Back to Journals » Journal of Inflammation Research » Volume 14
Molecular Examination of Differentially Expressed Genes in the Brains of Experimental Autoimmune Encephalomyelitis Mice Post Herceptin Treatment
Authors Al-Ani M, Elemam NM ![]() , Hachim IY, Raju TK, Muhammad JS
, Hachim IY, Raju TK, Muhammad JS ![]() , Hachim MY
, Hachim MY ![]() , Bendardaf R
, Bendardaf R ![]() , Maghazachi AA
, Maghazachi AA ![]()
Received 10 March 2021
Accepted for publication 22 May 2021
Published 17 June 2021 Volume 2021:14 Pages 2601—2617
DOI https://doi.org/10.2147/JIR.S310535
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Mena Al-Ani,1,2,* Noha Mousaad Elemam,1,2,* Ibrahim Y Hachim,1 Tom K Raju,2 Jibran Sualeh Muhammad,1,2 Mahmood Y Hachim,3 Riyad Bendardaf,1,4 Azzam A Maghazachi1,2
1Department of Clinical Sciences, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates; 2The Immuno-Oncology Group, Sharjah Institute for Medical Research (SIMR), University of Sharjah, Sharjah, United Arab Emirates; 3College of Medicine, Mohammed bin Rashid University of Medicine and Health Sciences, Dubai, United Arab Emirates; 4University Hospital Sharjah, Sharjah, United Arab Emirates
*These authors contributed equally to this work
Correspondence: Azzam A Maghazachi
Department of Clinical Sciences, College of Medicine, University of Sharjah, University City, Sharjah, 27272, United Arab Emirates
Email [email protected]
Objective: Herceptin (trastuzumab) is an approved drug for treating HER2+ breast cancer patients, but its use for other diseases is not established. We sought to investigate the effects of Herceptin on ameliorating experimental autoimmune encephalomyelitis (EAE) and to examine its effects on the expression of various genes.
Methods: We used in-silico analysis of publicly available data, qRT-PCR, and immunohistochemistry (IHC) to determine the expression of HER2+ cells in the brains of EAE mice. IHC was also utilized to determine the anti-inflammatory effects of Herceptin. The ability of Herceptin to alleviate the EAE clinical score was measured in these mice. Bioinformatics analysis of publicly available data and qRT-PCR were performed to investigate the differentially expressed genes that were either up-regulated or down-regulated during the high clinical score (HCS) of the disease.
Results: We observed that HER2/Erbb2, the receptor for Herceptin is upregulated in the brains of EAE mice when the brains were examined at the HCS stage. Further, we demonstrated that Herceptin ameliorates the EAE disease, increasing re-myelination, reducing brain inflammation, CD3+ T cell accumulation, and HER2+ cells in the brains of these mice. Molecular analysis demonstrated the expression of different genes that were either up-regulated or down-regulated during the HCS of the disease. Our combined bioinformatics and qRT-PCR analyses show increased mRNA expression of Atp6v0d2, C3, C3ar1, Ccl3, Ccl6, Cd74, Clec7a, Cybb, H2-Aa, Hspb1, Lilr4b, Lilrb4a, Mpeg1, Ms4a4a, Ms4a6c, Saa3, Serpina3n and Timp1, at HCS. Except for the mRNA levels of Cd74 and Clec7a which were increased at HCS when Herceptin was used in both prophylactic and therapeutic regimens, the levels of other described mRNAs were reduced.
Conclusion: These novel findings show that Herceptin ameliorates the clinical score in EAE mice and are the first to investigate in detail the differential gene expression post-treatment with the drug.
Keywords: herceptin, EAE, prophylactic, therapeutic, high clinical score, bioinformatics, qRT-PCR, immunohistochemistry, differential gene expression
Introduction
Multiple sclerosis (MS) is an autoimmune inflammatory disease that results from damage to myelin, the coating protective layer of neurons in the brain and spinal cord.1,2 This disrupts the communication signals of the nervous system, leading to a variety of signs and symptoms.3 MS patients suffer from neurological symptoms that appear according to the location of the damages within the nervous system. These include loss of sensation associated with numbness, muscle weakness, difficulty with coordination and balance (ataxia), as well as speech disturbance.4–8
In some MS patients, there are phases of severe and worsening clinical symptoms that are called relapses or exacerbations. These are usually followed by quiet periods where clinical symptoms are ameliorated, ie, remissions.9 So far, the available therapies of MS aim at improving body function and preventing the development of new lesions.10
It is known that the mechanism of the disease is a result of myelin-producing cell damage by the immune system.9 Notably, the hallmark of this disease asides from demyelination, is the inflammatory status that is caused by autoreactive T cells which penetrate through the blood-brain barrier (BBB), recognizing and destroying the myelin sheath.1,10 Additionally, multiple genetic variations have been associated with the development of MS.11,12 Experimental autoimmune encephalomyelitis “EAE” is the mouse model for human MS since it is quite similar to pathologies as observed in the central nervous system (CNS) inflammation and demyelination, and is commonly used for the evaluation of potential MS treatments.13
The epidermal growth factor “EGF” family signaling proteins are important for cell multiplication and migration. This family comprises EGFR “Erbb1/HER1”, “Erbb2/HER2”, “Erbb3/HER3”, and “Erbb4/HER4”.14 The antibody Trastuzumab “Herceptin™” binds to human Erbb2/HER2. Approximately 20–25% of invasive breast cancers exhibit overexpression of the HER2 tyrosine kinase receptor.15,16 Herceptin is often used to treat HER2+ breast cancer patients either at the early or advanced stage of the disease. The mechanisms by which Trastuzumab induces regression of HER2-overexpressing tumors are still being elucidated, but several molecular and cellular effects have been reported.17,18 It is worth mentioning that EGF family members were previously investigated in MS. For instance, blocking the EGF ligand was found to induce differentiation of various cell types in the CNS that are involved in the pathophysiology of MS.19,20 In this study, we aimed at examining the plausible effects of Herceptin for treating EAE. Furthermore, we sought to investigate whether there is a differential gene expression in the brains of EAE mice compared to healthy counterparts that may act as a diagnostic or prognostic biomarker for EAE and whether Herceptin might modulate such expression.
Materials and Methods
Induction of EAE Using Female Swiss Jim Lambert “SJL” Mice
All animal studies were reviewed and approved by the University of Sharjah animal care and use committee “ACUC”; approval number UOS-ACUC-16-003, and following the UoS research policy and procedures for animal care. For in vivo experiments, female SJL/J mice (H-2ˢ, purchased from Jackson Laboratories, Bar Harbor, ME, USA), aged 4–6 weeks, were used. These mice were injected subcutaneously (SC) with 200 μg of myelin proteolipid protein “PLP” 139–151 peptide (Abbiotec, San Diego, CA, USA) emulsified in complete Freund’s adjuvant containing 1 mg Mycobacterium tuberculosis (Sigma-Aldrich, Darmstadt, Germany). A 200 ng of Bordetella pertussis toxin (Sigma-Aldrich) was injected intraperitoneally “IP” at a similar time of PLP injection, and again after 48 h. The animals were independently monitored and checked daily, and the EAE clinical score was measured according to the following scoring protocol: 0 = no clinical disease, 1 = tail flaccidity, 2 = hind limb weakness, 3 = hind limb paralysis, 4 = forelimb paralysis, and 5 = moribund or death, as described.21
Treatment of EAE Mice with Herceptin
Herceptin was obtained from Hoffmann-La Roche Ltd. (Basel, Switzerland). EAE mice were divided into four groups, where each group consisted of 5 mice. The first group was considered as the disease control without treatment. The second group is called prophylactic, which includes mice that were injected IP with 5 μg Herceptin every 3 days since the initiation of the disease and until termination. The third group, designated as therapeutic, includes mice that were injected IP with 5 μg Herceptin every 3 days starting from the day of the appearance of symptoms (usually between 8–10 of the initiation of the diseases), and until termination. The last group includes EAE mice that were injected with 1 μg of isotype IgG control antibody (R&D Systems, Minneapolis, MN, USA) IP every 3 days, until termination.21
Identification of Top DEGs in EAE Brains
To identify EAE-specific differentially expressed genes “DEGs”, publicly available transcriptomic datasets (regardless of the studied mouse model) were retrieved from Gene Expression Omnibus “GEO” (https://www.ncbi.nlm.nih.gov/geo/) and reanalyzed as described.22 GSE99300 dataset was retrieved as it was designed to determine the distinct CNS surrogate biomarkers between two different mouse strains which develop primary progressive EAE. Microarray analysis was conducted using brain samples collected from EAE mice at the disease peak. GEOquery and limma R packages through the GEO2R tool were used to identify DEGs that showed p-value <0.05 and more than 5-fold change between EAE and the control un-diseased mice. The top DEGs between EAE and control brains of mice were illustrated using Heat Mapper software.
RNA Extraction and qRT-PCR Analysis
For RNA extraction, mice brains were collected and preserved under −80°C immediately after dissection. Total RNA from the whole brain was extracted using All prep DNA/RNA mini kit (Qiagen, Hilden, Germany), after crushing the brains using mortar and pestle on dry ice. The quantity and purity of RNA were checked using NanoDrop 2000 spectrophotometer (ThermoScientific, Waltham, MA, USA). About 1000 ng of total RNA was converted to cDNA with high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR “qRT-PCR” was performed using the GoTaq qPCR Master Mix (Promega, San Luis Obispo, CA, USA), along with the QuantStudio3 system (Applied Biosystems, Foster City, CA, USA), using the designed primers as listed in Table 1. Gene expression was calculated using the 2−ΔΔCT method with PPIA as a housekeeping gene, with 3 mice per group done in triplicates.
 |
Table 1 Primer Sequences of Mouse Genes That Were Investigated Using qRT-PCR |
Immunohistochemistry Analysis of Brain Tissues
Mice brains were fixed in 10% neutral buffered formalin (Thermo Scientific, Waltham, MA, USA) for a minimum of 24 h, and then embedded in paraffin and sectioned at 4 μm. The paraffin-embedded tissue slides were deparaffinized, then rehydrated with graded ethanol and distilled water. One set of the slides was used for hematoxylin and eosin “H&E” staining for morphological analysis with Shandon Harris hematoxylin (ThermoScientific, Waltham, MA, USA), and counterstained with Shandon eosin (ThermoScientific, Waltham, MA, USA). Slides stained with rabbit anti-mouse CD3 antibody (ThermoScientific, Waltham, MA, USA, Cat. no: MA5-14524), or rabbit anti-mouse HER2 (Abcam, Cambridge, UK, Cat. no: ab245336), were subjected to antigen retrieval by steaming the slides in Tris-EDTA buffer at pH 9.0 for 20 min, followed by blocking with 5% goat serum for 30 min. The anti-CD3 antibody used at 1:150 dilution, and anti-HER2 used at 1:200 dilution were incubated overnight at 4°C, followed by 1 h incubation with the secondary antibody goat anti-rabbit IgG HRP (Abcam, Cambridge, UK) at a dilution of 1:10,000 for anti-CD3, or 1:2000 for anti-HER2 in 1% BSA solution. The color was developed by 3,3′-diaminobenzidine (Abcam, Cambridge, UK), and counterstained with Shandon eosin. Other slides were used for histological myelin staining by incubating with Luxol fast blue solution (Abcam, Cambridge, UK) overnight at 37°C. The stain was differentiated using lithium carbonate solution, ethanol, and distilled water and counterstained with Cresyl Echt violet (Abcam, Cambridge, UK). The images were captured with Olympus DP74 microscope digital camera attached to a BX43 microscope (Olympus Life Sciences, Tokyo, Japan), at various magnification powers.
Statistical Analysis
All statistical analysis was done using GraphPad Prism 6 (San Diego, CA, USA). The non-parametric Mann Whitney U-test was used when comparing two groups. Also, the repeated measures ANOVA followed by Sidak’s or Dunnet’s multiple comparison’s tests was used when comparing more than two groups. P-value <0.05 was considered statistically significant.
Results
HER2/Erbb2 mRNA Expression in EAE Mice
While performing bioinformatics analysis, we observed that there was a significant elevation in HER2 expression in EAE mice as compared to normal mice (Figure 1A). In order to validate this finding, HER2 gene expression was assessed in the brains of EAE and normal mice. In these experiments, we examined the expression of HER2/Erbb2 mRNA in mice during high clinical score (HCS=day 18) and observed an up-regulation in the EAE untreated mice as compared to normal mice (P<0.05, Figure 1B). Upon administration of Herceptin whether prophylactically or therapeutically, there was a significant reduction in the mRNA level expression of HER2 during HCS (P<0.05, Figure 1B).
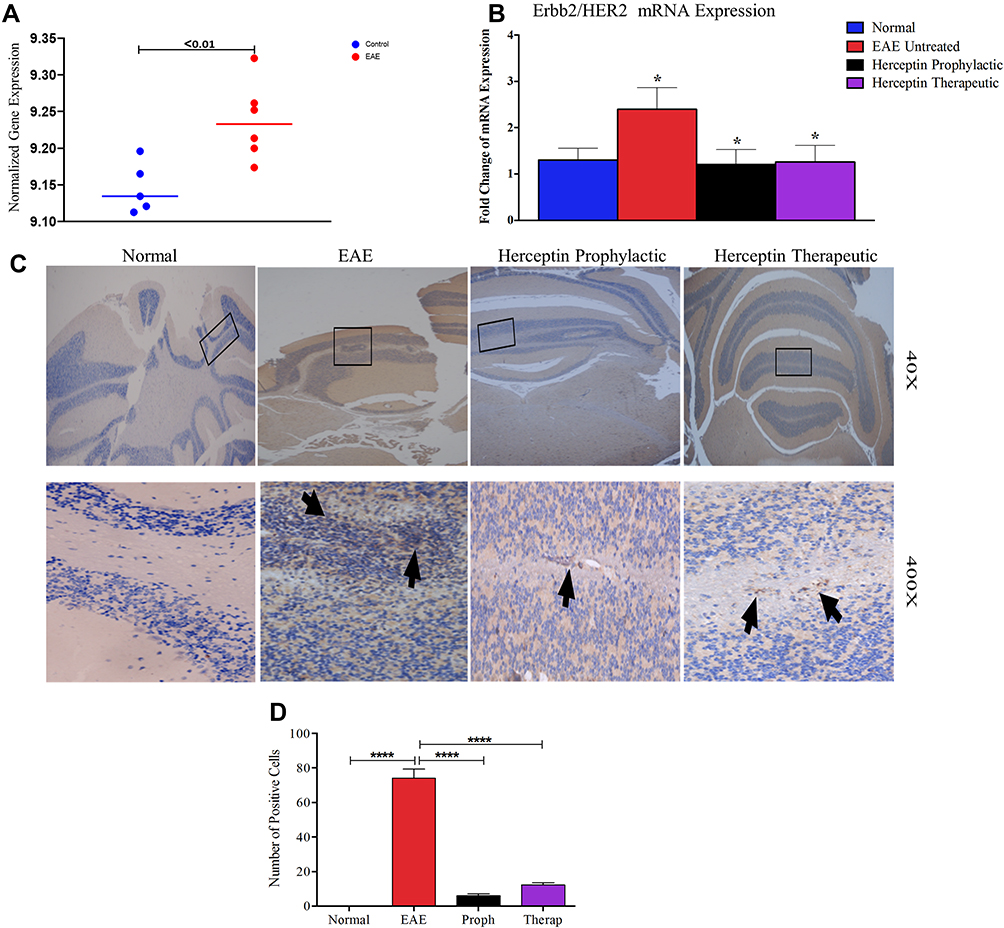 |
Figure 1 In silico and ex vivo gene expression of HER2/Erbb2 in normal, EAE untreated and Herceptin treated groups. (A) Expression of HER2/Erbb2 using publicly available data in EAE (red dots; n=5) compared to normal mice (blue dots, n=5). (B) HER2/Erbb2 mRNA expression was validated using qRT-PCR in normal, EAE untreated at the high clinical score (HCS), as well as after Herceptin prophylactic and therapeutic treatment regimens. P values indicate the significance compared to EAE untreated mice. *P<0.05. (C) Immunohistochemical analysis of the presence of HER2+ cells in the brains of normal mice, EAE untreated mice, or mice treated with Herceptin in a prophylactic or therapeutic regimen. Arrows indicate the presence of HER2+ positive cells. Rectangles (40 X) show the part of the brains that were enlarged (400 X). (D) Quantification of HER2+ cells in the brains of normal, EAE untreated or Herceptin treated mice. ****P<0.0001. The results are represented as mean ± SEM (n=3). Abbreviation: NS, not significant. |
To ascertain that HER2 might be present in the brains of EAE mice, we stained these brains with an anti-HER2 antibody and compared the expression of HER2 protein among these mice. Whereas normal mice brains were devoid of HER2+ cells, the brains of EAE mice show extensive accumulation of such cells (arrows in Figure 1C). Treatment with Herceptin reduced this accumulation but did not completely ablate it, since HER2+ cells were still present whether the mice were treated with Herceptin prophylactically or therapeutically (arrows in Figure 1C). Quantitatively, HER2+ cells were absent from the brains of normal mice but significantly increased in EAE mice (P<0.0001, Figure 1D). Intriguingly, brains of mice treated with Herceptin either by prophylactic or therapeutic regimen, have significantly lower numbers of HER+ cells (P<0.0001 as compared to untreated EAE mice).
Herceptin Ameliorates EAE Clinical Score
Based on the results showing that HER2 mRNA and protein are significantly increased in the brains of EAE mice, and that Herceptin alleviates this expression during HCS, we sought to examine whether Herceptin might ameliorate the EAE clinical score. All groups of mice were monitored for 40 days during which EAE clinical score and mouse body weights were measured. As shown, there was a reduction in the clinical score of mice treated prophylactically (P<0.0001), or therapeutically (P<0.01) with Herceptin compared to the EAE untreated group during the high clinical score “HCS at day 18ʹ (Figure 2A and B). However, there was no difference between the IgG isotype control group and EAE untreated mice at that time. While, there were significant differences between IgG treated group and Herceptin treated groups, whether prophylactically (P<0.001) or therapeutically (P<0.05). This was further supported by body weight changes as prophylactic (P<0.0001), or therapeutic (P<0.01) regimens of Herceptin-treated mice displayed an increase in their body weight compared to untreated groups as recorded daily (Figure 2C). IgG-treated mice show no significant differences with untreated mice (Figure 2C).
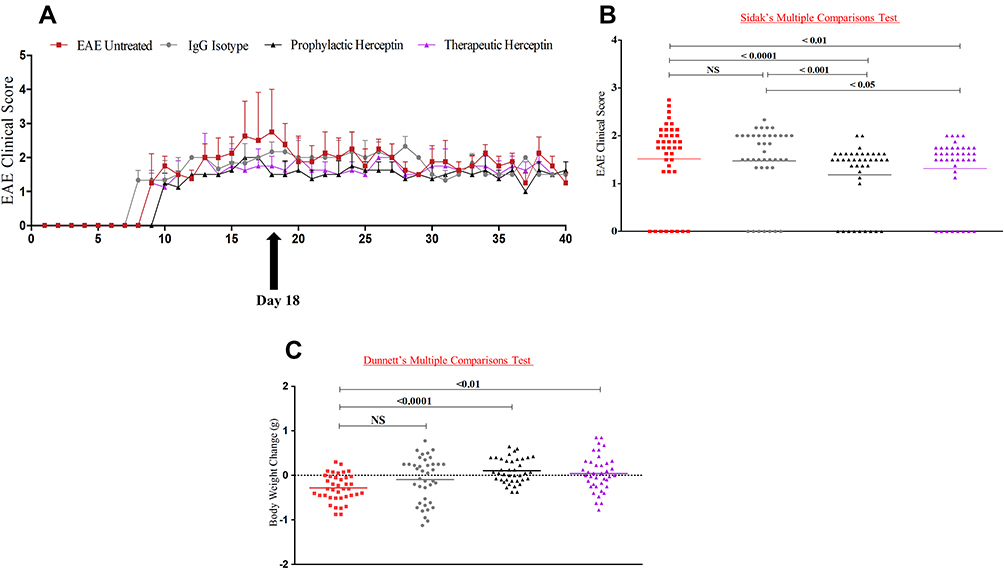 |
Figure 2 Herceptin reduces EAE high clinical score when used as a prophylactic or therapeutic drug. (A) Clinical score was recorded for 40 days in four groups of mice: untreated EAE mice (n=5, red line), IgG isotype control EAE mice (n=5, grey line), Herceptin prophylactic (n=5, black line), and Herceptin therapeutic (n=5, purple line). Also shown by the arrow, the time of recording the high clinical score (HCS, day 18 after initiation of the disease). (B) P values comparing the clinical score among the different groups. (C) Bodyweight change was recorded for each group. P values indicate the significance compared to EAE untreated mice. Abbreviation: NS, not significant. |
Histopathological Evaluation of the CNS Inflammation in EAE Mice
Normal mouse brains show lack of inflammation (Figure 3A). In contrast, the brains of EAE untreated or IgG treated mice exhibit strong inflammation, as determined by H&E staining (Figure 3B and C, respectively). Upon prophylactic Herceptin treatment, there was a reduction of inflamed perivascular space “PVS” when compared to untreated EAE or IgG treated mice (Figure 3D). Furthermore, CD3+ T cells were decreased in the PVS of Herceptin-treated mice, particularly after prophylactic treatment, since some CD3+ T cells were still observed in the brains of EAE mice treated therapeutically with Herceptin. Additionally, there was a remarkable re-myelination, as determined by Luxol fast staining when Herceptin was administered in EAE mice as prophylactic, and to a lesser extent as a therapeutic drug (Figure 3E).
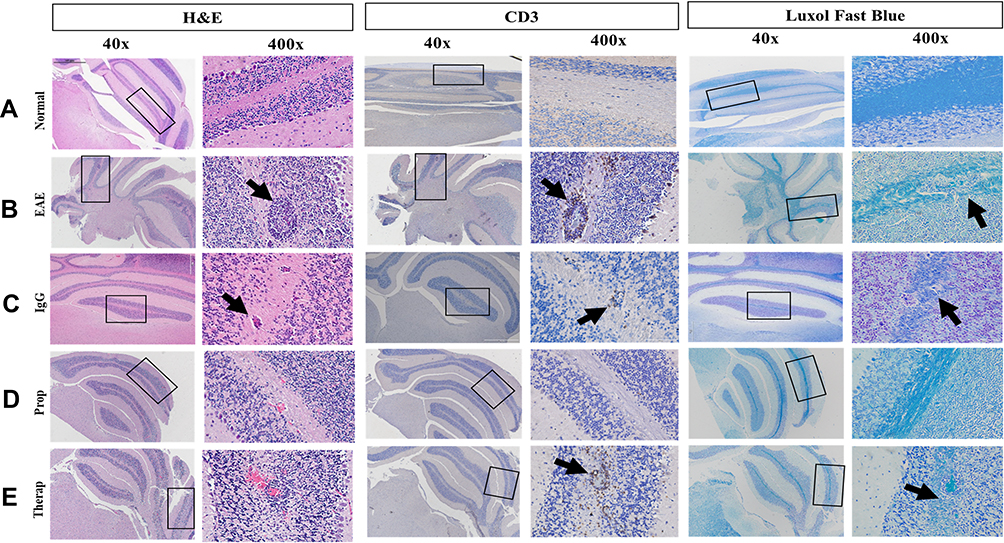 |
Figure 3 Immunohistochemical examination of the mice brains (A) Brains of normal mice. (B) Brains of EAE mice. (C) Brains of IgG-treated mice. (D) Brains of EAE mice prophylactically treated with Herceptin. (E) Brains of EAE mice therapeutically treated with Herceptin. Both 40X and 400 X magnifications are shown. Rectangles (40 X) show the part of the brains that were enlarged (400 X). H&E staining shows the inflammation determined by arrows. Infiltration of CD3+ cells (anti-CD3 staining) is also demonstrated by black arrows. Similarly, high demyelination is detected by Luxol Fast Blue staining (black arrows). |
Messenger RNA Expression for Differentially Expressed Genes in EAE Mice Brains
In silico analysis of publicly available data revealed top DEGs in EAE and normal mice. These genes were classified into 5 different categories: a. inflammatory mediators; b. chemokines; c. immunoregulatory signaling molecules; d. antigen presentation; and e. immuno-modulatory receptors (Figure 4A). Being a relapsing-remitting mouse model, it was interesting to investigate these genes at a heightened disease stage, which could resemble the relapses observed in human MS patients. As shown in Figure 4B, mRNAs levels for Lilrb4a, Saa3, H2-Aa, Cybb, Cd74, Timp1, Lilr4b, C3, Serpina3n, Mpeg1, Atp6v0d2, Ms4a4a, Hspb1, Ccl6, Ms4a6c, C3ar1, Clec7a, Ccl3, and Erbb2/Her2, were significantly upregulated in the brains of EAE untreated mice at high clinical score compared to normal mice brains. Other genes although detected using in silico analysis, qRT-PCR experiments did not show any detection in the mRNA of these genes which include C1qc, Clec4e, H2-Eb1, Igsf6, Wfdc17, Lyz2, Gpnmb, Cxcl13, and Lcn2. As they were not detected by qRT-PCR, they were not studied further.
 |
Figure 4 In silico and ex vivo analysis of differentially expressed genes “DEGs” between EAE and normal mice. (A) Heat map of DEGs in EAE untreated and normal mice. (B) Validation using RT-qPCR for mRNA expression in EAE mice at HCS compared to normal mice brains. P values show the significance compared to normal mice. Abbreviation: NS, not significant. |
Assessment of Inflammatory Mediators and Chemokines mRNA Expression in EAE Untreated or Herceptin Treated Mice
Since the genes described above and shown in Figure 4 are differentially expressed in EAE mice, it was interesting to determine whether there is any change in their mRNA expression upon treatment with Herceptin. Among the genes are the inflammatory mediators C3, Saa3, Serpina3n, and Hspb1. The mRNA expression for complement component C3 was significantly increased in EAE mice brains during HCS as compared to normal mice (P<0.001, Figure 5A). Intriguingly, it was downregulated upon Herceptin prophylactic (P<0.05), or therapeutic (P<0.05) regimen. Also, the mRNA for human serum amyloid A3 “Saa3” was one of the most upregulated in EAE mice (P<0.001, Figure 5B). Similar to C3, it was downregulated upon Herceptin prophylactic (P<0.01) or therapeutic treatment (P<0.001). Serpina3n (Figure 5C), and Hspb1 (Figure 5D) mRNA levels also showed a significant down-regulation upon administration of Herceptin by prophylactic or therapeutic regimens.
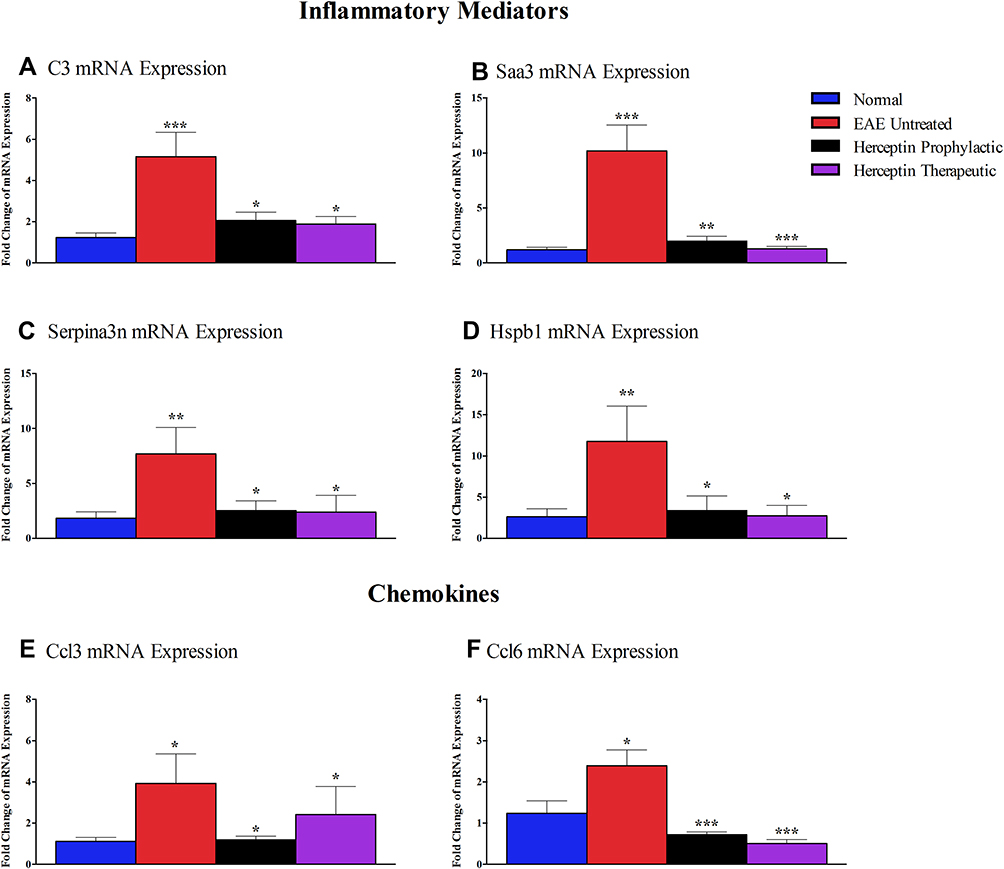 |
Figure 5 Messenger RNA expression of inflammatory mediators and chemokines in EAE untreated and Herceptin-treated mice. Brain-extracted mRNA expression of (A) C3, (B) Saa3, (C) Serpina3n, (D) Hspb1, (E) Ccl3, and (F) Ccl6 was assessed in EAE untreated mice, Herceptin prophylactically treated mice, and Herceptin therapeutically treated mice at HCS. Blue columns show the values in normal mice. P values indicate the significance of treated groups compared to EAE untreated mice. *P<0.05, **P<0.01, ***P<0.001. The results are represented as mean ± SEM (n=3). |
Similarly, the mRNA for the chemokine Ccl3 was elevated in EAE mice (P<0.05, Figure 5E). Upon Herceptin treatment regimens, it was down-regulated (P<0.05, Figure 5E). Ccl6 mRNA was also upregulated in EAE mice as compared to control mice (P<0.05, Figure 5F), and was significantly reduced upon prophylactic or therapeutic Herceptin administration (P<0.001, Figure 5F).
Assessment of Immuno-Regulatory Signaling and Antigen Presentation Molecules mRNA Expression
Based on the bioinformatics data, it was of interest to investigate the immuno-regulatory signaling genes, which include Cybb, Atp6vod2, and Timp1. As shown, mRNA for Cybb was upregulated in EAE mice as compared to its expression in normal mice (P<0.001, Figure 6A), and that such expression was reduced upon therapeutic (P<0.01), or prophylactic (P<0.01) Herceptin treatment regimen. Similarly, Atp6vod2 mRNA level was reduced upon Herceptin prophylactic or therapeutic administration (P<0.05, Figure 6B). The mRNA level of Timp1 was significantly increased in EAE mice (P<0.01, Figure 6C), but showed a trend of upregulation, although not statistically significant, upon Herceptin prophylactic or therapeutic administration (Figure 6C).
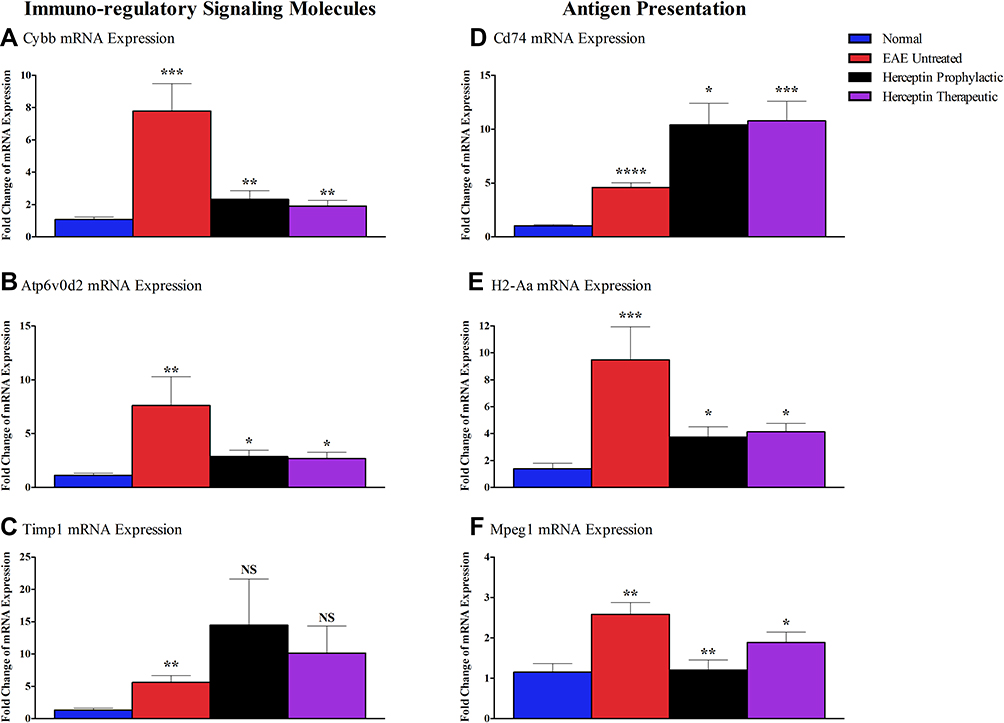 |
Figure 6 Messenger RNA of immuno-regulatory signaling and antigen presentation molecules in EAE untreated and Herceptin treated mice. Brain-extracted mRNA expression of immunoregulatory molecules: (A) Cybb, (B) Atp6vOd2, (C) Timp1, as well as the mRNA expression of antigen presentation molecules: (D) Cd74, (E) H2-Aa, and (F) Mpeg1, assessed in EAE untreated mice, Herceptin prophylactically treated mice, and Herceptin therapeutically treated mice at HCS. Bluecolumns show the values in normal mice. P values indicate the significance of treated groups compared to EAE untreated mice. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. The results are represented as mean ± SEM (n=3). Abbreviation: NS, not significant. |
The mRNA for antigen presentation genes including Cd74, H2-Aa, and Mpeg1, were assessed in EAE untreated, Herceptin prophylactic, or therapeutic treated mice. The mRNA levels of all three genes were increased in EAE mice as compared to normal mice (P<0.0001, P<0.001, and P<0.01, Figure 6D–F, respectively). Upon administration of Herceptin prophylactically or therapeutically, there was an elevation in the expression of Cd74 mRNA level (P<0.05 and P<0.001, respectively, Figure 6D). In contrast, there was a significant down-regulation of H2-Aa upon prophylactic or therapeutic administration of Herceptin (P<0.05 and P<0.05, respectively, Figure 6E). Similarly, Mpeg1 mRNA was reduced in mice treated with Herceptin used as either prophylactic or therapeutic drug (P<0.01 and P<0.05, respectively, Figure 6F).
Assessment of Immune Modulatory Receptors mRNA Expression Levels
As demonstrated by the in-silico analysis, the immune modulatory receptor genes including Clec7a, Lilrb4a, Ms4a4a, C3ar1, Lilr4b, and Ms4a6c were elevated in EAE mice (Figure 4). Clec7a mRNA expression was upregulated when Herceptin was used (prophylactic: P<0.05 or therapeutic: P<0.05, Figure 7A). In contrast, the mRNA level of Lilrb4a was significantly downregulated after prophylactic (P<0.01), or therapeutic (P<0.01) Herceptin treatment regimen (Figure 7B). As illustrated in Figure 7C, Ms4a4a mRNA levels were slightly reduced after Herceptin prophylactic or therapeutic treatment; however, this change was not statistically significant. Regarding C3ar1 (Figure 7D), Lilr4b (Figure 7E) and Ms4a6c (Figure 7F), mRNA levels were reduced when Herceptin was used prophylactically (P<0.05), or therapeutically (P<0.05).
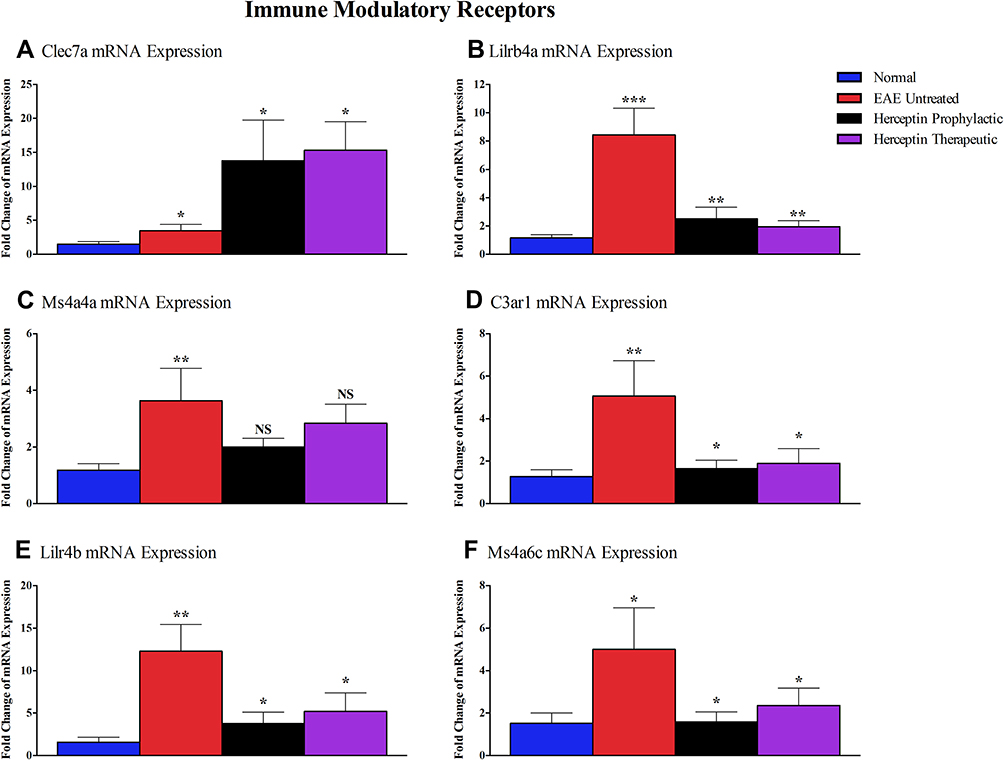 |
Figure 7 Messenger RNA of immune-modulatory receptors in EAE untreated and Herceptin treated mice. Messenger RNA expressions of (A) Clec7a, (B) Lilrb4a, (C) Ms4a4a, (D) C3ar1, (E) Lilr4b, and (F) Ms4a6c were measured in EAE untreated mice, Herceptin prophylactically treated mice, and Herceptin therapeutically treated mice at HCS. Blue columns show the values in normal mice. P values indicate the significance of treated groups compared to EAE untreated mice. *P<0.05, **P<0.01, ***P<0.001. The results are represented as mean ± SEM (n=3). Abbreviation: NS, not significant. |
Control IgG Treatment Did Not Affect mRNA Expression of All Examined DEGs
In order to ascertain that the observed differences are due to the specific effects of Herceptin and not to non-specific binding, IgG control antibody was injected into EAE mice after which the brains were evaluated by qRT-PCR to determine if there is any differential expression between mRNA expression of untreated EAE and IgG treated EAE mice. As shown, there were no significant differences in the mRNA expression of all investigated genes among EAE untreated and IgG treated mice, whether the IgG was used in a prophylactic or therapeutic regimen (Figure S1).
Discussion
Disease-modifying therapies “DMTs” of MS have been shown to significantly limit and delay relapses in MS patients. These therapies have shown positive results in relapsing-remitting MS patients, corroborated with improving cognitive dysfunctions.23,24 Normally, the health of patients improves drastically during the remitting period. The factors that differentiate the causes of relapse versus remission are not clear. EAE mouse model is universally used to investigate the therapeutic efficacies of DMTs.13 We noticed in SJL mice affected with EAE that there are periods where the clinical score of the disease is high. This period was called HCS, which could, to some extent, resemble the relapse period in MS patients. Subsequently, we examined in detail various gene expressions during this period.
Despite being an autoimmune disease, MS seems to have an association with specific genetic factors. For instance, studies reported that one of the strongest associations with MS is the gene for HLA-DRB1*15.25,26 In addition, many other immuno-regulatory genes were reported to be associated with MS where the immune system attacks the brain and spinal cord.25,26 Our in silico analysis demonstrated that there are differentially expressed genes regardless of the mouse model. Surprisingly, HER2/Erbb2 gene was differentially upregulated in untreated EAE when compared to normal mice, and this was supported at the mRNA and protein levels. Similarly, it was reported that abnormally high levels of EGF were found in the CNS of relapsing remitting MS (RRMS) patients causing dysregulation of the immune system and exacerbation of the disease.27 Previous studies showed that blocking EGF function using inhibitors improved the survival and function of neuronal cells,18 whereas high levels of EGF caused cell damage.27 Based on these observations, we hypothesized that blocking the EGF receptor HER2 using the drug Herceptin might possess therapeutic efficacy for treating mice affected with EAE. HER2 is present on 25–30% of breast cancer tissues and is used as a therapeutic target for these patients.28,29 The mechanism of action of Herceptin is plausibly due to blocking HER2 antigen, as well as antibody-dependent cellular cytotoxicity “ADCC”.30–32 Herceptin was not previously used for the treatment of autoimmune diseases, and specifically MS or EAE.
Here we observed that upon Herceptin treatment whether used in prophylactic or therapeutic regimen, HER2 mRNA expression was downregulated in the treated groups at the HCS. This result was corroborated with immunohistochemistry data where the accumulation of HER2+ cells was inhibited upon Herceptin treatment. This could be a feedback mechanism where the cells reduce the transcription of HER2 upon blocking its surface protein by the drug.33,34 Subsequently, we aimed to investigate the effects of injecting mice with Herceptin on the day of EAE induction (prophylactic regimen), as well as on the day of the appearance of clinical symptoms (therapeutic regimen). Herceptin prophylactic treatment was found to reduce the EAE high clinical score when compared to EAE untreated mice. Similarly, administration of Herceptin therapeutically significantly decreased the HCS severity of EAE when compared to EAE untreated group. There was a concomitant increase in the body weight for both treated groups, indicating a better health status of these mice. Furthermore, immunohistochemical staining revealed that both prophylactic and therapeutic treatment regimens of Herceptin decreased EAE inflammatory features which include perivascular inflammation, infiltration of CD3+ cells, and reversal of de-myelination, although the prophylactic regimen was more robust in reducing the signs of inflammation. Therefore, Herceptin possesses the ability to ameliorate EAE disease.
In addition, the in silico analysis revealed several genes to be differentially expressed in EAE during HCS, which were classified into 5 different classes. Inflammatory mediators are defined as soluble secreted diffusible particles that are stimulated at sites of damaged tissues and injury and can act at distant sites to manage inflammation.35 Among these is the C3, which was found to be increased in EAE untreated mice and is reduced upon Herceptin prophylactic or therapeutic administration. Several studies have previously reported that glial cells and neurons produce complement proteins inside the CNS at a higher extent during inflammation.36–38 Furthermore, the immunopathology of MS indicates precipitation of C3 in the brains of MS patients,39 which could be an essential factor in the EAE development.40–43 Another crucial inflammatory mediator is the serum amyloid A3 “Saa3”, an isoform of Saa that was found to be significantly elevated in untreated EAE at HCS. This supports a study showing that Saa levels are expanded within the peripheral blood of patients with RRMS, indicating an inflammatory state.44 Furthermore, Saa knockout mice show resistance to EAE induction,45 exhibiting the critical role of Saa in EAE. To this end, it should be stressed that Saa supports cell migration, cell growth, and angiogenesis, which are hallmarks of chronic inflammation.46 Therefore, it was of interest that Herceptin significantly reduced Saa3 levels at HCS when used as a prophylactic or therapeutic drug. We can conclude that Saa3 may be a potential biomarker for EAE/MS diagnosis as well as a prognostic marker for therapeutic efficacy as well as a target for anti-Saa therapy.
Our data revealed Serpin “serine protease” inhibitor family A member 3 “Serpina3n” is also increased in EAE untreated mice and is reduced upon Herceptin treatment, particularly at the heightened disease stage. Our findings match those reported earlier where Serpina3n level was significantly increased within the spinal cord and brains of EAE mice and diminished over time.47 It was also suggested that it might play a role in the neurodegenerative process within the CNS during EAE or MS.47 However, the role of Serpina3n in EAE or MS remains controversial, as other reports stated that Serpina3n did not interfere with the infiltration of immune cells into the CNS and could even decrease axonal damage.48
Stressed oligodendrocytes were known to produce small heat-shock protein αB-crystallin “Hspb5” which has a possible link to the progression of MS lesions.49 Similarly, MS lesions were found to have a high expression of heat shock protein β-1 “Hspb1”,50 supporting our data where Hspb1 level was elevated in untreated EAE mice at HCS. In our hands, Herceptin reduced the mRNA level of Hspb1 when used as a prophylactic or therapeutic drug.
Chemokines and their receptors have numerous functions in regulating the immune system, whether in physiological or pathological conditions such as MS.51–55 CCL3, is involved in the pathophysiology of MS,55–57 and is expressed within the CNS of EAE mice.58 We confirmed these findings and observed that Ccl3 mRNA was increased in untreated EAE mice. Studies also reported that treatment with anti-Ccl3 antibodies hindered EAE onset and decreased the aggregation of mononuclear cells within the CNS.59 In our hands, Herceptin therapy reduced Ccl3 mRNA levels in the treated groups when used prophylactically or therapeutically, which could explain the observed reduction of inflammation. Another macrophage-associated chemokine is CCL6, which is released by microglia and macrophages and acts in an autocrine fashion as a strong chemoattractant for macrophages,60 monocytes,55,61 and T cells.55 However, to the best of our knowledge, no previous data regarding Ccl6 was reported in EAE. Ccl6 mRNA was found to be upregulated in the brains of untreated mice in HCS and was reduced by the administration of Herceptin used in either prophylactic or therapeutic treatment regimen.
Several immuno-regulatory signaling molecules are required for the function of the immune system.62 Among these molecules is the cytochrome b-245, beta chain “Cybb”. Our results show that Cybb was upregulated in the brains of untreated EAE mice at HCS and was reduced upon treatment with Herceptin. These observations agree with a study reporting a significant upregulation of Cybb in CD4+ T cells in relapsing-remitting MS patients.63 Metalloproteinases “MMPs” and their inhibitors also play a crucial role in the pathology of many diseases including MS and EAE,64–66 as excessive MMP activity was recognized to be associated with the disease severity.66,67 Moreover, another study reported that MMP inhibitors such as Timp1 halted the blood brain barrier disturbance and progression of the clinical condition.68,69 Timp1 deficient mice have more leukocyte penetration and spinal cord demyelination.70 However, high levels of Timp1 were found to surround lesions in EAE, which supports our findings where Timp1 was increased, although not statistically significant in EAE mice treated with Herceptin.65
Antigen presentation is a key process involving multiple arms of the immune system, where any imbalance could lead to autoimmunity. Histocompatibility antigens in mice such as Cd74 and H2-Aa contribute to antigen presentation. In this study, Cd74 mRNA level was found to be upregulated in EAE untreated mice at HCS, with a further increase in its expression upon Herceptin treatment whether used as a prophylactic or therapeutic drug. This contradicts a previous study where CD74 was downregulated in blood B cells of early MS patients. However, this discrepancy could be due to the different tissues investigated, sites, or cell types.71 Likewise, H2-Aa was found to be significantly upregulated in EAE mice when the clinical score was measured at heightened disease. These findings highlight the potential role of H2-Aa in antigen presentation in EAE mice and possibly in MS patients as suggested previously by Falcão et al.72 Upon Herceptin administration as a prophylactic or therapeutic drug, there was a down-regulation in H2-Aa expression, supporting the findings that Herceptin reduced inflammation at sites of damage in EAE. Another mouse antigen-presenting molecule, macrophage-expressed gene 1 “Mpeg1”, was not previously investigated in EAE. Like the other antigen presentation markers, it was also significantly upregulated in EAE mice at HCS but reduced upon Herceptin prophylactic or therapeutic treatment regimen. Mpeg1 is the gene coding for pore-forming protein “perforin-2” whose excessive accumulation can lead to high inflammation and autoimmunity.73 Our results showing that Herceptin reduced the mRNA level of this molecule upon treating EAE mice confirm and support the findings that Herceptin is a novel anti-inflammatory drug.
Immuno-modulatory receptors mediate signaling pathways that allow immune and non-immune cells to function properly. For instance, Dectin-1 “Clec7a” which is expressed on myeloid and natural killer cells, was found to play a role in EAE as Dectin-1 deficient mice had severe EAE due to expansion of Th17 within the draining lymph nodes, as well as a reduction in regulatory T cells.74 Our findings indicate that Clec7a mRNA was upregulated in EAE mice at HCS. Further work is needed to better understand the role of Clec7a in EAE pathogenesis. The complement component 3a receptor 1 “C3ar1” was previously observed in regions of inflammation in MS and bacterial meningitis, indicating its role in autoimmunity and inflammation.75 Indeed, C3ar1 is upregulated in EAE untreated mice at the HCS stage, and its expression was restored back to normal upon prophylactic and therapeutic Herceptin treatment at heightened disease stage.
The leukocyte immunoglobulin-like receptors that include leukocyte immunoglobulin-like receptor, subfamily B, member 4A “Lilrb4a” and member 4 “Lilr4b”, which are expressed on monocytes, B cells, dendritic cells, and natural killer cells,76 were also examined in this study. In EAE untreated mice, we found both genes to be upregulated at HCS, indicating a possible increase in the infiltration of such immune cells at inflamed sites in the brains of EAE mice. Recently, members of the membrane-spanning 4 domains have been associated with autoimmune diseases. For instance, the subfamily A member 4A “Ms4a4a” and member 6c “Ms4a6c” are significantly upregulated in EAE mice at HCS, where Ms4a6c mRNA levels decreased upon Herceptin prophylactic and therapeutic treatment regimens. Ms4a4a gene was recently associated with another autoimmune disease, rheumatoid arthritis, highlighting its potential role as a biomarker in various autoimmune diseases.77 Noticeable, Ms4a6c gene was previously reported to associate with demyelinating injury in mice.78
In conclusion, this is the first study that examines the efficacy of Herceptin/Trastuzumab in treating EAE mice. It seems that HER2 might be involved in the pathogenesis of EAE disease. Further, we have dissected the expression of mRNA for various genes that are upregulated or downregulated during heightened EAE disease, designated here as high clinical score, which could resemble the relapse stage of the disease. We also examined the effects of Herceptin on the expression of DEGs at mRNA level in EAE mice (summarized in Table 2). Several genes were reported to be affected at the sites of inflammation, ie, brains of EAE mice, including inflammatory mediators, chemokines, immuno-regulatory signaling molecules, antigen presentation, and immune-modulatory receptors. We hypothesize that Herceptin might bind to HER2 via its variable Fab region while the Fc portion would stimulate natural killer cells to perform their cytolytic activity on the pathogenic HER2+ cells through ADCC. Future studies should deeply investigate the possible mechanism of action of Herceptin in ameliorating EAE clinical symptoms. These novel findings could form the basis for using these genes as potential molecular diagnostic or prognostic biomarkers, which can also be utilized to assess the efficacy of a particular treatment in EAE or MS disease.
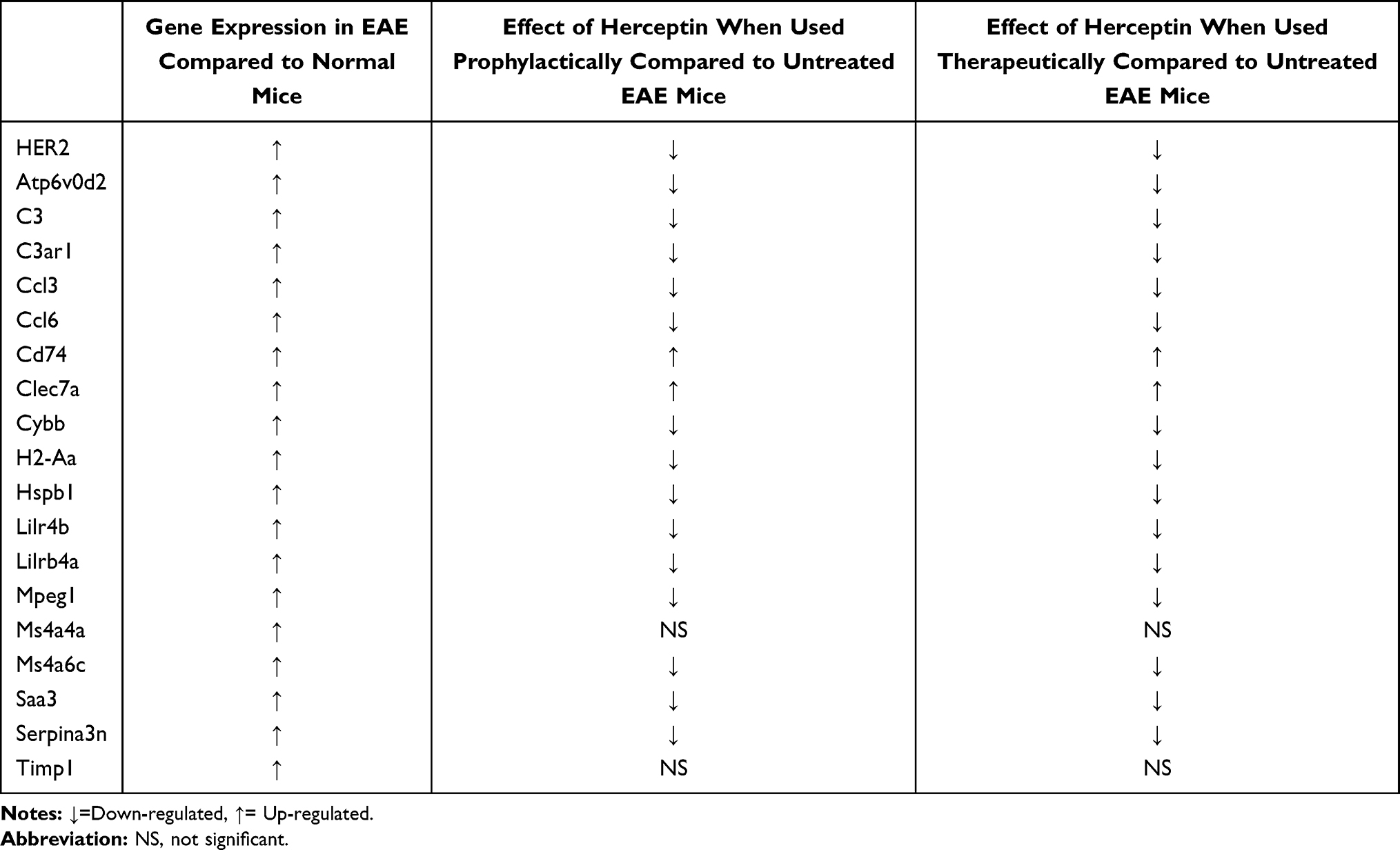 |
Table 2 Messenger RNA Expression Pattern of All Detected Genes in EAE Compared to Normal Mice, as Well as Gene Expression Upon Herceptin Treatment When Used Prophylactically or Therapeutically in Comparison to Untreated EAE Mice at the High Clinical Score (HCS) |
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; Atp6v0d2, ATPase, H+ transporting, lysosomal V0 subunit D2; BBB, blood-brain barrier; C1qc, complement component 1, q subcomponent, C chain; C3, complement component 3; C3ar1, complement component 3a receptor 1; Ccl3, chemokine (C-C motif) ligand 3; Ccl6, chemokine (C-C motif) ligand 6; Cd74, Cd74 antigen (invariant polypeptide of major histocompatibility complex, class II antigen-associated); cDNA, complementary DNA; Clec4e, C-type lectin domain family 4, member; Clec7a, C-type lectin domain family 7, member a; CNS, central nervous system; Cxcl13, chemokine (C-X-C motif) ligand 13; Cybb, cytochrome b-245, beta polypeptide; DEGs, differentially expressed genes; EAE, experimental autoimmune encephalomyelitis; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; GEO, gene expression omnibus; Gpnmb, glycoprotein (transmembrane) nmb; H&E, hematoxylin and eosin; H2-Aa, histocompatibility 2, class II antigen A, alpha; H2-Eb1, histocompatibility 2, class II antigen E beta; HCS, high clinical score; HER2/Erbb2, human epidermal growth factor receptor 2/Erythroblastic oncogene B; Hspb1, heat shock protein 1; Igsf6, immunoglobulin superfamily, member 6; IP, intraperitoneal; Lcn2, lipocalin 2; Lilr4b, leukocyte immunoglobulin-like receptor, subfamily B, member 4B; Lilrb4a, leukocyte immunoglobulin-like receptor, subfamily B, member 4A; Lyz2, lysozyme 2; MMPs, metalloproteinases; Mpeg1, macrophage expressed gene 1; MS, multiple sclerosis; Ms4a4a, membrane-spanning 4-domains, subfamily A, member 4A; Ms4a6c, membrane-spanning 4-domains, subfamily A, member 6C; PLP, proteolipid protein; RNA, ribonucleic acid; RT-qPCR, reverse transcription quantitative PCR; Saa3, serum amyloid A 3; SC, subcutaneously; Serpina3n, serine (or cysteine) peptidase inhibitor, clade A, member 3N; SJL, Swiss Jim Lambert; Timp1, tissue inhibitor of metalloproteinase 1; Wfdc17, WAP four-disulfide core domain 17.
Funding
This work was supported by grants from Terry Fox Foundation and the Sharjah Research Academy. Mena Al-Ani is a recipient of the L’Oréal-UNESCO For Women in Science Middle East Young Talents Programme.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi:10.1016/S0140-6736(08)61620-7
2. Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi:10.1146/annurev.neuro.30.051606.094313
3. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(11):2705–2712. doi:10.1093/brain/awh641
4. De Sa JC, Airas L, Bartholome E, et al. Symptomatic therapy in multiple sclerosis: a review for a multimodal approach in clinical practice. Ther Adv Neurol Disord. 2011;4(3):139–168. doi:10.1177/1756285611403646
5. Ghasemi N, Razavi S, Nikzad E. Multiple sclerosis: pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017;19(1):1–10. doi:10.22074/cellj.2016.4867
6. Fassbender K, Schmidt R, Mössner R, et al. Mood disorders and dysfunction of the hypothalamic-pituitary-adrenal axis in multiple sclerosis: association with cerebral inflammation. Arch Neurol. 1998;55(1):66–72. doi:10.1001/archneur.55.1.66
7. Nakajima H, Hosokawa T, Sugino M, et al. Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol. 2010;10(1):45. doi:10.1186/1471-2377-10-45
8. Sakai RE, Feller DJ, Galetta KM, Galetta SL, Balcer LJ. Vision in multiple sclerosis: the story, structure-function correlations, and models for neuroprotection. J Neuroophthalmol. 2011;31(4):362–373. doi:10.1097/WNO.0b013e318238937f
9. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society advisory committee on clinical trials of new agents in multiple sclerosis. Neurology. 1996;46(4):907–911. doi:10.1212/wnl.46.4.907
10. Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221–1231. doi:10.1016/S0140-6736(02)08220-X
11. Dyment DA, Ebers GC, Sadovnick AD. Genetics of multiple sclerosis. Lancet Neurol. 2004;3(2):104–110. doi:10.1016/s1474-4422(03)00663-x
12. Muñoz-Culla M, Irizar H, Otaegui D. The genetics of multiple sclerosis: review of current and emerging candidates. Appl Clin Genet. 2013;6:63–73. doi:10.2147/TACG.S29107
13. McCarthy DP, Richards MH, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol. 2012;900:381–401. doi:10.1007/978-1-60761-720-4_19
14. Koprivica V, Cho KS, Park JB, et al. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310(5745):106–110. doi:10.1126/science.1115462
15. Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712. doi:10.1126/science.2470152
16. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi:10.1056/NEJM200103153441101
17. Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;89(10):4285–4289. doi:10.1073/pnas.89.10.4285
18. Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–1684. doi:10.1056/NEJMoa052122
19. Douglas MR, Morrison KC, Jacques SJ, et al. Off-target effects of epidermal growth factor receptor antagonists mediate retinal ganglion cell disinhibited axon growth. Brain 132. 2009;132:3102–3121. doi:10.1093/brain/awp240
20. Gonzalez-Perez O, Quiñones-Hinojosa A. Dose-dependent effect of EGF on migration and differentiation of adult subventricular zone astrocytes. Glia. 2010;58(8):975–983. doi:10.1002/glia.20979
21. Al-Ani MR, Raju TK, Hachim MY, et al. Rituximab prevents the development of experimental autoimmune encephalomyelitis (EAE): comparison with prophylactic, therapeutic or combinational regimens. J Inflamm Res. 2020;13:151–164. doi:10.2147/JIR.S243514
22. Elemam NM, Hachim MY, Hannawi S, Maghazachi AA. Differentially expressed genes of natural killer cells can distinguish rheumatoid arthritis patients from healthy controls. Genes (Basel). 2020;11(5):492. doi:10.3390/genes11050492
23. Goldschmidt C, McGinley MP. Advances in the treatment of multiple sclerosis. Neuro Clin. 2001;39(1):21–33. doi:10.1016/j.ncl.2020.09.002
24. Förster M, Küry P, Aktas O, et al. Managing risks with immune therapies in multiple sclerosis. Drug Safety. 2019;42(5):633–647. doi:10.1007/s40264-018-0782-8
25. Handunnetthi L, Ramagopalan SV, Ebers GC. Multiple sclerosis, vitamin D, and HLA-DRB1*15. Neurology. 2010;74(23):1905–1910. doi:10.1212/WNL.0b013e3181e24124
26. Irizar H, Muñoz-Culla M, Zuriarrain O, et al. HLA-DRB1*15:01 and multiple sclerosis: a female association? Mult Scler. 2012;18(5):569–577. doi:10.1177/1352458511426813
27. Levy YA, Fainberg KM, Amidror T, Regev K, Auriel E, Karni A. High and dysregulated secretion of epidermal growth factor from immune cells of patients with relapsing-remitting multiple sclerosis. J Neuroimmunol. 2013;257(1–2):82–89. doi:10.1016/j.jneuroim.2013.01.012
28. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi:10.1126/science.3798106
29. Ménard S, Pupa M, Campiglio M, Tagliabue E. Biologic and therapeutic role of HER in cancer. Oncogene 22. 2003;22:6570–6578. doi:10.1038/sj.onc.1206779
30. Klapper LN, Waterman H, Sela M, Yarden Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000;60(13):3384–3388.
31. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi:10.1038/74704
32. Arnould L, Gelly M, Penault-Llorca F, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–267. doi:10.1038/sj.bjc.6602930
33. Shak S. Overview of the trastuzumab (Herceptin) anti-HER2 monoclonal antibody clinical program in HER2-overexpressing metastatic breast cancer. Herceptin Multinational Investigator Study Group. Semin Oncol. 1999;26(4 Suppl 12):71–77.
34. Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18(6):977–984. doi:10.1093/annonc/mdl475
35. Abdulkhaleq LA, Assi MA, Abdullah R, Zamri-Saad M, Taufiq-Yap YH, Hezmee MNM. The crucial roles of inflammatory mediators in inflammation: a review. Vet World. 2018;11(5):627–635. doi:10.14202/vetworld.2018.627-635
36. Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000;49(1–2):171–186. doi:10.1016/s0162-3109(00)80302-1
37. Levi‐Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol. 1987;139(7):2361–2366.
38. Morgan BP, Gasque P. Expression of complement in the brain: role in health and disease. Immunol Today. 1996;17(10):461–466. doi:10.1016/0167-5699(96)20028-f
39. Lumsden CE. The immunogenesis of the multiple sclerosis plaque. Brain Res. 1997;28(3):365–390. doi:10.1016/0006-8993(71)90052-7
40. Mead RJ, Singhrao SK, Neal JW, Lassmann H, Morgan BP. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol. 2002;168(1):458–465. doi:10.4049/jimmunol.168.1.458
41. Mead RJ, Neal JW, Griffiths MR, et al. Deficiency of the complement regulator CD59a enhances disease severity, demyelination and axonal injury in murine acute experimental allergic encephalomyelitis. Lab Invest. 2004;84(1):21–28. doi:10.1038/labinvest.3700015
42. Piddlesden SJ, Storch MK, Hibbs M, Freeman AM, Lassmann H, Morgan BP. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody‐mediated demyelinating experimental allergic encephalomyelitis. J Immunol. 1994;152(11):5477–5484.
43. Szalai AJ, Hu X, Adams JE, Barnum SR. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol Immunol. 2007;44(12):3132–3136. doi:10.1016/j.molimm.2007.02.002
44. Ristori G, Laurenti F, Stacchini P, et al. Serum amyloid A protein is elevated in relapsing-remitting multiple sclerosis. J Neuroimmunol. 1998;88(1–2):9–12. doi:10.1016/s0165-5728(98)00037-x
45. Lee JY, Hall JA, Kroehling L, et al. Serum Amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell. 2020;183(7):2036–2039. doi:10.1016/j.cell.2020.12.008
46. Connolly M, Marrelli A, Blades M, et al. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol. 2010;184(11):6427–6437. doi:10.4049/jimmunol.0902941
47. Aslam MS, Yuan L. Serpina3n: potential drug and challenges, mini review. J Drug Target. 2020;28(4):368–378. doi:10.1080/1061186X.2019.1693576
48. Haile Y, Carmine-Simmen K, Olechowski C, et al. Granzyme B-inhibitor serpina3n induces neuroprotection in vitro and in vivo. J Neuroinflammation. 2015;12:157. doi:10.1186/s12974-015-0376-7
49. van Noort JM, Bsibsi M, Gerritsen WH, et al. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010;69(7):694–703. doi:10.1097/NEN.0b013e3181e4939c
50. Peferoen LA, Gerritsen WH, Breur M, et al. Small heat shock proteins are induced during multiple sclerosis lesion development in white but not grey matter. Acta Neuropathol Commun. 2015;3:87. doi:10.1186/s40478-015-0267-2
51. Rolin J, Maghazachi AA. Implications of chemokines, chemokine receptors, and inflammatory lipids in atherosclerosis. J Leukoc Biol. 2014;95(4):575–585. doi:10.1189/jlb.1113571
52. Maghazachi AA, Al-Aoukaty A. Chemokines activate natural killer cells through heterotrimeric G-proteins: implications for the treatment of AIDS and cancer. FASEB J. 1998;12:913–924. doi:10.1096/fasebj.12.11.913
53. Rolin J, Maghazachi AA. Implications of chemokine receptors and inflammatory lipids in cancer. Immunotargets Ther. 2013;3:9–18. doi:10.2147/ITT.S32049
54. Elemam NM, Hannawi S, Maghazachi AA. Role of chemokines and chemokine receptors in rheumatoid arthritis. Immunotargets Ther. 2020;9:43–56. doi:10.2147/ITT.S243636
55. Dhaiban S, Al-Ani M, Elemam NM, Maghazachi AA. Targeting chemokines and chemokine receptors in multiple sclerosis and experimental autoimmune encephalomyelitis. J Inflamm Res. 2020;13:619–633. doi:10.2147/JIR.S270872
56. Baba T, Mukaida N. Role of macrophage inflammatory protein (MIP)-1α/CCL3 in leukemogenesis. Mol Cell Oncol. 2014;1(1):e29899. doi:10.4161/mco.29899
57. Cui LY, Chu SF, Chen NH. The role of chemokines and chemokine receptors in multiple sclerosis. Int Immunopharmacol. 2020;83:106314. doi:10.1016/j.intimp.2020.106314
58. Zhang GX, Baker CM, Kolson DL, Rostami AM. Chemokines and chemokine receptors in the pathogenesis of multiple sclerosis. Mult Scler. 2000;6(1):3–13. doi:10.1177/135245850000600103
59. Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Millerm SD. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155(10):5003–5010.
60. Asensio VC, Lassmann S, Pagenstecher A, Steffensen SC, Henriksen SJ, Campbell IL. C10 is a novel chemokine expressed in experimental inflammatory demyelinating disorders that promotes recruitment of macrophages to the central nervous system. Am J Pathol. 1999;154(4):1181–1191. doi:10.1016/S0002-9440(10)65370-9
61. LaFleur AM, Lukacs NW, Kunkel SL, Matsukawa A. Role of CC chemokine CCL6/C10 as a monocyte chemoattractant in a murine acute peritonitis. Mediators Inflamm. 2004;13(5–6):349–355. doi:10.1155/S0962935104000511
62. de la Fuente H, Cibrián D, Sánchez-Madrid F. Immunoregulatory molecules are master regulators of inflammation during the immune response. FEBS Lett. 2012;586(18):2897–2905. doi:10.1016/j.febslet.2012.07.032
63. Cardamone G, Paraboschi EM, Soldà G, Duga S, Saarela J, Asselta R. Genetic association and altered gene expression of CYBB in multiple sclerosis patients. Biomedicines. 2018;6(4):117. doi:10.3390/biomedicines6040117
64. Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2(7):502–511. doi:10.1038/35081571
65. Pagenstecher A, Stalder AK, Kincaid CL, Shapiro SD, Campbell IL. Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am J Pathol. 1998;152(3):729–741.
66. Pagenstecher A, Wussler EM, Opdenakker G, Volk B, Campbell IL. Distinct expression patterns and levels of enzymatic activity of matrix metalloproteinases and their inhibitors in primary brain tumors. J Neuropathol Exp Neurol. 2001;60(6):598–612. doi:10.1093/jnen/60.6.598
67. Gijbels K, Masure S, Carton H, Opdenakker G. Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol. 1992;41(1):29–34. doi:10.1016/0165-5728(92)90192-n
68. Gijbels K, Galardy RE, Steinman L. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J Clin Invest. 1994;94(6):2177–2182. doi:10.1172/JCI117578
69. Liedtke W, Cannella B, Mazzaccaro RJ, et al. Effective treatment of models of multiple sclerosis by matrix metalloproteinase inhibitors. Ann Neurol. 1998;44(1):35–46. doi:10.1002/ana.410440110
70. Crocker SJ, Whitmire JK, Frausto RF, et al. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am J Pathol. 2006;169(6):2104–2116. doi:10.2353/ajpath.2006.060626
71. Rijvers L, Melief MJ, van der Vuurst de Vries RM, et al. The macrophage migration inhibitory factor pathway in human B cells is tightly controlled and dysregulated in multiple sclerosis. Eur J Immunol. 2018;48(11):1861–1871. doi:10.1002/eji.201847623
72. Falcão AM, van Bruggen D, Marques S, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24(12):1837–1844. doi:10.1038/s41591-018-0236-y
73. McCormack R, Podack ER. Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. J Leukoc Biol. 2015;98(5):761–768. doi:10.1189/jlb.4MR1114-523RR
74. Chengkai Y, Tang N, Guo H, Zhang J. C-Type lectin receptor dectin-1 suppresses the development of experimental autoimmune encephalomyelitis. J Immunol. 2020;204(1 Supplement):
75. Gasque P, Singhrao SK, Neal JW, et al. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol. 1998;160(7):3543–3554.
76. Borges L, Hsu ML, Fanger N, Kubin M, Cosman D. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J Immunol. 1997;159(11):5192–5196.
77. Seyhan AA, Gregory B, Cribbs AP, et al. Novel biomarkers of a peripheral blood interferon signature associated with drug-naïve early arthritis patients distinguish persistent from self-limiting disease course. Sci Rep. 2020;10(1):8830. doi:10.1038/s41598-020-63757-3
78. Hammond TR, Dufort C, Dissing-Olesen L, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50(1):253–271.e6. doi:10.1016/j.immuni.2018.11.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.