Back to Journals » Infection and Drug Resistance » Volume 16
Molecular Epidemiology of Clinical Mycobacterium tuberculosis Isolates from Southern Xinjiang, China Using Spoligotyping and 15-Locus MIRU-VNTR Typing
Authors Yin C, Mijiti X, Liu H ![]() , Wang Q, Cao B, Anwaierjiang A, Li M, Liu M, Jiang Y, Xu M, Wan K, Zhao X, Li G, Xiao H
, Wang Q, Cao B, Anwaierjiang A, Li M, Liu M, Jiang Y, Xu M, Wan K, Zhao X, Li G, Xiao H
Received 12 October 2022
Accepted for publication 21 February 2023
Published 8 March 2023 Volume 2023:16 Pages 1313—1326
DOI https://doi.org/10.2147/IDR.S393192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Chunjie Yin,1,* Xiaokaiti Mijiti,2,* Haican Liu,3,* Quan Wang,2 Bin Cao,3,4 Aiketaguli Anwaierjiang,5 Machao Li,3 Mengwen Liu,1 Yi Jiang,3 Miao Xu,2 Kanglin Wan,3 Xiuqin Zhao,3 Guilian Li,3 Hui Xiao1
1School of Public Health, Xinjiang Medical University, Urumqi, People’s Republic of China; 2The Eighth Affiliated Hospital of Xinjiang Medical University, Urumqi, People’s Republic of China; 3State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, People’s Republic of China; 4School of Public Health, University of South China, Hengyang, People’s Republic of China; 5College of Xinjiang Uyghur Medicine, Hetian, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guilian Li; Hui Xiao, Email [email protected]; [email protected]
Background: In the last decades, the molecular epidemiological investigation of Mycobacterium tuberculosis has significantly increased our understanding of tuberculosis epidemiology. However, few such studies have been done in southern Xinjiang, China. We aimed to clarify the molecular epidemic characteristics and their association with drug resistance in the M. tuberculosis isolates circulating in this area.
Methods: A total of 347 isolates obtained from southern Xinjiang, China between Sep, 2017 and Sep, 2019 were included to characterize using a 15-locus MIRU-VNTR (VNTR-15China) typing and spoligotyping, and test for drug susceptibility profiles. Then the lineages and clustering of the isolates were analyzed, as well as their association with drug resistance.
Results: Spoligotyping results showed that 60 spoligotype international types (SITs) containing 35 predefined SITs and 25 Orphan or New patterns, and 12 definite genotypes were found, and the top three prevalent genotypes were Beijing genotype (207, 59.7%), followed by CAS1-Delhi (46, 13.6%), and Ural-2 (30, 8.6%). The prevalence of Beijing genotype infection in the younger age group (≤ 30) was more frequent than the two older groups (30~59 and ≥ 60 years old, both P values < 0.05). The Beijing genotype showed significantly higher prevalence of resistance to isoniazid, rifampicin, ethambutol, multi-drug or at least one drug than the non-Beijing genotype (All P values ≤ 0.05). The estimated proportion of tuberculosis cases due to transmission was 18.4% according to the cluster rate acquired by VNTR-15China typing, and the Beijing genotype was the risk factor for the clustering (OR 9.15, 95% CI: 4.18– 20.05).
Conclusion: Our data demonstrated that the Beijing genotype is the dominant lineage, associated with drug resistance, and was more likely to infect young people and contributed to tuberculosis transmission in southern Xinjiang, China. These findings will contribute to a better understanding of tuberculosis epidemiology in this area.
Keywords: MIRU-VNTR, spoligotyping, Beijing genotype, drug resistance, genetic diversity, Mycobacterium tuberculosis
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis complex, poses a great threaten to people’s health. In 2020, it was estimated that 9.9 million people fell ill with TB and 1.3 million died from TB among HIV-negative people worldwide.1 China had 842000 new cases which accounted for 8.5% of all estimated incident cases worldwide, and ranks the second of the 30 high TB burden countries in 2020.1 The emergence and spread of rifampicin-/multidrug-resistant TB (RR-/MDR-TB) poses a dilemma for TB control in the world and China. The World Health Organization (WHO) reported that close to half a million people developed RR-TB, of which 78% had MDR-TB globally.2 A national survey on drug-resistant TB in China showed that 5.7% of new cases had MDR-TB which was twice of the global average.3 Another report showed that the number of RR-TB cases increased year by year in China, from 10,019 in 2015 to18623in 2019, respectively.4 Tracking transmission dynamics of clinical isolates enables TB control program to effectively identify transmission hotspots and employ targeted intervention strategies.5 This is particularly important for controlling the spread of drug-resistant TB.
Molecular genotyping methods were universally applied in TB epidemiology and transmission chain study and played great roles in determining whether two or more TB cases were linked within a transmission chain.6 Classical genotyping methods involved IS6110-based restriction fragment length polymorphism (RFLP) analysis, mycobacterial interspersed repetitive-unit-variable numbers of tandem repeat (MIRU-VNTR) typing and spacer oligonucleotide typing (spoligotyping).7–10 Among these methods, IS6110-RFLP is very cumbersome and inconvenient11 and has been substituted by MIRU-VNTR and spoligotyping. MIRU-VNTR and spoligotyping both based on PCR are rapid and simple and have been widely used globally. Moreover, the results were demonstrated as digital, which can be used for inter-laboratory comparisons. Recently, the whole genome sequence (WGS) based single nucleotide polymorphisms (SNP) typing or core genome multi locus sequence typing (cgMLST) has been widely used in investigating the transmission chains and provide more clear insights into the molecular epidemiology of M. tuberculosis complex.12–14 However, WGS is expensive and needs professional technicians, and not readily available for resource limited setting and remote areas.
Xinjiang Uyghur Autonomous Region (Xinjiang, for short) locates in the northwestern of China and can be divided geographically into the southern and northern regions. According to a surveillance data between 2011 and 2015 in Xinjiang, the majority of the cases (71.8%) were reported from the southern region, pulmonary TB incidence in the southern Xinjiang region increased from 257.8 cases in 2011 to 312.7 cases in 2015 per 100,000 people.15 However, the genetic diversity of M. tuberculosis in Xinjiang especially in southern Xinjiang were grossly understudied. In this study, we applied 15-loci MIRU-VNTR typing and spoligotyping methods to characterize genetic diversity and analyze its association with drug resistance patterns in M. tuberculosis isolates from southern Xinjiang.
Materials and Methods
Patients and Mycobacterial Isolates
From Sep, 2017 to Sep, 2019, a total of 352 TB patients with M. tuberculosis isolates were consecutively collected from the TB specialist hospitals in charge of TB control in southern region of Xinjiang, including the Eighth Affiliated Hospital of Xinjiang Medical University, Kashgar Pulmonary Hospital, Kuqa County Infectious Disease Hospital and Wushi County People’s Hospital. These patients with M. tuberculosis isolates were in compliance with the TB diagnosis criteria issued by the National Health and Family Planning Commission of the People’s Republic of China.16 The HIV states of patients were not tested in this study.
The reference strains Mycobacterium bovis BCG were provided by Institute of Biological Product Control, National Institutes for Food and Drug Control, Beijing, China and M. tuberculosis H37Rv were from ATCC (27,294). The sub-culture, collection and inactivation of all strains were performed in a Biosafety level 3 (BSL-3) laboratory in Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention (NICDC, China CDC). Demographical data of TB cases were collected using patients’ clinical records.
Genomic DNA Extraction
DNA was extracted from mycobacterial culture according to the standard CTAB/NaCl method.17 Briefly, colonies from culture positive isolates and the standard strains of H37Rv and M. bovis were suspended in 1.5 mL Eppendorf tube with 400 µL of 1XTE buffer, and then heated for 30 min at 80 °C to kill the bacteria. The samples were then mixed with 50 µL 10 mg/mL lysozyme and incubated at 37°C for 16–24h. Then 5 µL 10 mg/mL proteinase K and 70 µL 10% SDS was added to the mixture and incubated for 10 min at 65 °C. After adding 5 M NaCl, CTAB-NaCl (4.1% NaCl and 10% CTAB) and chloroform-isoamyl alcohol (24:1 [vol/vol]), the tubes were centrifuged for 15 min at 13,000 × g in an Eppendorf centrifuge, the aqueous phase was transferred to another tube and mixed with isopropanol at −20 °C for 30 min and then centrifuged for 15 min at 13,000 × g. The DNA pellet was dissolved in 200–300 µL of TE buffer and stored at −20°C for further molecular analysis.
Spoligotyping
A total of 352 isolates were analyzed by spoligotyping. H37Rv and BCG were used as positive controls and distilled water as a negative control. Spoligotyping was performed following a standard protocol described previously.18 Briefly, the direct repeat (DR) regions were amplified with a pair of primers (forward:5’- Biotin- GGTTTTGGGTCTGACGAC −3’, reverse: 5’- GGTTTTGGGTCTGACGAC −3’), and the resulting PCR products were hybridized to a set of 43 spacer-specific oligonucleotide probes, which were immobilized on the membrane. The hybridized products were identified by enhanced chemiluminescence system (ECL, Amersham, UK). The spoligotype results were entered into an Excel spreadsheet in binary format and analyzed using the SITVIT2 database (http://www.pasteur-guadeloupe.fr:8081/SITVIT2/index.jsp) of the Pasteur Institute of Guadeloupe which contained more than 90,000 patterns from more than 160 countries of patient origin to determine the spoligotype international types (SITs) and genotypes.19
Mycobacterial Interspersed Repetitive-Unit-Variable Numbers of Tandem Repeat Typing
We performed the 15-loci MIRU-VNTR typing as described by Wan et al.20 The 15-loci set was selected by Wan et al15 and called VNTR-15China to facilitate efficient genotyping of large number of isolates with a limited panel of loci that would correctly cluster the bacteria into the main clades/genotype families in China. These loci included seven mycobacterial interspersed repetitive unit (MIRU) loci (MIRU10, MIRU16, MIRU23, MIRU26, MIRU27, MIRU39 and MIRU40), five exact tandem repeat (ETR) loci (ETR-A, ETR-B, ETR-C, ETR-D [alias MIRU04] and ETR-E [alias MIRU31]) and 3 Mtub loci (Mtub21, Mtub30 and Mtub39), and all of these 15 loci were also included in the VNTR-24 scheme described by Supply et al.21 In short, PCR products using primer pairs specific for the flanking regions of each locus were acquired, then verified by electrophoresis in 1.5% agarose gels and photographed with Bio-Rad ChemiDoc XRS+ imaging system (Bio-Rad, Hercules, CA, USA). The sizes of the amplicons and the number of the repeated unit of each locus were then determined by the Quantity one software (Bio-Rad, Hercules, CA, USA). The repeated units of all isolates at the 15 loci were used to perform cluster analysis with BioNumerics software (version 7.6, Applied Maths, Belgium).
Drug Susceptibility Testing
Three hundred out of the 352 M. tuberculosis isolates were used to perform drug susceptibility testing (DST). The DST was performed as described in our previous study.22 The isolates were first used to perform DST with 7H9 liquid medium based 96-well plate DST kit (Encode Medical Engineering Co., Ltd, Zhuhai, China) in the Eighth Affiliated Hospital of Xinjiang Medical University. For the isolates with inconsistent phenotypic and genotypic susceptibility results (the results were not shown in the present study), the traditional proportional method with Lowenstein-Jensen (L-J) medium was applied to confirm the susceptibility profiles in the NICDC, China CDC. The critical concentrations for the four studied drugs indicate resistance with 96-well plate DST kit were as following: isoniazid (INH), 0.4 µg/mL; rifampicin (RMP), 4.0 µg/mL; streptomycin (STR), 4.0 µg/mL; ethambutol (EMB), 5.0 µg/mL, while with proportional method were as following: INH, 0.2 µg/mL; RMP, 40.0 µg/mL; STR, 4.0 µg/mL; EMB, 2.0 µg/mL.23
Data Analysis
Statistical analysis of data was done with SPSS (version 21.0, SPSS Inc., Chicago, IL, United States). Association between categorical variables was evaluated using Chi-square test. Odds ratio (OR) with a 95% confidence interval (CI) was determined by logistic regression models. Multivariable logistic regression model was also used to study the associations between the clustering of the M. tuberculosis isolates based on MIRU-VNTR typing or spoligotyping and demographic and drug-resistance characteristics as applicable. P values ≤ 0.05 were considered statistically significant. The cluster analysis or generation of minimum spanning tree (MST) were performed by BioNumerics software (version 7.6, Applied Maths, Belgium). A cluster was defined as two or more isolates sharing identical spoligotype and/or VNTR-15China patterns,24 and the clustering rate was calculated using the formula, Rc = (nc - c)/n, in which n is the total number of isolates in the sample, nc is the total number of clustered isolates (size two or greater) and c is the number of clusters (genotypes represented by at least two cases).25 The minimum spanning tree20,26 was constructed by with the following options: (i) in case of equivalent solutions in terms of calculated distances, the selected tree was the one containing the highest number of links between genotypes differing at only one locus (“Highest number of single locus variants” option); (ii) the creation of hypothetical types (missing links) reducing the total length of the tree was allowed.
Results
Demographic Characteristics of the Patients
In the present study, due to five out of 352 isolates failed to obtain the VNTR typing results, we finally included 347 patients and their isolates for analysis. The demographic characteristics of the 347 TB patients are summarized in Table 1. The patients comprised of 46.4% (161/347) males and 53.6% (186/347) females. The ages of the patients ranged from 17 to 95 years. Of the 347 patients, 170 (49.0%) were new cases and 177 (51.0%) were previously treated cases. Geographically, the majority of patients were from Akesu (n = 154; 44.4%), followed by from Kashgar (93, 26.8%), Hetian (67, 19.3%) and other areas (33, 9.5%).
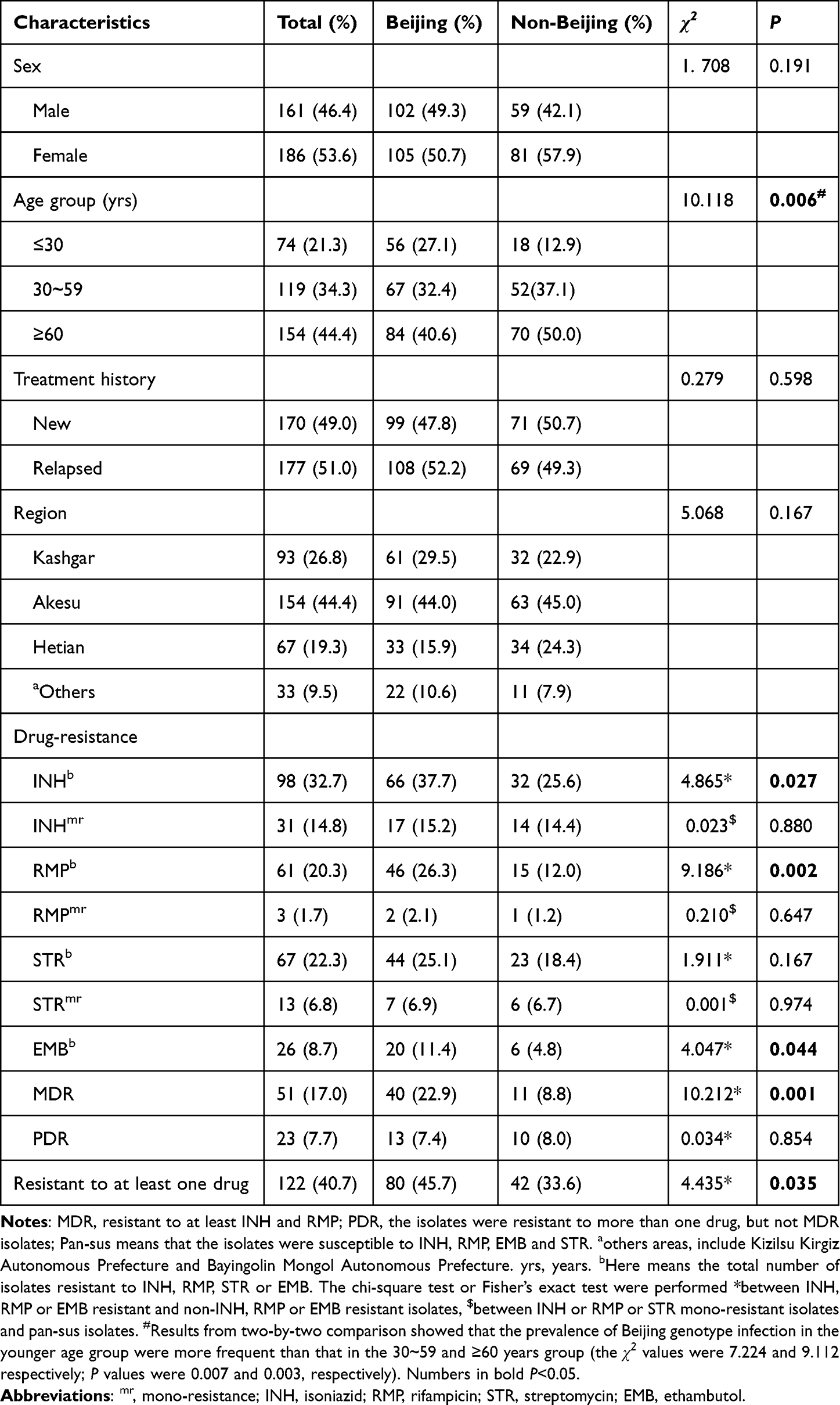 |
Table 1 Demographic and Drug-Resistance Characteristics of Patients with Beijing and Non-Beijing Family Isolates |
Drug-Resistance Characteristics of the Isolates
Of the 347 isolates, only 300 had confirmed DST results. As showed in Table 1, the drug-resistance profiles of the 300 isolates showed that INH owned the highest resistance rate (32.7%, 98/300), followed by STR (22.3%, 67/300), RMP (20.3%, 61/300), and EMB (8.7%, 26/300). In all, 122 (40.7%) isolates were resistant to at least one of the four drugs, 51 (17.0%) were MDR, 23 (7.7%) were resistant to more than one drug but not MDR and defined as poly-drug resistant (PDR). On the other hand, we observed that INH mono-resistant rate was 14.8% (31/300), RMP mono-resistant rate was 1.7% (3/300), STR mono-resistant rate was 6.8% (13/300) and EMB mono-resistant rate was 0.3% (1/300).
Spoligotype Distribution of the Mycobacterium tuberculosis Isolates
The spoligotyping data of the 347 M. tuberculosis isolates were analyzed in the SITVIT2 database and 60 distinct spoligo patterns containing 35 predefined SITs and 25 Orphan or New patterns were found (Table 2 and Supplemental Figure S1). The 347 isolates were classified into Beijing family (207, 59.7%), CAS (47, 13.5%), Ural-2 (30, 8.6%), EAI (11, 3.2%), H (3, 0.9%), LAM9 (2, 0.6%), T (21, 6.1%) and unknown or not defined (orphan or new) (26, 7.5%). Among the 207 Beijing genotype isolates, 189 carried classical type (SIT1) and 7 nonclassical types (SIT1162, SIT1311, SIT190, SIT260, SIT265, SIT269, or SIT632). Of the 46 CAS1-Delhi-type strains, the SITs were 25 (n = 15), 26 (n = 13), 357 (n = 14), 381 (n = 2), 1203 (n = 1) and 952 (n = 1). Of the 30 Ural-2 family isolates, SIT127 was the most common sub-genotype (29/30). T family strains covered four sub-lineages of T (1, 0.3%), T1 (12, 3.5%), T2 (5, 1.4%) and T3 (3, 0.9%), and divided into 15 different SITs (Table 2). In total, 321 were classified into 7 predefined lineages or 12 predefined sub-lineages.
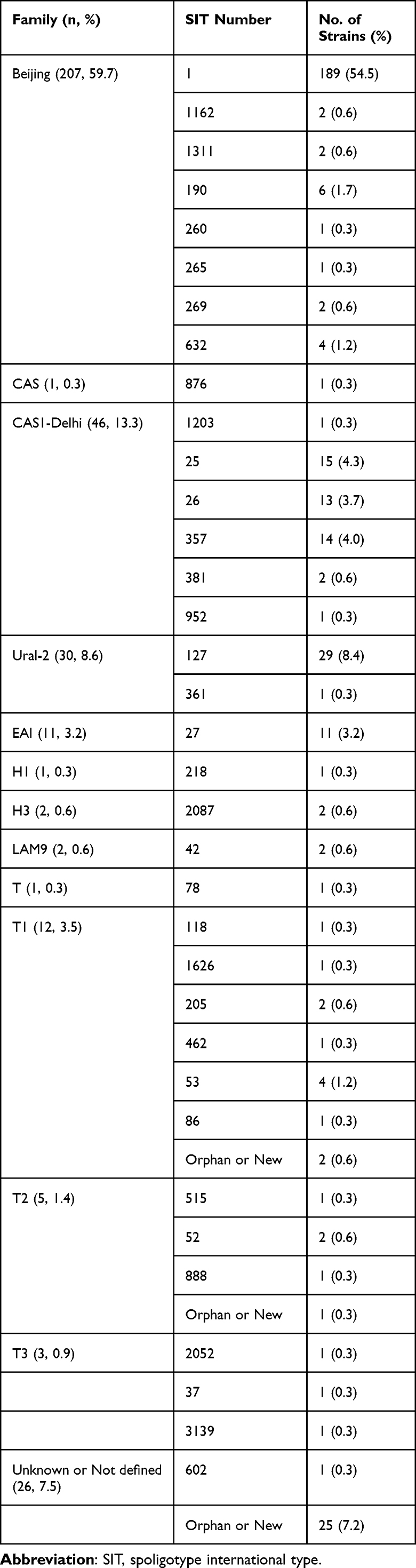 |
Table 2 The Distribution of Spoligotypes of 347 Mycobacterium tuberculosis Isolates |
Clustering analysis on the spoligotyping binary codes showed that 313 (90.2%) isolates formed 22 clusters, 34 (9.8%) isolates did not form clusters, the clustering rate was 83.9% (Supplemental Figure S1). Of the 22 clusters, the six most predominant clusters included SIT 1 (Beijing, 189 isolates), SIT 127 (Ural-2, 29 isolates), SIT 25 (CAS1-Delhi, 15 isolates), SIT 357 (CAS1-Delhi, 14 isolates), SIT 26 (CAS1-Delhi, 13 isolates) and SIT 27 (EAI, 11 isolates), these isolates account for 78.1% (271/347) of all isolates.
We further analyzed the factors contributing to the clusters, however, none of the demographic characteristics and drug-resistance profiles was found to be associated with the clustering based on spoligotyping binary codes (Supplemental Table S1).
Associations Between Drug-Susceptibility Patterns or Demographic Characteristics and Mycobacterium tuberculosis Genotypes
Statistical analysis on the 300 isolates tested for drug susceptibility revealed that the Beijing genotype showed higher proportions with INH, RMP or EMB resistance, or MDR or at least one drug resistance than non-Beijing genotype (all P values ≤0.05, Table 1). We also analyzed the distribution differences of Beijing genotype between mono-INH, mono-RMP or mono-STR resistant and pan-susceptible isolates, however, no significance difference was found. Of the total 347 cases, 74 cases were ≤30 years, 119 cases were between 30~59 years and 154 cases were ≥60 years, of which 56 (27.1%), 67 (32.4%) and 84 (40.6%) infected with Beijing genotype, respectively. The distribution of the Beijing genotype among the age groups showed statistically significant difference (P <0.05). We then performed a two-by-two comparison and found that the prevalence of Beijing genotype infection in the younger age group (≤30 years) were more frequent than the two older groups (both P values <0.05). The genotype distributions among groups with distinct sex, treatment histories or origin regions were not observed with statistically significant difference (all P values > 0.05). The results were shown in Table 1.
VNTR-15china Typing Results
Of the total 347 cases, the VNTR-15China typing revealed 283 different genotypes: 254 (73.20%) isolates had a unique VNTR profile and 93 (26.80%) isolates were grouped into 29 clusters. The largest cluster contained eight isolates of the Beijing genotype (SIT1). Whilst the other 28 clusters were composed of two to six isolates as following: four with six isolates, one with five isolates, one with four isolates, eight with three isolates, and 14 clusters with two isolates (Supplemental Figure S2). The clustering rate was 18.4%. We also explored the factors that contributed to the clustering of M. tuberculosis isolates based on VNTR-15China typing by Chi-square test, and found that the clustering rate was strongly associated with Beijing genotype isolates (OR 9.94, 95% CI: 4.79–20.63), MDR (OR 2.34, 95% CI: 1.21–4.52) and RMP resistance (OR 2.66, 95% CI: 1.11–6.35) (Table 3). Then the genotype and resistance were included in the multivariable logistic regression model for further analysis. The results showed that only Beijing genotype was the risk factor for the clustering of M. tuberculosis isolates (OR 9.15, 95% CI: 4.18–20.05) (Table 4).
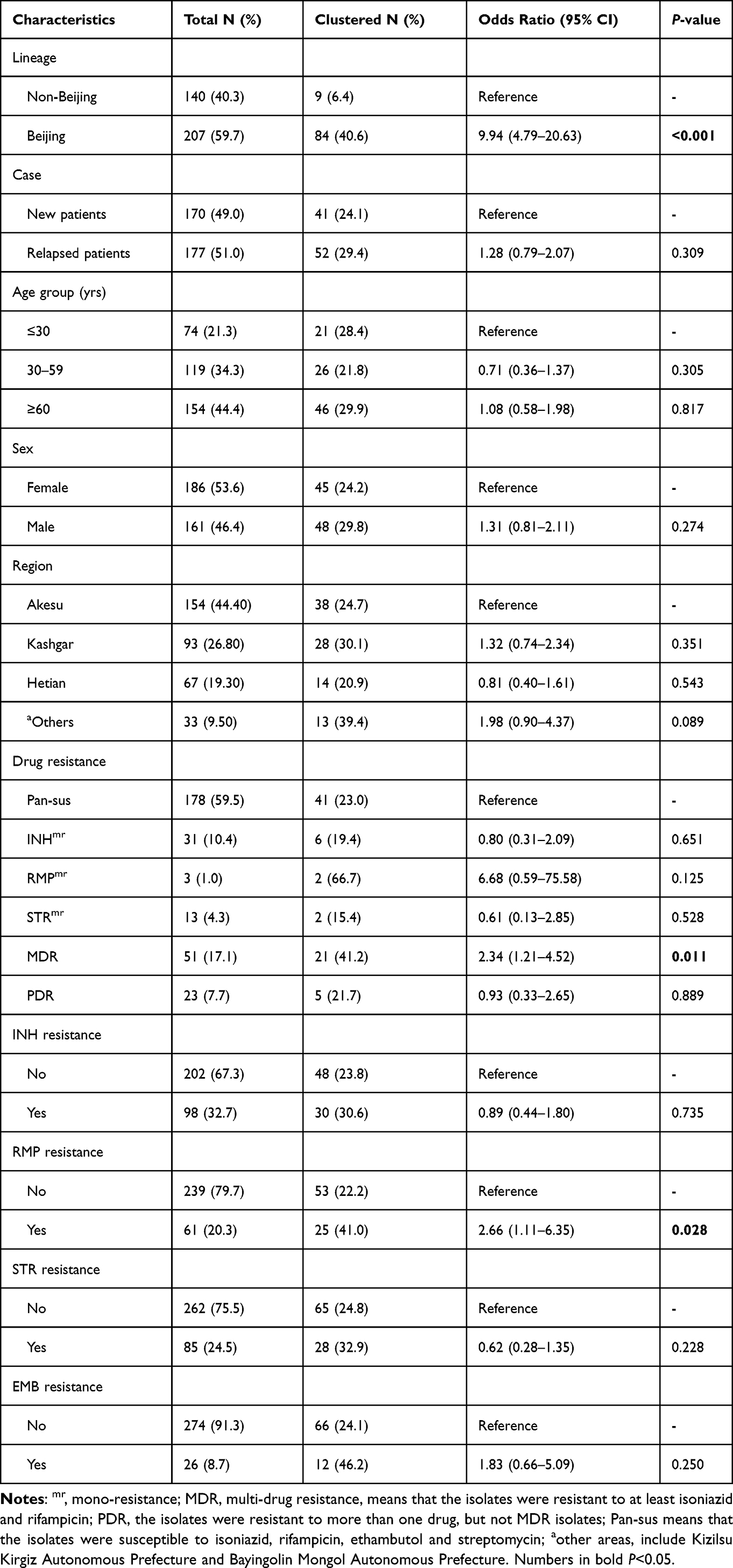 |
Table 3 Association Between the Clustering of the Mycobacterium tuberculosis Isolates Based on VNTR-15China Typing and Various Factors |
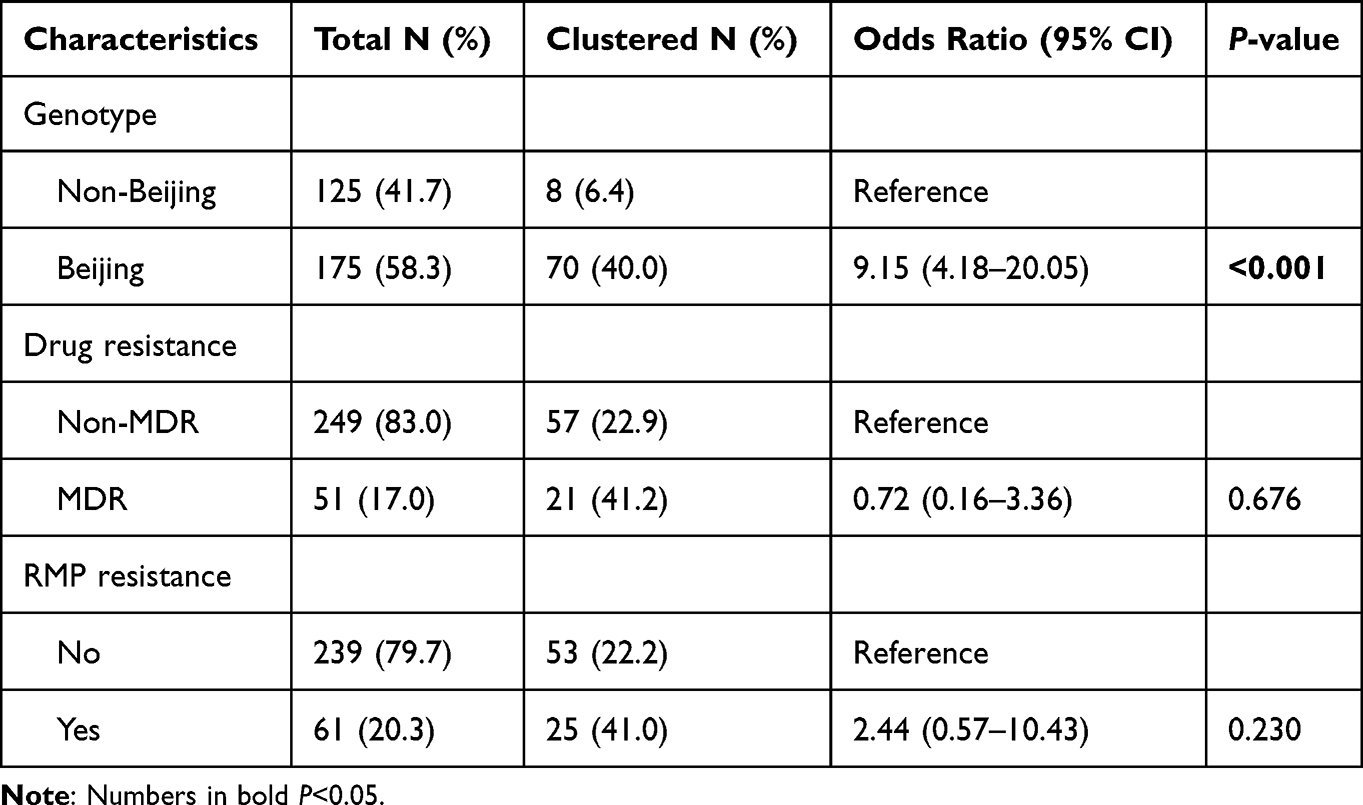 |
Table 4 Multivariable Logistic Regression Analysis on the Association Between the Clustering of Mycobacterium tuberculosis Isolates Based on VNTR-15China Typing and Various Factors |
In the present study, we tried to performed clustering analysis on the combined results of VNTR-15China typing and 43 binary codes of spoligotyping. However, we found that the clustering result on the combined data was the same as that on the VNTR-15China typing data (Supplemental Figure S3).
To visualize and demonstrate the genetic linkages among the clusters, we constructed a MST based on the VNTR-15China typing data and 43 binary codes of spoligotyping using the BioNumerics software (Figure 1). From the MST, it was evident that most of the clusters of the same genotypes were close to each other. It was also apparent that one strikingly large cluster belonging to the Beijing family. The second large cluster belonging to the CAS1-Delhi. On the other hand, Ural-2, T1, T2, T3, H3, LAM9, T, H1 and H3 were close to each other that may be categorized as a cluster. Moreover, many isolates with Orphan or New patterns were scattered in the MST.
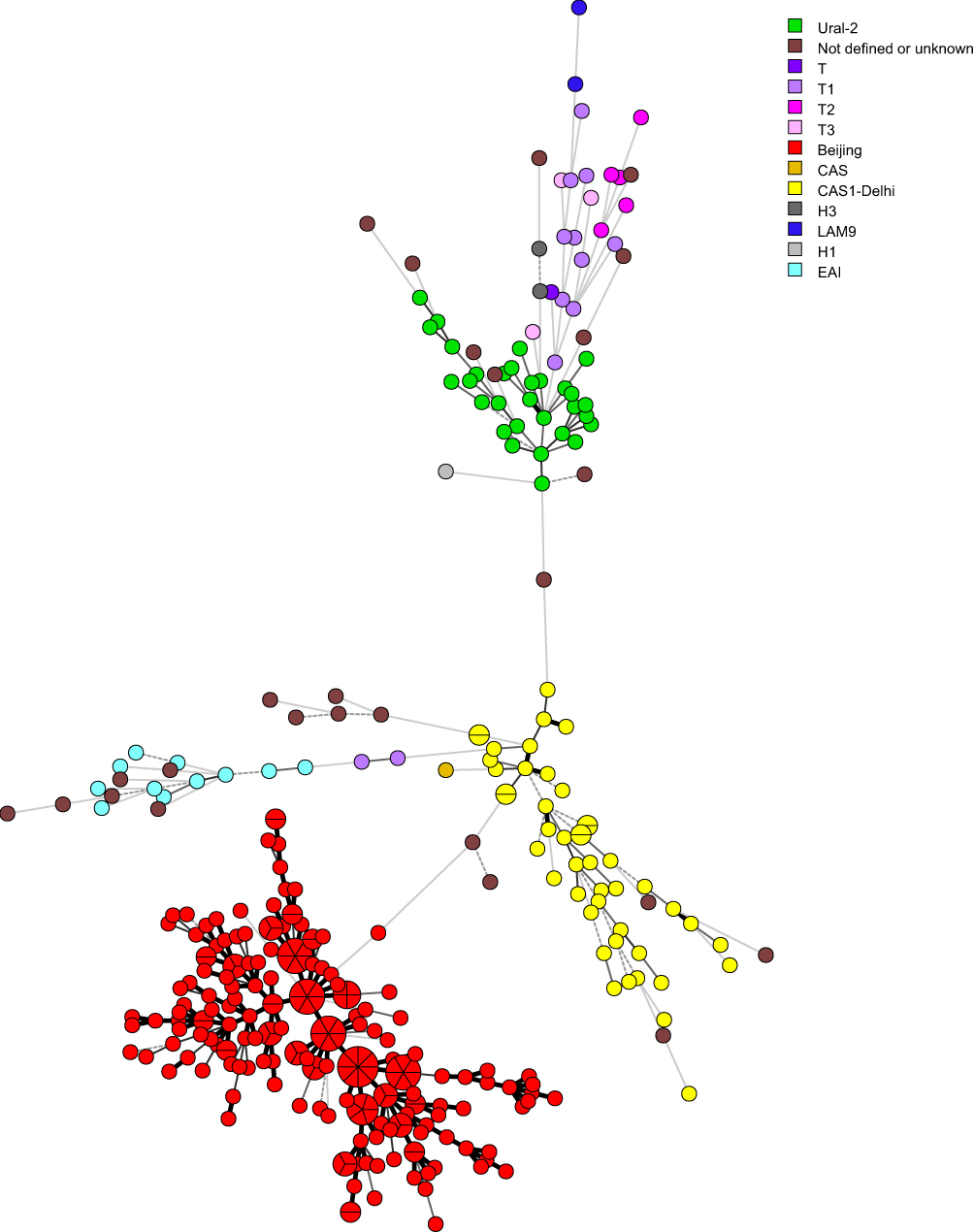 |
Figure 1 Minimum spanning tree (MST) based on the spoligotyping data of 347 M. tuberculosis isolates. Lines between nodes indicate genetic distance between genotypes. Portions in the circle divided by the lines indicated the number of the isolates belonging to a particular genotype. Classification of the isolates into different phylogenetic lineages is visualized by color coding. |
Discussion
Molecular epidemiology tools have been rarely applied in the control of TB in southern Xinjiang, China. In this study, we applied spoligotyping and VNTR-15China typing to characterize the genetic diversity of M. tuberculosis isolated from southern Xinjiang, China and explored its association with drug resistance and demographical characteristics. This is the first in-depth analysis on the molecular epidemiology of M. tuberculosis in this region and the results will provide clues for the TB control there.
Previous studies showed that the Beijing genotype of M. tuberculosis is the most prevalent in the national wide of China varied from 58.4% to 90.5%.22,27–33 The percentage of Beijing genotype isolates in the south of China was lower than that in the north.28 In this study, the spoligotyping results showed that the Beijing genotype is the predominant lineage in southern Xinjiang, accounting for 59.7% of the isolates, which is in line with the data from the south of China (53.2%), but lower than that from the northern China (76.5%).28 So, as a northwestern region, why southern Xinjiang showed similar prevalence frequency of Beijing genotype with the south of China needs further research. Several previous studies from Vietnam reported that the infection of Beijing genotype strains were more prevalent in young people,34,35 which was consistent with the results found in our study. For the higher prevalence of Beijing genotype in the younger people in southern Xinjiang, we speculated that compared with the elder people, the youngers were more active during work and life and have more chance to be infected with the Beijing genotype by contacting with people from other areas of China where Beijing genotype were prevalent. The second frequent family in our study was the CAS-type family (n=47, 13.6%), which was followed by Ural-2 family (n=30, 8.6%). The EAI, H1, H3, LAM9, T, T1, T2 and T3 families were found with less frequencies in this region. In previous reports,36–38 the CAS family was found as the predominant genotype in the countries of Afghanistan, Pakistan and India. Xinjiang located in northwestern China, is a vast area composed of a variety of ethnic communities such as Uyghur, Han, Kazakh and Other ethnic groups. Meanwhile, Xinjiang shares geographic borders with Russia, Pakistan, India, Mongolia, Kyrgyzstan, Kazakhstan, Afghanistan, Tajikistan, and so on, and has frequent trading, tourism and/or migration between countries. All of such features may subsequently lead to the genotypic diversity of M. tuberculosis in southern Xinjiang.
In the present study, we also analyzed the association of Beijing genotype with drug resistance in M. tuberculosis isolates from southern Xinjiang, and found that the Beijing genotype has higher proportion of isolates resistant to INH (37.7% Vs 25.6%), RMP (26.3% Vs 12.0%), EMB (11.4% Vs 4.8%), MDR (22.9% Vs 8.8%) or to at least one of four drugs (45.7% Vs 33.6%) than the non-Beijing genotype. The association between Beijing genotype and MDR has been shown in many settings.39–41 Study from Hunan, China reported that Beijing genotype had a significantly higher proportion of MDR than the non-Beijing genotype (OR 3.28, 95% CI: 1.01–10.37), and also showed a higher risk for developing drug resistance to all four first-line drugs (OR 5.97, 95% CI 1.05–44.33).39 Studies from Nepal40 and Bangladesh41 found that the Beijing genotype had an important role in transmitting MDR-TB. Another systematic review also found that in Pakistan and Iran the Beijing genotype exhibited a strong and statistically significant association with drug resistance.42 However, studies from Beijing43 and Chongqing44 of China both reported that no significant association was observed between the drug resistance and Beijing genotype. To exclude the possible confounding effects of MDR or PDR in a isolate, we analyzed the association of Beijing genotype with INH, RMP or STR resistance among the mono-INH, mono-RMP or mono-STR resistant and pan-susceptible isolates, however, no significant association was found. Even though, the sample size of mono-resistant isolates in the present study was small, and further studies with larger samples are needed. One limitation of the present study is that no second line drug resistance profiles of the isolates was acquired and we failed to find the association between the second line drug resistance and Beijing genotype. Anyway, the exact mechanism under the association of the Beijing genotype and drug resistance remains unresolved. Future studies should be conducted to determine why Beijing genotype accelerated the acquisition of drug resistance in some areas.
In our study, 347 M. tuberculosis isolates from southern Xinjiang were also analyzed by VNTR-15China typing, and 283 genotypes were obtained. There were 254 isolates with unique genotypes. Previous literature showed that isolates with unique genotypes were considered as unrelated isolates and patients with these isolates were always recognized resulting from independent infection or endogenous recurrence, whilst isolates that shared the same genotype were considered clustered and were assumed to be epidemiologically linked, although the link may be indirect.45 The clustering rates can vary depending on study design and setting or any link to an outbreak, which complicates any comparison of clustering and transmission rate consistency across studies. Our data revealed that the clustering rate of M. tuberculosis isolates by VNTR-15China was 18.4%, while that from Xinjiang, China by VNTR-9 scheme and Deng et al was 7.39%,46 and by VNTR-15China and Liu et al was 37.30%.47 The differences in the clustering rates in Xinjiang, China may be due to the sample coverage and collection periods, the isolates of the other two reports maybe mainly from the northern Xinjiang region with few from the southern Xinjiang according to the isolate source hospital’s location and function: Deng et al46 collected isolates from May 2015 to April 2018 while Liu et al47 collected isolates from 2006 and 2011, we collected the data from southern Xinjiang from 2017 to 2019. Other reports showed that the clustering rate from Yunnan Province, China by 12-locus VNTR was 23.6%.48 The clustering rate in the present study suggested that there was a relative high level of transmission in southern Xinjiang, however, the true transmission rate maybe lower if more loci were applied. In 18 out of the 29 clusters by the VNTR-15China typing, some patients in each cluster were further confirmed to own the same geographical origin, suggesting that genotyping combined with traditional tracing method will be an effective approach in identifying the transmission chain of M. tuberculosis isolates. We further found that the clustering rate was strongly associated with Beijing genotype (OR 9.15, 95% CI: 4.18–20.05), which was in line with previous studies from Peru.47–49
In the present study, the clustering rate of VNTR-15China typing was far lower than that of spoligotyping (18.4% Vs 83.9%), and the addition of the 43 binary codes of spoligotyping to the VNTR-15China typing data did not change the clustering rate of the latter. This result confirms that MIRU-VNTR has higher discriminatory than spoligotyping, which was in line with previous studies.50–52 The discriminatory level of spoligotyping is relatively low, and the genotypic clustering cannot be used to estimate TB transmission rates. Meanwhile, a previous study pointed out that the formation of clusters by MIRU-VNTR typing or spoligotyping was only an indicative of TB transmission, and the real TB transmission should be further confirmed by epidemiological linkage. It should be noticed that the clustering rates are not only due to the genotyping method, but also to the study subjects based on sampling or population, sample coverage and study duration. For example, the unique isolates might be clustered if some of the missing isolates had been available (eg, other cases with the same strain moved or located outside the study area were included). Though the clusters of M. tuberculosis by spoligotyping or MIRU-VNTR typing is an indicative of transmission, we could not uncover the timing of transmission events within a given cluster. Meehan et al14 reported that clusters based on spoligotyping could encompass transmission events that occurred almost 200 years prior to sampling, while 24-locus MIRU-VNTR typing often represented three decades of transmission and WGS based genotyping applying low SNP or cgMLST allele thresholds allows for determination of recent transmission events, eg in timespans of up to 10 years for a 5 SNP/allele cut-off. In 11 out of the 29 clusters by the VNTR-15China typing in the present study, we found that the isolates from the same cluster were from different geographical locations, similar phenomena have also been found in some earlier studies,41,53 further proving that clusters by MIRU-VNTR typing could not represent recent transmission and remote transmission maybe identified in a cluster. When classical genotyping methods are employed, contact tracing and/or WGS were suggested to imply to investigate potential transmission hotspots.14
In conclusion, our results revealed that Beijing genotype is the predominant genotype, associated with drug resistance, and was more likely to infect young people in southern Xinjiang, China. Meanwhile, the data showed the genotypic diversity of non-Beijing genotype in this region. In addition, we observed that Beijing genotype was more likely to contribute to the TB transmission in southern Xinjiang. The genotyping data are useful to map the population structure of M. tuberculosis in southern Xinjiang and to the TB Control Program in this region.
Abbreviations
BSL-3, Biosafety level 3; cgMLST, core genome multi locus sequence typing; CTAB, Cetyltrimethylammoniumbromide; CI, confidence interval; DST, drug susceptibility testing; EMB, ethambutol; INH, isoniazid; IS6110-RFLP, IS6110-based restriction fragment length polymorphism; MIRU-VNTR, mycobacterial interspersed repetitive-unit-variable numbers of tandem repeat; MDR-TB, multidrug-resistant Tuberculosis; MST, minimum spanning tree; NICDC, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention; OR, Odds ratio; PDR, poly-drug resistant; RR-TB, rifampicin resistant tuberculosis; RMP, rifampicin; SNP, single nucleotide polymorphisms; STR, streptomycin; TB, tuberculosis; WHO, World Health Organization; WGS, whole genome sequence.
Data Sharing Statement
The original contributions presented in the study are included in the article and Supplementary Materials, further inquiries can be directed to the corresponding authors.
Ethics Approval and Informed Consent
The experimental protocol was established, according to the ethical guidelines of the Helsinki Declaration and was approved by the Human Ethics Committee of the Eighth Affiliated Hospital of Xinjiang Medical University (XJMU8HEC-20161215). Written informed consent was obtained from individuals.
Acknowledgments
The authors thank all staffs working in the tuberculosis hospitals or institutes for tuberculosis control of Xinjiang Uygur Autonomous Region Chest Hospital, Kashgar, Kuqa and Wushi for supplying the clinical M. tuberculosis isolates and collecting data. Chunjie Yin, Xiaokaiti Mijiti and Haican Liu are co-first authors for this study.
Funding
This study was supported by grants from the Major Science and Technology Project of Xinjiang Uygur Autonomous Region (The research and application of key technologies for tuberculosis prevention and treatment in southern Xinjiang, 2017A03006-3) and the Ministry of Science and Technology, China (Mega Projects of Research on the Prevention and Control of HIV/AIDS, Viral Hepatitis Infectious Diseases, 2018ZX10103001-003-012).
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Global Tuberculosis Report 2021. World Health Organization; 2021.
2. World Health Organization. Global Tuberculosis Report 2020. World Health Organization; 2020.
3. Zhao Y, Xu S, Wang L, et al. National survey of drug-resistant tuberculosis in China. N Engl J Med. 2012;366(23):2161–2170. doi:10.1056/NEJMoa1108789
4. Su W, Ruan YZ, Li T, Du X, Jiang JW, Li RZ. Characteristics of rifampicin-resistant tuberculosis detection in China, 2015–2019. Infect Dis Poverty. 2021;10(1):99. doi:10.1186/s40249-021-00883-8
5. Kendall EA, Fofana MO, Dowdy DW. Burden of transmitted multidrug resistance in epidemics of tuberculosis: a transmission modelling analysis. Lancet Respir Med. 2015;3(12):963–972. doi:10.1016/S2213-2600(15)00458-0
6. Merker M, Kohl TA, Niemann S, Supply P. The evolution of strain typing in the mycobacterium tuberculosis complex. Adv Exp Med Biol. 2017;1019:43–78 doi:10.1007/978-3-319-64371-7_3.
7. Cavusoglu C, Turhan A, Akinci P, Soyler I. Evaluation of the Genotype MTBDR assay for rapid detection of rifampin and isoniazid resistance in Mycobacterium tuberculosis isolates. J Clin Microbiol. 2006;44(7):2338–2342. doi:10.1128/JCM.00425-06
8. Cerezo I, Jiménez Y, Hernandez J, Zozio T, Murcia MI, Rastogi N. A first insight on the population structure of Mycobacterium tuberculosis complex as studied by spoligotyping and MIRU-VNTRs in Bogotá, Colombia. Infect Genet Evol. 2012;12(4):657–663. doi:10.1016/j.meegid.2011.07.006
9. Mokrousov I, Narvskaya O, Limeschenko E, Vyazovaya A, Otten T, Vyshnevskiy B. Analysis of the allelic diversity of the mycobacterial interspersed repetitive units in Mycobacterium tuberculosis strains of the Beijing family: practical implications and evolutionary considerations. J Clin Microbiol. 2004;42(6):2438–2444. doi:10.1128/JCM.42.6.2438-2444.2004
10. Sola C, Filliol I, Legrand E, et al. Genotyping of the Mycobacterium tuberculosis complex using MIRUs: association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics. Infect Genet Evol. 2003;3(2):125–133. doi:10.1016/S1567-1348(03)00011-X
11. Moström P, Gordon M, Sola C, Ridell M, Rastogi N. Methods used in the molecular epidemiology of tuberculosis. Clin Microbiol Infect. 2002;8(11):694–704. doi:10.1046/j.1469-0691.2002.00460.x
12. He W, Tan Y, Liu C, et al. Drug-resistant characteristics, genetic diversity, and transmission dynamics of rifampicin-resistant Mycobacterium tuberculosis in Hunan, China, revealed by whole-genome sequencing. Microbiol Spectr. 2022;10(1):e0154321. doi:10.1128/spectrum.01543-21
13. Galagan JE. Genomic insights into tuberculosis. Nat Rev Genet. 2014;15(5):307–320 doi:10.1038/nrg3664.
14. Meehan CJ, Moris P, Kohl TA, et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine. 2018;37:410–416. doi:10.1016/j.ebiom.2018.10.013
15. He X, Cao M, Mahapatra T, et al. Burden of tuberculosis in Xinjiang between 2011 and 2015: a surveillance data-based study. PLoS One. 2017;12(11):e0187592. doi:10.1371/journal.pone.0187592
16. China NHaFPCotPsRo. Diagnosis of Tuberculosis, WS 288–2017. Beijing: People’s Medical Publishing House; 2017.
17. van Embden JD, Cave MD, Crawford JT, et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31(2):406–409. doi:10.1128/jcm.31.2.406-409.1993
18. Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35(4):907–914. doi:10.1128/jcm.35.4.907-914.1997
19. Couvin D, David A, Zozio T, Rastogi N. Macro-geographical specificities of the prevailing tuberculosis epidemic as seen through SITVIT2, an updated version of the Mycobacterium tuberculosis genotyping database. Infect Genet Evol. 2019;72:31–43. doi:10.1016/j.meegid.2018.12.030
20. Wan K, Liu J, Hauck Y, et al. Investigation on Mycobacterium tuberculosis diversity in China and the origin of the Beijing clade. PLoS One. 2011;6(12):e29190. doi:10.1371/journal.pone.0029190
21. Supply P, Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006;44(12):4498–510 doi:10.1128/JCM.01392-06.
22. Anwaierjiang A, Wang Q, Liu H, et al. Prevalence and molecular characteristics based on whole genome sequencing of Mycobacterium tuberculosis resistant to four anti-tuberculosis drugs from Southern Xinjiang, China. Infect Drug Resist. 2021;14:3379–3391. doi:10.2147/IDR.S320024
23. World Health Organization. Updated Interim Critical Concentrations for First-Line and Second-Line DST (as for May 2012). World Health Organization; 2012.
24. Shah Y, Poudel A, Maharjan B, et al. Genetic diversity of Mycobacterium tuberculosis Central Asian Strain isolates from Nepal and comparison with neighboring countries. Trans R Soc Trop Med Hyg. 2019;113(4):203–211. doi:10.1093/trstmh/try136
25. Torkaman MR, Nasiri MJ, Farnia P, Shahhosseiny MH, Mozafari M, Velayati AA. Estimation of recent transmission of Mycobacterium Tuberculosis strains among Iranian and afghan immigrants: a cluster-based study. J Clin Diagn Res. 2014;8(9):Dc05–8 doi:10.7860/JCDR/2014/8886.4864.
26. Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186(5):1518–1530. doi:10.1128/JB.186.5.1518-1530.2004
27. Guo YL, Liu Y, Wang SM, et al. Genotyping and drug resistance patterns of Mycobacterium tuberculosis strains in five provinces of China. Int J Tuberc Lung Dis. 2011;15(6):789–794. doi:10.5588/ijtld.10.0403
28. Pang Y, Zhou Y, Zhao B, et al. Spoligotyping and drug resistance analysis of Mycobacterium tuberculosis strains from national survey in China. PLoS One. 2012;7(3):e32976. doi:10.1371/journal.pone.0032976
29. Li Y, Cao X, Li S, et al. Characterization of Mycobacterium tuberculosis isolates from Hebei, China: genotypes and drug susceptibility phenotypes. BMC Infect Dis. 2016;16:107. doi:10.1186/s12879-016-1441-2
30. Wang J, Liu Y, Zhang CL, et al. Genotypes and characteristics of clustering and drug susceptibility of Mycobacterium tuberculosis isolates collected in Heilongjiang Province, China. J Clin Microbiol. 2011;49(4):1354–1362. doi:10.1128/JCM.02274-10
31. Dong H, Liu Z, Lv B, et al. Spoligotypes of Mycobacterium tuberculosis from different Provinces of China. J Clin Microbiol. 2010;48(11):4102–4106. doi:10.1128/JCM.00549-10
32. Li WM, Wang SM, Li CY, et al. Molecular epidemiology of Mycobacterium tuberculosis in China: a nationwide random survey in 2000. Int J Tuberc Lung Dis. 2005;9(12):1314–9.
33. Zhao LL, Chen Y, Chen ZN, et al. Prevalence and molecular characteristics of drug-resistant Mycobacterium tuberculosis in Hunan, China. Antimicrob Agents Chemother. 2014;58(6):3475–3480. doi:10.1128/AAC.02426-14
34. Anh DD, Borgdorff MW, Van LN, et al. Mycobacterium tuberculosis Beijing Genotype Emerging in Vietnam. Emerg Infect Dis. 2000;6 (3) :302–5 doi:10.3201/eid0603.000312.
35. Buu TN, Huyen MN, Lan NT, et al. The Beijing genotype is associated with young age and multidrug-resistant tuberculosis in rural Vietnam. Int J Tuberc Lung Dis. 2009;13(7):900–6.
36. Hasan Z, Tanveer M, Kanji A, Hasan Q, Ghebremichael S, Hasan R. Spoligotyping of Mycobacterium tuberculosis isolates from Pakistan reveals predominance of Central Asian Strain 1 and Beijing isolates. J Clin Microbiol. 2006;44(5):1763–1768. doi:10.1128/JCM.44.5.1763-1768.2006
37. Merza MA, Farnia P, Salih AM, Masjedi MR, Velayati AA. The most predominant spoligopatterns of Mycobacterium tuberculosis isolates among Iranian, Afghan-Immigrant, Pakistani and Turkish tuberculosis patients: a comparative analysis. Chemotherapy. 2010;56(3):248–257. doi:10.1159/000316846
38. Poonawala H, Kumar N, Peacock SJ. A review of published spoligotype data indicates the diversity of Mycobacterium tuberculosis from India is under-represented in global databases. Infect Genet Evol. 2020;78:104072 doi:10.1016/j.meegid.2019.104072.
39. Shi J, Zheng D, Zhu Y, et al. Role of MIRU-VNTR and spoligotyping in assessing the genetic diversity of Mycobacterium tuberculosis in Henan Province, China. BMC Infect Dis. 2018;18(1):447. doi:10.1186/s12879-018-3351-y
40. Maharjan B, Nakajima C, Isoda N, et al. Genetic diversity and distribution dynamics of multidrug-resistant Mycobacterium tuberculosis isolates in Nepal. Sci Rep. 2018;8(1):16634. doi:10.1038/s41598-018-34306-w
41. Rahman SMM, Rahman A, Nasrin R, et al. Molecular epidemiology and genetic diversity of multidrug-resistant Mycobacterium tuberculosis isolates in Bangladesh. Microbiol Spectr. 2022;10(1):e0184821. doi:10.1128/spectrum.01848-21
42. Hoffner S, Sahebi L, Ansarin K, Sabour S, Mohajeri P. Mycobacterium tuberculosis of the Beijing genotype in Iran and the World Health Organization Eastern Mediterranean Region: a meta-analysis. Microb Drug Resist. 2018;24(6):693–698. doi:10.1089/mdr.2017.0160
43. Liu Y, Jiang X, Li W, Zhang X, Wang W, Li C. The study on the association between Beijing genotype family and drug susceptibility phenotypes of Mycobacterium tuberculosis in Beijing. Sci Rep. 2017;7(1):15076. doi:10.1038/s41598-017-14119-z
44. Zhang D, An J, Wang Y, Pang Y. Genetic diversity of multidrug-resistant tuberculosis in a resource-limited region of China. Int J Infect Dis. 2014;29:7–11. doi:10.1016/j.ijid.2014.05.020
45. Kato-Maeda M, Metcalfe JZ, Flores L. Genotyping of Mycobacterium tuberculosis: application in epidemiologic studies. Future Microbiol. 2011;6(2):203–216. doi:10.2217/fmb.10.165
46. Wei D, Xiaohong Z, Zihan X, et al. Genotypic diversity of Mycobacterium tuberculosis isolates and its association with drug-resistance status in Xinjiang, China. Tuberculosis. 2021;128:102063 doi:10.1016/j.tube.2021.102063.
47. Liu J, Li J, Liu J, et al. Genotypic diversity of Mycobacterium tuberculosis clinical isolates in the multiethnic area of the Xinjiang Uygur autonomous Region in China. Biomed Res Int. 2017;2017:3179535 doi:10.1155/2017/3179535.
48. Chen L, Pang Y, Ma L, et al. First insight into the molecular epidemiology of Mycobacterium tuberculosis Isolates from the minority enclaves of Southwestern China. Biomed Res Int. 2017;2017:2505172 doi:10.1155/2017/2505172.
49. Iwamoto T, Grandjean L, Arikawa K, et al. Genetic diversity and transmission characteristics of Beijing family strains of Mycobacterium tuberculosis in Peru. PLoS One. 2012;7(11):e49651. doi:10.1371/journal.pone.0049651
50. Wondale B, Keehwan K, Medhin G, et al. Molecular epidemiology of clinical Mycobacterium tuberculosis complex isolates in South Omo, Southern Ethiopia. BMC Infect Dis. 2020;20(1):750. doi:10.1186/s12879-020-05394-9
51. Ali S, Beckert P, Haileamlak A, et al. Drug resistance and population structure of M. tuberculosis isolates from prisons and communities in Ethiopia. BMC Infect Dis. 2016;16(1):687. doi:10.1186/s12879-016-2041-x
52. Tadesse M, Abebe G, Bekele A, et al. The predominance of Ethiopian specific Mycobacterium tuberculosis families and minimal contribution of Mycobacterium bovis in tuberculous lymphadenitis patients in Southwest Ethiopia. Infect Genet Evol. 2017;55:251–259. doi:10.1016/j.meegid.2017.09.016
53. Dantas NG, Suffys PN, Carvalho Wda S, et al. Genetic diversity and molecular epidemiology of multidrug-resistant Mycobacterium tuberculosis in Minas Gerais State, Brazil. BMC Infect Dis. 2015;15:306. doi:10.1186/s12879-015-1057-y
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Whole Genomic Analysis Revealed High Genetic Diversity and Drug-Resistant Characteristics of Mycobacterium tuberculosis in Guangxi, China
Liang D, Song Z, Liang X, Qin H, Huang L, Ye J, Lan R, Luo D, Zhao Y, Lin M
Infection and Drug Resistance 2023, 16:5021-5031
Published Date: 3 August 2023
Phenotypic Drug Resistance Pattern and Mutation Characteristics of Mycobacterium tuberculosis from Different Body Fluids Among Extra Pulmonary Patients Presented in Selected Hospitals in Addis Ababa, Ethiopia
Alehegn E, Gebreyohanns A, Berhane BW, Wright JA, Hundie GB, Geremew RA, Gorems K, Gebreyohannes Z, Amare M, Abebaw Y, Diriba G, Zerihun B, Gebremichael AW, Kassa M, Gize A
Infection and Drug Resistance 2023, 16:5511-5522
Published Date: 22 August 2023
Comparative Study on Tuberculosis Drug Resistance and Molecular Detection Methods Among Different Mycobacterium Tuberculosis Lineages
He CJ, Wan JL, Luo SF, Guo RJ, Paerhati P, Cheng X, Duan CH, Xu AM
Infection and Drug Resistance 2023, 16:5941-5951
Published Date: 7 September 2023
Mycobacterium tuberculosis Sub-Lineage 4.2.2/SIT149 as Dominant Drug-Resistant Clade in Northwest Ethiopia 2020–2022: In-silico Whole-Genome Sequence Analysis
Mekonnen D, Munshea A, Nibret E, Adnew B, Getachew H, Kebede A, Gebrewahid A, Herrera-Leon S, Amor Aramendia A, Benito A, Abascal E, Jacqueline C, Aseffa A, Herrera-Leon L
Infection and Drug Resistance 2023, 16:6859-6870
Published Date: 26 October 2023
Correlation Between Xpert MTB/RIF Results and rpoB Mutations within Probe-Targeted Regions in Mycobacterium tuberculosis Isolates from Sichuan Basin
Li K, Li L
Infection and Drug Resistance 2025, 18:5161-5171
Published Date: 29 September 2025