (all purchased from BioLegend, USA). To characterize the NK92 cells’ chemokine receptor expression, NK92 cells were stained with Zombie aqua, and antibodies against CCR2 (clone KO36C2), CCR6 (G034E3), CCR7 (G043H7), CD62L (DREG-56), CX3CR1 (2A9-1), CXCR3 (G025H7), and CXCR4 (12G5) (purchased from BioLegend). Flow cytometry was performed on a BD FACS Celesta flow cytometer (BD Biosciences, USA) and analyzed using FlowJo 10.8.1 software (BD Biosciences).
Quantification of HIV Reactivation, bnAb Binding, Neutralization, and Viral p24 Expression
To quantify the effect of the free agents and NDs on HIV reactivation, bnAb binding, and soluble p24 expression, ACH-2 cells were cultured in media alone or incubated with vehicle (PBS), TNF-α, 3BNC117, TNF-α+3BNC117, Blank NDs, TNF-α-NDs, 3BNC117-NDs, or TNF-α-3BNC117-NDs for 24 h at a dose of 4 ng/mL TNF-α and 22 ng/mL 3BNC117, based on the cumulative release kinetics of the encapsulated agents. Then, intracellular and cell surface staining was performed to assess viral reactivation and 3BNC117 binding, respectively. To measure intracellular capsid protein, ACH-2 cells were washed in PBS and re-suspended in Fixable Viability Stain 660 (BioLegend). Intracellular capsid staining was performed by incubating the ACH-2 cells in 100 µL Fixation/Permeabilization buffer, washing the cells in 200 µL 1X Permeabilization/Wash buffer (BD Biosciences), and staining with a fluorescent anti-capsid antibody (clone KC57) (Beckman Coulter, USA). To quantify 3BNC117 binding to the surface of ACH-2 cells, ACH-2 cells were fixed with BD Fixation buffer and stained with HRP-labeled goat anti-human IgG (gamma chain) cross-adsorbed secondary antibody (Invitrogen, USA). ACH-2 cells were visualized on a CytoFLEX flow cytometer (Beckman Coulter) and analyzed in FlowJo 10.8.1 software for HIV reactivation (p24 expression) and 3BNC117 binding (IgG secondary antibody binding). In separate experiments assessing the functional stability of these agents over time, the PLGA NDs or free agents were first pre-incubated in PBS at 37 °C for 72 h before proceeding with their addition to ACH-2 in culture, staining, and flow cytometric analysis of reactivation and 3BNC117 binding. Neutralization assays using Tzm-bl reporter cells (NIH AIDS Reagent Program) were conducted using viral cell culture supernatant from ACH-2 cells reactivated with 20 ng/mL TNF-α. Virus was incubated in the presence of each agent (0–10 µg/mL) at 37°C for 90 min in 96-well clear-bottom black culture plate (Greiner Bio-One, USA). Tzm-bl cells (10,000) were added to the plate with 20 mg/mL DEAE-Dextran (Sigma-Aldrich, USA). After 48 h, half of the media was removed from each well and replaced with an equivalent volume of SpectraMax Glo Steady-Luc reporter assay reagent (Molecular Devices). Luminescence was measured using a SpectraMax i3x multimode detection platform (Molecular Devices). To quantify extracellular levels of viral capsid protein p24, 100,000 ACH-2 cells were treated with PBS, TNF-α (500 ng/mL), 3BNC117 (5 µg/mL), TNF-α+3BNC117, Blank NDs, or TNF-α-3BNC117-NDs, in the absence or presence of NK92 cells (10,000 cells) at a 1:10 E:T ratio. Cell-free supernatant was collected from the cell culture by centrifugation at 2000 ×g for 10 minutes and analyzed by p24 ELISA (Abcam, UK) per the manufacturer’s instructions.
Quantification of NK Cell-Mediated Cytotoxicity
To evaluate the effect of the free agents and NDs in combination with NK cells on ACH-2 cell viability, ACH-2 cells were plated in 96-well plates (Corning, USA) at 1 million cells per mL of ACH-2 cell media. Immediately after plating, ACH-2 cells were treated with vehicle (PBS), Blank NDs, TNF-α-3BNC117-NDs, or free TNF-α and 3BNC117 at equivalent doses (30 ng/mL TNF-α and 2 µg/mL 3BNC117, to ensure robust reactivation and ample antibody in the context of co-culture) determined by ND release at 24 h. At 19 h, 40,000 NK92 cells were added to designated conditions of ACH-2 culture to achieve a 1:2.5 effector: target (E:T) cell ratio. This ratio was selected based on co-culture studies evaluating ACH-2 cell viability after 24 h of co-incubation with NK92 cells at varying E:T ratios so as to identify an E:T ratio that would permit a suitable dynamic range of cytotoxicity to be assessed when the NK92 cells were combined with free or PLGA-encapsulated agents. NK92 cells were stimulated 72 h prior with 500 IU/mL IL-2 (Clinigen Group). After 24 h from the time of plating, the cell cultures were incubated with Fc Block, and stained with Fixable Viability Stain 660 and fluorescent antibodies against CD56 and CD5 (BioLegend). A fixed volume (80 μL of 100 μL) was acquired for every condition on a CytoFLEX S flow cytometer (Beckman Coulter). The number of live ACH-2 cells was determined by viability staining on the CD56-/CD5+ population using FlowJo 10.8.1 software. Percent killing was then calculated based on the ratio of live ACH-2 cells in the condition of interest to live untreated ACH-2 cells. (n≥3/group.)
Results
PLGA NDs Encapsulate and Release LRA and bnAb
PLGA NDs were formulated to encapsulate either vehicle (PBS; Blank NDs), TNF-α (TNF-α-NDs), 3BNC117 (3BNC117-NDs), or both TNF-α and 3BNC117 (TNF-α-3BNC117-NDs). All NDs exhibited monodisperse size distributions (PDI < 0.2) and mean hydrodynamic diameters between 200 and 250 nm (Figure S1). The NDs remained stable when stored in deionized water at 4 °C over 21 days, as demonstrated by consistent size distributions measured using DLS (Figure 1A). The negative surface charge (zeta potential) of the NDs was maintained over 21 days after encapsulation of TNF-α and/or 3BNC117 (Figure 1B), an important characteristic for formulation stability.22,30 TNF-α-3BNC117-NDs exhibited spherical morphology as visualized by scanning electron microscopy (SEM) (Figure 1C). The average encapsulation efficiencies for TNF-α were 98.2 ± 1.1% (TNF-α-ND) and 99.3 ± 0.23% (TNF-α-3BNC117-ND) (Figure 1D), and the average encapsulation efficiencies for 3BNC117 were 89.8 ± 12.1% (3BNC117-ND) and 94.2 ± 6.9% (TNF-α-3BNC117-ND) (Figure 1E), indicating a robust and effective synthesis and encapsulation scheme. Thus, size, stability, and negative surface charge of the NDs remained consistent regardless of the contents encapsulated.
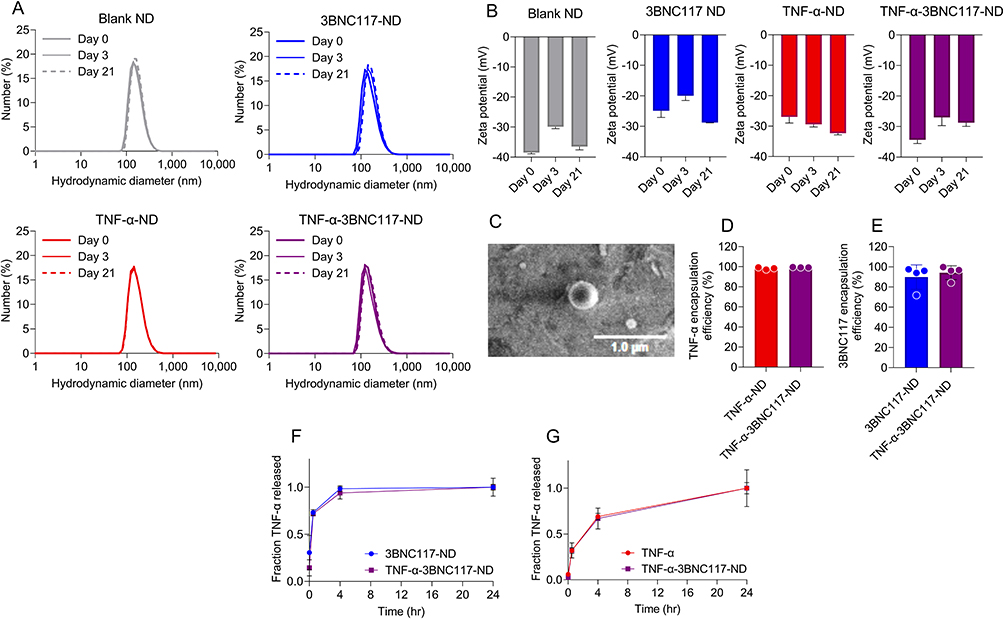 |
Figure 1 Stable and monodisperse PLGA NDs encapsulate and release TNF-α and 3BNC117. (A) ND hydrodynamic diameters were measured over 21 days by DLS. Gray: Blank NDs, Blue: 3BNC117-NDs, Red: TNF-α-NDs, Purple: TNF-α-3BNC117-NDs. (B) ND Zeta potential was measured by electrophoretic light scattering over 21 days. (C) TNF-α-3BNC117-ND visualized by SEM. The encapsulation efficiency of (D) TNF-α and (E) 3BNC117 was determined by ELISA. (F and G) TNF-α-3BNC117-NDs were incubated in PBS at 37 °C. Fractional release of (F) TNF-α and (G) 3BNC117 into the solution at the times indicated was analyzed by ELISA. Values represent means ± st. dev (A–E) or SEM (F and G). (n≥3/group). |
To determine the release of TNF-α and 3BNC117 from PLGA, NDs were incubated in PBS at physiologic conditions (pH 7.4, temperature 37°C) at 1:10 or 1:50 dilutions. At predetermined time intervals, the solution was centrifuged, and supernatants were analyzed for TNF-α and 3BNC117 content by ELISA. Both TNF-α (Figure 1F) and 3BNC117 (Figure 1G) were efficiently released from PLGA NDs, with the majority of each payload releasing in the initial hours of incubation and continuing over the course of 24 h. Rapid initial release that slows over time is broadly consistent with the well-characterized biphasic release pattern of PLGA nanoparticle drug delivery systems.31,32 These data illustrate the stability and consistency of the synthesized NDs, and their ability to release encapsulated agents over time in physiological conditions. The controlled release kinetics observed in these studies may confer advantages to the PLGA NDs over free administration of TNF-α and 3BNC117.20–22
TNF-α and 3BNC117 NDs Do Not Dramatically Impact NK Cell Phenotype
Since the proposed therapeutic platform relies on the co-administration of both NDs and NK92 cells, we investigated the effect of the NDs and their encapsulated agents on NK92 cell viability and phenotype. To quantify the expression of activation markers, we analyzed the expression of the transduced Fc receptor CD16; the activation markers CD25 and CD69; the post-activation marker TIM-3; the exhaustion marker PD-1; and the activating receptors NKp46 and NKG2D. To confirm a sustained NK cell homing phenotype, we analyzed the expression of the chemokine receptors CCR2, CCR5, CCR6, CCR7, CD62L, CX3CR1, CXCR3, and CXCR4. We have demonstrated the biocompatibility of PLGA NDs encapsulating other agents in the context of primary NK cells in previously published literature,22 and therefore hypothesized that these NDs would similarly not impact NK92 cell phenotype significantly. PLGA itself was non-toxic to NK92 cells (Figure S2), as was free TNF-α, even at concentrations greater than that required for viral reactivation (Figure S3 and S4). TNF-α was also found not to perturb expression of multiple activating and inhibitory receptors (Figure S4A), with the exceptions of modest TIM-3 and CD69 upregulation at the highest dose (400 ng/mL TNF-α). Increases in TIM-3 and CD69 expression have been observed in response to multiple different pro-inflammatory cytokines.33 TNF-α downregulated CXCR4, a chemokine receptor associated with the retention of NK cells in bone marrow,34 but did not significantly affect the expression of any other chemokine receptor evaluated (Figure S4B). PLGA NDs loaded with either TNF-α and/or 3BNC117 and dose-matched free agents likewise had minimal effect on NK92 cell viability and expression of the same activating and inhibitory markers (Figure 2A and B) and chemokine receptors described above (Figure 2C and D). Nevertheless, incubation with TNF-α-3BNC117 NDs was associated with minor but statistically significant increases in the expression of CD25, TIM-3, and CCR5, and a minor decrease in CD16 expression. These data suggest that co-administering NK92 cells with LRA- and bnAb-NDs does not substantially alter the basal characteristics of NK92 cell surface marker expression, and thus may be biocompatible as a combination therapy with NK92 cells.
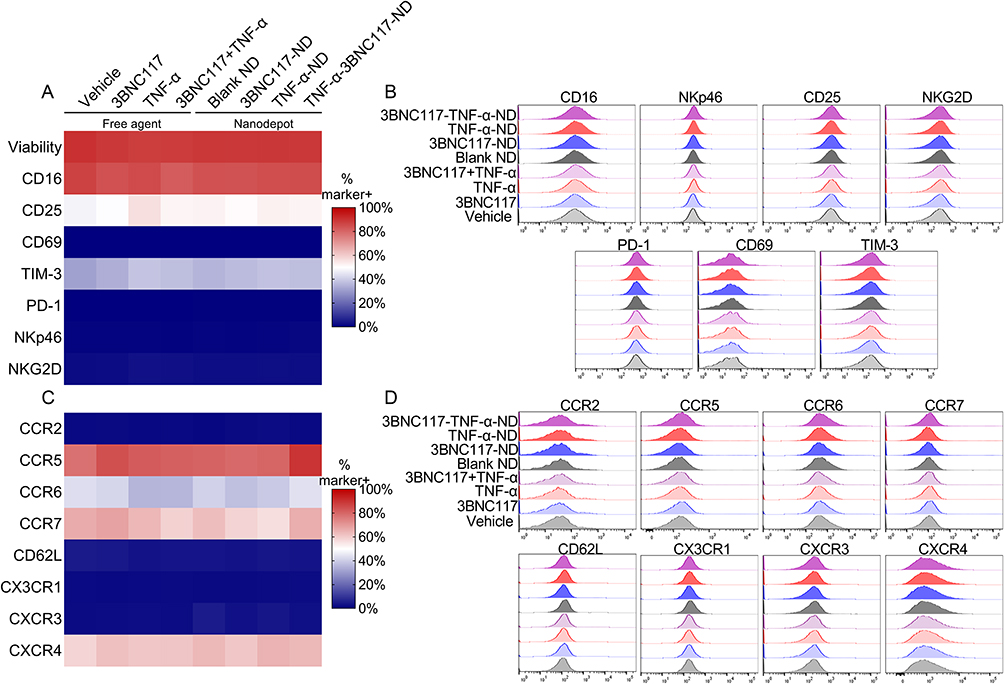 |
Figure 2 TNF-α, 3BNC117, and PLGA NDs minimally impact NK cell phenotype. NK92 cells were cultured with NDs or their constituent controls. After 24 h, NK92 cell viability and surface marker expression were quantified by flow cytometry. (A and B) Heat map and histograms depicting (A) frequency of marker-positive cells or (B) mean fluorescence intensity values for activating or inhibitory receptors. (C and D) Heat map and histograms depicting (C) frequency of marker-positive cells or (D) mean fluorescence intensity values for chemokine receptors. (n≥3/group). |
TNF-α and 3BNC117 NDs Reactivate Latent HIV and Bind to ACH-2 Cells
Next, we investigated the effect of the NDs on the latently infected ACH-2 cell line, which exhibits reactivation of latently integrated provirus upon TNF-α treatment (Figure S3).23,35 We incubated ACH-2 cells with NDs and measured cell viability, reactivation of HIV, bnAb binding to the cell surface, neutralization of reactivated virus, and extracellular p24 (gating strategy illustrated in Figure S5A). Treatment with free TNF-α, 3BNC117, blank NDs, or NDs encapsulating TNF-α, 3BNC117, or both TNF-α+3BNC117 sustained ACH-2 viability >80% after 24 hours (Figure 3A and S5B). TNF-α from both the TNF-α-NDs and the TNF-α-3BNC117-NDs significantly reactivated HIV expression in target ACH-2 cells as measured by intracellular capsid protein p24 staining (Figure 3B and S5C). Intracellular p24 expression in ACH-2 cells was 3.4-fold higher after TNF-α-ND treatment (87.5% p24+) and 3.0-fold higher after TNF-α-3BNC117-ND treatment (77.5% p24+) compared to basal expression levels (25.8% p24+), indicative of effective latency reversal by TNF-α. Complementary to evaluating viral reactivation, we also examined whether 3BNC117 from NDs could be detected at the surface of (reactivated) ACH-2 cells, presumably binding gp120 on budding virions. To visualize this, ACH-2 cells were stained with a fluorescent secondary antibody (anti-IgG) to quantify primary 3BNC117 binding. Free 3BNC117 administered in combination with free TNF-α bound 34.5% of ACH-2 cells, while 3BNC117 released from TNF-α-3BNC117-NDs bound 57.5% of ACH-2 cells (Figure 3C and S5D), illustrating the capacity of the TNF-α and 3BNC117 combination and the TNF-α-3BNC117-NDs to reactivate HIV from latency and facilitate bnAb binding to newly exposed viral epitopes on the target cells. Given the short half-life of TNF-α at physiological temperature and pH (Figure S6A), reactivation and gp120 binding were also quantified after first pre-incubating the NDs or loading-dose-matched free agents over multiple days at 37°C. After 72 h in cell media (FBS+), free agents and PLGA NDs both reactivated ACH-2 HIV (Figure S6B) and targeted ACH-2 cell surface with IgG (3BNC117) (Figure S6C). However, when pre-incubations were performed in PBS in the absence of stabilizing proteins from serum or other cell media components, free TNF-α failed to reactivate ACH-2 cells as reflected in a baseline frequency of capsid+ ACH-2 cells after 24 h of incubation time allotted for reactivation to occur. By contrast, TNF-α-NDs appeared to preserve LRA function, promoting an approximate 2-fold increase in reactivated (capsid+) ACH-2 cells, and suggesting a beneficial stabilizing or protective effect of encapsulating TNF-α within NDs (Figure S6D). Interestingly, free TNF-α reactivation function was rescued in the presence of 3BNC117 antibody. However, the degree of antibody binding associated with NDs was increased relative to free co-administration (Figure S6E and F).
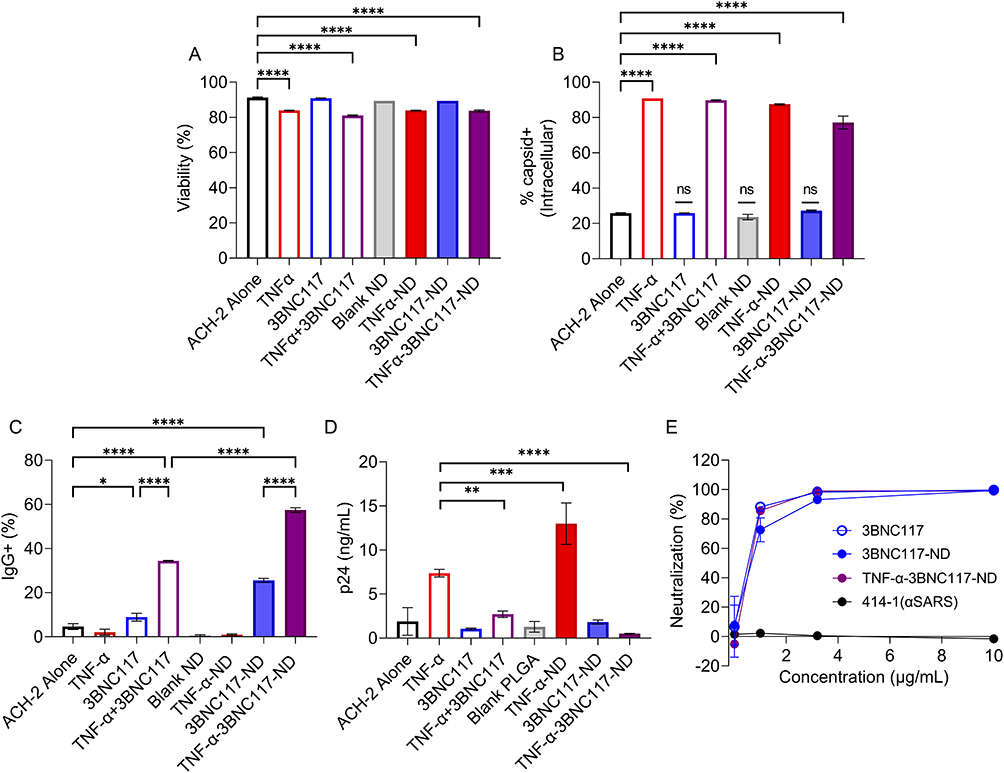 |
Figure 3 NDs encapsulating TNF-α and 3BNC117 retain multiple anti-viral functions. ACH-2 cells were cultured with NDs or free controls. After 24 h, (A) viability and (B) intracellular expression of capsid protein was analyzed as a measure of latent HIV reactivation by flow cytometry. (C) 3BNC117 binding to total live lymphocytes was analyzed by detecting secondary antibody (IgG) binding via flow cytometry (n=3/group). (D) Extracellular p24 detected via p24 ELISA after 24 h incubation with NDs or free controls. (E) Percent neutralization of reactivated ACH-2 HIV as determined by Tzm-bl assay. *p < 0.05 **p < 0.01 ***p < 0.001 ****p < 0.0001. Significance was determined by one-way ANOVA with Dunnett’s multiple comparisons test (A–C) or Sidak’s multiple comparisons test (D). Values represent means ± st. dev. (n≥2/group). |
The binding of 3BNC117 to virus at the surface of infected primary cells was recently demonstrated to not only neutralize the nascent virions, but also tether them to host cell membranes, forming aggregates and reducing extracellular levels of p24.36 To test this antiviral function of 3BNC117 in our platform, we measured p24 in the supernatants of ACH-2 cells cultured in media alone or in the presence of free TNF-α, 3BNC117, TNF-α and 3BNC117, blank NDs or TNF-α-3BNC117-NDs. Treatment of ACH-2 cells with TNF-α induced a 6.7-fold increase in extracellular p24. The combined administration of 3BNC117 with TNF-α significantly abrogated the efflux of p24 (2.5-fold p24 increase over baseline) (Figure 3D). Importantly, the TNF-α-3BNC117-NDs induced reactivation without any attendant increase in extracellular p24, which was observed to remain at baseline (un-reactivated) levels. This suggests a benefit of the ND approach, perhaps due to the sustained release of the encapsulated agents, enhanced bNAb contact with viral gp120 in the ND formulation, or through the PLGA ND itself binding soluble p24, as has been documented in the literature with other proteins.37–39 Additionally, both 3BNC117 and 3BNC117-NDs, but not non-specific anti-SARs antibodies, effectively neutralized ACH-2 HIV-1 virus from the supernatant of reactivated ACH-2 cultures as determined by a standard Tzm-bl assay (Figure 3E). Together, these data illustrate the functionality of TNF-α-3BNC117-NDs, wherein the encapsulated agents function in concert to reactivate target cells while simultaneously promoting neutralization of reactivated virus and enhanced trapping of the soluble products of reactivation. Thus, the TNF-α-3BNC117-NDs may mitigate the potential risk of increased transmission and virulence concomitant with latency reversal.
TNF-α and 3BNC117 NDs Enhance NK92 Cell-Mediated Cytotoxicity Toward Target ACH-2 Cells
Finally, we sought to illustrate the proof-of-concept feasibility of LRA- and bnAb-containing NDs to enhance NK92 cell cytotoxicity against latently infected cells. First, we incubated ACH-2 cells with NK92 cells at various E:T (NK92:ACH-2) cell ratios to quantify basal intrinsic NK92 cell cytotoxicity against ACH-2 cells. ACH-2 cells were killed by NK92 cells in a dose-dependent manner (Figure 4A), with ACH-2 cell viability lowest for the 2:1 E:T ratio (52% ACH-2 cell death) and highest for the 1:32 E:T ratio (9% ACH-2 cell death). Based on these determined ranges, we then incubated ACH-2 cells with TNF-α-3BNC117-NDs or dose-matched free agent controls (Figure S7) and evaluated ACH-2 viability after a 5 h co-culture with NK92 cells at an E:T ratio of 1.2.5. NK92 cells alone were associated with 35.5% ACH-2 cell death. As anticipated, Blank NDs did not significantly change NK92 cell-mediated cytotoxicity toward ACH-2 cells (34.6% ACH-2 cell death). Treatment of ACH-2 cells with NK92 cells in combination with TNF-α and 3BNC117 significantly enhanced NK92 cell-mediated killing (58.4% ACH-2 cell death). Critically, treatment of ACH-2 cells with NK92 cells in combination with TNF-α-3BNC117-NDs treatment significantly enhanced NK92 cell-mediated killing (68.5% ACH-2 cell death), suggesting the efficacy of the proposed platform (Figure 4B and S8).
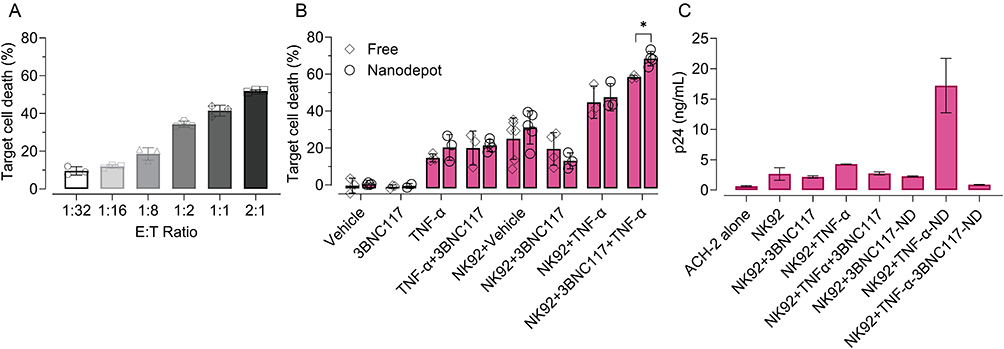 |
Figure 4 TNF-α and 3BNC117-containing NDs enhance NK cell cytotoxicity toward ACH-2 cells and reduce extracellular p24. (A) NK92 cells were co-cultured with ACH-2 cells for 24 h at varied E:T ratios. ACH-2 viability was measured by viability staining and flow cytometry. (B) ACH- 2 cells were incubated for 24 h with NDs or their constituent controls (30 ng/mL TNF-α, 2 μg/mL 3BNC117). At 19 h, for select conditions ACH-2 cells were co-cultured for the final 5 h with NK cells at an E:T ratio of 1:2.5, where indicated. After 24 h, the number of live ACH-2 cells was determined by flow cytometry. Percent killing was calculated based on the ratio of remaining live ACH-2 in the condition of interest to live ACH-2 untreated. Significance was determined by unpaired two-tailed t-test. (C) p24 detected after 24 h of ACH-2 cell incubation with NDs or free controls, with the addition of NK cells at a 1:2.5 E:T ratio for the final 5 h (co-culture). The data in panel (C) was collected and analyzed in the same assay corresponding to Figure 3D. Therefore, the amount of extracellular p24 in the ACH-2 alone condition is represented again here for the sake of comparison. Significance was determined by one-way ANOVA with Sidak’s multiple comparisons test. All values represent means ± st. dev. **p < 0.01 ***p < 0.001 ****p < 0.0001. |
To test the capability of the TNF-α-3BNC117-NDs to reduce extracellular p24 in the setting of an NK92 and ACH-2 cell co-culture, we measured p24 in the supernatants of treated cells. Extracellular p24 was significantly increased in the supernatants of ACH-2 and NK92 cell co-cultures in response to treatment with TNF-α (from a basal level of 0.64 ng/mL to 4.3 ng/mL p24), indicative of effective latency reversal in ACH-2 cells. Extracellular p24 decreased to 2.7 ng/mL when 3BNC117 was administered in combination with TNF-α, but still remained significantly higher than baseline p24 expression (0.6 ng/mL) (Figure 4C). Only the administration of TNF-α-3BNC117-NDs in combination with NK92 cells to ACH-2 cells was associated with near-baseline levels of extracellular p24 (0.90 ng/mL; Figure 4C), suggestive of the functionality of TNF-α-3BNC117-NDs to generate enhanced killing of latently infected targets while mitigating p24 release from ACH-2 cells. PLGA itself did not interfere with the detection of p24 by ELISA (Figure S9).
These data illustrate the potency of TNF-α-3BNC117-NDs to function in concert with NK92 cells, wherein TNF-α reactivates HIV in latently infected target ACH-2 cells, exposing bnAb binding sites on ACH-2 cells for 3BNC117 binding, which in turn facilitates enhanced NK92 cell-mediated cytotoxicity. To our knowledge, this demonstrates the first evidence of a PLGA ND-mediated system to enhance eradication of the latent HIV reservoir by combining the effects of NK cells via co-encapsulated LRA and bnAb. These data provide rationale for future testing of alternate LRA/bnAb combinations alone and in combination with NK cells (eg, NK92 cells, allogeneic or autologous primary NK cells) for HIV control applications. Further studies are warranted in samples sourced from people living with HIV (PLWH) on ART in parallel with in vivo studies.
Discussion
PLGA nanoparticles with similar size distributions to the NDs described here have been demonstrated to reach lymph nodes – the predominate anatomical niche of HIV reservoir – by transport across lymphatic endothelial cells.40–42 NK cells are likewise able to reach lymph nodes by chemotaxis, and have been shown in an allogeneic in vivo setting to traffic to lymph nodes as well as other secondary lymphoid organs.43 NK92 cells were found to express chemokine receptors that impact migration to the lymph nodes, such as CCR7. In vivo, the ability to coordinate the delivery of the LRA with the immediate presence of the bNAb may increase the efficiency by which newly reactivated virions are neutralized. This comparative advantage over free administration of these agents is difficult to illustrate directly in vitro, where the control condition offered for comparison with the LRA-bNAb-ND is simply the simultaneous co-addition of both free LRA and bNAb into the same well or tube. In vitro, both agents are artificially co-localized, whereas in vivo their co-accumulation in lymph nodes or other anatomical niches to therapeutic concentrations is not guaranteed. By contrast, we have shown that the LRA-bNAb-NDs co-release both LRA and bNAb together, which may be critical for efficient immune-mediated clearance of reactivated targets.
However, the present version of this strategy – utilizing TNF-α and 3BNC117 – is presumably insufficient for thorough elimination of viral reservoirs in vivo, where multiple barriers to efficacy are manifest. These include the challenges of addressing the vast patient-to-patient heterology in HIV infections,44 the difficulty of accessing small numbers of latently infected cells in anatomical sites of reservoir;45 the interference and uptake of the PLGA NDs from the mononuclear phagocyte system (MPS) and their clearance from filtering organs;46 and the necessity to achieve spatiotemporal coordination of all three components of the platform described herein. There are also limitations associated with the impressive but imperfect breadth and potency of any LRA or bNAb. The orchestrated effect of these agents, while complementary, is ultimately dependent on and limited to the efficacies of each individual agent. More effective LRAs and bNAbs are undoubtedly needed to maximize the efficacy of any combination approach. Nevertheless, a ND which co-encapsulates multiple different combinations of LRAs and bNAbs in combination with promising NK cell adoptive therapy may represent a uniquely efficacious combination approach with the potential to overcome these obstacles.
Conclusion
We have presented the proof-of-concept feasibility of a novel tripartite nanoparticle-based strategy that co-localizes the effects of an LRA, bnAb, and NK92 cells to promote killing of latently HIV-infected targets in vitro. Stable PLGA NDs co-encapsulated and released TNF-α and 3BNC117 and which efficiently reactivated and neutralized latent HIV in vitro as measured by increased expression of p24 in ACH-2 cells and inhibition of viral infection in Tzm-bl cells. When combined with NK92 cells, TNF-α-3BNC117-NDs enhanced the cytotoxicity of NK92 cells against ACH-2 cells by priming them for augmented NK92 cell-mediated cytolysis, suggesting the potential of our approach in controlling HIV.
Abbreviations
LRA, latency reversing agent; bnAb, broadly neutralizing antibody; PLGA, poly lactic-co-glycolic acid; ND, nanodepot.
Ethics
The research described in this study utilizes human cell lines purchased from the vendors or sources described in the Materials and Methods section. The authors do not have access to any identifying patient information (eg, name, social security number, medical record number, pathology accession number, or any other code) that would permit samples used in this study to be linked to individually identifiable living individuals. Hence, the studies have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. Further, based on the decision tool for human subjects research by The Office of Extramural Research of the National Institutes of Health, United States, the study is not considered human subjects research.
Acknowledgments
Research reported in this publication was supported in part by the George Washington Cancer Center and the National Institute of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R33AI136102, with additional support (to JG) under Award Number T32 AI158105. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are grateful to Dr. Alberto Bosque and Dr. Sanjay Maggirwar for helpful discussions, and we thank the GWU Flow Cytometry Core and Nanofabrication and Imaging Core Facilities for expert technical assistance.
Disclosure
Dr Conrad Russell Y Cruz reports equity ownership, consultation fees from Mana Therapeutics, sponsored research agreement, also signed as a consultant but no consulting has been done yet from Catamaran Bio, outside the submitted work; In addition, Dr Conrad Russell Y Cruz has a patent NK cells pending to Children’s National Hospital, a patent HIV specific T cells pending to Children’s National Hospital, a patent Antibody secreting T or NK cells pending to Children’s National Hospital, a patent Use of nanoparticles and immune cells pending to Children’s National Hospital. Dr Rohan Fernandes reports grants from National Institute of Allergy and Infectious Diseases, National Institutes of Health, non-financial support, George Washington pays the salary of its employees, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Chun TW, Stuyver L, Mizell SB, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. 1997;94(24):13193–13197.
2. Chun TW, Davey RT Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401(6756):874–875.
3. Sension MG. Long-Term suppression of HIV infection: benefits and limitations of current treatment options. J Assoc Nurses AIDS Care. 2007;18(1 Suppl):S2–10.
4. Margolis AM, Heverling H, Pham PA, Stolbach A. A review of the toxicity of HIV medications. J Med Toxicol. 2014;10(1):26–39.
5. Mikulak J, Oriolo F, Zaghi E, Di Vito C, Mavilio D. Natural killer cells in HIV-1 infection and therapy. Aids. 2017;31(17):2317–2330.
6. Breunig M, Lungwitz U, Liebl R, Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc Natl Acad Sci U S A. 2007;104(36):14454–14459.
7. Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163–194.
8. Brandstadter JD, Yang Y. Natural killer cell responses to viral infection. J Innate Immun. 2011;3(3):274–279.
9. Ahmad R, Sindhu ST, Toma E, et al. Evidence for a correlation between antibody-dependent cellular cytotoxicity-mediating anti-HIV-1 antibodies and prognostic predictors of HIV infection. J Clin Immunol. 2001;21(3):227–233.
10. Baum LL, Cassutt KJ, Knigge K, et al. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J Immunol. 1996;157(5):2168–2173.
11. Forthal DN, Landucci G, Daar ES. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. J Virol. 2001;75(15):6953–6961.
12. Nag P, Kim J, Sapiega V, et al. Women with cervicovaginal antibody-dependent cell-mediated cytotoxicity have lower genital HIV-1 RNA loads. J Infect Dis. 2004;190(11):1970–1978.
13. Gooneratne SL, Richard J, Lee WS, Finzi A, Kent SJ, Parsons MS. Slaying the Trojan horse: natural killer cells exhibit robust anti-HIV-1 antibody-dependent activation and cytolysis against allogeneic T cells. J Virol. 2015;89(1):97–109.
14. Lambotte O, Ferrari G, Moog C, et al. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. Aids. 2009;23(8):897–906.
15. Lewis GK, Pazgier M, Evans DT, et al. Beyond Viral Neutralization. AIDS Res Hum Retroviruses. 2017;33(8):760–764.
16. Cheng J, Teply BA, Sherifi I, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–876.
17. Li Y, Pei Y, Zhang X, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Control Release. 2001;71(2):203–211.
18. Gdowski A, Ranjan A, Mukerjee A, Vishwanatha J. Development of biodegradable nanocarriers loaded with a monoclonal antibody. Int J Mol Sci. 2015;16(2):3990–3995.
19. Langer R, Folkman J. Polymers for the sustained release of proteins and other macromolecules. Nature. 1976;263(5580):797–800.
20. Hines DJ, Kaplan DL. Poly(lactic-co-glycolic) acid-controlled-release systems: experimental and modeling insights. Crit Rev Ther Drug Carrier Syst. 2013;30(3):257–276.
21. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397.
22. Sweeney EE, Balakrishnan PB, Powell AB, et al. PLGA nanodepots co-encapsulating prostratin and anti-CD25 enhance primary natural killer cell antiviral and antitumor function. Nano Res. 2020;13(3):736–744.
23. Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc Natl Acad Sci U S A. 1989;86(15):5974–5978.
24. Shingai M, Nishimura Y, Klein F, et al. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature. 2013;503(7475):277–280.
25. Danhier F, Ansorena E, Silva JM, Coco R, Le breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522.
26. Martinez Rivas CJ, Tarhini M, Badri W, et al. Nanoprecipitation process: from encapsulation to drug delivery. Int J Pharm. 2017;532(1):66–81.
27. Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17(3):247–289.
28. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6:368.
29. Clouse KA, Powell D, Washington I, et al. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J Immunol. 1989;142(2):431–438.
30. Platel A, Carpentier R, Becart E, Mordacq G, Betbeder D, Nesslany F. Influence of the surface charge of PLGA nanoparticles on their in vitro genotoxicity, cytotoxicity, ROS production and endocytosis. J Appl Toxicol. 2016;36(3):434–444.
31. Pourtalebi Jahromi L, Ghazali M, Ashrafi H, Azadi A. A comparison of models for the analysis of the kinetics of drug release from PLGA-based nanoparticles. Heliyon. 2020;6(2):e03451.
32. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18.
33. Ndhlovu LC, Lopez-Verges S, Barbour JD, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119(16):3734–3743.
34. Bernardini G, Sciume G, Bosisio D, Morrone S, Sozzani S, Santoni A. CCL3 and CXCL12 regulate trafficking of mouse bone marrow NK cell subsets. Blood. 2008;111(7):3626–3634.
35. Poli G, Kinter A, Justement JS, et al. Tumor necrosis factor alpha functions in an autocrine manner in the induction of human immunodeficiency virus expression. Proc Natl Acad Sci U S A. 1990;87(2):782–785.
36. Dufloo J, Planchais C, Fremont S, et al. Broadly neutralizing anti-HIV-1 antibodies tether viral particles at the surface of infected cells. Nat Commun. 2022;13(1):630.
37. Gossmann R, Fahrlander E, Hummel M, Mulac D, Brockmeyer J, Langer K. Comparative examination of adsorption of serum proteins on HSA- and PLGA-based nanoparticles using SDS-PAGE and LC-MS. Eur J Pharm Biopharm. 2015;93:80–87.
38. Sempf K, Arrey T, Gelperina S, et al. Adsorption of plasma proteins on uncoated PLGA nanoparticles. Eur J Pharm Biopharm. 2013;85(1):53–60.
39. Ndumiso M, Buchtova N, Husselmann L, et al. Comparative whole Corona fingerprinting and protein adsorption thermodynamics of PLGA and PCL nanoparticles in human serum. Colloids Surf B Biointerfaces. 2020;188:110816.
40. Kobayashi H, Kawamoto S, Bernardo M, Brechbiel MW, Knopp MV, Choyke PL. Delivery of gadolinium-labeled nanoparticles to the sentinel lymph node: comparison of the sentinel node visualization and estimations of intra-nodal gadolinium concentration by the magnetic resonance imaging. J Control Release. 2006;111(3):343–351.
41. McCright J, Skeen C, Yarmovsky J, Maisel K. Nanoparticles with dense poly(ethylene glycol) coatings with near neutral charge are maximally transported across lymphatics and to the lymph nodes. Acta biomaterialia. 2022;145:146–158.
42. Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release. 2006;112(1):26–34.
43. Olson JA, Zeiser R, Beilhack A, Goldman JJ, Negrin RS. Tissue-specific homing and expansion of donor NK cells in allogeneic bone marrow transplantation. J Immunol. 2009;183(5):3219–3228.
44. Siemieniuk RA, Beckthold B, Gill MJ. Increasing HIV subtype diversity and its clinical implications in a sentinel North American population. Can J Infect Dis Med Microbiol. 2013;24(2):69–73.
45. Ho YC, Shan L, Hosmane NN, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–551.
46. Bourguignon T, Torrano AA, Houel-Renault L, Machelart A, Brodin P, Gref R. An original methodology to study polymeric nanoparticle-macrophage interactions: nanoparticle tracking analysis in cell culture media and quantification of the internalized objects. Int J Pharm. 2021;610:121202.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.