Back to Journals » Drug Design, Development and Therapy » Volume 15
MYC/NBS1-Mediated DNA Damage Response is Involved in the Inhibitory Effect of Hydroxysafflor Yellow A on Glioma Cells
Authors Tang D, Huang T, Tian Q, Wang J
Received 26 October 2020
Accepted for publication 30 January 2021
Published 28 April 2021 Volume 2021:15 Pages 1749—1763
DOI https://doi.org/10.2147/DDDT.S288841
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Dongfang Tang,1 Tao Huang,2 Qilong Tian,2 Julei Wang2
1Department of Neurosurgery, Henan Provincial People’s Hospital, Central China Fuwai Hospital, Central China Fuwai Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China; 2Department of Neurosurgery, The Second Affiliated Hospital of Air Force Military Medical University, Xi’an City, People’s Republic of China
Correspondence: Julei Wang
Department of Neurosurgery, The Second Affiliated Hospital of Air Force Military Medical University, No. 569 Xinsi Road, Baqiao District, Xi’an City, Shaanxi Province, 710038, People’s Republic of China
Tel +86-29-84777777
Email [email protected]
Background: The role of Hydroxysafflor Yellow A (HSYA) in glioma is less studied, this research determined the effect of HSYA on glioma cells.
Methods: The expressions of MYC and NBS1 in glioma tissues were detected by bioinformatics analysis and verified by RT-qPCR. The target relationship between MYC and NBS1 was predicted by bioinformatics. After treating the cells with HSYA, silenced MYC, or overexpressed NBS1, the viability, apoptosis, proliferation, invasion, migration, and DNA damage of the glioma cells were detected by MTT, flow cytometry, colony formation, transwell, wound healing, and γH2AX immunofluorescence assays, respectively. IC50 of HSYA in glioma cells was analyzed by Probit regression analysis. The expressions of MYC, NBS1, factors related to migration, invasion, apoptosis, and DNA damage of the glioma cells were determined by Western blot or RT-qPCR.
Results: MYC and NBS1 were high-expressed in glioma, and NBS1 was targeted by MYC. HSYA and siRNA targeting MYC inhibited the cell viability, proliferation, invasion, migration, and induced the cell apoptosis of glioma cells. HSYA upregulated the expressions of MYC, γH2AX, E-Cadherin, Bax, and Cleaved-PARP1, stimulated the activation of NBS1, MRE11, RAD50, and ATM, and downregulated the expressions of N-Cadherin and Bcl2 in glioma cells. SiMYC decreased the IC50 of HSYA in the glioma cells, enhanced the sensitivity of glioma cells to HSYA, and inhibited the activation of NBS1 and ATM. NBS1 overexpression reversed the effect of siRNA targeting MYC on glioma cells.
Conclusion: MYC silencing inhibited the DNA damage response via regulation of NBS1, leading to DNA repair deficiency, and subsequently enhanced the sensitivity of glioma cells to HSYA.
Keywords: glioma, hydroxysafflor yellow A, MYC, NBS1, drug sensitivity, DNA repair
Introduction
Glioma, a common primary tumor in the central nervous system, mostly occurs at the neuroectoderm. The incidence of glioma accounts for about 45–50% of intracranial tumors.1–3 Currently, glioma is mainly treated by surgery combined with multiple treatments, such as radiotherapy, chemotherapy, and tumor biological activation immune method,4 but the treatment outcome of glioma is far from satisfactory.5 Studies showed that many natural plants contain anti-tumor active ingredients with a variety of component capable of improving human immune functions and inhibiting the biological functions of tumor cells.6–8
Safflower (Carthamus tinctorius L.) is a traditional Chinese medicine and has the effects of activating blood circulation, removing blood stasis, clearing menstruation, and relieving pain.9 Safflor yellow is a chalcone compound of safflower, and Hydroxysafflor Yellow A (HSYA) is the most important active ingredient in safflower yellow.10 The pharmacological effects of HSYA, such as improving myocardial ischemia and neurological deficits, resisting oxidative stress, and reducing inflammatory reactions, have been extensively studied.10–15 Furthermore, research has proved that HSYA has an anticancer effect on different types of cancers, for instance, HSYA inhibits the angiogenesis of liver cancer by inactivating MAPK,16 and HSYA suppresses the proliferation and metastasis of lung cancer cells by regulating PI3K/AKT pathway.17 However, whether HSYA had an anticancer effect on glioma remained unclear.
MYC transcription factor is an oncogene with a regulatory effect on many cancers, including glioma.18,19 In addition, researches have reported that MYC is involved in the drugs resistance of cancer cells, for example, MYC drives Nab-Paclitaxel resistance of pancreatic ductal carcinoma cells,20 and modulates the drug resistance of leukemia stem cells through ERK/MSK pathway.21 Moreover, MYC also mediates the PARP inhibitor resistance of glioblastoma.22 Therefore, MYC becomes an important target in anticancer research. The expression of MYC can be regulated by HSYA in human HUVEC cells and human liver cancer cells.23,24 However, whether HSYA has a regulatory effect on MYC in glioma should be further examined and studied.
Based on the above background, the present research attempted to determine the role of HSYA in glioma and to explore whether the role of HSYA in glioma was related to the function of MYC.
Methods
Ethics Statement
Cancer tissues and adjacent tissues of glioma patients (n=50) were obtained from Fuwai Central China Cardiovascular Hospital. The use of the clinical tissues was approved by the Ethics Committee of Fuwai Central China Cardiovascular Hospital (F2020030510N), in accordance with the Declaration of Helsinki, and informed consent was provided by all the patients.
Bioinformatics Analysis
The expression of MYC in glioma tissues and normal tissues was analyzed through GEPIA2 (http://gepia2.cancer-pku.cn/#index). The correlation between MYC and NBS1 in glioma was analyzed through StarBase (http://starbase.sysu.edu.cn). The target relationship between MYC with NBS1 was predicted using CISTROME database (http://cistrome.org/db/) and Cister database (zlab.bu.edu).
Cell Culture
Normal human astrocytes cell lines including HEB (BNCC283657, BNBIO, Beijing, China) and NHA (BFN60808805, BLUEFBIO, Shanghai, China), glioma cell lines including A172 (BNCC317373, BNBIO), LN-229 (BNCC306188, BNBIO), T98G (BNCC302929, BNBIO), and U-251MG (Tongpai Biotechnology, Shanghai, China, http://www.shtpbio.com/tongpai-Products-10947704/), and human HEK293T cells (CL-0005, PROCELL, Wuhan, China) were grown in complete medium, which consists of DMEM medium (30-2002, ATCC, Manassas, Virginia, USA) with 10% FBS (10437010, Gibco, Waltham, Massachusetts, USA) and 1% Penicillin-Streptomycin (15140163, Gibco). All the cells were cultured in a 37°C environment with 5% CO2.
Drugs Treatment
For HSYA treatment, the cells were cultured in a medium containing 25, 50, or 100 μM of HSYA (N1526, APExBIO, Houston, Texas, USA) for 24, 48, or 72 h (Figure S1A and B). Then, the cells were collected for later use. For 10058-F4 (inhibitor of MYC) treatment, the cells were in a medium containing 25 μM of 10058-F4 (A1169, APExBIO) for 48 h. Then, the cells were collected for later use.
Cell Transfection
The siRNA1 targeting MYC (Figure S1C; siMYC-1; target sequence: 5ʹ-CCGGTTTTCGGGGCTTTATCTAA-3ʹ), siRNA2 targeting MYC (siMYC-2; target sequence: 5ʹ-CTGCTTAGACGCTGGATTTTTTT-3ʹ), siRNA3 targeting MYC (siMYC-3; target sequence: 5ʹ-GAGCAAAAGCTCATTTCTGAAGA-3ʹ), siRNA negative control (siNC; 5ʹ-AATTCTCCGAACGTGTCACGT-3ʹ), plasmids overexpressing NBS1 were cloned into pcDNA3.1 vector (Genepharma, Shanghai, China). Before cell transfection, LN-229 and U-251MG cells were, respectively, cultured in a 6-well plate to 80% confluence. Then, the siRNA or plasmids were transfected into the cells using 3 μL lipofectamine 3000 (L3000015, Invitrogen, Waltham, Massachusetts, USA). After 48-h transfection, the cells were harvested for drug treatment or later use.
Dual-Luciferase Reporter Assay
After the promoter of NBS1 was predicted to be targeted by MYC, the dual-luciferase reporter assay was performed to verify the prediction following a previous report.25 To examine the binding of MYC to the promoter region of NBS1, NBS1 promoter wild-type sequence (wt-NBS1) and mutant-type sequence containing MYC binding site A and/or B (mut-NBS1) were respectively cloned into the promoter region of the pGL3-Basic vector (VT1554, YouBio, Hunan, China). Then, the HEK293 cells were transfected with the vectors and MYC for 48 h. After the cells were administrated with the Dual-Luciferase Reporter Assay System Kit (E1960, Promega, Shanghai, China), the luciferase activity was determined using a SpectraMax reader (Molecular Devices, Shanghai, China).
MTT Assay
The LN-229 and U-251MG cells in 96-well plates were treated with different concentrations of HSYA or 10058-F4 for indicated times. Then, the cultured media were replaced with 100 μL MTT solution (C0037, Beyotime, Shanghai, China), and the cells were further cultured for 4 h. Then, the MTT solution was discarded, and 100 μL of DMSO (ST038, Beyotime) was added into the wells to dissolve formazan. Finally, the absorbance under 570 nm was determined using an Imark microplate reader (Bio-Rad, CA, USA).
Colony Formation Assay
After being transfected with siRNA targeting MYC or further treated with HSYA, 1 × 103 LN-229 and U-251MG cells were resuspended in a 2-mL culture medium and placed into a 6-well plate to grow for 14 days. Then, the cell colonies in the 6-well plate were incubated with fixative (P885233, MREDA, Beijing, China) for 10 min and dyed with purple crystal buffer (C0121, Beyotime) for 10 min. After washing with PBS (C0221A, Beyotime) for three times, the stained colonies were recorded using a camera (D-LUX7, Leica, Wetzlar, Germany) and the colony numbers were quantified using the Image J 1.8.0 software.
Transwell Assays
After being transfected with siRNA targeting MYC or further treated with HSYA, 1 × 105 LN-229 and U-251MG cells were resuspended in 200 μL medium without FBS and, respectively, placed into a transwell chamber (354234, Corning, NY, USA), which were inserted into the 24-well plate. Each well of the 24-well plate contained a 700-μL complete medium. After cell incubation for 48 h, the invaded cells were fixed with fixative and dyed with purple crystal buffer for 15 min. Finally, the stained cells were recorded using a DM4M optical microscope (Leica, Solms, Germany) and analyzed using the Image J 1.8.0 software.
Wound Healing Assays
After cell transfection with siRNA targeting MYC or further treatment with HSYA, 1 × 105 LN-229 and U-251MG cells were resuspended in 2 mL of complete medium and added into each well of a 6-well plate. The cells were cultured for 16 h (100% confluence), a straight wound was created in each well, and then the cells were cultured in a medium without FBS for 24 h. The picture of each wound at 0 and 24 h was recorded by a DMLA optical microscope. Image J 1.8.0 software was applied to analyze the data.
Flow Cytometry
After cell transfection or further treatment with drugs, the LN-229 and U-251MG cells were collected and washed with PBS twice. Then, the cells were stained with Annexin V-FITC (D17360, OKA) and Propidium iodide (D17360, OKA) for 15 min. Finally, the cell apoptosis signal was analyzed by the FACS-LSR II flow cytometer system (Becton-Dickinson, NJ, USA).
Immunofluorescence
After cell transfection with siRNA targeting MYC or treatment with HSYA, the LN-229 and U-251MG cells were fixed by fixative and permeabilized by 0.5% Triton X-100 (T8787, Merck, St. Louis, Missouri, USA). Then, the cells were incubated with the γH2AX antibody (ab195188, Abcam, Cambridge, UK) overnight at 4°C. After washing with PBS, the cells were further dyed with DAPI (C1002, Beyotime) for 20 min. Finally, the cell images were observed and recorded under a BSF-40 fluorescence microscope (Batuo Instrument, Shanghai, China) at the magnification × 100.
RT-qPCR
The mRNA in the clinical tissues and cultured cells were extracted. Briefly, the tissue and cell samples were mixed with Trizol (R21086, OKA, Beijing, China) and chloroform (A23482, OKA) for 30-min centrifugation (14,000 × g) to collect the supernatant. Then, the supernatant was mixed with isopropanol (E15794, OKA) and further centrifuged (14,000 × g) for 15 min. After the mRNA was collected from the precipitations, it was synthesized into cDNA using BeyoRT III cDNA synthesis regent (D7178L, Beyotime). Finally, the cDNA amplification was performed by mixing the cDNA with the Supermix (AQ601-01, TransGen, Beijing, China) and primers of target genes in the QuantStudio6 system (Applied Biosystems, CA, USA). After the amplifying reaction, the target gene expressions were quantified using the 2−ΔΔCt method. The primers used in the experiments were as follows: MYC-F: 5ʹ-GGCTCCTGGCAAAAGGTCA-3ʹ, MYC-R: 5ʹ-CTGCGTAGTTGTGCTGATGT-3ʹ; NBS1-F: 5ʹ-GACTGGCGTTGAGTACGTTGT-3ʹ, NBS1-R: 5ʹ-TGATTTCGGCTGATCGACTGA-3ʹ; MRE11-F: 5ʹ-TCCGTGAGGCTATGACCAGG-3ʹ, MRE11-R: 5ʹ-TTGGTTGCTGCTGAGATGCTAT-3ʹ; β-actin-F: 5ʹ-CCACGAAACTACCTTCAACTCC-3ʹ, β-actin-R: 5ʹ-GTGATCTCCTTCTGCATCCTGT-3ʹ.
Western Blot Assays
Total proteins from the cultured cells were extracted using NP-40 (P0013F, Beyotime) and nucleus proteins in the cells were extracted using Nucleus Protein Extraction Kit (P0027, Beyotime). The protein concentration was determined by the BCA kit (P0009, Beyotime). Then, 25 μg of proteins was separated by the SDS-PAGE gel (P0052A, Beyotime) and transferred to the surface of PVDF membranes (FFP39, Beyotime). After membrane block with 5% milk without FBS, the membranes were soaked in 5 μL relative primary antibodies at 4°C for 16 h. The next day, the membrane was incubated with rabbit second-antibody (1:10,000, ab205718, Abcam) or mouse second-antibody (1:10,000, ab205719, Abcam) for 2 h at room temperature. Finally, after the membrane was covered with 200 μL of detection solution (P0019, Beyotime), and the image signal was detected using the Image Lab 3.0 Software (Bio-Rad, CA, USA). The relative primary antibodies were as follows: MYC (1:1000, ab32072, Abcam), E-Cadherin (1:2000, ab231303, Abcam), N-Cadherin (1:1500, ab18203, Abcam), Bcl-2 (1:800, ab59348, Abcam), Bax (1:3000, ab32503, Abcam), Cleaved PARP1 (1:1500, ab32064, Abcam), PARP1 (1:5000, ab32138, Abcam), NBS1 (1:1000, ab181729, Abcam), p-NBS1 (1:1000, ab109453, Abcam), MRE11 (1:800, ab208020, Abcam), p-MRE11 (1:1000, #4859, CST, Boston, Massachusetts, USA), RAD50 (1:3000, ab124682, Abcam), p-RAD50 (1:1000, #14223, CST), ATM (1:2000, ab199726, Abcam), p-ATM (1:10,000, ab81292, Abcam), γH2AX (1:1500, ab81299, Abcam), Histone H3 (1:2500, ab1791, Abcam), β-actin (1:5000, ab8226, Abcam), and GAPDH (1:5000, ab8245, Abcam).
Statistical Analysis
The expressions of MYC and NBS1 in the clinical cancer tissues and adjacent tissues were analyzed by pared-samples t-test. Comparison between multiple groups was conducted using one-way ANOVA. IC50 was analyzed using Probit regression analysis based on the data of MTT assays. All statistical analyses were implemented using Graphpad 8.0 software. Mean ± SD was used to present the statistical data. P < 0.05 was regarded as statistically significant.
Results
MYC Was High-Expressed in Glioma Tissues and Cells
The analysis in GEPIA2 showed that the expression of MYC in glioma tissues was higher than that in normal tissues (P < 0.05, Figure 1A). For further verification, the expression of MYC in clinical glioma tissues and adjacent normal tissues were determined again, as shown in Figure 1B, the expression of MYC was high-expressed in glioma tissues (P < 0.001). Meanwhile, the high expression of MYC was also discovered in the cultured glioma cell lines as compared with the normal cells (P < 0.01), indicating that MYC might have a regulatory effect on the development of glioma.
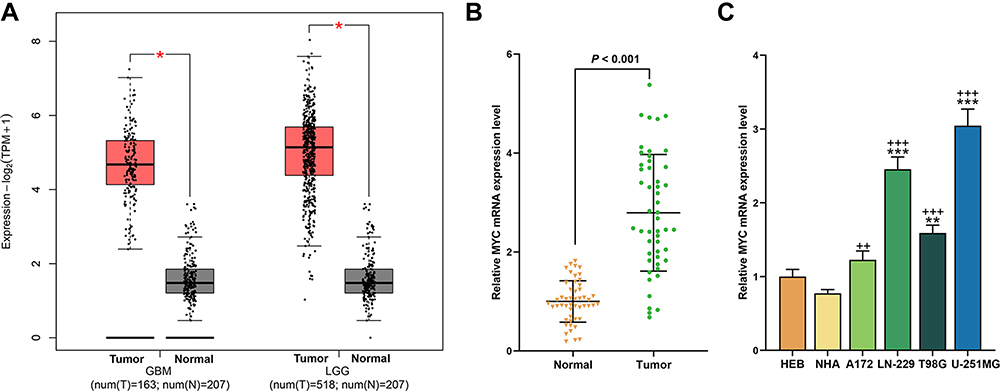 |
Figure 1 MYC was high-expressed in glioma tissues and cells. (A) The expression of MYC in glioma tissues and normal tissues was analyzed through GEPIA2 (*P < 0.05, vs Normal). (B) The expression of MYC in clinical glioma tissues and adjacent tissues was analyzed by RT-qPCR, β-actin was an internal control. N=50. (C) The expression of MYC in glioma cells and normal cells was analyzed by RT-qPCR, β-actin was an internal control N=3 (**P < 0.01, ***P < 0.001, vs HEB; ++P < 0.01, +++P < 0.001, vs NHA). |
HSYA and siRNA Targeting MYC Regulated the Cell Viability, Proliferation, Migration, Invasion, and Apoptosis, and siRNA Targeting MYC Enhanced the Sensitivity of Glioma Cells to HSYA
Then, we observed that the viabilities of LN-229 and U-251MG cells were reduced (Figure 2A) by HSYA after treatment with different doses of HSYA for 24 h (P < 0.001) and further reduced by siRNA targeting MYC as compared with the siNC (P < 0.01). Meanwhile, the IC50 of HSYA in the two cells was calculated, and it was found that siRNA targeting MYC decreased the IC50 of HSYA in the two cells (P < 0.001, Figure 2B). As shown in Figure 2C, the proliferation of the LN-229 and U-251MG cells was inhibited by HSYA and further reduced by siRNA targeting MYC as compared with the siNC (P < 0.05). Subsequently, the abilities of the LN-229 and U-251MG cells to migrate and invade after silencing MYC or treatment with HYSA were evaluated. As presented in Figure 3, the invasion (Figure 3A) and the migration (Figure 3B) of LN-229 and U-251MG cells were inhibited by HYSA (P < 0.05), and MYC silence further reduced the invasion and migration of HYSA-treated cells (P < 0.01).
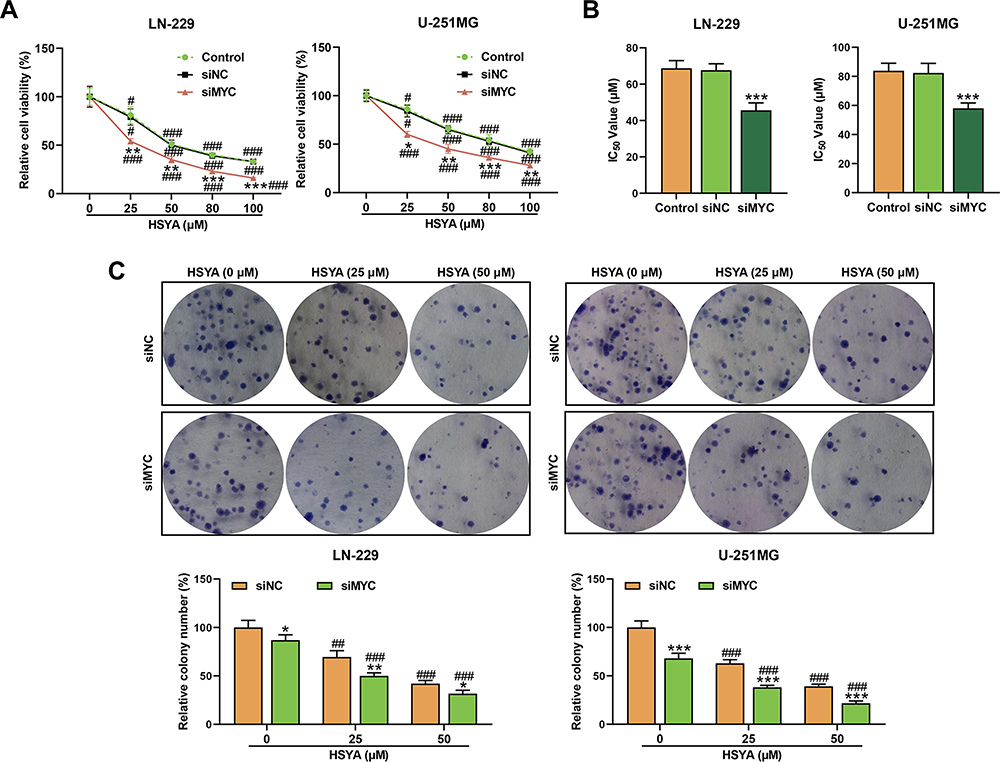 |
Figure 2 HSYA and siRNA targeting MYC inhibited the cell viability and proliferation, and siRNA targeting MYC enhanced the sensitivity of glioma cells. (A) The viabilities of LN-229 and U-251MG cells after transfection with siRNA targeting MYC or treatment with different doses of HSYA for 48 h were detected by MTT assays. (B) The IC50 of HSYA in LN-229 and U-251MG cells was calculated through Probit regression analysis. (C) The proliferation of LN-229 and U-251MG cells after transfection with siRNA targeting MYC or treatment with HSYA for 48 h was detected by colony formation assays. N=3 (*P < 0.05, **P < 0.01, ***P < 0.001, vs siNC; #P < 0.05, ##P < 0.01, ###P < 0.001, vs 0 μM). Abbreviations: HSYA, Hydroxysafflor Yellow A; siMYC, siRNA targeting MYC; siNC, siRNA negative control. |
 |
Figure 3 HSYA and siRNA targeting MYC inhibited the cell migration and invasion, and siRNA targeting MYC enhanced the sensitivity of glioma cells. (A) The invasion of LN-229 and U-251MG cells after transfection with siRNA targeting MYC or treatment with HSYA were detected by transwell assays. (B) The migration of LN-229 and U-251MG cells after being transfected with siRNA targeting MYC or treatment with HSYA were detected by wound healing assays. N=3 (*P < 0.05, **P < 0.01, ***P < 0.001, vs siNC; #P < 0.05, ##P < 0.01, ###P < 0.001, vs 0 μM). Abbreviations: HSYA, Hydroxysafflor Yellow A; siMYC, siRNA targeting MYC; siNC, siRNA negative control. |
We used 25 μM HSYA in the apoptosis detection and later experiments. As illustrated in Figure 4A, the apoptosis of the two cells was induced by both HSYA and siRNA targeting MYC (P < 0.001). After co-treatment with siRNA targeting MYC and HSYA, the effect of HSYA on promoting cell apoptosis was enhanced by siRNA targeting MYC (P < 0.001). To further verify these above discoveries, the expressions of EMT markers and apoptosis markers were determined. The results (Figure 4B) exhibited that HSYA and siRNA targeting MYC upregulated the expressions of E-Cadherin, Bax, and Cleaved PARP1 (P < 0.001), but downregulated the expressions of N-Cadherin and Bcl2 (P < 0.05). Furthermore, the effect of HSYA on these factors was promoted by siRNA targeting MYC (P < 0.001). These results indicated that the effect of HSYA on glioma cells might be realized by regulating MYC. All these results indicated that HSYA and siRNA targeting MYC inhibited cell viability and proliferation, and that siRNA targeting MYC enhanced the sensitivity of glioma cells to HSYA by inhibiting cell viability and proliferation.
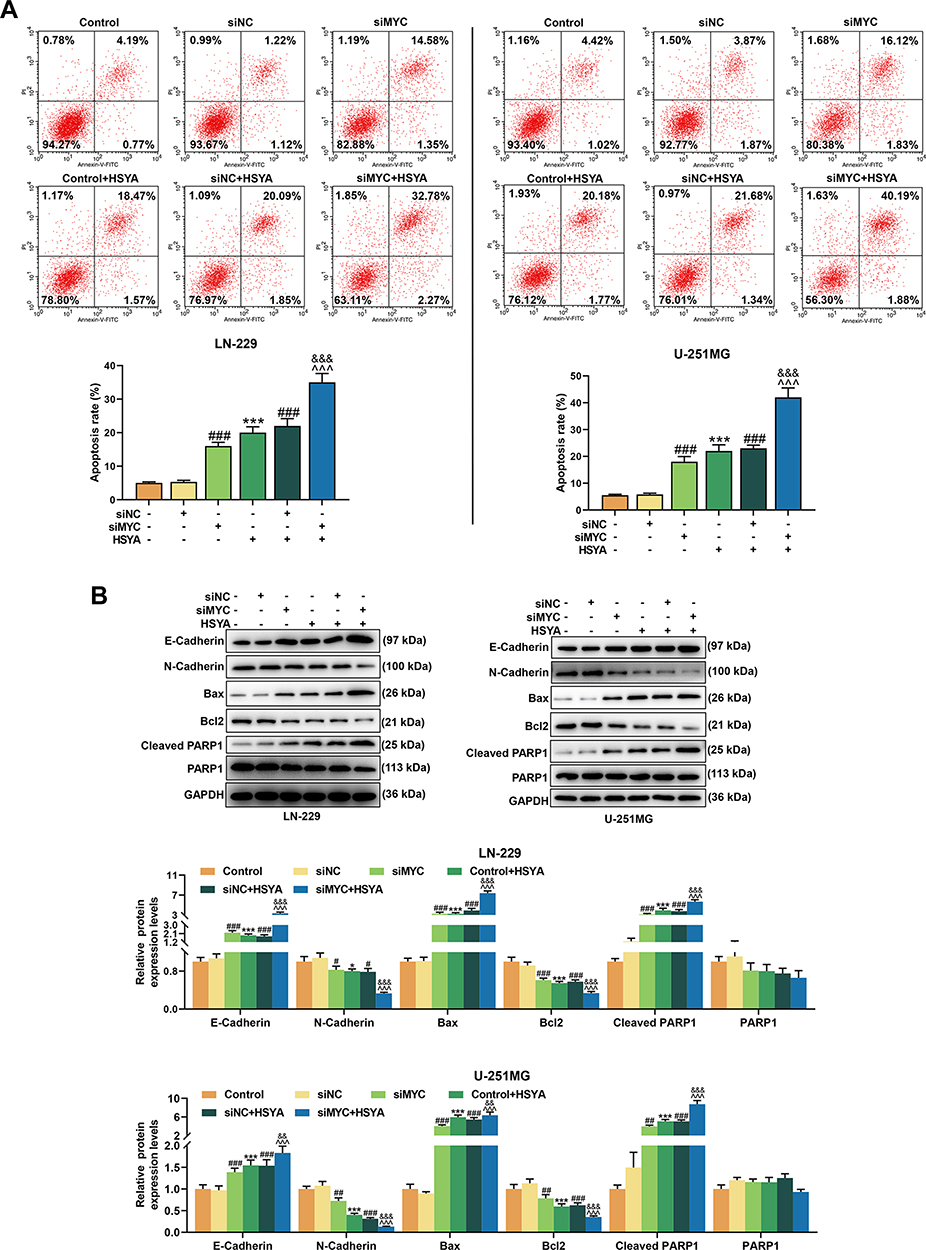 |
Figure 4 HSYA and siRNA targeting MYC induced the apoptosis, and siRNA targeting MYC further enhanced the effect of HSYA on apoptosis of glioma cells, siRNA targeting MYC enhanced the sensitivity of glioma cells. (A) The apoptosis of LN-229 and U-251MG cells after transfection with siRNA targeting MYC or treatment with HSYA were detected by flow cytometry. (B) The expressions of E-Cadherin, N-Cadherin, Bax, Bcl2, Cleaved PARP1, and PARP1 in LN-229 and U-251MG cells after transfection with siRNA targeting MYC or treatment with HSYA were detected by Western blot assay, GAPDH was an internal control. N=3 (*P < 0.05, ***P < 0.001, vs Control; #P < 0.05, ##P < 0.01, ###P < 0.001, vs siNC; ^^^P < 0.001, vs siMYC; &&P < 0.01, &&&P < 0.001, vs siNC+HSYA). Abbreviations: HSYA, Hydroxysafflor Yellow A; siMYC, siRNA targeting MYC; siNC, siRNA negative control. |
HSYA and siRNA Targeting MYC Promoted the DNA Damage of Expressions of Glioma Cells and siRNA Targeting MYC Further Enhanced the Effect of HSYA on DNA Damage
As the apoptosis of the cells was induced by HSYA and γH2AX is a biomarker of DNA Double-strand breaks (DSBs),26 thus, the γH2AX expression was determined in the LN-229 and U-251MG cells after silencing MYC or treatment with HYSA. As depicted in Figure 5, the expression of γH2AX in the LN-229 and U-251MG cells was promoted by siRNA targeting MYC and HSYA, moreover, the effect of HSYA on enhancing γH2AX expression was further promoted by siRNA targeting MYC. This result suggested that the effect of HSYA on glioma cells was involved in DNA damage and repair.
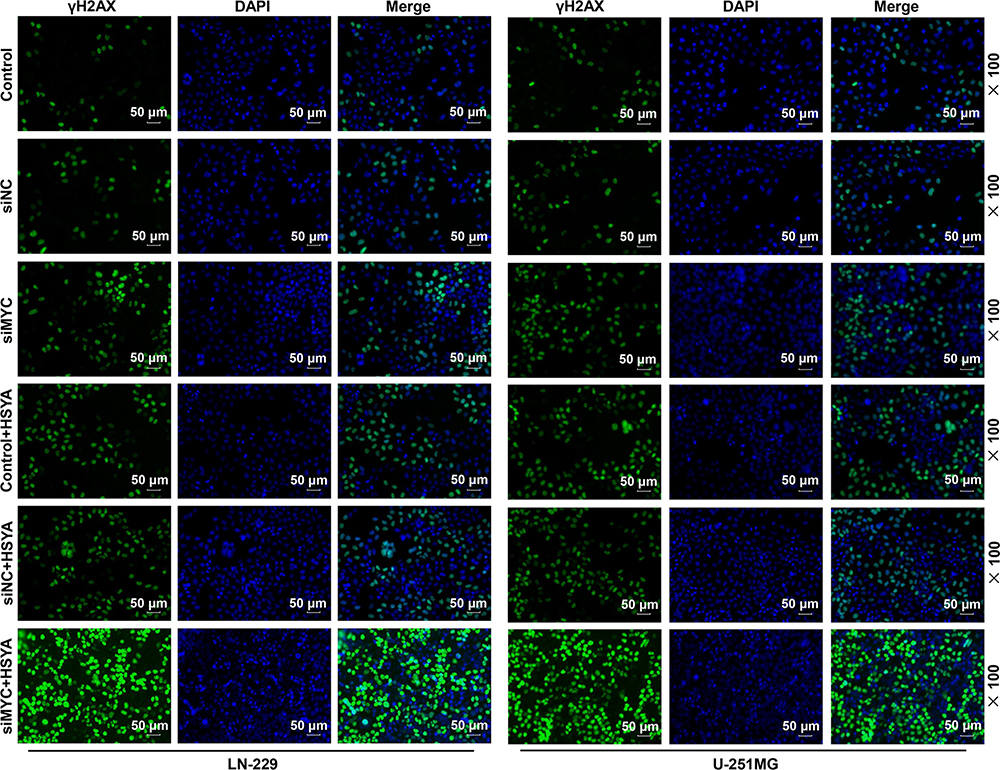 |
Figure 5 HSYA and siRNA targeting MYC promoted the DNA damage of expression of glioma cells and siRNA targeting MYC further enhanced the effect of HSYA on DNA damage. The expression of γH2AX in LN-229 and U-251MG cells after transfection with siRNA targeting MYC or treatment with HSYA was detected by Immunofluorescence. N=3. Abbreviations: HSYA, Hydroxysafflor Yellow A; siMYC, siRNA targeting MYC; siNC, siRNA negative control. |
MYC Targeted High-Expressed NBS1 and the Two Genes Were Positively Correlated in Glioma Tissues
Considering that NBS1 and MRE11 were closely associated with the DSBs,27 the expressions of NBS1 and MRE11 in the LN-229 and U-251MG cells were detected (Figure 6A). The transfection of siRNA targeting MYC downregulated the levels of MYC and NBS1 (P < 0.001), and the expression of MRE11 did not change. Meanwhile, the up-regulated expression of NBS1 was found in glioma tissues (P < 0.001, Figure 6B), and we also found a positive correlation between NBSI with MYC in glioma tissues by RT-qPCR detection (Figure 6C) and starBase prediction (Figure 6D). In addition, it was predicted that the promoter region of NBS1 was targeted by MYC (Figure 6E). Furthermore, a dual-luciferase reporter assay was performed to confirm the prediction. As shown in Figure 6F, the luciferase activity was increased after the co-transfection of MYC and wt-NBS1 and the co-transfection of MYC and mut-NBS1 (site A and/or B), as compared with the Control (P < 0.001). However, as compared with the cells co-transfected with MYC and wt-NBS1, the luciferase activity of the cells co-transfected with MYC and mut-NBS1 (site A and/or B) was reduced (P < 0.001). These findings indicated that MYC targeted NBS1 and was positively correlated with NBS1 in glioma.
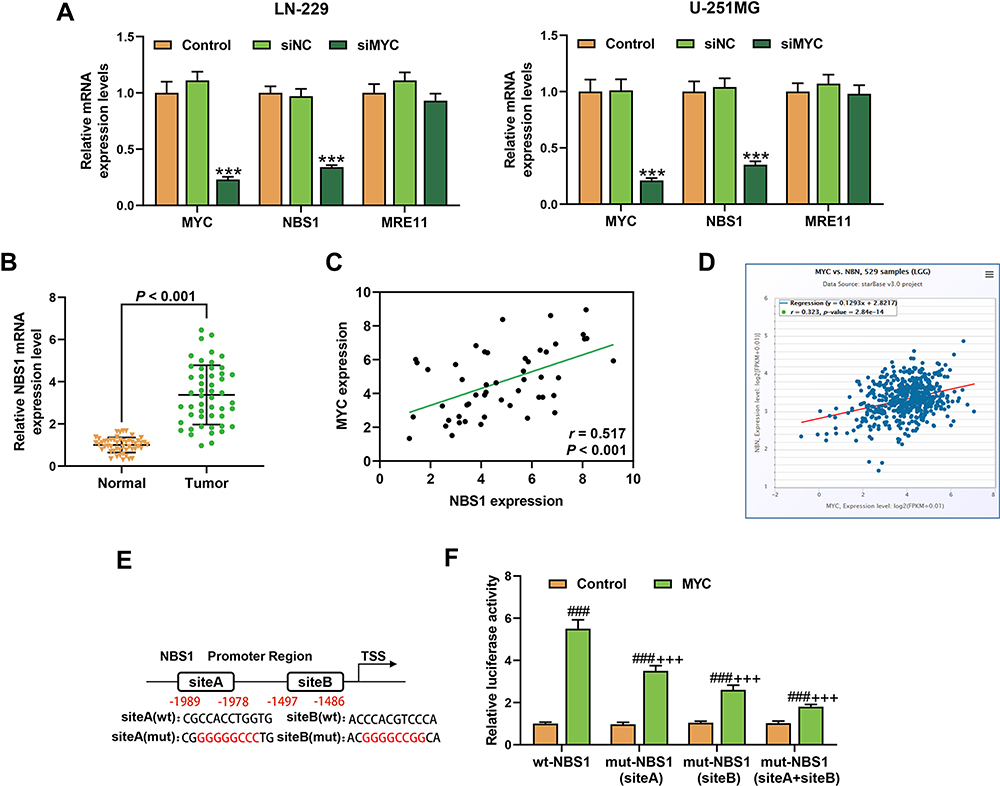 |
Figure 6 MYC targeted high expressed NBS1 and the two genes shared positive correlation in glioma tissues. (A) The expressions of MYC, NBS1, and MRE11 in LN-229 and U-251MG cells after transfection with siRNA targeting MYC were detected by RT-qPCR, β-actin was an internal control. N=3. (***P < 0.001, vs Control). (B) The expression of NBS1 in glioma tissues were detected by RT-qPCR, β-actin was an internal control. N=50. (C) The correlation between MYC with NBS1 in glioma tissues was detected by Pearson (N=50) (C) and predicted by starBase (D). (E) The binding sites in the NBS1 promoter with MYC were predicted through CISTROME and Cister. (F) The target relationship of MYC and NBS1 was verified by dual-luciferase reporter assays. N=3 (###P < 0.001, vs Control; +++P < 0.001, vs wt-NBS1). Abbreviations: siMYC, siRNA targeting MYC; siNC, siRNA negative control. |
siRNA Targeting MYC Caused DNA Repair Deficiency in HSYA-Treated Glioma Cells by Inhibiting the Transduction of Signaling Related to DNA Repair
Then, we further detected the signaling related to DNA damage response in the LN-229 and U-251MG cells after transfection with siRNA targeting MYC or further treatment with HSYA. As shown in Figure 7, the expression of MYC was upregulated by HSYA and downregulated by siRNA targeting MYC (P < 0.001), and the expressions of γH2AX was upregulated by HSYA and siRNA targeting MYC (P < 0.001). In addition, the ratios of p-NBS1/NBS1, p-MRE11/MRE11, p-RAD50/RAD50, and p-ATM/ATM were increased by HSYA (P < 0.001), but the ratios of p-NBS1/NBS1 and p-ATM/ATM were decreased by siRNA targeting MYC (P < 0.001), moreover, siRNA targeting MYC had no effect on the ratios of p-MRE11/MRE11 or p-RAD50/RAD50. Also, the effect of HSYA on these factors was counteracted by siRNA targeting MYC (P < 0.05). The results indicated that the DNA damage response (DNA repair signaling) in the HSYA-damages cells was normally regulated, but siRNA targeting MYC caused DNA repair deficiency.
 |
Figure 7 siRNA targeting MYC caused DNA repair deficiency in HSYA-treated glioma cells by inhibiting the transduction of signaling related to DNA repair. The expressions of MYC, NBS1, p-NBS1, MRE11, p-MRE11, RAD50, p-RAD50, ATM, p-ATM, and γH2AX in LN-229 and U-251MG cells after transfection with siRNA targeting MYC or further treatment with HSYA were detected by Western blot, Histone H3 was an internal control. N=3 (*P < 0.05, **P < 0.01, ***P < 0.001, vs Control; ##P < 0.01, ###P < 0.001, vs siNC; ^^P < 0.01, ^^^P < 0.001, vs siMYC; &P < 0.05, &&P < 0.01, vs siNC+HSYA). |
NBS1 Overexpression Induced the HSYA Resistance in MYC-Silenced Glioma Cells
To further determined whether NBS1 s was responsible for the increased sensitivity to HSYA in MYC silenced cell, the LN-229 and U-251MG were overexpressed with NBS1 (Figure 8A) and the inhibitor of MYC (10058-F4) was used to silence MYC. Then, the viability of the cells was determined (Figure 8B), and we found that the viabilities of both LN-229 and U-251MG were promoted by NBS1 overexpression (P < 0.05) but inhibited by 10058-F4 (P < 0.001). However, after co-treatment with NBS1 overexpression plasmids and 10058-F4, the effects of 10058-F4 on cell viability were reversed by NBS1 overexpression. Furthermore, the sensitivity of MYC-silenced cells to HSYA was found to be reduced by NBS1 overexpression, because as shown in Figure 8C, IC50 of HSYA in MYC-silenced glioma cells was increased by NBS1 overexpression (P < 0.001). Moreover, the apoptosis induced by HSYA in MYC silenced cells was reduced by NBS1 overexpression (Figure 9A). Then, the expressions of factors related to DNA damage and repair were determined (Figure 9B), and the results showed that the expressions of NBS1, p-NBS1, and p-ATM, which were downregulated by MYC silencing, were upregulated by NBS1 overexpression (P < 0.05). Moreover, NBS1 overexpression also downregulated the expression of γH2AX. All these results proved that NBS1 overexpressing induced HSYA resistance in MYC-silenced glioma cells, indicating NBS1 signaling was involved in the MYC and glioma chemosensitivity.
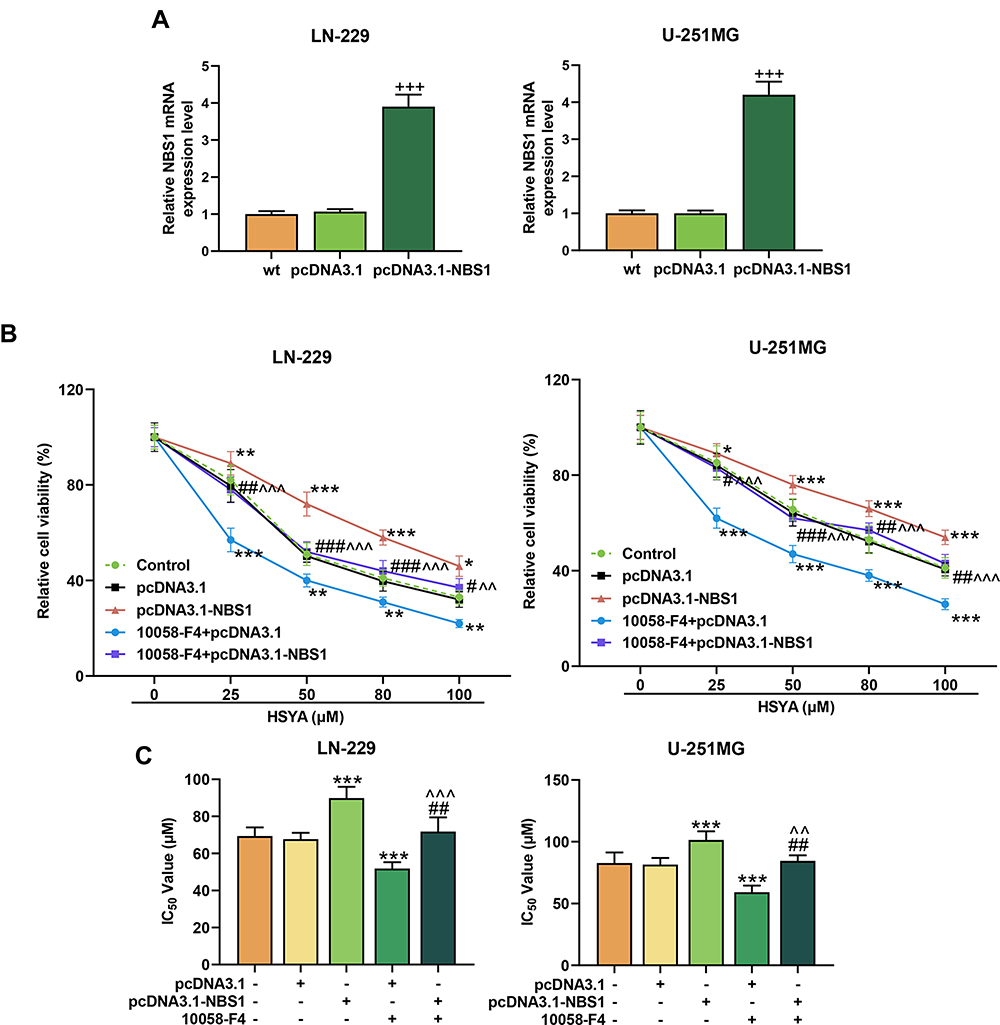 |
Figure 8 NBS1 overexpression induced the HSYA resistance in MYC-silenced glioma cells. (A) The overexpression efficiency of NBS1 in LN-229 and U-251MG cells were detected by RT-qPCR, β-actin was an internal control (+++P < 0.001, vs pcDNA3.1). (B) The cell viability of LN-229 and U-251MG cells after treatment with MYC inhibitor or overexpressed NBS1 was detected by MTT assays. (C) The IC50 of HSYA in LN-229 and U-251MG cells after treatment with MYC inhibitor or overexpressed NBS1 was calculated through Probit regression analysis. N=3 (*P < 0.05, **P < 0.01, ***P < 0.001, vs pcDNA3.1; #P < 0.05, ##P < 0.01, ###P < 0.001, vs pcDNA3.1-NBS1; ^^P < 0.01, ^^^P < 0.001, vs pcDNA3.1-NBS1+10058-F4). Abbreviation: HSYA, Hydroxysafflor Yellow A. |
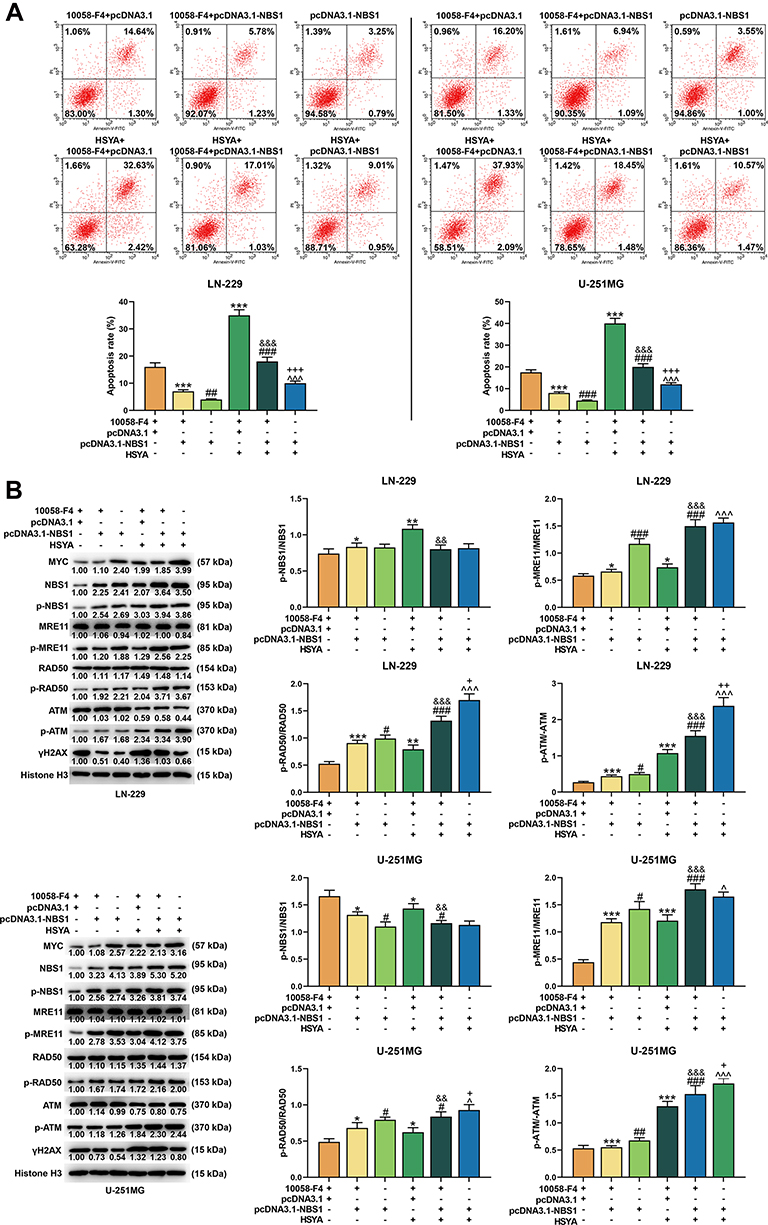 |
Figure 9 NBS1 overexpression reduced apoptosis in MYC-silenced glioma cells, reversed the effects of HSYA on cell apoptosis, and disrupted the signaling of DNA damage and repair in MYC silenced glioma cells. (A) The apoptosis of NBS1 in LN-229 and U-251MG cells were detected by flow cytometry. (B) The expressions of MYC, NBS1, p-NBS1, MRE11, p-MRE11, RAD50, p-RAD50, ATM, p-ATM, and γH2AX in LN-229 and U-251MG cells were detected by Western blot, Histone H3 was an internal control. N=3 (*P < 0.05, **P < 0.01, ***P < 0.001, vs 10058-F4+pcDNA3.1; #P < 0.05, ##P < 0.01, ###P < 0.001, vs 10058-F4+pcDNA3.1-NBS1; &&P < 0.01, &&&P < 0.001, vs 10058-F4+pcDNA3.1+HYSA; ^P < 0.05, ^^^P < 0.001, vs pcDNA3.1-NBS1; +P < 0.05, ++P < 0.01, +++P < 0.001, vs 10058-F4+pcDNA3.1-NBS1+HYSA). Abbreviation: HSYA, Hydroxysafflor Yellow A. |
Discussion
Apart from the pharmacological effects such as anti-myocardial ischemia, anti-oxidation, and anti-inflammation,10–15 HSYA also has an anti-cancer effect.16,17 Similarly, the anticancer effect of HSYA on glioma was also confirmed in the present study, as we found that HSYA suppressed the viability, proliferation, migration, and invasion, induced the apoptosis of glioma cells.
We predicted and verified that MYC is high-expressed in glioma tissues and cell, and such a finding is consistent with a previous study.28 In addition, MYC can be regulated by HSYA in human HUVEC cells and liver cancer cells.23,24 To explore whether the effect of HSYA on glioma was associated with MYC, the expression of MYC in glioma cells was detected after treatment with HSYA. We discovered that HSYA stimulated the expression of MYC (Figure S1), which might be related to tumor adaptation and drug resistance and commonly caused in cancers. MYC is involved in the drugs resistance of cancer cells, for example, MYC drives Nab-Paclitaxel resistance in pancreatic ductal carcinoma cells20 and modulates the drug resistance of leukemia stem cells through ERK/MSK pathway.21 Moreover, MYC also mediates the PARP inhibitor resistance in glioblastoma.22 We then speculated that MYC might be involved in the resistance of glioma cells to HSYA. Therefore, the biological activities of glioma cells after treatment with HSYA and silencing MYC were further evaluated, and the results showed that siRNA targeting MYC reduced the IC50 of HSYA in glioma cells and enhanced the inhibitory effect of HSYA on glioma cells, revealing that siRNA targeting MYC increased the sensitivity of glioma cells to HSYA.
Meanwhile, γH2AX is a marker of DSBs and can initiate the DNA repair response.26 The results showed that the effect of MYC on HSYA-induced glioma cells was associated with the DNA repair because we found that the level of γH2AX in glioma cells was upregulated. NBS1 is an important factor in DNA repair and can be directly targeted by MYC.29,30 Consistently, in this study, the targeted relationship was verified by bioinformatics prediction and experiments confirmation. For the first time, NBS1 was verified to be high-expressed and shared a positive correlation with MYC in glioma. During the DNA repair after DSBs, NBS1 bindings with MRE11 and RAD50 in the nucleus then form the MRN (MRE11-RAD50-NBS1) complex, which directly binds to damaged DNA and initiates homologous recombination-dependent repair.27,31,32 The MRN complex also recruits and activates ATM, which is a vital kinase in the DNA damage response signaling network.33 The activation of ATM (p-ATM) modulates hundreds of substrates involved in the cell cycle, DNA repair, and apoptosis.34–36 To further explore the regulation of HSYA and MYC on DNA repair in glioma cells, the expressions of these DNA repair factors were examined. We discovered that the activation of NBS1, MRE11, RAD50, and ATM was promoted by HSYA, and that siRNA targeting MYC reduced the activation of NBS1 and ATM without affecting MRE11 and RAD50. These phenomena indicated that the MYC silencing inhibited the MRN-ATM, resulting in DNA repair deficiency, and subsequently enhanced the sensitivity of glioma cells to HSYA.
To further verify whether NBS1 was responsible for the increased sensitivity to HSYA in MYC silencing glioma cells, NBS1 was overexpressed in the cells. Then, we detected that the NBS1 overexpression reversed the inhibitory effect on cell viability and the promoting effect on cell apoptosis, also, the sensitivity of MYC-silenced cells to HSYA was reduced by NBS1 overexpression. Mechanically, the inhibited MRN-ATM pathway was promoted by NBS1 overexpression, demonstrating that the effect of MYC silencing on promoting the sensitivity of glioma cells to HSYA was realized by regulating NBS1.
Our research revealed that HSYA and MYC silencing had an inhibitory effect on glioma cells. HSYA could promote the expression of MYC in glioma cells and MYC silencing could enhance the sensitivity of glioma cells to HSYA, which could be explained by that both HSYA and MYC silencing could cause the DNA damage of glioma cells. In HSYA-treated glioma cells, the DNA repair response was normal, but MYC silencing could inhibit the MRN-ATM via regulating NBS1 to cause DNA repair deficiency, and subsequently enhanced the sensitivity of glioma cells to HSYA.
Funding
This work was supported by the Key Research and Development Plan of Shaanxi Province [2019SF-058].
Disclosure
The authors declare no conflicts of interest for this work.
References
1. van Iersel L, van Santen HM, Potter B, et al. Clinical impact of hypothalamic-pituitary disorders after conformal radiation therapy for pediatric low-grade glioma or ependymoma. Pediatr Blood Cancer. 2020;67(12):e28723. doi:10.1002/pbc.28723
2. Wirsching HG, Weller M. Does neuronal activity promote glioma progression? Trends Cancer. 2020;6(1):1–3. doi:10.1016/j.trecan.2019.11.002
3. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284–297. doi:10.1007/s13311-017-0519-x
4. Taal W, Bromberg JE, van den Bent MJ. Chemotherapy in glioma. CNS Oncol. 2015;4(3):179–192. doi:10.2217/cns.15.2
5. Hao Z, Hu S, Liu Z, Song W, Zhao Y, Li M. Circular RNAs: functions and prospects in glioma. J Mol Neurosci. 2019;67(1):72–81. doi:10.1007/s12031-018-1211-2
6. Shafabakhsh R, Asemi Z. Quercetin: a natural compound for ovarian cancer treatment. J Ovarian Res. 2019;12(1):55. doi:10.1186/s13048-019-0530-4
7. Li Z, Xu W, Ren X, Xu J, Chen J. Puerarin promotes DUSP1 expression by regulating miR‑133a‑3p in breast cancer. Mol Med Rep. 2019;19(1):205–212. doi:10.3892/mmr.2018.9682
8. Abrams SL, Follo MY, Steelman LS, et al. Abilities of berberine and chemically modified berberines to inhibit proliferation of pancreatic cancer cells. Adv Biol Regul. 2019;71:172–182. doi:10.1016/j.jbior.2018.10.003
9. Zheng M, Guo X, Pan R, Gao J, Zang B, Jin M. Hydroxysafflor Yellow A alleviates ovalbumin-induced asthma in a guinea pig model by attenuating the expression of inflammatory cytokines and signal transduction. Front Pharmacol. 2019;10:328. doi:10.3389/fphar.2019.00328
10. Lee M, Li H, Zhao H, Suo M, Liu D. Effects of Hydroxysafflor Yellow A on the PI3K/AKT pathway and apoptosis of pancreatic β-cells in Type 2 diabetes mellitus rats. Diabetes Metab Syndr Obes. 2020;13:1097–1107. doi:10.2147/dmso.s246381
11. Liu J, Yue S, Yang Z, et al. Oral hydroxysafflor yellow A reduces obesity in mice by modulating the gut microbiota and serum metabolism. Pharmacol Res. 2018;134:40–50. doi:10.1016/j.phrs.2018.05.012
12. Yang G, Wang N, Seto SW, Chang D, Liang H. Hydroxysafflor yellow a protects brain microvascular endothelial cells against oxygen glucose deprivation/reoxygenation injury: involvement of inhibiting autophagy via class I PI3K/Akt/mTOR signaling pathway. Brain Res Bull. 2018;140:243–257. doi:10.1016/j.brainresbull.2018.05.011
13. Zhou D, Ding T, Ni B, Jing Y, Liu S. Hydroxysafflor Yellow A mitigated myocardial ischemia/reperfusion injury by inhibiting the activation of the JAK2/STAT1 pathway. Int J Mol Med. 2019;44(2):405–416. doi:10.3892/ijmm.2019.4230
14. Sheng C, Peng W, Xia Z, Wang Y. Plasma and cerebrospinal fluid pharmacokinetics of hydroxysafflor yellow A in patients with traumatic brain injury after intravenous administration of Xuebijing using LC-MS/MS method. Xenobiotica. 2020;50(5):545–551. doi:10.1080/00498254.2019.1668983
15. Jin M, Wang L, Wu Y, Zang BX, Tan L. Protective effect of hydroxysafflor yellow A on bleomycin-induced pulmonary inflammation and fibrosis in rats. Chin J Integr Med. 2018;24(1):32–39. doi:10.1007/s11655-017-2094-z
16. Zhang J, Li J, Song H, Xiong Y, Liu D, Bai X. Hydroxysafflor yellow A suppresses angiogenesis of hepatocellular carcinoma through inhibition of p38 MAPK phosphorylation. Biomed Pharmacother. 2019;109:806–814. doi:10.1016/j.biopha.2018.09.086
17. Jiang M, Zhou LY, Xu N, An Q. Hydroxysafflor yellow A inhibited lipopolysaccharide-induced non-small cell lung cancer cell proliferation, migration, and invasion by suppressing the PI3K/AKT/mTOR and ERK/MAPK signaling pathways. Thorac Cancer. 2019;10(6):1319–1333. doi:10.1111/1759-7714.13019
18. Chen X, Yang F, Zhang T, et al. MiR-9 promotes tumorigenesis and angiogenesis and is activated by MYC and OCT4 in human glioma. J Exp Clin Cancer Res. 2019;38(1):99. doi:10.1186/s13046-019-1078-2
19. Zhang G, Dong Z, Prager BC, et al. Chromatin remodeler HELLS maintains glioma stem cells through E2F3 and MYC. JCI Insight. 2019;4(7). doi:10.1172/jci.insight.126140
20. Parasido E, Avetian GS, Naeem A, et al. The sustained induction of c-MYC drives Nab-Paclitaxel resistance in primary pancreatic ductal carcinoma cells. Mol Cancer Res. 2019;17(9):1815–1827. doi:10.1158/1541-7786.mcr-19-0191
21. Zhang Y, Chen HX, Zhou SY, et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol Cancer. 2015;14(1):56. doi:10.1186/s12943-015-0326-0
22. Ning JF, Stanciu M, Humphrey MR, et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun. 2019;10(1):2910. doi:10.1038/s41467-019-10993-5
23. Wang J, Wang J, Wang X, et al. Molecular mechanism of inhibition of the abnormal proliferation of human umbilical vein endothelial cells by hydroxysafflor-yellow A. Pharm Biol. 2016;54(9):1800–1807. doi:10.3109/13880209.2015.1129541
24. Yang F, Li J, Zhu J, Wang D, Chen S, Bai X. Hydroxysafflor yellow A inhibits angiogenesis of hepatocellular carcinoma via blocking ERK/MAPK and NF-κB signaling pathway in H22 tumor-bearing mice. Eur J Pharmacol. 2015;754:105–114. doi:10.1016/j.ejphar.2015.02.015
25. Zhou W, Zhang S, Li J, Li Z, Wang Y, Li X. lncRNA TINCR participates in ALA-PDT-induced apoptosis and autophagy in cutaneous squamous cell carcinoma. J Cell Biochem. 2019;120(8):13893–13902. doi:10.1002/jcb.28662
26. Siddiqui MS, François M, Fenech MF, Leifert WR. Persistent γH2AX: a promising molecular marker of DNA damage and aging. Mutat Res Rev Mutat Res. 2015;766:1–19. doi:10.1016/j.mrrev.2015.07.001
27. Bian L, Meng Y, Zhang M, Li D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment. Mol Cancer. 2019;18(1):169. doi:10.1186/s12943-019-1100-5
28. Wang J, Wang H, Li Z, et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3(11):e3769. doi:10.1371/journal.pone.0003769
29. Chiang YC, Teng SC, Su YN, Hsieh FJ, Wu KJ. c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. J Biol Chem. 2003;278(21):19286–19291. doi:10.1074/jbc.M212043200
30. Wan R, Wu J, Baloue KK, Crowe DL. Regulation of the Nijmegen breakage syndrome 1 gene NBS1 by c-myc, p53 and coactivators mediates estrogen protection from DNA damage in breast cancer cells. Int J Oncol. 2013;42(2):712–720. doi:10.3892/ijo.2012.1757
31. Her J, Bunting SF. How cells ensure correct repair of DNA double-strand breaks. J Biol Chem. 2018;293(27):10502–10511. doi:10.1074/jbc.TM118.000371
32. Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. 2018;87(1):263–294. doi:10.1146/annurev-biochem-062917-012415
33. Li D, Ye L, Lei Y, Wan J, Chen H. Downregulation of FoxM1 sensitizes nasopharyngeal carcinoma cells to cisplatin via inhibition of MRN-ATM-mediated DNA repair. BMB Rep. 2019;52(3):208–213. doi:10.5483/BMBRep.2019.52.3.249
34. Roger E, Gout J, Arnold F, et al. Maintenance therapy for ATM-deficient pancreatic cancer by multiple DNA damage response interferences after platinum-based chemotherapy. Cells. 2020;9(9):2110. doi:10.3390/cells9092110
35. Vančevska A, Ahmed W, Pfeiffer V, Feretzaki M, Boulton SJ, Lingner J. SMCHD1 promotes ATM-dependent DNA damage signaling and repair of uncapped telomeres. EMBO J. 2020;39(7):e102668. doi:10.15252/embj.2019102668
36. Wu W, Gou H, Dong J, et al. Usnic acid inhibits proliferation and migration through ATM mediated DNA damage response in RKO colorectal cancer cell. Curr Pharm Biotechnol. 2020;21. doi:10.2174/1389201021666201002155955
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.