Back to Journals » Journal of Hepatocellular Carcinoma » Volume 10
N6-Methyladenosine-Modified ATP8B1-AS1 Exerts Oncogenic Roles in Hepatocellular Carcinoma via Epigenetically Activating MYC
Authors Tan C, Huang Y, Huang Z, Ning Y, Huang L, Wu X, Lu Y, Wei H, Pu J
Received 1 April 2023
Accepted for publication 6 July 2023
Published 6 September 2023 Volume 2023:10 Pages 1479—1495
DOI https://doi.org/10.2147/JHC.S415318
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ahmed Kaseb
Chuan Tan,1,* Yanyan Huang,2,* Zheng Huang,2,* Yuanjia Ning,2 Lizheng Huang,2 Xianjian Wu,1 Yuan Lu,1 Huamei Wei,3 Jian Pu1
1Department of Hepatobiliary Surgery, Affiliated Hospital of Youjiang Medical University for Nationalities, Baise, People’s Republic of China; 2Graduate College of Youjiang Medical University for Nationalities, Baise, People’s Republic of China; 3Department of Pathology, Affiliated Hospital of Youjiang Medical University for Nationalities, Baise, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Pu, Department of Hepatobiliary Surgery, Affiliated Hospital of Youjiang Medical University for Nationalities, No. 18 Zhongshan Two Road, Baise, Guangxi, People’s Republic of China, Email [email protected]
Purpose: N6-methyladenosine (m6A) modification has shown critical roles in regulating mRNA fate. Non-coding RNAs also have important roles in various diseases, including hepatocellular carcinoma (HCC). However, the potential influences of m6A modification on non-coding RNAs are still unclear. In this study, we identified a novel m6A-modified ATP8B1-AS1 and aimed to investigate the effects of m6A on the expression and role of ATP8B1-AS1 in HCC.
Methods: qPCR was performed to measure the expression of related genes. The correlation between gene expression and prognosis was analyzed using public database. m6A modification level was measured using MeRIP and single-base elongation- and ligation-based qPCR amplification method. The roles of ATP8B1-AS1 in HCC were investigated using in vitro and in vivo functional assays. The mechanisms underlying the roles of ATP8B1-AS1 were investigated by ChIRP and ChIP assays.
Results: ATP8B1-AS1 is highly expressed in HCC tissues and cell lines. High expression of ATP8B1-AS1 is correlated with poor overall survival of HCC patients. ATP8B1-AS1 is m6A modified and the 792 site of ATP8B1-AS1 is identified as an m6A modification site. m6A modification increases the stability of ATP8B1-AS1 transcript. m6A modification level of ATP8B1-AS1 is increased in HCC tissues and cell lines, and correlated with poor overall survival of HCC patients. ATP8B1-AS1 promotes HCC cell proliferation, migration, and invasion, which were abolished by the mutation of m6A-modified 792 site. Mechanistic investigation revealed that m6A-modified ATP8B1-AS1 interacts with and recruits m6A reader YTHDC1 and histone demethylase KDM3B to MYC promoter region, leading to the reduction of H3K9me2 level at MYC promoter region and activation of MYC transcription. Functional rescue assays showed that depletion of MYC largely abolished the oncogenic roles of ATP8B1-AS1.
Conclusion: m6A modification level of ATP8B1-AS1 is increased and correlated with poor prognosis in HCC. m6A-modified ATP8B1-AS1 exerts oncogenic roles in HCC via epigenetically activating MYC expression.
Keywords: hepatocellular carcinoma, N6-methyladenosine, noncoding RNA, histone methylation, MYC signaling
Introduction
Liver cancer is one of the most common malignancies worldwide, which has relatively poor prognosis.1 According to global cancer statistics 2020, liver cancer is the six most commonly diagnosed cancer, following breast, lung, colorectal, prostate, and stomach cancers.2 However, liver cancer is the third leading cause of cancer death, following lung and colorectal cancers.2 Hepatocellular carcinoma (HCC) is the major histological subtype of liver cancer.3 Until now, the efficient therapy for HCC is still surgical resection.4 Unlike other common cancers, such as lung and breast cancers, liver cancer is not sensitive to commonly used targeted therapy and immunotherapy.5–8 Thus, further revealing the molecular mechanisms underlying HCC initiation and progression is urgently needed.
Many molecules and signaling pathways aberrations have been found in HCC, such as P53, Wnt–β-catenin signaling, TGF-β signaling, IL6–JAK–STAT signaling, AKT signaling, MET signaling, MYC signaling, and et al.9–13 Underlying these signaling pathways, many genetic and epigenetic aberrations have been identified.14 Classical genetic aberrations found in HCC include chromosome 17p loss, TP53 mutation, TERT promoter mutation, CTNNB1 mutation, AXIN1 mutation.3 Compared with genetic aberrations, more epigenetic aberrations have also been identified in HCC, such as DNA methylation, histone acetylation, and histone methylation.15 Aberrant epigenetic modifications regulate the expression of target genes, which contributes to more frequently found gene expression dysregulations in HCC.16
Recently, apart from epigenetic modifications, epitranscriptomic modifications also have been shown to play important roles in gene expression modulation.17 Among epitranscriptomic modifications, N6-methyladenosine (m6A) modification is the most common internal modification in eukaryotic RNA.18 m6A is reversibly added by writers, such as the methyltransferases METTL3, METTL14, WTAP, and METTL16, and removed by erasers, such as the demethylases FTO and ALKBH5.19–22 m6A-modified RNAs were recognized by readers, such as YTHDC1/2, YTHDF1/2/3, IGF2BP1/2/3, and HNRNPG/C.23,24 m6A has critical roles in RNA metabolisms and fates, such as RNA splicing, nuclear export, translation, and stability.25 Until now, most m6A modifications were found in messenger RNAs (mRNAs).26 Long noncoding RNA (lncRNA) is another class of long RNAs with limited protein-encoding potential.27–30 Dysregulated expressions and critical roles of lncRNAs in various diseases, including cancers have been frequently revealed.31–36 Several studies have revealed m6A modification in lncRNAs.37,38 METTL3 installed m6A modification in lncKCNQ1OT1, which increased the stability and expression of lncKCNQ1OT1.37 However, METTL16-induced m6A modification in lncRNA RAB11B-AS1 decreased the stability and expression of RAB11B-AS1.38 The other potential influences of m6A modification on the fates of lncRNAs need further investigation.
A previous study has identified several m6A modification and prognosis-related lncRNAs in HCC via analyzing The Cancer Genome Atlas (TCGA) Liver Hepatocellular Carcinoma (LIHC) data.39 Among these lncRNAs, we found that not only the expression level of ATP8B1-AS1 (also annotated as AC027097.1 or RP11-35G9.3), but also the m6A modification level of ATP8B1-AS1 was increased in HCC and associated with poor prognosis of HCC patients. Thus, we focused on ATP8B1-AS1 in this study. ATP8B1-AS1 is located in chromosome 18q21.31. ATP8B1-AS1 has 3 exons and its antisense strand is ATP8B1. In this study, we further investigated the contribution of m6A modification to the expression, role, and clinical relevance of ATP8B1-AS1 in HCC. We identified the classical oncogene MYC as the critical downstream target of m6A-modified ATP8B1-AS1. MYC has been shown to be increased and play oncogenic roles in HCC.40,41
Materials and Methods
HCC Samples
The correlation between ATP8B1-AS1 expression and overall survival of HCC patients based on TCGA-LIHC data was analyzed using the online tool GEPIA (http://gepia.cancer-pku.cn/). The TCGA-LIHC RNA-seq data and corresponding clinical information were downloaded from https://portal.gdc.cancer.gov/. Furthermore, 79 pairs of HCC tissues and matched adjacent noncancerous liver tissues were randomly obtained from HCC patients who received surgery at the Affiliated Hospital of Youjiang Medical University for Nationalities. Written informed consents were acquired from all participants. This study was performed following the Helsinki Declaration and with the approval from Youjiang Medical University for Nationalities Institutional Review Board.
Cell Culture
Human immortalized liver cell-line THLE-2 (cat. no. CRL-2706) and HCC cell line SNU-398 (cat. no. CRL-2233) were acquired from American Type Culture Collection (ATCC, Manassas, VA, USA). Human HCC cell lines HuH-7 (cat. no. SCSP-526) and SK-HEP-1 (cat. no. TCHu109) were acquired from the Chinese Academy of Sciences Cell Bank (Shanghai, China). THLE-2 was maintained in the BEGM Bullet Kit (cat. no. CC-3170, Lonza). SNU-398 was maintained in RPMI 1640 medium (Invitrogen Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen). HuH-7 was cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% FBS. SK-HEP-1 was maintained in Eagle’s Minimum Essential Medium (Invitrogen) supplemented with 10% FBS. All cells were cultured at 37°C containing 5% CO2 and routinely tested as mycoplasma-free.
Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was isolated from indicated tissues and cells using the RNA isolater Total RNA Extraction Reagent (cat. no. R401, Vazyme, Nanjing, China). The RNA was subjected to reverse transcription using the HiScript III RT SuperMix for qPCR (cat. no. R323, Vazyme) to generate the first strand complementary DNA (cDNA). The cDNA was further subjected to quantitative polymerase chain reaction (qPCR) using the ChamQ Universal SYBR qPCR Master Mix (cat. no. Q711, Vazyme) on QuantStudio Real-Time PCR Instrument (Applied biosystems, Foster City, CA, USA) with the primers 5’-AGTTTCACTCTTGTTGCCC-3’ (sense) and 5’-TCTTGTCCCTCTGTCCTT-3’ (antisense) for ATP8B1-AS1, 5’-TGCTGTGGCAGAAAAGAAG-3’ (sense) and 5’-TCAGACCAGAGACTAACGAAC-3’ (antisense) for METTL3, 5’-ACTGTGGAAGAAGATGGAGG-3’ (sense) and 5’-TGAAACGATGTCTGTGAGGT-3’ (antisense) for FTO, 5’-CTTCCCCTACCCTCTCAA-3’ (sense) and 5’-CGATTTCTTCCTCATCTTCT-3’ (antisense) for MYC, 5’-ATCAAGTAGTGCCTCCAG-3’ (sense) and 5’-CTTCCTCCTCATTCTCAG-3’ (antisense) for YTHDC1, 5’-GCCTCCAACAACAAAACC-3’ (sense) and 5’-CCATCACCATCTCCTTCAC-3’ (antisense) for KDM3B, 5’-GTCGGAGTCAACGGATTTG-3’ (sense) and 5’-TGGGTGGAATCATATTGGAA-3’ (antisense) for GAPDH. GAPDH was used as endogenous control. Relative quantification was calculated using the comparative Ct method.
Western Blot
Total protein was isolated from indicated cells using the RIPA Lysis Buffer (Beyotime, Shanghai, China), which was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by being transferred onto nitrocellulose membrane. After block using 5% nonfat milk, the membranes were incubated with primary antibodies specific for METTL3 (cat. no. ab195352, 1:1000, Abcam, Cambridge, MA, USA), FTO (cat. no. ab126605, 1:1000, Abcam), MYC (cat. no. ab32072, 1:1000, Abcam), or GAPDH (cat. no. ab8245, 1:5000, Abcam). The second antibodies used were Goat anti-Mouse IgG H&L (IRDye® 680RD) preadsorbed (cat. no. ab216776, 1:10,000, Abcam) or Goat anti-Rabbit IgG H&L (IRDye® 800CW) preadsorbed (cat. no. ab216773, 1:10,000, Abcam). Lastly, the membranes were scanned on an Odyssey infrared scanner ((Li-Cor, Lincoln, NE, USA).
Vectors Construction, Transfection, and Stable Cell Lines Construction
The cDNA encoding ATP8B1-AS1 was PCR-amplified with the PrimeSTAR Max DNA Polymerase (cat. no. R045Q, Takara, Tokyo, Japan) and the primers 5’-TTGGTACCGAGCTCGGATCCATTTCCTACTTGGTCCTGTTC-3’ (sense) and 5’-GTGCTGGATATCTGCAGAATTCTTCTAAGCAGAATTGAAATTTTTAT-3’ (antisense), followed by being cloned into the BamH I and EcoR I sites of the pcDNA3.1(+) vector (Invitrogen) using the NovoRec plus One step PCR Cloning Kit (cat. no. NR005, Novoprotein, Suzhou, China) to construct ATP8B1-AS1 expression vector. The m6A-modified 792 site mutated ATP8B1-AS1 expression vector was constructed using the Fast Mutagenesis System (cat. no. FM111, TransGen, Beijing, China) with the primers 5’-TCAAAGCACTGGAAGGTCAGAGGGACAA-3’ (sense) and 5’-ACCTTCCAGTGCTTTGACATTTGGTGGG-3’ (antisense). Two pairs of cDNA oligonucleotides targeting ATP8B1-AS1 were synthesized and cloned into the shRNA lentivirus expressing vector (GenePharma, Shanghai, China) to generate shRNA lentivirus targeting ATP8B1-AS1. Scrambled non-targeting shRNA lentivirus were used as negative control (NC). The sequences of shRNA oligonucleotides were as follows: 5’-GATCCGCAAAGGAAACGGCAGCTATATTCAAGAGATATAGCTGCCGTTTCCTTTGCTTTTTTG-3’ (sense) and 5’-AATTCAAAAAAGCAAAGGAAACGGCAGCTATATCTCTTGAATATAGCTGCCGTTTCCTTTGCG-3’ (anti-sense) for shRNA-ATP8B1-AS1-1, 5’-GATCCGCTGCCATGTTTCACAATTACTTCAAGAGAGTAATTGTGAAACATGGCAGCTTTTTTG-3’ (sense) and 5’-AATTCAAAAAAGCTGCCATGTTTCACAATTACTCTCTTGAAGTAATTGTGAAACATGGCAGCG-3’ (antisense) for shRNA-ATP8B1-AS1-2, 5’-GATCCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACTTTTTTG-3’ (sense) and 5’-AATTCAAAAAAGTTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAACG-3’ (antisense) for shRNA-NC. METTL3 and FTO expression vectors were obtained from GenePharma. ON-TARGETplus Human YTHDC1 siRNA SMART Pool (cat. no. L-015332-02-0010), ON-TARGETplus Human KDM3B siRNA SMART Pool (cat. no. L-020378-01-0010), and ON-TARGETplus Human MYC siRNA SMART Pool (cat. no. L-003282-02-0010) were purchased from Horizon Discovery (Cambridge, England). The transfection of vectors and siRNAs was conducted using the GP-transfect-Mate (GenePharma).
To construct HCC cells with wild-type or m6A-modified 792 site mutated ATP8B1-AS1 stable overexpression, ATP8B1-AS1 expression vector or m6A-modified 792 site mutated ATP8B1-AS1 expression vector was transfected into SNU-398 and HuH-7 cells. Forty-eight hours later, the cells were treated with 1000 µg/mL neomycin for four weeks to select cells with stable wild-type or m6A-modified 792 site mutated ATP8B1-AS1 overexpression. To construct HCC cells with ATP8B1-AS1 stable depletion, shRNA lentivirus targeting ATP8B1-AS1 were infected into SNU-398 and SK-HEP-1 cells. Ninety-six hours later, the cells were treated with 2 µg/mL puromycin for four weeks to select cells with stable ATP8B1-AS1 depletion.
m6A Modification Detection
m6A-modified transcripts were enriched by the methylated RNA immunoprecipitation (MeRIP) assays using the Magna MeRIP m6A Kit (cat. no. 17–10499, Millipore, Billerica, MA, USA). The enriched RNAs were detected by qPCR as described above. The specific m6A modification site was predicted by the online tool SRAMP (http://www.cuilab.cn/sramp). m6A modification level of 792 site of ATP8B1-AS1 was measured using the previously described single-base elongation- and ligation-based qPCR amplification method (termed “SELECT”).42 Concurrent detection of non-modified 788 site was carried out and served as input. The sequences of probes used were as follows: 5’-tagccagtaccgtagtgcgtgGGGCCATCCCTCTTGTCCCTCTG-3’ (up) and 5’-5phos/CCTTCCAGTGCTTTGACATTTGGTGcagaggctgagtcgctgcat-3’ (down) for 792 site, 5’-tagccagtaccgtagtgcgtgCATCCCTCTTGTCCCTCTGTCCT-3’ (up) and 5’-5phos/CCAGTGCTTTGACATTTGGTGGGcagaggctgagtcgctgcat-3’ (down) for 788 site. The primers for qPCR of SELECT were 5’-ATGCAGCGACTCAGCCTCTG-3’ (sense) and 5’-TAGCCAGTACCGTAGTGCGTG-3’ (antisense).42
Cell Proliferation, Migration, and Invasion Assays
Cell proliferation was evaluated using Glo cell viability assay and 5-ethynyl-2’-deoxyuridine (EdU) incorporation assay. Glo cell viability assay was performed using the CellTiter-Glo Luminescent Cell Viability Assay (cat. no. G7572, Promega, Madison, WI, USA) after culture for 48h as previously reported.43 The luminescence was used to indicate cell viability. EdU incorporation assay was conducted using the Cell-Light EdU Apollo567 In Vitro Kit (cat. no. C10310-1, RiboBio, Guangzhou, China) following the provided protocol. Briefly, after culture for overnight, indicated HCC cells were treated with 50 μM EdU for 2h. Next, the treated cells were fixed in 4% paraformaldehyde for 30 min and permeabilized in 0.5% Triton X-100 for 10 min. The cells were further stained with Apollo dye solution (RiboBio) and the cell nucleuses were stained with DAPI. The number of EdU-positive cells was assessed using the fluorescence microscope (Carl Zeiss, Oberkochen, Germany). Cell migration and invasion were evaluated by transwell migration and invasion assays, respectively. Briefly, indicated cells resuspended in 200μL serum-free medium were loaded in the upper chambers of the Millicell Hanging Inserts (8-μm pore, Millipore) precoated with or without Matrigel (BD Biosciences, San Jose, CA, USA). Complete medium supplemented with 10% FBS was loaded into the lower chambers. After culture for 24h, the cells remaining on the upper chambers were removed. The cells invading into the bottom side of the inserts were fixed, stained, and quantified under a microscope by counting at least five random high-power fields.
Subcutaneous Tumor Growth Assay
Indicated HCC cells were subcutaneously inoculated into the flank of 5-week-old male BALB/C athymic mice, which were purchased from Shanghai SLAC Laboratory Animal Co. and fed in specific pathogen-free condition. Subcutaneous tumor volumes were measured every week and calculated using the formula 0.5×length×width2. At the 28th day after inoculation, subcutaneous tumors were resected, weighed, and photographed. This study was approved by Affiliated Hospital of Youjiang Medical University for Nationalities Institutional Review Board.
Chromatin Isolation by RNA Purification (ChIRP) Assay
ChIRP assay was conducted in SNU-398 cells using the EZ-Magna ChIRP RNA Interactome Kit (cat. no. 17–10495, Millipore). The sequences of ATP8B1-AS1 antisense DNA probes were as follows: 1, 5’-ccacgaacaggaccaagtag-3’; 2, 5’-aagcctgttatgaggagcaa-3’; 3, 5’-aattggtcttatctctccag-3’; 4, 5’-ggacggaagatgctttttct-3’; 5, 5’-cattgtgcagggaagcatat-3’; 6, 5’-tgggagctacagctattgaa-3’; 7, 5’-attagcccttctttttacag-3’; 8, 5’-aagtttttaggtgtgttggt-3’; 9, 5’-ggtttacggtttatgtcagt-3’; 10, 5’-tacattgggggaattaccga-3’; 11, 5’-gcagaacagaagttgccatt-3’. The enriched DNA was measured by qPCR with the primers: 5’-CTTCTTTCCTCCACTCTCCC-3’ (sense) and 5’-ACCCTCTCCCTTTCTCTGC-3’ (antisense) for MYC promoter; 5’-GACGCTTTCTTTCCTTTCGC-3’ (sense) and 5’-CTGCCCATTCATTTCCTTCC-3’ (antisense) for GAPDH promoter.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was conducted in SNU-398 cells using the EZ-Magna ChIP A/G Chromatin Immunoprecipitation Kit (cat. no. 17–10086, Millipore) and a KDM3B antibody (cat. no. 5377, Cell Signaling Technology, Danvers, MA, USA) or an H3K9me2 antibody (cat. no. ab1220, Abcam). The enriched DNA was measured by qPCR with the primers: 5’-CTTCTTTCCTCCACTCTCCC-3’ (sense) and 5’-ACCCTCTCCCTTTCTCTGC-3’ (antisense) for MYC promoter.
Statistical Analysis
The GraphPad Prism v6.0 Software (San Diego, CA, USA) was used to carry out all statistical analyses. Mann–Whitney test, Wilcoxon matched-pairs signed rank test, Log rank test, one-way ANOVA followed by Dunnett’s multiple comparisons test, Spearman correlation analysis, or two-tailed unpaired t-test were performed as shown in figure legends. P < 0.05 was considered statistically significant.
Results
ATP8B1-AS1 is Highly Expressed and Correlated with Poor Overall Survival in HCC
The clinical correlation of ATP8B1-AS1 in HCC was first analyzed using the TCGA-LIHC data. The results showed that high expression of ATP8B1-AS1 was correlated with short overall survival time (Figure 1A). ATP8B1-AS1 was highly expressed in HCC tissues compared with that in normal liver tissues (Figure 1B). Furthermore, the expression of ATP8B1-AS1 was higher in HCC tissues with poor differentiation than those with good differentiation (Figure 1C). The TCGA-LIHC data also revealed that the expression of ATP8B1-AS1 was positively correlated with high alpha-fetoprotein (AFP) level (Figure 1D). To further investigate the clinical relevance of ATP8B1-AS1 in HCC, we collected 79 pairs of HCC tissues and matched adjacent noncancerous liver tissues and measured ATP8B1-AS1 expression in these tissues. The results showed that ATP8B1-AS1 was also highly expressed in HCC tissues compared with that in matched adjacent noncancerous liver tissues (Figure 1E). Increased expression of ATP8B1-AS1 was correlated with short overall survival time in our HCC cohort (Figure 1F). The expression of ATP8B1-AS1 in immortalized liver cell-line THLE-2 and HCC cell lines HuH-7, SNU-398, and SK-HEP-1 were measured. The results showed that consistent with the expression pattern in HCC tissues, ATP8B1-AS1 was also highly expressed in HCC cell lines compared with that in immortalized liver cell line (Figure 1G). Therefore, these data suggested that ATP8B1-AS1 is highly expressed and correlated with poor overall survival in HCC.
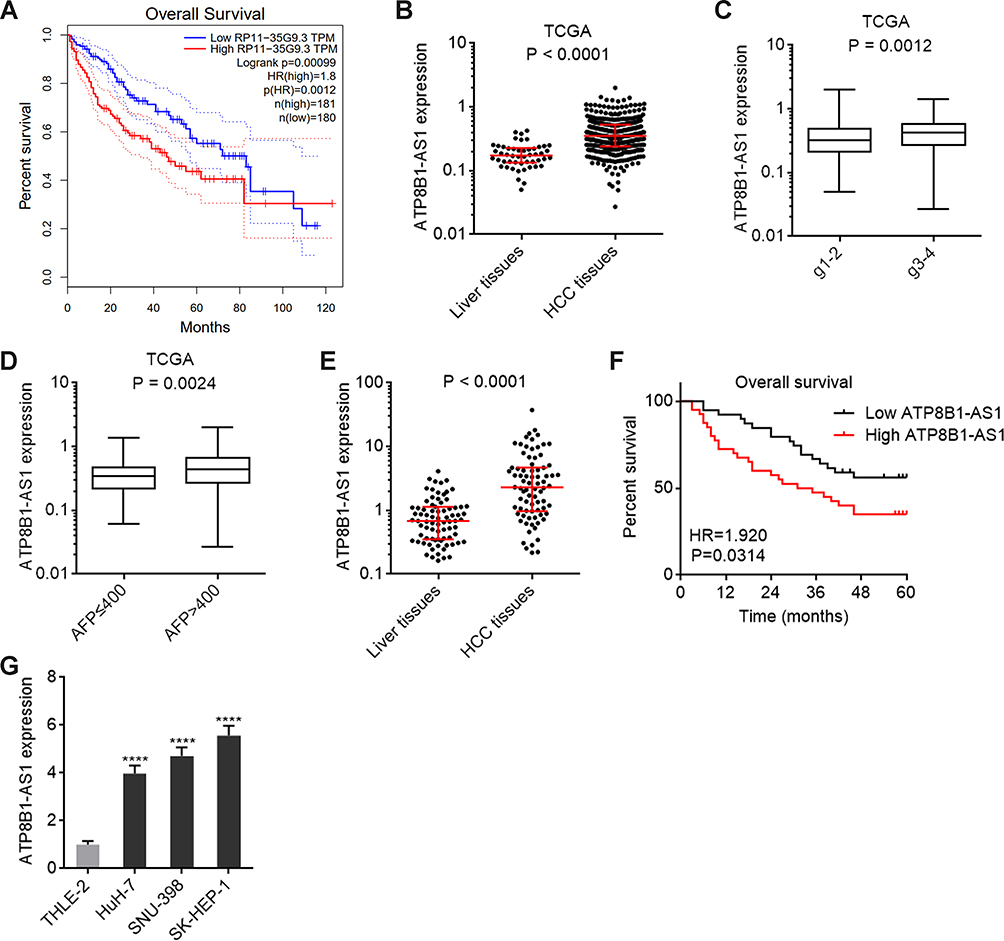 |
Figure 1 ATP8B1-AS1 is upregulated and correlated with poor overall survival in HCC. (A) The correlation between ATP8B1-AS1 (RP11-35G9.3) expression and overall survival in HCC based on TCGA-LIHC RNA-seq data, analyzed by GEPIA. (B) ATP8B1-AS1 expression in HCC tissues (n = 371) and normal liver tissues (n = 50), based on TCGA-LIHC RNA-seq data. (C) ATP8B1-AS1 expression in HCC tissues with grade 1–2 (n = 232) or grade 3–4 (n = 134), based on TCGA-LIHC RNA-seq data. (D) ATP8B1-AS1 expression in HCC tissues with AFP level ≤ 400 (n = 213) or > 400 (n = 65), based on TCGA-LIHC RNA-seq data. For (B–D), P values were calculated by Mann–Whitney test. (E) ATP8B1-AS1 expression in 79 pairs of HCC tissues and matched adjacent liver tissues was measured by qPCR. P values were calculated by Wilcoxon matched-pairs signed rank test. (F) Kaplan-Meier survival curve of our HCC cohort stratified by ATP8B1-AS1 expression level. Median ATP8B1-AS1 expression level was used as cut-off. n = 79, HR = 1.920, P = 0.0314 by Log rank test. (G) ATP8B1-AS1 expression in immortalized liver cell line THLE-2 and HCC cell lines HuH-7, SNU-398, and SK-HEP-1 was detected by qPCR. Results are presented as mean ± SD based on three independent experiments. ****P < 0.0001 by one-way ANOVA followed by Dunnett’s multiple comparisons test. |
m6A Modification Increases the Stability of ATP8B1-AS1 Transcript
To investigate the potential correlation between m6A modification and ATP8B1-AS1, we first analyzed the correlation between the expression of ATP8B1-AS1 and m6A methylases in HCC tissues using the TCGA-LIHC data. The results showed that the expression of ATP8B1-AS1 was positively correlated with the expression of m6A methylases METTL3 and WTAP (Figure 2A and B), implying that ATP8B1-AS1 may be modulated by m6A modification. RIP assays using m6A antibody showed that m6A modification of ATP8B1-AS1 was found in all tested HCC cell lines, including HuH-7, SNU-398, and SK-HEP-1 (Figure 2C). The online tool SRAMP (http://www.cuilab.cn/sramp) predicted the 792 site of ATP8B1-AS1 as the very high confident m6A modification site (Figure 2D). Next, we used the previously reported single-base elongation- and ligation-based qPCR amplification method (SELECT) to detect the m6A modification level of 792 site of ATP8B1-AS1 (Figure 2E).42 The results showed that the 792 site of ATP8B1-AS1 was really m6A modified in both HuH-7 and SNU-398 cells (Figure 2F). The m6A modification level of 792 site of ATP8B1-AS1 was increased by m6A methylase METTL3 and was decreased by m6A demethylase FTO in both HuH-7 and SNU-398 cells (Figures S1 and F). RNA stability assays showed that ectopic expression of METTL3 elongated the half-life of ATP8B1-AS1 transcript, and while ectopic expression of FTO shortened the half-life of ATP8B1-AS1 transcript in both HuH-7 and SNU-398 cells (Figure 2G and H), suggesting that m6A modification increases the stability of ATP8B1-AS1 transcript.
 |
Figure 2 m6A modification increases the stability of ATP8B1-AS1 transcript. (A) The correlation between ATP8B1-AS1 expression and METTL3 expression, based on TCGA-LIHC RNA-seq data. r = 0.4780, P < 0.0001 by Spearman correlation analysis. (B) The correlation between ATP8B1-AS1 expression and WTAP expression, based on TCGA-LIHC RNA-seq data. r = 0.2894, P < 0.0001 by Spearman correlation analysis. (C) MeRIP assays using m6A specific antibody, followed by qPCR to detect m6A-modified ATP8B1-AS1. (D) The m6A modification site of ATP8B1-AS1, predicted by SRAMP. (E) Schematic of SELECT to detect m6A modification level. (F) The m6A modification level of ATP8B1-AS1 in METTL3 or FTO overexpressed HuH-7 and SNU-398 cells was detected by SELECT. (G and H) The stability of ATP8B1-AS1 transcript over time in METTL3 or FTO overexpressed HuH-7 (G) and SNU-398 (H) cells was detected after blocking new RNA synthesis using α-amanitin (50 µM) and normalized to 18S rRNA (a product of RNA polymerase I that is unchanged by α-amanitin). For (C) and (F–H), results are presented as mean ± SD based on three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA followed by Dunnett’s multiple comparisons test. |
m6A Modification Level of ATP8B1-AS1 is Increased and Correlated with Poor Survival in HCC
We next measured the m6A modification level of ATP8B1-AS1 in 79 pairs of HCC tissues and matched adjacent noncancerous liver tissues using SELECT. The results showed that m6A modification level of ATP8B1-AS1 was increased in HCC tissues compared with that in matched adjacent noncancerous liver tissues (Figure 3A). Kaplan–Meier survival analyses showed that increased m6A modification level of ATP8B1-AS1 was correlated with short overall survival time in HCC (Figure 3B). Furthermore, m6A modification level of ATP8B1-AS1 was positively correlated with ATP8B1-AS1 expression level in these 79 HCC tissues (Figure 3C), supporting the positive regulation of ATP8B1-AS1 transcript stability by m6A modification. m6A modification level of ATP8B1-AS1 was also increased in HCC cell lines compared with that in immortalized liver cell line (Figure 3D). Consistent with m6A modification level of ATP8B1-AS1, the expression of m6A methylase METTL3 was increased, and while the expression of m6A demethylase FTO was decreased in HCC cell lines compared with that in immortalized liver cell line (Figure S2A and B). Collectively, these data suggested that consistent with the expression level, the m6A modification level of ATP8B1-AS1 is also increased and correlated with poor survival in HCC.
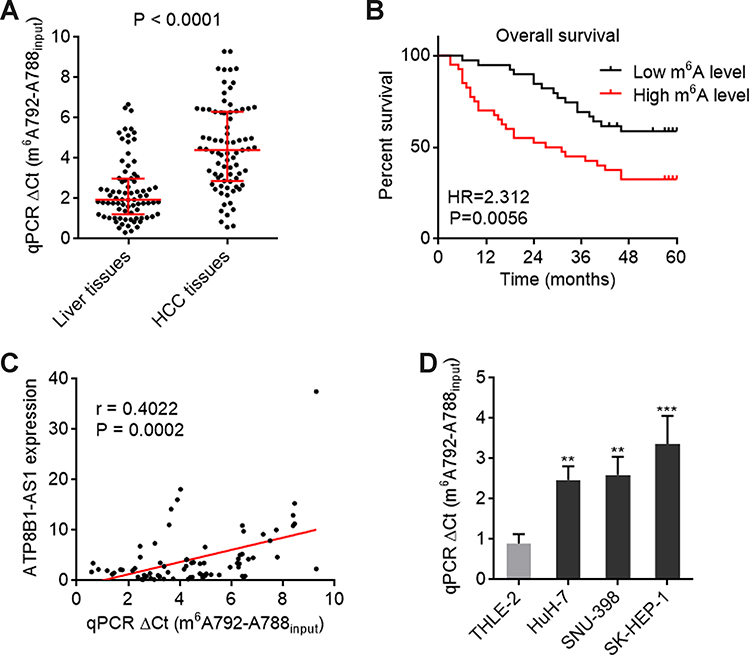 |
Figure 3 m6A modification level of ATP8B1-AS1 is upregulated and correlated with poor overall survival in HCC. (A) m6A modification level of ATP8B1-AS1 in 79 pairs of HCC tissues and matched adjacent liver tissues was detected by SELECT. P < 0.0001 by Wilcoxon matched-pairs signed rank test. (B) Kaplan-Meier survival curve of our HCC cohort stratified by m6A modification level of ATP8B1-AS1. Median m6A modification level of ATP8B1-AS1 was used as cut-off. n = 79, HR = 2.312, P = 0.0056 by Log rank test. (C) The correlation between m6A modification level of ATP8B1-AS1 and ATP8B1-AS1 expression level in HCC tissues, n = 79, r = 0.4022, P = 0.0002 by Spearman correlation analysis. (D) m6A modification level of ATP8B1-AS1 in immortalized liver cell line THLE-2 and HCC cell lines HuH-7, SNU-398, and SK-HEP-1 was detected by SELECT. Results are presented as mean ± SD based on three independent experiments. **P < 0.01, ***P < 0.001 by one-way ANOVA followed by Dunnett’s multiple comparisons test. |
ATP8B1-AS1 Promotes HCC Cell Proliferation, Migration, and Invasion in an m6A Modification Dependent Manner
To investigate the roles of ATP8B1-AS1 in HCC and the potential contribution of m6A modification to the roles of ATP8B1-AS1, we stably overexpressed wild-type and the 792 site mutated ATP8B1-AS1 with similar overexpression efficiencies in SNU-398 and HuH-7 cells (Figure 4A and B). Glo cell viability assays showed that ectopic expression of ATP8B1-AS1 increased cell viability of both SNU-398 and HuH-7 cells, which was abolished by the mutation of m6A-modified 792 site of ATP8B1-AS1 (Figure 4C and D). EdU incorporation assays further confirmed that ectopic expression of ATP8B1-AS1 promoted cell proliferation of both SNU-398 and HuH-7 cells, which was abolished by the mutation of m6A-modified 792 site of ATP8B1-AS1 (Figure 4E). Transwell migration and invasion assays showed that ectopic expression of ATP8B1-AS1 promoted cell migration and invasion of both SNU-398 and HuH-7 cells, which were abolished by the mutation of m6A-modified 792 site of ATP8B1-AS1 (Figure 4F and G). Therefore, these data suggested that ATP8B1-AS1 promotes HCC cellular proliferation, migration, and invasion in an m6A modification dependent manner.
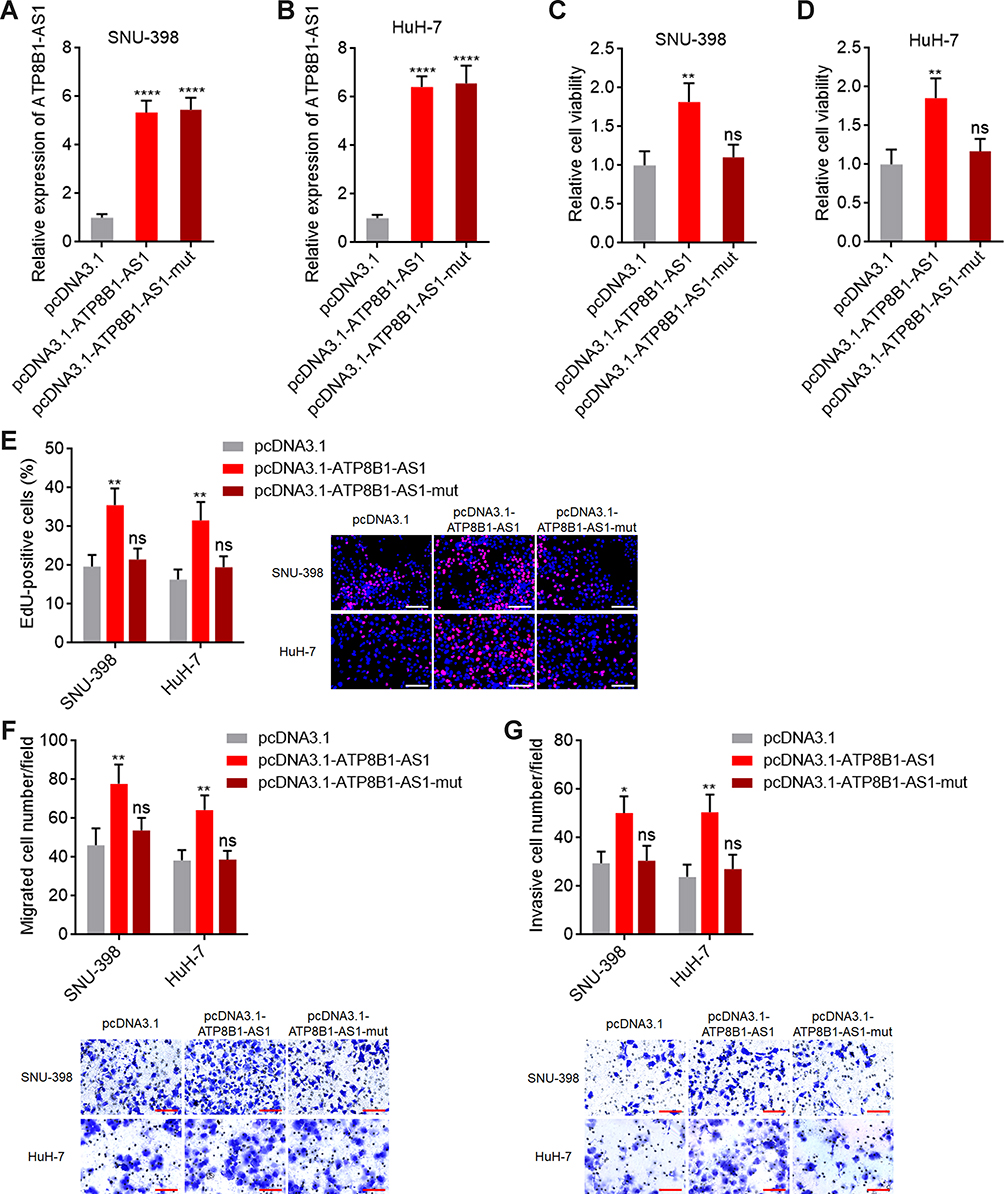 |
Figure 4 ATP8B1-AS1 exerts oncogenic roles in HCC in an m6A dependent manner. (A and B) ATP8B1-AS1 expression in SNU-398 (A) and HuH-7 (B) cells with wild-type or m6A-modified 792 site mutated ATP8B1-AS1 overexpression was measured by qPCR. (C and D) Cell proliferation of SNU-398 (C) and HuH-7 (D) cells with wild-type or mutated ATP8B1-AS1 overexpression was measured by Glo cell viability assay. (E) Cell proliferation of SNU-398 and HuH-7 cells with wild-type or mutated ATP8B1-AS1 overexpression was measured by EdU incorporation assays. Red color represents EdU-positive and proliferative cells. Scale bars = 100 µm. (F) Cell migration of SNU-398 and HuH-7 cells with wild-type or mutated ATP8B1-AS1 overexpression was measured by transwell migration assays. Scale bars = 100 µm. (G) Cell invasion of SNU-398 and HuH-7 cells with wild-type or mutated ATP8B1-AS1 overexpression was measured by transwell invasion assays. Scale bars = 100 µm. Results are presented as mean ± SD based on three independent experiments. *P < 0.05, **P < 0.01, ****P < 0.0001, by one-way ANOVA followed by Dunnett’s multiple comparisons test. Abbreviation: ns, not significant. |
Depletion of ATP8B1-AS1 Repressed HCC Cell Proliferation, Migration, and Invasion
To further confirm the oncogenic roles of ATP8B1-AS1 in HCC, we stably silenced ATP8B1-AS1 expression in SNU-398 and SK-HEP-1 cells (Figure 5A and B). Glo cell viability assays showed that SNU-398 and SK-HEP-1 cells with ATP8B1-AS1 silencing had decreased cell viability compared with control SNU-398 and SK-HEP-1 cells, respectively (Figure 5C and D). EdU incorporation assays also showed that SNU-398 and SK-HEP-1 cells with ATP8B1-AS1 silencing had repressed cell proliferation compared with control SNU-398 and SK-HEP-1 cells (Figure 5E). Subcutaneous xenograft assays showed that SNU-398 cells with ATP8B1-AS1 silencing formed smaller tumors and had slower tumor growth than control SNU-398 cells (Figure 5F), suggesting that depletion of ATP8B1-AS1 also repressed HCC tumor growth in vivo. Transwell migration and invasion assays showed that SNU-398 and SK-HEP-1 cells with ATP8B1-AS1 silencing had reduced cell migration and invasion compared with control SNU-398 and SK-HEP-1 cells (Figure 5G and H). Therefore, these data suggested that depletion of ATP8B1-AS1 repressed HCC cell proliferation, migration, and invasion.
 |
Figure 5 Depletion of ATP8B1-AS1 exerts tumor suppressive roles in HCC. (A and B) ATP8B1-AS1 expression in SNU-398 (A) and SK-HEP-1 (B) cells with ATP8B1-AS1 depletion was measured by qPCR. (C and D) Cell proliferation of SNU-398 (C) and SK-HEP-1 (D) cells with ATP8B1-AS1 depletion was measured by Glo cell viability assay. (E) Cell proliferation of SNU-398 and SK-HEP-1 cells with ATP8B1-AS1 depletion was measured by EdU incorporation assays. Red color represents EdU-positive and proliferative cells. Scale bars = 100 µm. (F) Tumor volume, weight, and photograph of subcutaneous tumors formed by SNU-398 cells with ATP8B1-AS1 depletion. (G) Cell migration of SNU-398 and SK-HEP-1 cells with ATP8B1-AS1 depletion was measured by transwell migration assays. Scale bars = 100 µm. (H) Cell invasion of SNU-398 and SK-HEP-1 cells with ATP8B1-AS1 depletion was measured by transwell invasion assays. Scale bars = 100 µm. Results are presented as mean ± SD based on three independent experiments (A–E, G and H) or n=5 mice in each group (F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one-way ANOVA followed by Dunnett’s multiple comparisons test (A–E, G and H) or Mann–Whitney test (F). |
m6A-Modified ATP8B1-AS1 Epigenetically Activates MYC Expression via Decreasing H3K9me2 Level at MYC Promoter
To reveal the mechanisms underlying the oncogenic roles of ATP8B1-AS1 in HCC, we performed Gene Set Enrichment Analysis (GSEA) using the TCGA-LIHC data. According to ATP8B1-AS1 expressing level, the TCGA-LIHC samples were divided into ATP8B1-AS1 high expression group and ATP8B1-AS1 low expression group. GSEA showed that genes from liver cancer subclass S2 were significantly enriched in ATP8B1-AS1 high expression group (Figure 6A). Liver cancer subtype S2 indicates HCC with proliferation, MYC and AKT1 activation.44 GSEA of the TCGA-LIHC data further showed that the genes bound by MYC were also significantly enriched in ATP8B1-AS1 high expression group (Figure 6B). Genes upregulated by MYC were significantly enriched in ATP8B1-AS1 high expression group, and while genes downregulated by MYC were significantly enriched in ATP8B1-AS1 low expression group (Figure 6C). Thus, the TCGA-LIHC data suggested that ATP8B1-AS1 may be positively correlated with MYC signaling activation.
 |
Figure 6 GSEA shows the correlation between ATP8B1-AS1 expression and MYC signaling activation. TCGA-LIHC samples were divided into ATP8B1-AS1 high expression and low expression group according to median ATP8B1-AS1 expression level. GSEA shows the positive correlation between ATP8B1-AS1 high expression and liver cancer subclass S2 gene signatures (A). MYC-bound genes were enriched in ATP8B1-AS1 high expression group (B). Genes upregulated by MYC were enriched in ATP8B1-AS1 high expression group, and genes downregulated by MYC were enriched in ATP8B1-AS1 low expression group (C). Abbreviation: NES, normalized enrichment score. |
Next, we investigated the potential influence of ATP8B1-AS1 on MYC. The TCGA-LIHC RNA-data revealed positive correlation between the expression of ATP8B1-AS1 and MYC (Figure 7A). Consistent with m6A modification level and the expression of ATP8B1-AS1, the expression of MYC was also increased in HCC cell lines compared with that in immortalized liver cell line (Figure S3A). Ectopic expression of ATP8B1-AS1 upregulated MYC mRNA and protein levels, which were abolished by the mutation of m6A-modified 792 site of ATP8B1-AS1 (Figure 7B and S3B). Conversely, silencing of ATP8B1-AS1 downregulated MYC mRNA and protein levels (Figure 7C and S3C). These findings suggested that ATP8B1-AS1 may regulate MYC transcription in an m6A modification dependent manner. Recently, m6A modified RNA was revealed to bind m6A reader YTHDC1, which further binds and recruits the histone H3 lysine 9 di-methylation (H3K9me2) demethylase KDM3B to target gene regions, inducing H3K9me2 demethylation and gene activation.45 H3K9me2 is strongly associated with transcriptional repression.46 In order to investigate whether m6A modified ATP8B1-AS1 regulates MYC expression in such a manner, we first detected whether ATP8B1-AS1 binds MYC promoter using ChIRP assays. The results showed that ATP8B1-AS1 specifically bound to MYC promoter region (Figure 7D). Next, we performed ChIP assays to investigate whether ATP8B1-AS1 influences the binding of KDM3B to MYC promoter region and the H3K9me2 level at MYC promoter region. The results showed that the ectopic expression of ATP8B1-AS1 promoted the binding of KDM3B to MYC promoter region and reduced the H3K9me2 level at MYC promoter region, which were both abolished by the mutation of m6A-modified 792 site of ATP8B1-AS1 (Figure 7E). Silencing of ATP8B1-AS1 reduced the binding of KDM3B to MYC promoter region and increased the H3K9me2 level at MYC promoter region (Figure 7F). Depletion of YTHDC1 abolished the increased binding of KDM3B to MYC promoter region, and the reduced H3K9me2 level at MYC promoter region (Figures S3D and 7G), which suggested that the regulation of these epigenetic modifications at MYC promoter by m6A-modified ATP8B1-AS1 is dependent on the m6A reader YTHDC1. Consistently, depletion of YTHDC1 abolished the upregulation of MYC by m6A-modified ATP8B1-AS1 (Figure 7H). Depletion of KDM3B also abolished the upregulation of MYC by m6A-modified ATP8B1-AS1 (Figures S3E and 7I). Therefore, these data suggested that m6A-modified ATP8B1-AS1 epigenetically activates MYC expression via binding YTHDC1 and further interacting with and recruiting KDM3B to decrease H3K9me2 level at MYC promoter.
 |
Figure 7 ATP8B1-AS1 epigenetically activates MYC expression in an m6A dependent manner. (A) The correlation between ATP8B1-AS1 and MYC expression, based on TCGA-LIHC RNA-seq data. r = 0.3206, P < 0.0001 by Spearman correlation analysis. (B) MYC expression in SNU-398 cells with wild-type or m6A-modified 792 site mutated ATP8B1-AS1 overexpression was measured by qPCR. (C) MYC expression in SNU-398 cells with ATP8B1-AS1 depletion was measured by qPCR. (D) ChIRP assays with ATP8B1-AS1 antisense probes or control probes were conducted in SNU-398 cells to measure the binding of ATP8B1-AS1 to MYC promoter or GAPDH promoter. GAPDH promoter was used as negative control. (E) ChIP assays with KDM3B or H3K9me2 specific antibodies were conducted in SNU-398 cells with wild-type or m6A-modified 792 site mutated ATP8B1-AS1 overexpression to measure the binding of KDM3B to MYC promoter and H3K9me2 level at MYC promoter. (F) ChIP assays with KDM3B or H3K9me2 specific antibodies were conducted in SNU-398 cells with ATP8B1-AS1 depletion to measure the binding of KDM3B to MYC promoter and H3K9me2 level at MYC promoter. (G) ChIP assays with KDM3B or H3K9me2 specific antibodies were conducted in SNU-398 cells with wild-type ATP8B1-AS1 overexpression and YTHDC1 silencing to measure the binding of KDM3B to MYC promoter and H3K9me2 level at MYC promoter. (H) MYC expression in SNU-398 cells with wild-type ATP8B1-AS1 overexpression and YTHDC1 silencing was measured by qPCR. (I) MYC expression in SNU-398 cells with wild-type ATP8B1-AS1 overexpression and KDM3B silencing was measured by qPCR. Results are presented as mean ± SD based on three independent experiments. **P < 0.01, ***P < 0.001, ****P < 0.0001, by one-way ANOVA followed by Dunnett’s multiple comparisons test (B–F) or two-tailed unpaired t-test (G–I). Abbreviation: ns, not significant. |
Depletion of MYC Reverses the Oncogenic Roles of ATP8B1-AS1
To assess the potential contribution of MYC to the oncogenic roles of ATP8B1-AS1 in HCC, we silenced MYC expression in ATP8B1-AS1 stably overexpressed SNU-398 cells (Figure 8A). Glo cell viability assays showed that silencing of MYC reversed the increased cell viability caused by ATP8B1-AS1 overexpression (Figure 8B). EdU incorporation assays further showed that silencing of MYC reversed the increased cell proliferation caused by ATP8B1-AS1 overexpression (Figure 8C). Transwell migration and invasion assays showed that silencing of MYC reversed the increased cell migration and invasion caused by ATP8B1-AS1 overexpression (Figure 8D and E). Therefore, these data suggested that the oncogenic roles of ATP8B1-AS1 are dependent on the activation of MYC.
 |
Figure 8 Depletion of MYC reverses the oncogenic roles of ATP8B1-AS1 in HCC. (A) MYC expression in SNU-398 cells with ATP8B1-AS1 overexpression and MYC silencing was measured by qPCR. (B) Cell proliferation of SNU-398 cells with ATP8B1-AS1 overexpression and MYC silencing was measured by Glo cell viability assay. (C) Cell proliferation of SNU-398 cells with ATP8B1-AS1 overexpression and MYC silencing was measured by EdU incorporation assays. Red color represents EdU-positive and proliferative cells. Scale bars = 100 µm. (D) Cell migration of SNU-398 cells with ATP8B1-AS1 overexpression and MYC silencing was measured by transwell migration assays. Scale bars = 100 µm. (E) Cell invasion of SNU-398 cells with ATP8B1-AS1 overexpression and MYC silencing was measured by transwell invasion assays. Scale bars = 100 µm. Results are presented as mean ± SD based on three independent experiments. **P < 0.01, ***P < 0.001, by one-way ANOVA followed by Dunnett’s multiple comparisons test. Abbreviation: ns, not significant. |
Discussion
As importantly functional molecules in HCC, lncRNAs have been intensively investigated.47–49 Many HCC-related lncRNAs show aberrant expression and correlation with prognosis of HCC patients.50–52 However, the contribution of m6A modification to the expression and role of lncRNAs are still largely unclear.
In this study, we identified ATP8B1-AS1 as an m6A-related and prognosis-related lncRNA in HCC. High expression level of ATP8B1-AS1 was previously reported to be correlated with good prognosis in pancreatic adenocarcinoma and breast cancer, but with poor prognosis in HCC.39,53,54 However, the potential influences of m6A on the clinical significances of ATP8B1-AS1 are still unknown. Here, using public TCGA-LIHC data and our own cohort, we found ATP8B1-AS1 was increased in HCC tissues and cell lines and correlated with poor overall survival of HCC patients. Second, we identified ATP8B1-AS1 as an m6A-modified lncRNA and further confirmed the m6A modification site on ATP8B1-AS1. Third, we found that the m6A modification increased ATP8B1-AS1 transcript stability and upregulated ATP8B1-AS1 level. Fourth, we found that the m6A modification level of ATP8B1-AS1 was increased in HCC tissues and cell lines, and also correlated with poor overall survival of HCC patients. Moreover, the hazard ratio (HR) of m6A modification level of ATP8B1-AS1 is greater than the HR of ATP8B1-AS1 expression level, calculated by the same HCC cohort. Thus, these findings suggested that m6A modification level of lncRNAs may have more important clinical relevance than the expression of lncRNAs.
Previous reports have showed that m6A modification could regulate the splicing, stability, or translation of target mRNAs.55,56 m6A modification increased the stability of lncKCNQ1OT1, and while decreased the stability of RAB11B-AS1.37,38 In this study, we also found that m6A modification increases the stability of ATP8B1-AS1. The detailed mechanisms underlying these different effects of m6A modification on transcript stability need further investigation. Furthermore, in this study, we found that m6A modification modulates the function of ATP8B1-AS1. ATP8B1-AS1 shows oncogenic roles in HCC, including promoting HCC cell proliferation, migration, and invasion. The oncogenic roles of ATP8B1-AS1 in HCC are dependent on m6A modification, as mutation of the m6A modification site nearly completely abolished the oncogenic roles of ATP8B1-AS1.
Considering the importance of m6A modification in the function of ATP8B1-AS1, we hypothesized that the mechanisms of ATP8B1-AS1 are also dependent on m6A modification. Through performing GSEA of TCGA-LIHC data, we identified MYC signaling as the critical downstream target of ATP8B1-AS1. Further experimental detection showed that ATP8B1-AS1 directly activated MYC transcription in an m6A dependent manner. m6A-modified ATP8B1-AS1 bound to m6A reader YTHDC1, which further interacted with and recruited the H3K9me2 demethylase KDM3B. Further, we found that ATP8B1-AS1 directly bound to MYC promoter region. Thus, only m6A-modified ATP8B1-AS1 could recruit KDM3B to MYC promoter region, leading to the reducing of H3K9me2 level at MYC promoter region and MYC transcription activation. Our findings revealed a link between m6A-modified lncRNAs and epigenetic modulation and provided novel evidences for the roles of m6A modification in epigenetic modulation.45
Conclusions
In conclusion, this study identified m6A-modified ATP8B1-AS1 as an oncogenic molecular event in HCC. m6A modification level of ATP8B1-AS1 is increased and correlated with poor overall survival in HCC. m6A-modified ATP8B1-AS1 exerts oncogenic roles in HCC through epigenetically activating MYC. Targeting ATP8B1-AS1 or the m6A modification of ATP8B1-AS1 represent novel therapeutic strategy for HCC.
Abbreviations
m6A, N6-methyladenosine; HCC, hepatocellular carcinoma; mRNA, messenger RNA; lncRNA, long noncoding RNA; TCGA, The Cancer Genome Atlas; LIHC, liver hepatocellular carcinoma; FBS, fetal bovine serum; qPCR, quantitative polymerase chain reaction; cDNA, complementary DNA; NC, negative control; MeRIP, methylated RNA immunoprecipitation; EdU, 5-ethynyl-2’-deoxyuridine; ChIRP, chromatin isolation by RNA purification; ChIP, chromatin immunoprecipitation; AFP, alpha-fetoprotein; SELECT, single-base elongation- and ligation-based qPCR amplification method; GSEA, Gene Set Enrichment Analysis; H3K9me2, histone H3 lysine 9 di-methylation.
Ethics Approval and Consent to Participate
This study was performed following the Helsinki Declaration and with the approval from Youjiang Medical University for Nationalities Institutional Review Board. Written informed consents were acquired from all participants.
Funding
This work was supported by Guangxi science and technology project (2021AC20006) and high-level personnel project of Affiliated Hospital of Youjiang Medical University for Nationalities (R202210307).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst. 2017;109. doi:10.1093/jnci/djx030
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi:10.3322/caac.21660
3. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6. doi:10.1038/s41572-020-00240-3
4. Villanueva A. Hepatocellular Carcinoma. N Engl J Med. 2019;380:1450–1462. doi:10.1056/NEJMra1713263
5. Baretti M, Kim AK, Anders RA. Expanding the immunotherapy roadmap for hepatocellular carcinoma. Cancer Cell. 2022;40:252–254. doi:10.1016/j.ccell.2022.02.017
6. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19:222–232. doi:10.1038/s41590-018-0044-z
7. Zhou G, Boor PPC, Bruno MJ, Sprengers D, Kwekkeboom J. Immune suppressive checkpoint interactions in the tumour microenvironment of primary liver cancers. Br J Cancer. 2022;126:10–23. doi:10.1038/s41416-021-01453-3
8. Hajiev S, Allara E, Motedaysmallie CAL, et al. Impact of age on sorafenib outcomes in hepatocellular carcinoma: an international cohort study. Br J Cancer. 2021;124:407–413. doi:10.1038/s41416-020-01116-9
9. Sun Y, Wu L, Zhong Y, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184:404–421 e16. doi:10.1016/j.cell.2020.11.041
10. You H, Zhang N, Yu T, et al. Hepatitis B virus X protein promotes MAN1B1 expression by enhancing stability of GRP78 via TRIM25 to facilitate hepatocarcinogenesis. Br J Cancer. 2023;128:992–1004. doi:10.1038/s41416-022-02115-8
11. Li J, Li MH, Wang TT, et al. SLC38A4 functions as a tumour suppressor in hepatocellular carcinoma through modulating Wnt/beta-catenin/MYC/HMGCS2 axis. Br J Cancer. 2021;125:865–876. doi:10.1038/s41416-021-01490-y
12. Xu Q, Liao Z, Gong Z, et al. Down-regulation of EVA1A by miR-103a-3p promotes hepatocellular carcinoma cells proliferation and migration. Cell Mol Biol Lett. 2022;27:93. doi:10.1186/s11658-022-00388-8
13. Wen J, Huang Z, Wei Y, et al. Hsa-microRNA-27b-3p inhibits hepatocellular carcinoma progression by inactivating transforming growth factor-activated kinase-binding protein 3/nuclear factor kappa B signalling. Cell Mol Biol Lett. 2022;27:79. doi:10.1186/s11658-022-00370-4
14. Xing Y, Liu Y, Qi Z, Liu Z, Wang X, Zhang H. LAGE3 promoted cell proliferation, migration, and invasion and inhibited cell apoptosis of hepatocellular carcinoma by facilitating the JNK and ERK signaling pathway. Cell Mol Biol Lett. 2021;26:49. doi:10.1186/s11658-021-00295-4
15. Yan W, Han Q, Gong L, et al. MBD3 promotes hepatocellular carcinoma progression and metastasis through negative regulation of tumour suppressor TFPI2. Br J Cancer. 2022;127:612–623. doi:10.1038/s41416-022-01831-5
16. Zhao W, Mo H, Liu R, Chen T, Yang N, Liu Z. Matrix stiffness-induced upregulation of histone acetyltransferase KAT6A promotes hepatocellular carcinoma progression through regulating SOX2 expression. Br J Cancer. 2022;127:202–210. doi:10.1038/s41416-022-01784-9
17. Wu Y, Xu X, Qi M, et al. N(6)-methyladenosine regulates maternal RNA maintenance in oocytes and timely RNA decay during mouse maternal-to-zygotic transition. Nat Cell Biol. 2022;24:917–927. doi:10.1038/s41556-022-00915-x
18. Tang Q, Li L, Wang Y, et al. RNA modifications in cancer. Br J Cancer. 2023;2023:1. doi:10.1038/s41416-023-02275-1
19. Ito-Kureha T, Leoni C, Borland K, et al. The function of Wtap in N(6)-adenosine methylation of mRNAs controls T cell receptor signaling and survival of T cells. Nat Immunol. 2022;23:1208–1221. doi:10.1038/s41590-022-01268-1
20. Cai Z, Zhang Y, Yang L, et al. ALKBH5 in mouse testicular Sertoli cells regulates Cdh2 mRNA translation to maintain blood-testis barrier integrity. Cell Mol Biol Lett. 2022;27:101. doi:10.1186/s11658-022-00404-x
21. Zhang R, Qu Y, Ji Z, et al. METTL3 mediates Ang-II-induced cardiac hypertrophy through accelerating pri-miR-221/222 maturation in an m6A-dependent manner. Cell Mol Biol Lett. 2022;27:55. doi:10.1186/s11658-022-00349-1
22. Sun Z, Hong Q, Liu Y, et al. Oviduct transcriptomic reveals the regulation of mRNAs and lncRNAs related to goat prolificacy in the luteal phase. Animals. 2022;12. doi:10.3390/ani12202823
23. Paramasivam A, Priyadharsini JV. RNA N6-methyladenosine: a new player in autophagy-mediated anti-cancer drug resistance. Br J Cancer. 2021;124:1621–1622. doi:10.1038/s41416-021-01314-z
24. Weng H, Huang F, Yu Z, et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022;40:1566–1582 e10. doi:10.1016/j.ccell.2022.10.004
25. Lan J, Xu B, Shi X, Pan Q, Tao Q. WTAP-mediated N(6)-methyladenosine modification of NLRP3 mRNA in kidney injury of diabetic nephropathy. Cell Mol Biol Lett. 2022;27:51. doi:10.1186/s11658-022-00350-8
26. Cheng Y, Xie W, Pickering BF, et al. N(6)-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell. 2021;39:958–972 e8. doi:10.1016/j.ccell.2021.04.017
27. Yuan JH, Yang F, Wang F, et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi:10.1016/j.ccr.2014.03.010
28. Unfried JP, Ulitsky I. Substoichiometric action of long noncoding RNAs. Nat Cell Biol. 2022;24:608–615. doi:10.1038/s41556-022-00911-1
29. Zhang X, Jiang Q, Li J, et al. KCNQ1OT1 promotes genome-wide transposon repression by guiding RNA-DNA triplexes and HP1 binding. Nat Cell Biol. 2022;24:1617–1629. doi:10.1038/s41556-022-01008-5
30. de Jong JJ, Valderrama BP, Perera J, et al. Non-muscle-invasive micropapillary bladder cancer has a distinct lncRNA profile associated with unfavorable prognosis. Br J Cancer. 2022;127:313–320. doi:10.1038/s41416-022-01799-2
31. Yuan JH, Liu XN, Wang TT, et al. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat Cell Biol. 2017;19:820–832. doi:10.1038/ncb3538
32. Li Y, Ding T, Hu H, et al. LncRNA-ATB participates in the regulation of calcium oxalate crystal-induced renal injury by sponging the miR-200 family. Mol Med. 2021;27:143. doi:10.1186/s10020-021-00403-2
33. Li G, Kryczek I, Nam J, et al. LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nat Cell Biol. 2021;23:526–537. doi:10.1038/s41556-021-00672-3
34. Ouyang J, Zhong Y, Zhang Y, et al. Long non-coding RNAs are involved in alternative splicing and promote cancer progression. Br J Cancer. 2022;126:1113–1124. doi:10.1038/s41416-021-01600-w
35. Li R, Wang X, Zhu C, Wang K. lncRNA PVT1: a novel oncogene in multiple cancers. Cell Mol Biol Lett. 2022;27:84. doi:10.1186/s11658-022-00385-x
36. Wang X, Song Y, Shi Y, Yang D, Li J, Yin B. SNHG3 could promote prostate cancer progression through reducing methionine dependence of PCa cells. Cell Mol Biol Lett. 2022;27:13. doi:10.1186/s11658-022-00313-z
37. Zhou Z, Cao Y, Yang Y, Wang S, Chen F. METTL3-mediated m(6)A modification of lnc KCNQ1OT1 promotes doxorubicin resistance in breast cancer by regulating miR-103a-3p/MDR1 axis. Epigenetics. 2023;18:2217033. doi:10.1080/15592294.2023.2217033
38. Dai YZ, Liu YD, Li J, et al. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B-AS1 in an m(6)A-dependent manner. Cell Mol Biol Lett. 2022;27:41. doi:10.1186/s11658-022-00342-8
39. Wu X, Deng Z, Liao X, et al. Establishment of prognostic signatures of N6-methyladenosine-related lncRNAs and their potential functions in hepatocellular carcinoma patients. Front Oncol. 2022;12:865917. doi:10.3389/fonc.2022.865917
40. Lin Y, Xiao Y, Liu S, Hong L, Shao L, Wu J. Role of a lipid metabolism-related lncRNA signature in risk stratification and immune microenvironment for colon cancer. BMC Med Genomics. 2022;15:221. doi:10.1186/s12920-022-01369-8
41. Wu Y, Wang Y, Yao H, et al. MNX1-AS1, a c-Myc induced lncRNA, promotes the Warburg effect by regulating PKM2 nuclear translocation. J Exp Clin Cancer Res. 2022;41:337. doi:10.1186/s13046-022-02547-3
42. Xiao Y, Wang Y, Tang Q, Wei L, Zhang X, Jia G. An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N(6) -methyladenosine modification. Angew Chem Int Ed Engl. 2018;57:15995–16000. doi:10.1002/anie.201807942
43. Liu XN, Yuan JH, Wang TT, Pan W, Sun SH. An alternative POLDIP3 transcript promotes hepatocellular carcinoma progression. Biomed Pharmacother. 2017;89:276–283. doi:10.1016/j.biopha.2017.01.139
44. Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi:10.1158/0008-5472.CAN-09-1089
45. Li Y, Xia L, Tan K, et al. N(6)-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat Genet. 2020;52:870–877. doi:10.1038/s41588-020-0677-3
46. Methot SP, Padeken J, Brancati G, et al. H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity. Nat Cell Biol. 2021;23:1163–1175. doi:10.1038/s41556-021-00776-w
47. Farzaneh M, Ghasemian M, Ghaedrahmati F, et al. Functional roles of lncRNA-TUG1 in hepatocellular carcinoma. Life Sci. 2022;308:120974. doi:10.1016/j.lfs.2022.120974
48. Zhang L, Yang F, Yuan JH, et al. Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis. 2013;34:577–586. doi:10.1093/carcin/bgs381
49. Zheng M, Zhou H, Xie J, Zhang H, Shen X, Zhu D. Molecular typing and prognostic model of lung adenocarcinoma based on cuprotosis-related lncRNAs. J Thorac Dis. 2022;14:4828–4845. doi:10.21037/jtd-22-1534
50. Zhu XT, Yuan JH, Zhu TT, Li YY, Cheng XY. Long noncoding RNA glypican 3 (GPC3) antisense transcript 1 promotes hepatocellular carcinoma progression via epigenetically activating GPC3. FEBS J. 2016;283:3739–3754. doi:10.1111/febs.13839
51. Xia A, Yuan W, Wang Q, et al. The cancer-testis lncRNA lnc-CTHCC promotes hepatocellular carcinogenesis by binding hnRNP K and activating YAP1 transcription. Nat Cancer. 2022;3:203–218. doi:10.1038/s43018-021-00315-4
52. DiStefano JK. Long noncoding RNAs in the initiation, progression, and metastasis of hepatocellular carcinoma. Noncoding RNA Res. 2017;2:129–136. doi:10.1016/j.ncrna.2017.11.001
53. Wang J, Xiang J, Li X. Construction of a competitive endogenous RNA network for pancreatic adenocarcinoma based on weighted gene co-expression network analysis and a prognosis model. Front Bioeng Biotechnol. 2020;8:515. doi:10.3389/fbioe.2020.00515
54. Su X, Malouf GG, Chen Y, et al. Comprehensive analysis of long non-coding RNAs in human breast cancer clinical subtypes. Oncotarget. 2014;5:9864–9876. doi:10.18632/oncotarget.2454
55. Diao MN, Zhang XJ, Zhang YF. The critical roles of m6A RNA methylation in lung cancer: from mechanism to prognosis and therapy. Br J Cancer. 2023;2023:1–6. doi:10.1038/s41416-023-02246-6
56. Shimura T, Kandimalla R, Okugawa Y, et al. Novel evidence for m(6)A methylation regulators as prognostic biomarkers and FTO as a potential therapeutic target in gastric cancer. Br J Cancer. 2022;126:228–237. doi:10.1038/s41416-021-01581-w
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
CTB-193M12.5 Promotes Hepatocellular Carcinoma Progression via Enhancing NSD1-Mediated WNT10B/Wnt/β-Catenin Signaling Activation
Zhang S, Jiang M, Cao H, Xiong J, Xu J
Journal of Hepatocellular Carcinoma 2022, 9:553-569
Published Date: 7 June 2022
N6-Methyladenosine-Modified LEAWBIH Drives Hepatocellular Carcinoma Progression through Epigenetically Activating Wnt/β-Catenin Signaling
Wei H, Huang L, Lu Q, Huang Z, Huang Y, Xu Z, Li W, Pu J
Journal of Hepatocellular Carcinoma 2023, 10:1991-2007
Published Date: 6 November 2023