Back to Journals » International Journal of Nanomedicine » Volume 15
Multimodal Mesoporous Silica Nanocarriers for Dual Stimuli-Responsive Drug Release and Excellent Photothermal Ablation of Cancer Cells
Authors Tran VA, Vo VG ![]() , Shim K, Lee SW
, Shim K, Lee SW ![]() , An SSA
, An SSA ![]()
Received 17 March 2020
Accepted for publication 18 August 2020
Published 8 October 2020 Volume 2020:15 Pages 7667—7685
DOI https://doi.org/10.2147/IJN.S254344
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Vy Anh Tran1,2 *,* Van Giau Vo3,4 *,* Kyuhwan Shim,5 Sang-Wha Lee,1 Seong Soo A An6
1Department of Chemical and Biological Engineering, Gachon University, Seongnam, Republic of Korea; 2NTTHi-Tech Institute, Nguyen Tat Thanh University, Ho Chi Minh 700000, Vietnam; 3Institute of Research and Development, Duy Tan University, Danang 550000, Vietnam; 4Department of Industrial and Environmental Engineering, Graduate School of Environment, Gachon University, Seongnam 13120, Republic of Korea; 5Department of Neurology, Veterans Medical Research Institute, Veterans Health Service Medical Center, Seoul 05368, Republic of Korea; 6Department of BioNano Technology, Gachon University, Seongnam 13120, Republic of Korea
*These authors contributed equally to this work
Correspondence: Sang-Wha Lee; Seong Soo A An
Gachon University, San 65, Bokjeong-Dong, Sujeong-Gu, Seongnam 461-701, South Korea
Email [email protected]; [email protected]
Background: Core-shell types of mesoporous silica nanoparticles (MSNs) with multimodal functionalities were developed for bio-imaging, controlled drug release associated with external pH, and near-infrared radiation (NIR) stimuli, and targeted and effective chemo-photothermal therapeutics.
Materials and Methods: We synthesized and developed a core-shell type of mesoporous silica nanocarriers for fluorescent imaging, stimuli-responsive drug release, magnetic separation, antibody targeting, and chemo-photothermal therapeutics. Also, the biocompatibility, cellular uptake, cytotoxicity, and photothermal therapy on these FS3-based nanocarriers were systematically investigated.
Results: Magnetic mesoporous silica nanoparticles was prepared by coating a Fe3O4 core with a mesoporous silica shell, followed by grafting with fluorescent conjugates, so-called FS3. The resulting FM3 was preloaded with therapeutic cisplatin and coated with polydopamine layer, so-called FS3P/C. Eventually, graphene oxide-wrapped FS3P/C (FS3P-G/C) exhibited high sensitivity in the dual stimuli (pH, NIR)-responsive controlled release behavior. On the other hand, Au NPs-coated FS3P/C (FS3P-A/C) exhibited more stable release behavior, irrespective of pH changes, and exhibited much more enhanced release rate under the same NIR irradiation. Notably, FS3P-A/C showed strong NIR absorption, enabling photothermal destruction of HeLa cells by its chemo-photothermal therapeutic effects under NIR irradiation (808 nm, 1.5 W/cm2). The selective uptake of FS3-based nanocarriers was confirmed in cancer cell lines including HeLa (American Type Culture Collection - ATCC) and SHSY5Y (ATCC 2266) by the images obtained from confocal laser scanning microscopy, flow cytometry, and transmission electron microscopy instruments. Cisplatin-free FS3-based nanocarriers revealed good cellular uptake and low cytotoxicity against cancerous HeLa and SH-SY5Y cells, but showed no obvious toxicity to normal HEK293 (ATCC 1573) cell.
Conclusion: Along with the facile synthesis of FS3-based nanocarriers, the integration of all these strategies into one single unit will be a prospective candidate for biomedical applications, especially in chemo-photothermal therapeutics, targeted delivery, and stimuli-responsive controlled drug release against multiple cancer cell types.
Keywords: mesoporous silica NPs, Au NPs, stimuli-responsive drug release, chemo-photothermal therapy, cancerous cells
Introduction
Smart nanocarriers with controlled drug delivery have been actively developed for overcoming typical limitations of conventional free drugs, such as lack of selectivity, non-specific targeting, poor solubility, limited stability, and rapid release.1,2 Multifunctional mesoporous nanocarriers are normally constructed for controlled drug release in response to internal or external triggers such as polydopamine (PDA),3,4 graphene oxide (GO),5,6 and hydrogel matrix.7,8 However, the construction of internal trigger release systems, which are responding to subtle change in the surroundings, encountered significant challenges in cancer and inflammation processes, because of arbitrary pH changes during their evolution.9 Therefore, externally triggered release systems controlled by NIR irradiation, external pH changes, and electromagnetic field effects are intensively investigated.1,10,11
In recent years, mesoporous silica nanoparticles (MSNs) have been actively employed as smart nanocarriers for anticancer therapeutics due to its easy functionalization, large surface area for high drug loading, specific targeting cancer therapy, and bio-imaging agent.12–14 Furthermore, inorganic nanocrystals (Au, Ag, Fe3O4, MnO, CuS) are integrated with MSNs to create a multifunctional core-shell type of MSNs-based drug delivery system.15 Among them, gold and magnetic NPs are becoming important ingredients in the core-shell MSNs, because of their optical uniqueness and magnetic features in biomedical applications. These core-shell type MSNs can be promising theranostic nanocarriers with high loading capability, cell targeting specificity, and bio-imaging agents, as well as by adding stimuli-responsive drug release and hyperthermia treatment.16–20
Graphene oxide (GO) possesses advantageous biocompatibility, large specific surface area, and π-conjugated nanostructures which can confer excellent water solubility, physiological stability, and capacity for drug delivery.21,22 Likewise, polydopamine (PDA) possess good biocompatibility, unique chemical structure, and photothermal heating effects, which can be rapidly integrated into the construction of tumor-targeted drug delivery systems.23 In the meanwhile, plasmonic gold can absorb the near-infrared radiation (NIR) and convert it to thermal energy because of intense surface plasmon resonance (SPR) and low quantum yield.24,25 It is highly expected that plasmonic gold combined with PDA or GO nanosheets can produce synergistic photothermal heating effects on the cancer cell treatment.26–28 Additionally, the combination of hyperthermia and chemotherapy in one unit would be very effective in killing multiple cancer cells.29–31
In the case of nanocarriers without targeting moieties, tumor accumulation is dependent upon direct injection, selective tumor embolization, or passive targeting. Therefore, non-targeting nanocarriers usually cause harmful impacts of released drugs during the process of cancer therapy, such as side effects on the nearby cells (or tissues) and low accumulation of drugs in the lesion treatment.32,33 In this regard, core-shell type magnetic MSNs can be fully exploited as a promising targeted-delivery vehicle, wherein drugs are delivered to the specific location in vivo under an external magnetic field.34 A more advanced platform based on core-shell type MSNs can be actively targeted to cancer cells by surface functionalization with natural ligands for cell surface receptors, nucleic acids, peptides, non-immunoglobulin scaffolds, or antibodies.35 Among them, antibodies can play a vital role in modern cancer therapeutics as a specific targeting agent for cancer cells without significant side-effects and adequate plasma half-life.36
The aim of the current work is to overcome the limitations of conventional drug delivery systems, eg, poor solubility, limited stability, rapid metabolization of the drug, undesired side-effects, and lack of selectivity toward specific cells types.37,38 The core-shell type mesoporous MSNs were prepared by embedding a Fe3O4 core in a mesoporous silica shell, followed by grafting with fluorescent conjugates, and coating with a PDA/GO double layer, hereafter referred to as FS3-based NPs (or nanocarriers). Our FS3-based NPs combined multimodal functions onto one single unit, such as mesoporosity, for high drug loading, stimuli-responsive drug delivery, fluorescent bio-imaging, magnetic separation, photothermal heating effect, and specific antibody targeting. Notably, GO-coated FS3 exhibited high sensitivity in the dual stimuli (pH, NIR)-responsive release behavior, whereas Au Nps-deposited FS3 highlighted strong NIR absorption, enabling photothermal destruction of HeLa cells under NIR irradiation (808 nm, 1.5 W/cm2, 5–10 minutes). Finally, the biocompatibility, cellular uptake, cytotoxicity, and photothermal therapy on these FS3-based nanocarriers were systematically investigated for improved treatment of cancer cells.
Experimental Sections
Chemicals
FeCl3.6H20 (98.0–102%), FeCl2.4H20 (99.99%), NaOH (≥98%), tetraethyl orthosilicate (TEOS, 99%), cetyltrimethylammonium bromide (CTAB, 99%), NH4F (99.99%), dopamine hydrochloride (powders), 3-aminopropyltrimethoxy-silane (APTMS, 97%), fluorescein isothiocyanate (FITC, 90%), chloroform (≥99.5%), cisplatin (pharmaceutical secondary standard), dimethyl sulfoxide (DMSO, ≥99.9%), tris(hydroxymethyl)aminomethane (Tris, ≥99.8%), graphene oxide (GO, 4 mg/mL), 3,4-dihydroxyhydrocinnamic acid (hydrocaffeic acid, 98%), HAuCl4 (99.999%), 2-(N-morpholino) ethanesulfonic acid hydrate (MES, ≥99.5%), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC, crystalline), and N-hydroxysuccinimide (NHS, 98%) were purchased from Sigma-Aldrich (South Korea). Phosphate-buffered saline (PBS) was purchased from Bioneer (South Korea). A monoclonal anti-human epidermal growth factor receptor (EGFR) antibody, clone AT6E3 (EGFRab), was obtained from ATGen, Inc. Ethanol and water (high-performance liquid chromatography (HPLC) grade) were used as without further purification treatment. Glassware was cleaned by an aqua regia solution of HNO3:HCl=1:3 and then washed with deionized (DI) water more than three times.
Synthesis of Magnetic NPs (Fe3O4)
Fe3O4 magnetic NPs were synthesized using co-precipitation of ferric and ferrous salts under atmospheric pressure (760 mm of Hg). Both FeCl3.6H20 (7.5 g) and FeCl2.4H20 (3 g) were added into 160 mL of deoxygenated water by purging inert N2 gas. The solution was stirred for 60 minutes at constant temperature of 30°C. The chemical precipitation occurred at 30°C under vigorous stirring by injecting 40 mL of NaOH (2.0 M) slowly under the steady continuous purging with N2 gas. Even after the color changed from yellow to black, the solution was continuously stirred at 70°C for 6 hours. The solution reduced to room temperature (25–26°C, RT), and the precipitates were separated using a permanent magnet bar and washed with deoxygenated DI water to neutral pH. Finally, Fe3O4 was washed with acetone and dried in the freeze-dryer system.39
Synthesis of FS3
The synthesis procedures of conjugating fluorescein isothiocyanate (FITC) onto MSF were conducted under dark conditions, due to the light-sensitive property. FITC (3 mg) was mixed with 3-aminopropyltrimethoxysilane (APTMS, 140 μL) in 4 mL of ethanol for 6 hours in the dark. The resultant APTMS-FITC complex solution was stored at approximately 10°C. Fe3O4 nanocrystals (20 mg, ca. 15 nm) were dispersed in chloroform (2 mL). The Fe3O4 solution was poured into CTAB (7.5 mL, 0.16 M). The mixed solution was stirred vigorously for 1 hour, leading to an oil-in-water microemulsion brow milk solution. The mixture was heated up to 60°C and aged at the same temperature for 20 minutes to evaporate chloroform, forming a transparent black solution of Fe3O4/CTAB.40,41
NH4F (0.4 g) and water (70 mL) were heated to 70°C for 40 minutes to reach approximately pH 10, followed by the addition of Fe3O4/CTAB solution under constant stirring at 70°C for 30 minutes. After slowly adding TEOS (1 mL) into the mixed solution by droplet injection, a white-brown sol-gel solution was formed under vigorous stirring for 5 minutes. The APTMS-FITC complex was slowly added into the sol-gel solution. The synthetic procedure was carried out in the dark with reflux at 70°C for 12 hours. The products were centrifuged and washed with copious DI water and ethanol (thrice) to remove unreacted components. To extract the remaining CTAB, the isolated product was dispersed in ethanol (200 mL) and kept stirred under reflux at 70°C for 5 hours. The final product was centrifuged, washed with water, dried in a freeze dryer for 72 hours, and stored in the dark prior to the further characterization.41,42 To compare with FS3, only silica complex (S3) without Fe3O4 was synthesized by the same method. In addition, MSNs was also synthesized by the same method without grafting the APTMS-FITC complex.
Cisplatin Loading into FS3
Cisplatin was weakly acidic due to its monohydrated form at neutral pH.43 Cisplatin solubility was at 2.5 mg/mL and 4 mg/mL in water at 25°C and 35°C, respectively. However, the solubility of cisplatin in DMSO was notably high (25 mg/mL at 25°C. Due to the light susceptibility (or sensitivity) of cisplatin, all synthetic steps were conducted in the dark.
For loading cisplatin drug into mesoporous nanoparticles (NPs), FS3 (150 mg) was dispersed in DMSO (5 mL), until the concentration of cisplatin reached 0.033 M. The solution was continuously stirred for 24 hours at 25–26°C (RT) to load the maximal amounts of cisplatin. Cisplatin-loaded FS3 (FS3/C) was separated by centrifugation. In order to determine the loading amount of cisplatin, the supernatant was collected to calculate the difference between the initial amount of cisplatin and final remaining values in supernatant.44
PDA and GO Coating (FS3P-G)
To coat polydopamine (PDA) layer over the FS3-drug system, FS3/C (100 mg) was dispersed in Tris-HCl buffer (10 mL of 10 mM at pH 8.5), followed by adding dopamine hydrochloride (10 mg). The polymerization reaction of dopamine was carried out for 3 hours under continuous stirring in the dark. The PDA-coated FS3/C (FS3P/C) was centrifuged for 5 minutes and washed with DI water to remove unpolymerized residues. The PDA–coated FS3/C (FS3P/C) was stored at 4°C prior to the release test.43 To prepare a GO-coated FS3-drug system, FS3P/C (50 mg) was dispersed in GO solution (2mL). The mixed solution was stirred at RT for 2 hours to complete the GO wrapping process. The GO-coated FS3 (FS3P-G/C) was collected by centrifugation at 6,000 rpm and thoroughly washed with DI water to remove any residue.45
Gold Deposition (FS3P-A/C)
Catechol moieties were conjugated onto the FS3P/C using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) coupling reaction. Shortly, hydrocaffeic acid (5 mg in 1.5 mL of PBS solution at pH 5.0) was added into the FS3P/C solution (50 mg in 3 mL of PBS solution) containing EDC (8 mg). The reaction was carried out at pH 5.0 for 2 hours. The resulting FS3P/C-Catechol was isolated by centrifugation. To deposit gold NPs onto the FS3-drug system, FS3P/C-Catechol (50 mg) was added into HAuCl4 solution (6 mL of 0.5 mM), and the deposition reaction was continued for 2 hours at 25–26°C (RT) under continuous stirring. The product was collected by centrifugation (4500 x g, 5 minutes), and washed with DI water for a further utilization.
EGFR Antibody Conjugation (FS3P-G-E/C)
To conjugate antibodies onto the GO surfaces, EGFR antibody (10 μg) was dispersed in MES buffer (100 μL, pH 6). EDC (25 μL at 1 μL/mL) was annexed to actuate the carboxylic acid groups of EGFR antibody. This solution was quickly added to the particle (50 μg) in MES buffer (200 μL). NHS (30 μL of 1 mg/mL in MES) was added to the suspension. The suspension was dispersed using an orbital shaker machine for 5 hours and washed with DI water. The product was dried in a freeze-dryer system and stored at 4°C.
Determination of Encapsulation Efficiency (EE) and Loading Capacity (LC)
The EE and LC of cisplatin-loaded FS3 were determined by quantifying the absorption of the clear supernatant using UV-vis spectroscopy (NanoDrop, NanoDrop Technologies, Wilmington, DE, USA). The corresponding calibration curve was obtained by testing the supernatant of cisplatin. Tests were performed in triplicate for each sample. The absorbance value of cisplatin was measured using UV-vis spectroscopy in DMSO and PBS buffer at the wavelength of 310 nm and 301 nm, respectively.
The percentage of encapsulation efficiency and loading capacity of cisplatin in FS3, FS3P, FS3P-G, and FS3P-A were determined by the following equations,46 respectively: 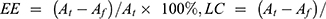
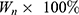 . Here, At is the total amount of cisplatin, Af was the amount of free cisplatin in the supernatant after centrifugation, and Wn is the weight of FS3 system for drug release. All measurements were made in triplicate, and the average value was reported.
. Here, At is the total amount of cisplatin, Af was the amount of free cisplatin in the supernatant after centrifugation, and Wn is the weight of FS3 system for drug release. All measurements were made in triplicate, and the average value was reported.
In vitro Drug Release Test of As-prepared Samples
The in vitro drug release tests were carried out as follows: 50 mg of each sample was dispersed in a definite volume (5 mL) of PBS at pH 7.4 at 37°C. The resulting suspension was placed in an incubator and stirred at 600 rpm for a definite period, and a 4-µL aliquot was taken out of the dissolution medium at appropriate time intervals. The concentration of released drug was measured using UV-vis spectroscopy (NanoDrop, USA) at 301 nm.46
Cellular Uptake and Intracellular Distribution
The HeLa cells were obtained from the American Type Culture Collection (ATCC CCL-2) and were grown in full medium to a final density of a 1×105 cells/well in 6-well plates with the presence of a glass coverslips. On the next day, the cells were treated with as-prepared NPs (SP/C, FS3P-G/C, FS3P-A/C, and FS3P-G-E/C) at 5 µg/mL for 4 hours. Cells were further washed with PBS three times and fixed with 4% paraformaldehyde in PBS for 10 minutes at RT. Cells on the slides were stained with 4,6 diamidino-2-phenylindole (DAPI, 1 μg/mL) in PBS for 20 minutes at RT to stain the nucleus cells after washing three times with PBS. The cells in each slide were mounted using the Eukitt® Quick-hardening mounting medium (Sigma-Aldrich) for observation under confocal microscopy. Lastly, the mounted cells were imaged at λ = 408 and 488 nm for detecting fluorescence intensity of DAPI and FITC by confocal microscopy (Nikon TE2000-E Eclipse C1si), respectively.
The distribution of FMAF-based NPs in cells was also tested by flow cytometry analysis. HeLa cells were seeded on a 6-well plate and cultured at 37°C under 5% CO2 on the day before. Then, the cells were treated with as-prepared NPs at 5 µg/mL. Following 4 hours and 24 hours co-incubation, cells were rinsed with PBS and collected by trypsinization. The cells were collected by centrifugation, and 1×PBS (500 μL) was added to re-suspend the cells for flow cytometry (LSRII, BD Biosciences). The FICT fluorescence of the four NPs was tested with approximately 10,000 gated cells. The normal cells without any fluorescence labeling were used. The FlowJo software was used to analyses flow cytometry data.
Furthermore, to visualize NP distributions within cells, the four different samples were added to SH-SY5Y cells cultured in 75-cm2 flasks at 5 μg/mL for 4 hours. Cells were rinsed with PBS (three times). Cells were scraped off the flask and centrifuged into a pellet. The pellet was set in resin, and micron-sized slices were cut. After drying the samples, transmission electron microscopy (TEM) was taken using a Hitachi H-7600 (Japan) microscope operated at 80 kV and a Tecnai G2 F30 (Germany) microscope operated at 300 kV.
Cytotoxicity Study
The HeLa, neuroblastoma (SH-SY5Y ATCC CRL 2266), and embryonic kidney (HEK293 ATCC CRL-1573) cell lines were cultured in a 96-well plate with a cell density of 2×104 cells per well. The day after, old medium was replaced with fresh medium containing different NP samples (S, S3, SP/C, FS3P-G/C, FS3P-A/C, and FS3P-G-E/C) at various concentrations (10, 5, 2.5, 1.25, and 0.625 μg/mL). After another incubation of 48 hours, old medium was removed, followed by washing with PBS three times, and fresh medium (100 μL) was added into each well. After another 30 minutes incubation to equilibrate to RT, 100 μL of CellTiter-Glo® Luminescent reagent (Promega) was added to each well to measure cell viability.47 After gently shaking for 10 minutes at RT, the luminescence signal was measured using a microplate reader (Perkin Elmer, Victor X5, USA). The percentage of cell viability was calculated based on luminescence absorbance changes by comparing absorbance values between test and control wells without any treatment. The experiments were repeated three times for statistical analysis.
In addition, FS3P-G/C and FS3P-A/C were further evaluated for efficacy of photothermal therapy in HeLa cells. Then 2×104 cells/well were grown on 96-well plates and incubated for 24 hours at 37°C with the samples at 5 µg/mL. After 4 hours of incubation, the cells were irradiated for 5, 10, or 15 minutes with or without 808 nm NIR laser at 1.5 W/cm2. The cell viability was measured using CellTiter-Glo® Luminescent after 48 hours incubation. To further evaluate the morphology of cells, a JuLI™ Stage Real-Time Cell History Recorder (NanoEntek, Korea) was used during the experiment.
Results and Discussion
Synthesis and Characterization of Multifunctional Mesoporous NPs
Synthetic Scheme of FS3-Based NPs
The objective of the present investigation was to synthesize core-shell Fe3O4-mesoporous silica NPs coated with PDA layer and Au NPs. According to the preparation strategy presented in Scheme 1, the initial step was to synthesize superparamagnetic NPs. The following step was to prepare the core-shell Fe3O4-mesoporous NPs grafted with APTMS-FITC fluorescent complex, hereafter referred to as FS3. The core-shell FS3 NPs were coated with biocompatible PDA with multiple amine groups. The PDA-coated FS3 (FS3P) NPs exhibited the highly positive zeta-potentials in aqueous solution (+21.1 mv) from the negative value of FS3 (−19.1 mv), and the hydrodynamic size of the NPs was approximately 100 nm by DLS instrument at the Smart Materials Research Center for IoT at Gachon University. The size of FS3P was smaller than that of the vascular leakage in tumor sites for their passage into the tumor, but larger than that of normal vascular pores. Furthermore, the combination of APTMS-FITC complex and PDA layer played as a gatekeeper, which produced a pore closure effect, resulting in sustained releases of small drug molecules through the porous channels of FS3-based NPs. After attaching hydrocaffeic acid onto the surface of FS3P through electrostatic interaction and hydrogen bonding between carboxylate groups of hydrocaffeic acid and amine groups of PDA, the catechol-enriched PDA surface acted as a highly reductive reservoir for the seed formation of Au and continuous growth into gold NPs.48 In addition, cross-linking reactions between oxidized quinones and amine groups of PDA produced additional anchoring gold NPs on the surface of FS3P through catechol-gold interactions.49,50 Notably, GO-wrapped FS3P/C (FS3P-G/C) improved the drug delivery performance through the consolidated double layer consisting of GO nanosheets and PDA layer, improving the absorption of FS3P into cells.51 More detailed descriptions about APTMS-FITC complex, polydopamine (PDA) layer, and electrostatic interactions between PDA and graphene oxide are provided in Supplementary Figure S1. Whole abbreviations of as-prepared FS3-based NPs are also listed in the bottom part of Scheme 1.
Porosity and Pore Structures – X-Ray Diffraction – Magnetic Property Analysis of FS3-Based NPs
The Brunauer-Emmett-Teller (BET) surface area and pore structures of as-prepared samples (S, S3, and FS3) were analyzed by N2 adsorption-desorption isotherm measurements. Then, S3 was the abbreviated word for MSNs grafted with APTMS-FITC complex. Figure 1A-1 shows the nitrogen adsorptions of S, S3, and FS3, displaying typical type II isotherms with two steps indicating the presence of mesoporous structures according to the International Union of Pure and Applied Chemistry (IUPAC) classification. A pore-filling adsorption isotherm was sharp in the relative pressure of 0.85–1.0 atm, confirming the narrow pore size distribution. The hysteresis loop observed around 0.9 atm was attributed to the inter-particle porosity, which was usually observed in mesoporous NPs system.16–18 Compared to that of MSNs, the adsorbed nitrogen amount of S3 significantly increased, but the shape of the hysteresis remained unchanged. The BET surface area and pore volume of MSN were 260 m2/g and 0.62 cm3/g, respectively. After grafting with APTMS-FITC complex, the BET surface area and pore volume of S3 significantly increased to 1,156 m2/g and 1.67 cm3/g, respectively (Supplementary Table S1). However, an average pore diameter decreased from 2.38 nm to 1.45 nm, and pore size reduced to nearly half from 10.2 nm to 5.6 nm (as summarized in Supplementary Table S1). These data indicate the grafting of the APTMS-FITC complex onto the pore walls of MSNs. After performing concurrent synthesis of core-shell Fe3O4-incorporated S3 (so-called FS3), the BET surface area and pore volume of FS3 were reduced to 846 m2/g and 1.30 cm3/g from the initial values of 1,156 m2/g and 1.67 cm3/g, respectively. On the contrary, average pore diameter slightly increased from 1.45 nm to 1.54 nm, as shown in Figure 1A-2. These results suggested that the incorporation of Fe3O4 NPs within the mesoporous silica matrix did not change the pore structure of the FS3 significantly. The APTMS-FITC complex were uniformly distributed on the interior pore walls and the exterior silica surface (as depicted in Scheme 1), which were beneficial for loading large amounts of drug molecules. After PDA was coated over the FS3 NPs, the BET surface area of PDA-coated FS3 (FS3P) significantly reduced from 846 to 16 m2/g. On the contrary, pore size increased dramatically from 6.2 nm to 23.1 nm, indicative of large void space generated by PDA coating process. These data finally indicated the full capsulation of mesoporous FS3 by PDA layer.
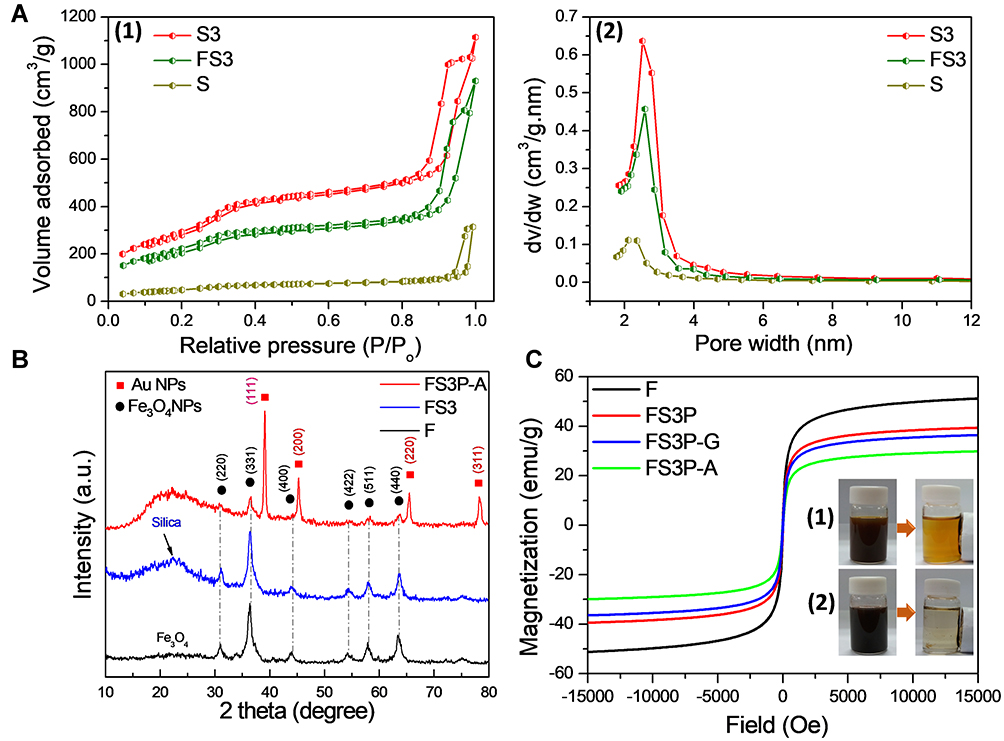 |
Figure 1 (A) Nitrogen adsorption/desorption isotherms (1) and pore size distribution (2) by the Barrett–Joyner–Halenda (BJH) analysis of S, S3, and FS3. (B) X-Ray Diffraction (XRD) intensity measurements of F, FS3, and FS3P-A. (C) Magnetic hysteresis loops of F, FS3P, FS3P-G, and FS3P-A; photographs of (1) Fe3O4 NPs and (2) FS3P-A NPs before and after separation with an external magnetic field. |
Powder X-ray powder diffraction (XRD) analysis confirmed the composition of the as-synthesized samples, as shown in Figure 1B. The powder diffraction pattern of the MSNs indicated a broad peak at 2θ=22.5º that revealed the amorphous nature of the silica matrix (ie, the absence of ordered crystalline structure).51 The XRD pattern of Fe3O4 NPs was also compared with those of FS3 and FS3P-A NPs. The diffraction peaks of FS3 and FS3P-A matched well with those of Fe3O4 NPs, confirming the incorporation of iron oxide in all the samples. The diffraction signatures at two-theta values of 30.9°, 36.4°, 43.9°, 54.2°, 57.9°, and 63.5° are corresponding to (220), (311), (400), (422), (511), and (440) planes for Fe3O4, respectively. After FS3P was anchored by gold NPs, FS3P-A exhibited an additional four peaks at 2θ values of 39.0°, 45.3°, 65.4°, and 78.4°, corresponding to (111), (200), (220), and (311) lattice planes of the face-centered cubic gold (space group FM3M, JCPDS File No. 89–3697), respectively.
Hysteresis loops of the magnetic NPs are shown in Figure 1C. All the samples incorporated with Fe3O4 NPs revealed a superparamagnetic behavior that was desirable for biomedical applications.51 Pure Fe3O4 exhibited a saturation magnetization at 51.2 emu/g. This value was reduced to 39.4 emu/g when Fe3O4. NPs was encapsulated with mesoporous silica shell, and grafting with APTMS-FITC complex and coating with polydopamine layer (FS3P). After wrapping by GO nanosheets, the magnetization of FS3P-G was slightly decreased to 36.4 emu/g. The magnetization was further decreased to 29.8 emu/g when gold NPs covered the surface of the sample (FS3P-A). The decrease in the magnetization values was due to the increase of nonmagnetic materials. Nonetheless, magnetic NPs have several advantages in drug delivery systems: i) facile separation by a simple magnetic manipulation, ii) magnetic-targeting capability, and iii) high NIR absorption and conversion into thermal energy.18,52
FTIR Analysis – SEM and TEM Images
FTIR spectrum was measured to characterize the surface modifications at each synthetic step. As shown in Figure 2A-1, the characteristic peaks corresponding to the antisymmetric and symmetric stretching vibrations of the Si-O-Si bond in oxygen-silica tetrahedron were clearly observed at 1,078 and 791 cm−1, respectively. The weak bands at 3,656 and 933 cm−1 were assigned to stretching vibrations of silanol groups and absorbed water, respectively. After grafting with APTMS-FITC complex, the FS3 showed new bands at 713 and 1,441 cm−1, corresponding to C-S and CO vibrations, respectively. The peaks at 3,145 and 3,662 cm−1 were assigned as NH stretch and NH symmetric from NH-CS-NH groups originated from APTMS-FITC complex, respectively, indicating the successful grafting APTMS-FITC complex onto the pore walls (Figure 2A-2). After the PDA layer formed over the FS3 NPs, a broad band appeared at ~3,660 cm−1 attributed to the stretching vibrations of OH and NH bonds in PDA. Additional peak also appeared at ~3,014 cm−1, which was assigned as the C-H asymmetric vibration of aliphatic CH2 in PDA. The peaks at 1,502 and 1,679 cm−1 also indicated the presence of C-N and C=C groups of the benzene ring (Figure 2A-3). After wrapping with GO nanosheets, the absorption band at 3,830 cm−1 was assigned to the O-H stretching vibrations. The absorption peak at 2,796 and 3,025 cm−1 could be assigned as the C=O stretching of carboxylic and/or carbonyl moiety functional groups (Figure 2A-4). All the FTIR spectra indicated the successful surface modifications of multifunctional FS3 during the consecutive fabrication steps, as described in Scheme 1.
 |
Figure 2 (A) FT-IR spectra of (1) S, (2) FS3, (3) FS3P, and (4) FS3P-G. (B) SEM image and inset TEM image of Fe3O4 (F) (1), SEM image of FS3 (2), SEM image of FS3P (3), SEM image and inset TEM image of FS3P-A (4); TEM images of S (5), and TEM images of FS3P (6). |
According to the scanning electron microscope (SEM) and transmission electron microscopy (TEM) (inset) images in Figure 2B-1, the Fe3O4 NPs exhibited a spherical shape with a diameter range of 10–20 nm. After the concurrent sol-gel reaction and grafting of APTMS-FITC complex, the Fe3O4-incorporated S3 (FS3) NPs exhibited uniform orbicular shape with an average diameter of ∼90 nm (Figure 2B-2). When the FS3 was coated with a thin PDA layer, the size of FS3P slightly increased by ~10 nm (Figure 2B-3). As shown in Figure 2B-4, small gold NPs anchored on the surface of FS3P NPs. Supplementary Figure S2A reveals the UV-vis absorption spectra of FS3P-A which was prepared by 1.5 mM of 1.0 wt% HAuCl4.53,54 The distinctly broader peak was observed at ~700 nm, due to the clustering effect of gold NPs. A SEM image of FS3P-G in Supplementary Figure S2B reveals the sheet-like wrapping of GO over the FS3P NPs. TEM images clearly depictthe two-dimensional pore arrangement of MSNs with perpendicular mesopores (Figure 2B-5). The TEM image of FS3P shows the yolk-shell structures, in which S3 shell uniformly coated onto the magnetic Fe3O4 cores. In addition, Figure 2B-6 displays the distinct coverage of PDA layer (~3 nm) on the periphery of NPs. The chemical compositions were examined using scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS). According to three major components (Au, Si, and Fe) shown in EDS spectra of Supplementary Figure S2C, core-shell type magnetic silica NPs were anchored with Au NPs, as previously shown in Figure 2B-4.
In vitro Drug Release Under Stimuli (pH, NIR) Operation
Photothermal Heating Effect by NIR Irradiation
To study the photothermal heating effect on the FS3-drug system, the temperature of FS3-drug solution was monitored under the irradiation of NIR light at 808 nm (1.5 W/cm2) (Figure 3). As shown in Figure 3A, the temperature of MSNs solution increased slightly from 24.1°C to 29.2°C upon the NIR irradiation for 10 minutes. However, the temperature of Fe3O4 solution significantly increased from 24.6°C to 45.9°C under the same irradiation condition. After PDA/GO double layer coating, FS3P-G/C exhibited a significant increase in the solution temperature by ~25°C (from 24.5°C to 49.0°C). In contrast, FS3P-G/C without Fe3O4 exhibited a lower increase in temperature by ~15°C (from 24.7°C to 39.7°C). Remarkably, gold-coated FS3 (FS3P-A/C) exhibited the maximal increase in the solution temperature, up to 59.1°C, probably due the excellent heating effect of plasmonic gold NPs.
 |
Figure 3 (A) Temperature-variation curves of S, S3P-G/C, FS3P-G/C, S3P-A/C, and FS3P-A/C, solutions during the exposure to an 808 nm laser at a power density of 1.5 W/cm2 for 10 minutes; (B) cisplatin release profiles from SP/C, FS3P-G/C, and FS3P-A/C in PBS at pH 7.4 and pH 5.5; (C) cumulative cisplatin release from FS3P-G/C and FS3P-A/C in PBS at pH 5.5 without and with NIR irradiation (808 nm laser, 1.5W/cm2) for 10 minutes; the dotted lines shown in (C) indicate the temperature change of in-vitro solution induced by the irradiation of NIR light for 10 minutes; (D) mechanism of stimuli pH/NIR responsive controlled release from FS3P-G/C (1) and FS3P-A/C (2). |
Magnetic Fe3O4 can enhance the photothermal heating efficiency of the FS3-drug system upon NIR irradiation through polarized radiation in Fe3O4.55–57 As shown in Figure 3A, S3P-A/C NPs displayed high photothermal efficiency and quickly reached 54.3°C, but the temperature rise was lower in comparison to that of FS3P-A/C (including Fe3O4). This result clearly indicates that a photothermal heating effect was additionally attributed to incorporated Fe3O4 upon NIR irradiation. In the case of FS3P-G/C, the PDA/GO double layers could have a synergy effect on the enhancement of photothermal conversion efficiency. PDA can obviously absorb the NIR light with a high photothermal efficiency and the photothermal heating effect was directly contributed by the degree of π conjugations in GO. Even though GO was highly oxidized with disrupted π conjugations, the chemical reduction of GO by PDA layer can restore the disrupted π conjugations, consequently leading to the increase of NIR absorption.22,58 Noteworthy, FS3P-A/C showed the highest photothermal effect due to the presence of gold NPs with plasmon resonance tenability and high photothermal conversion efficiency.59,60
To manifest the influence of NPs concentration and NIR power density on photothermal heating effect, six kinds of NPs were tested with two different operating conditions: i) standard power density of NIR and low concentration of NPs (1.5 W/cm2 and 2.5 mg/mL), ii) high power density of NIR and standard concentration of NPs (2.0 W/cm2 and 5 mg/mL). Then, standard power density and concentration were considered as the values of 1.5 W/cm2 and 5 mg/mL, respectively. In the case of the standard power density of NIR (1.5 W/cm2) and low concentration of NPs (2.5 mg/mL) shown in Supplementary Figure S3A, most NPs exhibited the lower photothermal efficiency in comparison to the standard case shown in Figure 3A. However, both FS3P-A/C and S3P-A/C NPs still exhibited a distinct photothermal effect due to the presence of plasmonic gold NPs, ie, the solution temperatures of both NPs increased to 56.1°C and 51.3°C, respectively, after 10 minutes irradiation of NIR light. In the case of the high power density of NIR (2.0 W/cm2) and the standard concentration of NPs (5 mg/mL) shown in Figure S3B, all the NPs exhibited the stronger photothermal effects in comparison to the standard case shown in Figure 3A, ie, all the solution temperatures increased over 50°C except for the MSN solution. As expected, Fe3O4 NPs induced the increase of solution temperature up to 63.4°C. Notably, FS3-based NPs with both gold and Fe3O4 (ie, FS3P-A) exhibited the highest solution temperature of 83.3°C after NIR irradiation (2.0 W/cm2).
pH-Controlled Release Effect
The release profiles of cisplatin from SP/C, FS3P-G/C, and FS3P-A/C were measured in PBS at pH 5.5 and pH 7.4, respectively. As seen from Figure 3B, the released fraction of cisplatin from SP/C was slightly higher at pH 7.4 than at pH 5.5, but the difference was not distinguishable. At 5 hours, ~21.2% of cisplatin released from the SP/C. At 25 hours, the released cisplatin fraction increased to ~51% and asymptotically approached ~73% at 90 hours. On the other hand, FS3P-G/C exhibited a much higher release fraction at pH 5.5 than that at pH 7.4, whereas FS3P-A/C did not show a clear difference in the release fractions between pH 7.4 and pH 5.5.
For the in-vitro release test of FS3P-A/C at 5 hours, the release fraction was 11.6% at pH 7.4, but the release fraction slightly increased to 13.5% at pH 5.5. The small disparity of release fraction between pH 5.5 and pH 7.4 was maintained up to 50 hours. Then, the release fraction (at pH 5.5) approached the saturation value of 34.0% at 80 hours, whereas the release fraction (at pH 7.4) more quickly reached an asymptotic value of 26.6% at 60 hours. For the in-vitro release test of FS3P-G/C at 10 hours, the release fraction increased to 24.0% at pH 5.5 when compared with the release fraction of 14.2% at pH 7.4. The divergence in the release fraction increased with release times. At 30 hours, the release fraction was 30.3% at pH 5.5 and 24.4% at pH 7.4, and finally reached a saturation value of 39% (pH 5.5) and 28.1% (at pH 7.4) after 80 hours.
Regarding the physicochemical structure of FS3P-G/C, amino groups in PDA tend to be deprotonated at neutral pH 7.4, but protonated at acidic pH 5.5. Thus, the electrostatic (or hydrogen-bonding) interactions between functional groups of PDA and APTMS-FITC complex would be weakened (or broken) under acidic pH condition, consequently leading to a faster release of cisplatin at pH 5.5 than at pH 7.4 (Figure 3D).22,61 In addition, acidic pH condition could induce more weakened interaction between GO nanosheets and PDA layer, resulting in more release of cisplatin at pH 5.5. Thus, the release behavior of FS3-drug system with PDA/GO double layers was strongly dependent on the pH of surrounding solution.62
Figure S4A showed the release profiles of FS3P/C and FS3P-A/C in PBS at pH 7.4 and pH 5.5, respectively. In general, the release rate of FS3P/C was faster than that of FS3P-A/C, and the release fraction at pH 5.5 was higher at pH 7.4. At pH 7.4, only 12.9% of cisplatin released from FS3P-A/C at 10 hours, but the release fraction of cisplatin from FS3P/C was 17.2%. At 30 hours, the release fraction reached 21.6% (FS3P-A/C) and 24.2% (FS3P/C), respectively, followed by a gradual increase to an asymptotic value of 26.8% (with Au NPs) and 30.7% (without Au NPs) at 70 hours. At pH 5.5, however, the release fractions were 15.2% (FS3P-A/C) and 21.2% (FS3P/C) at 10 hours. The release fractions further increased to 23.5% (with gold) and 28.3% (without gold) at 30 hours, finally approaching an asymptotic value of 32.8% (with gold) and 37.4% (without gold) at 70 hours.
In the case of FS3P-A/C, gold NPs were deposited on the outer surface of PDA layer. Thus, gold NPs played as a blocking layer which partially hindered the release of cisplatin from the FS3-drug system. As a result, the release rate of FS3P-A/C was lower than that of FS3P/C at both pH 7.4 and pH 5.5.
NIR Irradiation Controlled Release Effect
In vitro release tests were carried out with or without irradiation of NIR light, as shown in Figure 3C. The release fractions of cisplatin from FS3P-G/C and FS3P-A/C were significantly increased by periodic irradiation of NIR light (808 nm, 1.5 W/cm2) for 10 minutes at 10, 20, and 30 hours, respectively. The dotted lines shown in Figure 3C indicate the temperature variation of the solution by periodic NIR irradiation for 10 minutes.
In the case of FS3P-A/C at pH 5.5, the released cisplatin fraction slowly increased to 15.1% for 10 hours. At the first NIR irradiation at 10 hours, the cisplatin release fraction rapidly increased to 45.9% by three-fold. When a NIR laser system turned off, the solution temperature decreased to 31.2°C within 3 hours. After the first NIR irradiation, the release fraction of cisplatin slowly increased to 51.5% for 20 hours, but the release fraction was much higher than that of normal case without NIR irradiation (19.7%). At the second NIR irradiation at 20 hours, the release fraction again jumped to 69.0% within 10 minutes. Afterwards, NIR-induced release fraction gradually increased from 71.5% to 75.5% by the third NIR irradiation for 10 minutes. The release fraction of cisplatin gradually approached to an asymptotic value of 77.8% at 50 hours. On the other hand, the released cisplatin fraction without NIR irradiation slowly reached to 15.2% at 10 hours, 23.5% at 30 hours, and finally reached an asymptotical value of 34.0% at 80 hours.
Under the irradiation of NIR light at pH 5.5, GO-coated FS3 (FS3P-G/C) exhibited a slower release rate than that of gold-coated FS3 (FS3P-A/C). The disparity of release rate increased with periodic numbers of NIR irradiation. At the first irradiation of NIR: 40.6% (GO)<45.9% (Au NPs); at the second irradiation of NIR: 54.4% (GO)<69.0% (Gold); at the third irradiation of NIR: 61.2% (GO)<75.5% (Au NPs). The release fraction of cisplatin from FS3P-A/C at pH 5.5 without NIR irradiation was less than ~30% at 80 hours of release times.
In summary, GO-wrapped FS3 (FS3P-G/C) exhibited high sensitivity of release rate at acidic pH 5.5 (Figure 3D-1). On the other hand, Au NPs-coated FS3 (FS3P-A/C) exhibited more stable and sustained release at both pH 5.5 and pH 7.4, but exhibited extremely sensitive release behavior under NIR irradiation, as shown in Table 1. Thus, core-shell FS3-drug systems (eg, FS3P-G/C and FS3P-A/C) can be potentially applied for targeted tumor therapy as chemo-photothermal agents with highly sensitive stimuli (pH and NIR)-responsiveness (Figure 3D-2).
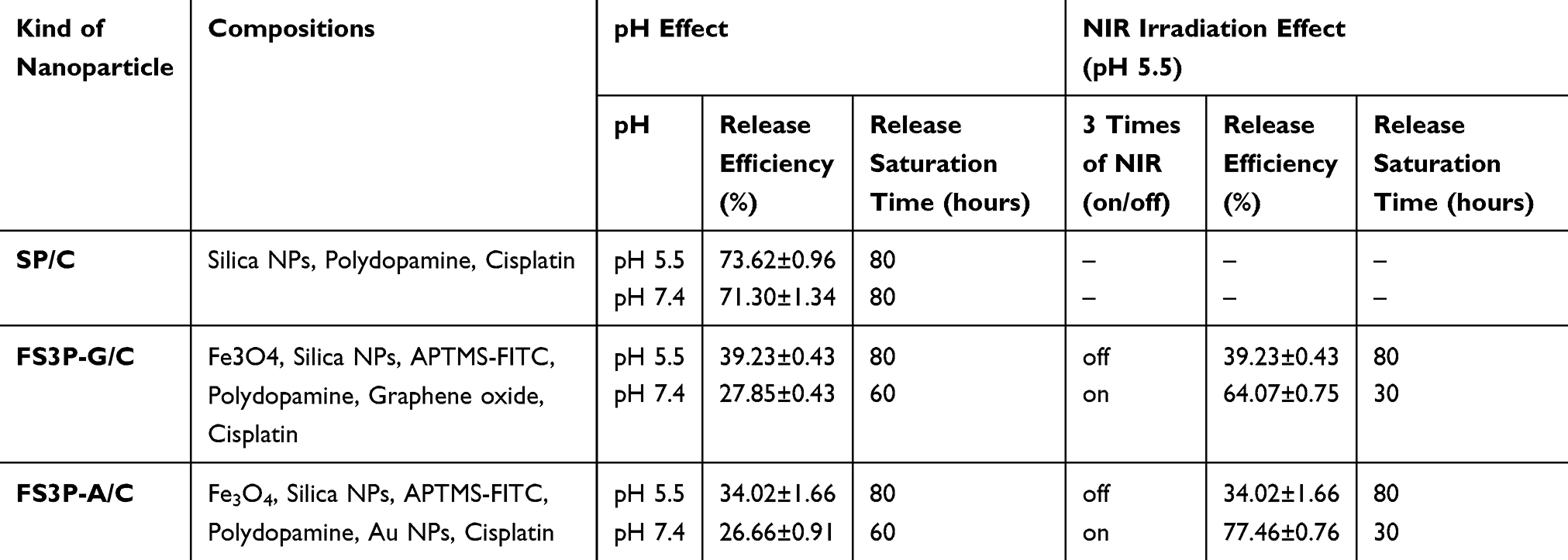 |
Table 1 Summary of Drug Release Data for Each Type of Particles Under Different Conditions of Time vs pH and NIR Irradiation |
Kinetic Model Applications to Cisplatin Release
The release kinetics was analyzed by an empirical Hill equation: 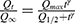
where Qmax is the maximum amount of drug release, Q1/2 is the time required for 50% drug release, and γ is a sigmoidicity factor. The Hill equation can suitably describe release profiles with sigmoid behavior.63
The cisplatin release profiles can also be fitted by a power-law equation, based on the Korsmeyer–Peppas (K-P) model, which is usually applied to predict the diffusion mechanism in anomalous drug release kinetics: 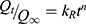
where Qt/Q∞ is the normalized fraction of the drug release, kR is the relaxation rate constant, and n is the release exponent. In the K-P model, the value of n characterizes the release mechanism. In the case of a spherical matrix, n=0.43 for Fickian diffusion, and 0.43<n<0.85 for non-Fickian transport.22
As shown in Supplementary Figure S4B, the cisplatin release from SP/C follows sigmoid behavior fitted by Hill Equation (1), in which a sigmoidicity factor was 2.1 (Supplementary Table S2). At the first stage of release fraction, it shows a slow release behavior below 5% of cisplatin release fraction. This slow stage is usually caused by mechanical lag processing including disaggregation of mesoporous NPs and/or disintegration of PDA coating. After then, SP/C without APTMS-FITC conjugates showed a rapid release of cisplatin, kind of a burst effect, up to a saturation value later, whereas FS3P/C showed a retarded release and finally reached saturation after a long-period of time (around 7 days). Thus, the release kinetics of FS3P/C were analyzed by the K-P model with a release exponent of n=0.64 (Table S2).
In the beginning, it may start off at a rapid rate and slow down (burst effect), start off immediately, or it may take time to build up (time lag).
According to Supplementary Figure S4B, the cisplatin release kinetics of FS3P-G/C and FS3P-A/C were fitted by the K-P model (2), in which a release fraction less than 60% was used. The fitted release exponents of FS3P-G/C and FS3P-A/C were n=0.62 and n=0.71, respectively, that were higher than 0.43, indicating that the release kinetics shows non-Fickian diffusion behavior. In summary, the release kinetics of FS3-based NPs can be controlled by APTMS-FITC internal conjugates with PDA layer, and further retarded by the additional diffusion barriers of external GO and/or Au NPs layer.
Fluorescent Images, In vitro Anticancer Efficacy, and Protein Corona Analysis
Cellular Uptake and Intracellular Distribution
Cellular uptake of SP/C, FS3P-G/C, FS3P-A/C, and FS3P-G-E/C was incubated with HeLa cells for 4 hours, and confocal fluorescence images (CLSM) were taken. All treated groups’ emitted green fluorescence in HeLa cells, indicating the direct cellular uptake of FS3-based nanocarriers, as shown in Figure 4, presenting good effectiveness of the NPs. Green (FITC) fluorescence either from NPs in the cell cytoplasm and blue fluorescence from DAPI in the cell nuclei were simultaneously observed. The merged image of blue and green fluorescences revealed that the majority of the NPs including SP/C (Figure 4A), FS3P-G/C (Figure 4B), FS3P-A/C (Figure 4C), and FS3P-G-E/C (Figure 4D) were significantly accumulated in HeLa cells, whereas no fluorescence was observed in HEK293 cells treatment with SP/C (Supplementary Figure S5A), FS3P-G/C (Supplementary Figure S5B), FS3P-A/C (Supplementary Figure S5C), and FS3P-G-E/C (Supplementary Figure S5D) under the same conditions. Remarkably, when compared to that of ab-unconjugated counterpart (Figure 4B), the internalization of EGF-conjugated FS3P-G/C (Figure 4D) was more retained in the treated cells, indicative of the effectiveness of ab on cancer cell targeting and treatment.
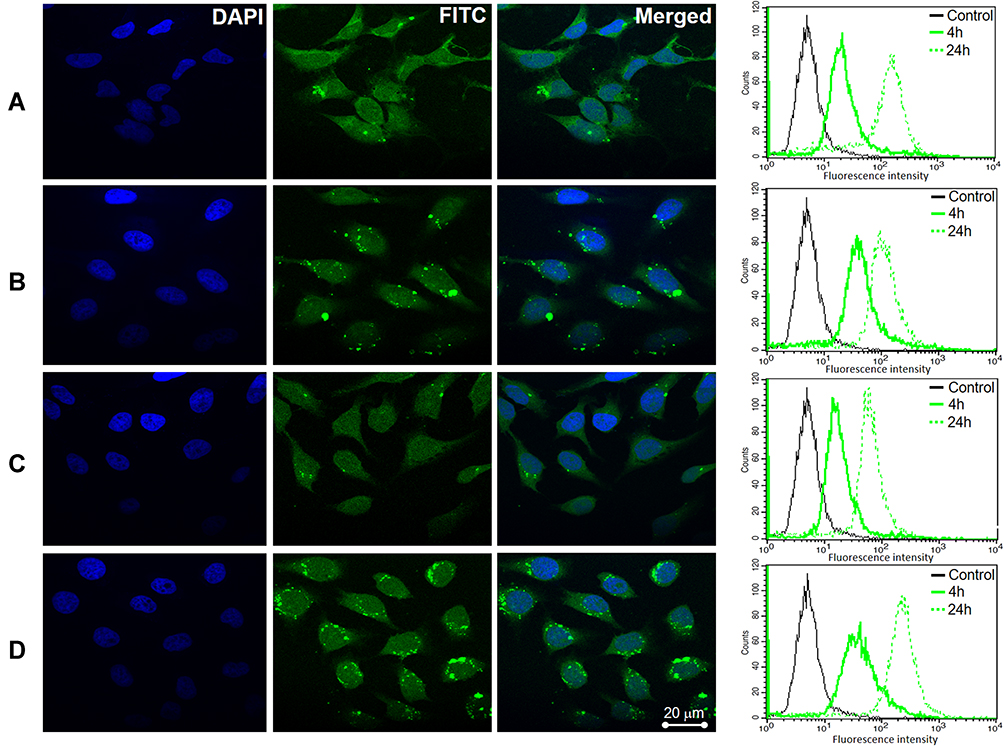 |
Figure 4 Confocal microscopy imaging and flow cytometry analysis of HeLa cells after incubation with as-prepared samples: (A) SP/C, (B) FS3P-G/C, (C) FS3P-A/C, and (D) FS3P-G-E/C. |
The cellular uptake of SP/C, FS3P-G/C, FS3P-A/C, and FS3P-G-E/C NP was also confirmed by flow cytometry analysis. The fluorescence intensity of single cell emission detected by flow cytometry that would be a good indication of the amount of NPs internalized by each cell. As shown in Figure 4 (right panel), the peak of the fluorescence intensity was dramatically increased between 4 hours and 24 hours incubation with the NPs, suggesting the promoted NP internalization by HeLa cells. Using flow cytometry and confocal microscopy, the results totally reveal that fluorescently labeled FITC NPs were significantly internalized in cancer cells, indicating the impressive efficiency of drug delivery.
Furthermore, to confirm the targeted delivery of NPs into the cells, as-prepared samples (such as SP/C, FS3P-G/C, FS3P-A/C, and FS3P-G-E/C were analyzed by TEM. According to the TEM images of cells treated with NPs, all three NPs were localized mainly in the cytoplasmic matrix covering the nucleus (orange arrows), while the same behavior was not observed in the control cells (Figure 5), indicating that three NPs were well trapped in cancerous cells but not inside the nucleus. The increased cellular uptake was usually accorded with surface properties of NPs. Compared to untreated cells (Figure 5A), some small aggregates were observed in the cells incubated with SP/C (Figure 5B), while a few large aggregates were localized nearby the nucleus of the cells incubated with GO-coated FS3 (Figure 5C), FS3P-A/C (Figure 5D), and EGFR ab-conjugated FS3 (Figure 5E). Correlating these TEM images with confocal images and flow cytometry analysis (Figure 4), the intracellular distribution of NPs seemed to be locally disrupted and discontinuous because of large aggregates of NPs that were capable of crossing cellular membranes. Supplementary Figure S6 supports these observations by providing detailed data of TEM images of the cells treated with the SP/C (Supplementary Figure S6A), FS3P-G/C (Supplementary Figure S6B), and FS3P-G-E/C (Supplementary Figure S6C).
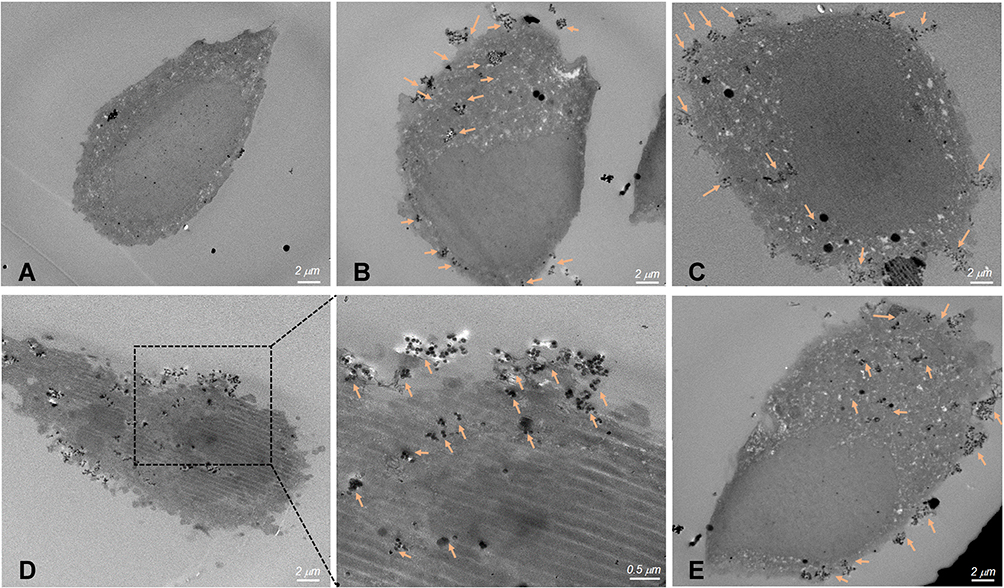 |
Figure 5 Thin-section TEM images of cell incubated with magnetic mesoporous silica nanoparticles. (A) Control cells without nanoparticles, (B) SP/C, (C) FS3P-G/C, (D) FS3P-A/C, and (E) FS3P-G-E/C. Arrows denote metal oxide particles or particulate matter. |
In vitro Anticancer Efficacy of the Various NPs
To evaluate the anticancer effects of those NPs, the CellTiter-Glo® Luminescent reagent was used to measure adenosine triphosphate (ATP) as an indication of cell viability47 of HeLa, SH-SY5Y, and kidney HEK293 cell lines after the treatment. As seen from Figure 6A, MSN and MAF did not affect the cell viability irrespective of the different surface modifications. Furthermore, these nanocarriers at high dosage of 10 µg/mL did not affect the cell viability, ie, HeLa, SH-SY5Y, and HEK293 were still viable up to more than 85%. This suggested that FS3-based nanocarriers revealed low cytotoxicity toward both cancerous cells and normal cells. The viability test showed that FS3 was a safe nanocarrier, hardly causing any cytotoxicity on its own. SP/C, FS3P-G/C, and FS3P-G-E/C displayed the discrepant anti-proliferation efficiency mediated by the drug release from the FS3 samples, demonstrating substantially enhanced cytotoxicity to both cancer cells, HeLa and SH-SY5Y, in a concentration-dependent manner. When the concentrations of SP/C, FS3P-G/C, and FS3P-G-E/C increased from 2.5 to 10 µg/mL, the viability of both HeLa and SH-SY5Y cancer cell lines decreased sharply from ~65% to ~10%, from ~60% to ~8%, and from ~40% to less than 7%, respectively. The surface groups of the SP/C samples induced cytotoxicity against both HeLa and SH-SY5Y cancer cell lines through the final intracellular delivery and biological function of the material, suggesting its potential clinical utility as an anticancer agent. Besides, GO can induce the generation of reactive oxygen species (ROS) in cells and the exposure of cancer cells to GO can lead to cancer cell killing by enhancing the cytotoxicity effect exerted through the induction of DNA damage.64
 |
Figure 6 (A) Cytotoxicity against HeLa, SH-SY5Y, and HEK293 cells lines induced by different nanoparticles S, S3, SP/C, FS3P-G/C, FS3P-A/C, and FS3P-G-E/C at their concentrations ranging from 0.625 to 10 µg/mL. (B) Cell viability of HeLa cells (the up panel) and their morphological observation (the down panel) incubated with or without FS3P-A/C (concentration 5 μg/mL) with or without 808 nm NIR laser irradiation at 1.5 W/cm2 for 5, 10, and 15 minutes. |
In addition, in the case of FS3P-A/C, the HeLa and SH-SY5Y cells were still viable up to more than 65% at concentration ranges from 0.625 to 5 µg/mL, and over 50% cells died at the highest concentration of 10 µg/mL. When compared to FS3P-G/C, gold-deposited FS3 showed a lower inhibition effect on the proliferation of HeLa and SH-SY5Y cancer cells at 5 µg/mL, probably due to the strong aggregation behavior of gold NPs.
Gold NPs were promising candidates for a number of consumer and food products due to their inertness at low doses. In general, the cytotoxicity of gold NPs depended on physicochemical properties such as particle size and surface chemistry. Many toxicity studies demonstrated that gold NPs were mostly inert after acute exposures (short-term treatment), as long as the NPs were above 4–5 nm in diameter.65–67 This acute toxicity could be ascribed to the use of very high concentrations and different cell type sensitivities.67–70 On the other hand, high-doses of FS3P-A/C may be more effective in cell death as an acute burst of exposure. On this basis, the concentrations of FS3P-A/C (10 μM), at which both cancers revealed 22% viability, were selected for further treatment-induced therapy. More importantly, the lower concentrations of FS3P-A/C (5 μg/mL) killed the vast majority of cells under 808 nm NIR laser irradiation at 1.5 W/cm2 for 5 minutes (Figure 6B, third upper panel). This figure was dramatically increased after irradiation for more than 10 minutes compared to the corresponding control experimental samples without laser irradiation, suggesting its photothermal ablation may be effective in the cancer cells. The cell morphology was further observed by light microscope. In the control group, there was no noticeable change with the exception of 15 minutes in the NIR-irradiated group (Figure 6B, lower panel) was observed. While approximately 55% dead cells were observed after treatment of 5 µg/mL of FS3P-A/C without 808 nm NIR laser irradiation, this figure was rapidly increased to over 70% under NIR laser irradiation at 1.5 W/cm2 for 5, 10, or 15 minutes (Figure 6B in the upper panel), indicating the strong effect of photothermal ablation on cancer cell cytotoxicity. Taken together, these results strongly suggest that the phototoxicity was mainly due to irradiation of extracellular FS3P-A/C that damaged cell membranes, indicating that the cytotoxic death effect on cancer cells was clearly promoted by photothermal ablation by FS3P-A/C under NIR irradiation. On the other hand, the potential ability of FS3P-G/C for laser-induced heating was also tested through the same model. As shown in Supplementary Figure S7, cell viability was gradually decreased (approximately 50%) when the irradiation time was increased up to 15 minutes only. However, the cell viability was also decreased by NIR light exposure followed by 15 minutes irradiation without treatments (Figure 6B and Supplementary Figure S7). Conclusively, with the excellent laser-induced heating properties, FS3P-A/C could be a novel promising agent for photothermal therapy.71
Figure 6A also reveals the viability of HEK293 cells treated with NPs at different dosages. Interestingly, almost all cells were still alive at the whole range of NP concentrations, indicating that drug-loaded FS3 showed no obvious cytotoxicity toward normal cells. The cell viability was more than 80% even at 10 µg/mL of NP concentration. These results suggested that the FS3-based nanocarriers were non-toxic materials at low concentrations and slightly toxic at high concentrations (10 µg/mL).72,73 Thus, FS3s as a drug platform to kill cancer cells were effective at concentrations lower than 10 µg/mL, implying their biocompatibility with normal cells and supporting new developments in the biotechnological, pharmaceutical, and biomedical industries.74,75
Conclusions
In this work, core-shell type mesoporous silica nanocarriers were developed for fluorescent imaging, stimuli-responsive drug release, magnetic separation, antibody targeting, and chemo-photothermal therapeutics. Firstly, magnetic mesoporous silica nanoparticles were prepared by coating a Fe3O4 core with a mesoporous silica shell, followed by grafting with fluorescent conjugates, so-called FS3. The resulting FS3 was preloaded with therapeutic cisplatin and coated with polydopamine (PDA) layer, so-called FS3P/C. Eventually, graphene oxide-wrapped FS3P/C (FS3P-G/C) exhibited high sensitivity in the dual stimuli (pH, NIR)-responsive controlled release behavior, ie, showing the increased release rate when the solution pH was changed from pH 7.4 to pH 5.5 and enhancement of release rate under NIR irradiation (808 nm, 1.5 W/cm2) for 10 minutes. On the other hand, gold-coated FS3P/C (FS3P-A/C) exhibited more stable release behavior, irrespective of pH changes, and exhibited a much more enhanced release rate under the same NIR irradiation, because of its high photothermal heating effect. Notably, FS3P-A/C showed strong NIR absorption, enabling photothermal destruction of HeLa cells by its chemo-photothermal therapeutic effects under NIR irradiation (808 nm, 1.5 W/cm2). The selective uptake of FS3-based nanocarriers was confirmed in various cancer cell lines by the images obtained from CLSM, flow cytometry, and TEM instruments. Cisplatin-free FS3-based nanocarriers revealed good cellular uptake and low cytotoxicity against cancerous HeLa and SH-SY5Y cells, but showed no obvious toxicity to normal HEK293 cells. Along with the facile synthesis of FS3-based nanocarriers, the integration of all these strategies into one single unit will be a prospective candidate for biomedical applications, especially in chemo-photothermal therapeutics, targeted delivery, and stimuli-responsive controlled drug release against multiple cancer cell types.
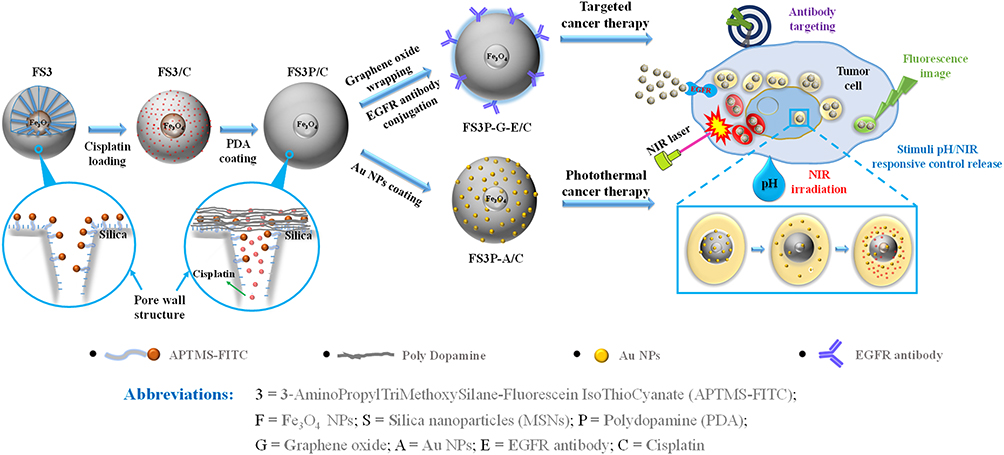 |
Scheme 1 Schematic illustration of core-shell FS3, double layer coating by PDA and Au coating (FS3P-A/C), Graphene oxide wrapping and EGFR antibody conjugating (FS3P-G-E/C); mechanism of stimuli pH/NIR responsive controlled release; and biomedical application through Au/Fe3O4/PDA photothermal therapy, the magnetically guided and EGFR antibody target. |
Funding
This study was supported financially by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry, and Energy (MOTIE) of the Republic of Korea [grant no. 20194030202440] and the National Research Foundation of Korea (NRF-2020R1A2B5B01002463).
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Rodzinski A, Guduru R, Liang P, et al. Targeted and controlled anticancer drug delivery and release with magnetoelectric nanoparticles. Sci Rep. 2016;6(1):20867. doi:10.1038/srep20867
2. Mo Y, Wang H, Liu J, et al. Controlled release and targeted delivery to cancer cells of doxorubicin from polysaccharide-functionalised single-walled carbon nanotubes. J Mater Chem B. 2015;3(9):1846–1855. doi:10.1039/C4TB02123A
3. Zhang Q, Liu J, Yuan K, Zhang Z, Zhang X, Fang X. A multi-controlled drug delivery system based on magnetic mesoporous Fe3O4 nanopaticles and a phase change material for cancer thermo-chemotherapy. Nanotechnology. 2017;28(40):405101. doi:10.1088/1361-6528/aa883f
4. Ho CC, Ding SJ. The pH-controlled nanoparticles size of polydopamine for anti-cancer drug delivery. J Mater Sci Mater Med. 2013;24(10):2381–2390. doi:10.1007/s10856-013-4994-2
5. Weaver CL, LaRosa JM, Luo X, Cui XT. Electrically controlled drug delivery from graphene oxide nanocomposite films. ACS Nano. 2014;8(2):1834–1843. doi:10.1021/nn406223e
6. He D, He X, Wang K, Zou Z, Yang X, Li X. Remote-controlled drug release from graphene oxide-capped mesoporous silica to cancer cells by photoinduced pH-jump activation. Langmuir. 2014;30(24):7182–7189. doi:10.1021/la501075c
7. Li J, Mooney DJ. Designing hydrogels for controlled drug delivery. Nat Rev Mater. 2016;1(12):16071.
8. Pimenta AFR, Ascenso J, Fernandes JCS, Colaco R, Serro AP, Saramago B. Controlled drug release from hydrogels for contact lenses: drug partitioning and diffusion. Int J Pharm. 2016;515(1–2):467–475. doi:10.1016/j.ijpharm.2016.10.047
9. Chen FM, Liu X. Advancing biomaterials of human origin for tissue engineering. Prog Polym Sci. 2016;53:86–168. doi:10.1016/j.progpolymsci.2015.02.004
10. Han R, Yi H, Shi J, et al. pH-responsive drug release and NIR-triggered singlet oxygen generation based on a multifunctional core–shell–shell structure. Phys Chem Chem Phys. 2016;18(36):25497–25503. doi:10.1039/C6CP05308D
11. Zeng X, Liu G, Tao W, et al. A drug-self-gated mesoporous antitumor nanoplatform based on pH-sensitive dynamic covalent bond. Adv Funct Mater. 2017;27(11):1605985. doi:10.1002/adfm.201605985
12. Baeza A, Manzano M, Colilla M, Vallet-Regí M. Recent advances in mesoporous silica nanoparticles for antitumor therapy: our contribution. Biomater Sci. 2016;4(5):803–813. doi:10.1039/C6BM00039H
13. Cheng W, Nie J, Gao N, et al. A multifunctional nanoplatform against multidrug resistant cancer: merging the best of targeted chemo/gene/photothermal therapy. Adv Funct Mater. 2017;27(45):1704135. doi:10.1002/adfm.201704135
14. Zhou Y, Quan G, Wu Q, et al. Mesoporous silica nanoparticles for drug and gene delivery. Acta Pharm Sin B. 2018;8(2):165–177. doi:10.1016/j.apsb.2018.01.007
15. Wang Y, Gu H. Core–shell-type magnetic mesoporous silica nanocomposites for bioimaging and therapeutic agent delivery. Adv Mater. 2015;27(3):576–585. doi:10.1002/adma.201401124
16. Gao Q, Xie W, Wang Y, et al. A theranostic nanocomposite system based on radial mesoporous silica hybridized with Fe3O4 nanoparticles for targeted magnetic field responsive chemotherapy of breast cancer. RSC Adv. 2018;8(8):4321–4328. doi:10.1039/C7RA12446E
17. Yang C, Guo W, Cui L, et al. pH-responsive magnetic core–shell nanocomposites for drug delivery. Langmuir. 2014;30(32):9819–9827. doi:10.1021/la501833u
18. Yu X, Zhu Y. Preparation of magnetic mesoporous silica nanoparticles as a multifunctional platform for potential drug delivery and hyperthermia. Sci Technol Adv Mater. 2016;17(1):229–238. doi:10.1080/14686996.2016.1178055
19. Ansari L, Malaekeh-Nikouei B. Magnetic silica nanocomposites for magnetic hyperthermia applications. Int J Hyperthermia. 2017;33(3):354–363. doi:10.1080/02656736.2016.1243736
20. Zeng X, Luo M, Liu G, et al. Polydopamine-modified black phosphorous nanocapsule with enhanced stability and photothermal performance for tumor multimodal treatments. Adv Sci. 2018;5(10):1800510. doi:10.1002/advs.201800510
21. Jiang JH, Pi J, Jin H, Cai JY. Functional graphene oxide as cancer-targeted drug delivery system to selectively induce oesophageal cancer cell apoptosis. Artif Cells Nanomed Biotechnol. 2018;46(sup3):S297–s307. doi:10.1080/21691401.2018.1492418
22. Tran AV, Shim K, Vo Thi TT, Kook JK, An SSA, Lee SW. Targeted and controlled drug delivery by multifunctional mesoporous silica nanoparticles with internal fluorescent conjugates and external polydopamine and graphene oxide layers. Acta Biomater. 2018;74:397–413. doi:10.1016/j.actbio.2018.05.022
23. Wang Z, Duan Y, Duan Y. Application of polydopamine in tumor targeted drug delivery system and its drug release behavior. J Control Release. 2018;290:56–74. doi:10.1016/j.jconrel.2018.10.009
24. Hogan NJ, Urban AS, Ayala-Orozco C, Pimpinelli A, Nordlander P, Halas NJ. Nanoparticles heat through light localization. Nano Lett. 2014;14(8):4640–4645. doi:10.1021/nl5016975
25. Karampelas IH, Liu K, Alali F, Furlani EP. Plasmonic nanoframes for photothermal energy conversion. J Phys Chem C. 2016;120(13):7256–7264. doi:10.1021/acs.jpcc.5b12743
26. GhavamiNejad A, SamariKhalaj M, Aguilar LE, Park CH, Kim CS. pH/NIR light-controlled multidrug release via a mussel-inspired nanocomposite hydrogel for chemo-photothermal cancer therapy. Sci Rep. 2016;6:33594. doi:10.1038/srep33594
27. Jiang W, Mo F, Jin X, et al. Tumor-targeting photothermal heating-responsive nanoplatform based on reduced graphene oxide/mesoporous silica/hyaluronic acid nanocomposite for enhanced photodynamic therapy. Adv Mater Interfaces. 2017;4(20):1700425. doi:10.1002/admi.201700425
28. Tao W, Zhu X, Yu X, et al. Black phosphorus nanosheets as a robust delivery platform for cancer theranostics. Adv Mater. 2017;29(1):1603276. doi:10.1002/adma.201603276
29. Ma Y, Liang X, Tong S, Bao G, Ren Q, Dai Z. Gold nanoshell nanomicelles for potential magnetic resonance imaging, light-triggered drug release, and photothermal therapy. Adv Funct Mater. 2013;23(7):815–822. doi:10.1002/adfm.201201663
30. Liu H, Chen D, Li L, et al. Multifunctional gold nanoshells on silica nanorattles: a platform for the combination of photothermal therapy and chemotherapy with low systemic toxicity. Angewandte Chemie. 2011;50(4):891–895. doi:10.1002/anie.201002820
31. Jiang W, Mo F, Lin Y, Wang X, Xu L, Fu F. Tumor targeting dual stimuli responsive controllable release nanoplatform based on DNA-conjugated reduced graphene oxide for chemo-photothermal synergetic cancer therapy. J Mater Chem B. 2018;6(26):4360–4367. doi:10.1039/C8TB00670A
32. NDong C, Tate JA, Kett WC, et al. Tumor cell targeting by iron oxide nanoparticles is dominated by different factors in vitro versus in vivo. PLoS One. 2015;10(2):e0115636. doi:10.1371/journal.pone.0115636
33. Rosen JE, Chan L, Shieh DB, Gu FX. Iron oxide nanoparticles for targeted cancer imaging and diagnostics. Nanomedicine:NBM. 2012;8(3):275–290. doi:10.1016/j.nano.2011.08.017
34. Wu M, Huang S. Magnetic nanoparticles in cancer diagnosis, drug delivery and treatment. Mol Clin Oncol. 2017;7(5):738–746.
35. Blanco E, Kessinger CW, Sumer BD, Gao J. Multifunctional micellar nanomedicine for cancer therapy. Exp Biol Med. 2009;234(2):123–131. doi:10.3181/0808-MR-250
36. Alibakhshi A, Abarghooi Kahaki F, Ahangarzadeh S, et al. Targeted cancer therapy through antibody fragments-decorated nanomedicines. J Control Release. 2017;268:323–334. doi:10.1016/j.jconrel.2017.10.036
37. Xiong J, Sun Q, Chen J, Li Z, Dou S. Ambient controlled synthesis of advanced core–shell plasmonic Ag@ZnO photocatalysts. CrystEngComm. 2016;18(10):1713–1722. doi:10.1039/C6CE00013D
38. Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. doi:10.1038/nrd2591
39. Rejeeth C, Vivek R, Kannan S. A novel magnetic drug delivery nanocomplex with a cisplatin-conjugated Fe3O4 core and a PEG-functionalized mesoporous silica shell for enhancing cancer drug delivery efficiency. RSC Adv. 2015;5(115):94534–94538. doi:10.1039/C5RA19274A
40. Kim J, Kim HS, Lee N, et al. Multifunctional uniform nanoparticles composed of a magnetite nanocrystal core and a mesoporous silica shell for magnetic resonance and fluorescence imaging and for drug delivery. Angewandte Chemie. 2008;47(44):8438–8441. doi:10.1002/anie.200802469
41. Tran VA, Lee SW. A prominent anchoring effect on the kinetic control of drug release from mesoporous silica nanoparticles (MSNs). J Colloid Interface Sci. 2018;510:345–356. doi:10.1016/j.jcis.2017.09.072
42. Lodha A, Lodha M, Patel A, et al. Synthesis of mesoporous silica nanoparticles and drug loading of poorly water soluble drug cyclosporin A. J Pharm Bioallied Sci. 2012;4(Suppl 1):S92–S94. doi:10.4103/0975-7406.94153
43. Andersson A, Hedenmalm H, Elfsson B, Ehrsson H. Determination of the acid dissociation constant for Cis–Diammineaquachloroplatinum(I I) ion. A hydrolysis product of cisplatin. J Pharm Sci. 1994;83(6):859–862. doi:10.1002/jps.2600830620
44. Vivero-Escoto JL, Elnagheeb M. Mesoporous silica nanoparticles loaded with cisplatin and phthalocyanine for combination chemotherapy and photodynamic therapy in vitro. Nanomaterials. 2015;5(4):2302–2316. doi:10.3390/nano5042302
45. Sreejith S, Ma X, Zhao Y. Graphene oxide wrapping on squaraine-loaded mesoporous silica nanoparticles for bioimaging. J Am Chem Soc. 2012;134(42):17346–17349. doi:10.1021/ja305352d
46. Raj V, Prabha G. Synthesis, characterization and in vitro drug release of cisplatin loaded Cassava starch acetate–PEG/gelatin nanocomposites. J Assoc Arab Univ Basic Appl Sci. 2016;21:10–16. doi:10.1016/j.jaubas.2015.08.001
47. Nguyen NH, Nguyen TT, Ma PC, Ta QTH, Duong TH, Vo VG. Potential antimicrobial and anticancer activities of an ethanol extract from bouea macrophylla. Molecules. 2020;25(8):1996. doi:10.3390/molecules25081996
48. Kohri M, Nannichi Y, Kohma H, et al. Size control of polydopamine nodules formed on polystyrene particles during dopamine polymerization with carboxylic acid-containing compounds for the fabrication of raspberry-like particles. Colloids Surf a Physicochem Eng Asp. 2014;449:114–120. doi:10.1016/j.colsurfa.2014.02.049
49. Lee Y, Park TG. Facile fabrication of branched gold nanoparticles by reductive hydroxyphenol derivatives. Langmuir. 2011;27(6):2965–2971. doi:10.1021/la1044078
50. Zhang M, Zheng J, Zheng Y, et al. Preparation, characterization and catalytic activity of core–satellite Au/Pdop/SiO2/Fe3O4 magnetic nanocomposites. RSC Adv. 2013;3(33):13818–13824. doi:10.1039/c3ra41537f
51. Dong F, Firkowska-Boden I, Arras MML, Jandt KD. Responsive copolymer–graphene oxide hybrid microspheres with enhanced drug release properties. RSC Adv. 2017;7(7):3720–3726. doi:10.1039/C6RA25353A
52. Shen L, Li B, Qiao Y. Fe₃O₄ nanoparticles in targeted drug/gene delivery systems. Materials. 2018;11(2):324. doi:10.3390/ma11020324
53. Amendola V, Meneghetti M. Size evaluation of gold nanoparticles by UV−vis spectroscopy. J Phys Chem C. 2009;113(11):4277–4285. doi:10.1021/jp8082425
54. Haiss W, Thanh NTK, Aveyard J, Fernig DG. Determination of size and concentration of gold nanoparticles from UV−vis spectra. Anal Chem. 2007;79(11):4215–4221. doi:10.1021/ac0702084
55. Zhao Y, Sadat ME, Dunn A, et al. Photothermal effect on Fe3O4 nanoparticles irradiated by white-light for energy-efficient window applications. Sol Energy Mater Sol Cells. 2017;161:247–254. doi:10.1016/j.solmat.2016.11.039
56. Dunn AW, Ehsan SM, Mast D, et al. Photothermal effects and toxicity of Fe3O4 nanoparticles via near infrared laser irradiation for cancer therapy. Mater Sci Eng C. 2015;46:97–102. doi:10.1016/j.msec.2014.09.043
57. Chen Y, Zhang F, Wang Q, et al. The synthesis of LA-Fe3O4@PDA-PEG-DOX for photothermal therapy–chemotherapy. Dalton Transactions. 2018;47(7):2435–2443. doi:10.1039/C7DT04080F
58. Robinson JT, Tabakman SM, Liang Y, et al. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J Am Chem Soc. 2011;133(17):6825–6831. doi:10.1021/ja2010175
59. Wang Y, Black KCL, Luehmann H, et al. Comparison study of gold nanohexapods, nanorods, and nanocages for photothermal cancer treatment. ACS Nano. 2013;7(3):2068–2077. doi:10.1021/nn304332s
60. Riley RS, Day ES. Gold nanoparticle-mediated photothermal therapy: applications and opportunities for multimodal cancer treatment. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(4):e1449. doi:10.1002/wnan.1449
61. Tong Y, Shao L, Li X, et al. Adhesive and stimulus-responsive polydopamine-coated graphene oxide system for pesticide-loss control. J Agric Food Chem. 2018;66(11):2616–2622. doi:10.1021/acs.jafc.7b05500
62. Zhao M, Wu W, Su B. pH-controlled drug release by diffusion through silica nanochannel membranes. ACS Appl Mater Interfaces. 2018;10(40):33986–33992. doi:10.1021/acsami.8b12200
63. Murzin DY, Heikkilä T. Modeling of drug dissolution kinetics with sigmoidal behavior from ordered mesoporous silica. Chem Eng Commun. 2014;201(5):579–592. doi:10.1080/00986445.2013.782290
64. Pelin M, Fusco L, Martin C, et al. Graphene and graphene oxide induce ROS production in human HaCaT skin keratinocytes: the role of xanthine oxidase and NADH dehydrogenase. Nanoscale. 2018;10(25):11820–11830. doi:10.1039/C8NR02933D
65. Soenen SJ, Rivera-Gil P, Montenegro J-M, Parak WJ, De Smedt SC, Braeckmans K. Cellular toxicity of inorganic nanoparticles: common aspects and guidelines for improved nanotoxicity evaluation. Nano Today. 2011;6(5):446–465. doi:10.1016/j.nantod.2011.08.001
66. Alkilany AM, Murphy CJ. Toxicity and cellular uptake of gold nanoparticles: what we have learned so far? J Nanopart Res. 2010;12(7):2313–2333. doi:10.1007/s11051-010-9911-8
67. Khlebtsov N, Dykman L. Biodistribution and toxicity of engineered gold nanoparticles: a review of in vitro and in vivo studies. Chem Soc Rev. 2011;40(3):1647–1671. doi:10.1039/C0CS00018C
68. Pernodet N, Fang X, Sun Y, et al. Adverse effects of citrate/gold nanoparticles on human dermal fibroblasts. Small. 2006;2(6):766–773. doi:10.1002/smll.200500492
69. Mironava T, Hadjiargyrou M, Simon M, Jurukovski V, Rafailovich MH. Gold nanoparticles cellular toxicity and recovery: effect of size, concentration and exposure time. Nanotoxicology. 2010;4(1):120–137. doi:10.3109/17435390903471463
70. Patra HK, Banerjee S, Chaudhuri U, Lahiri P, Dasgupta AK. Cell selective response to gold nanoparticles. Nanomedicine:NBM. 2007;3(2):111–119. doi:10.1016/j.nano.2007.03.005
71. Iturrioz-Rodríguez N, Correa-Duarte MA, Fanarraga ML. Controlled drug delivery systems for cancer based on mesoporous silica nanoparticles. Int J Nanomedicine. 2019;14:3389–3401. doi:10.2147/IJN.S198848
72. Hudson SP, Padera RF, Langer R, Kohane DS. The biocompatibility of mesoporous silicates. Biomaterials. 2008;29(30):4045–4055. doi:10.1016/j.biomaterials.2008.07.007
73. Chang B, Guo J, Liu C, Qian J, Yang W. Surface functionalization of magnetic mesoporous silica nanoparticles for controlled drug release. J Mater Chem. 2010;20(44):9941–9947. doi:10.1039/c0jm01237h
74. Iturrioz-Rodriguez N, Gonzalez-Dominguez E, Gonzalez-Lavado E, et al. A biomimetic escape strategy for cytoplasm invasion by synthetic particles. Angewandte Chemie. 2017;56(44):13736–13740. doi:10.1002/anie.201707769
75. Gonzalez-Dominguez E, Iturrioz-Rodriguez N, Padin-Gonzalez E, et al. Carbon nanotubes gathered onto silica particles lose their biomimetic properties with the cytoskeleton becoming biocompatible. Int J Nanomedicine. 2017;12:6317–6328. doi:10.2147/IJN.S141794
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.