Back to Journals » Infection and Drug Resistance » Volume 13
Molecular Characterization of Hepatitis C Virus for Developed Antiviral Agents Resistance Mutations and New Insights into in-silico Prediction Studies
Authors El-Sokkary MMA ![]() , Gotina L, Al-Sanea MM
, Gotina L, Al-Sanea MM ![]() , Pae AN, Elbargisy RM
, Pae AN, Elbargisy RM ![]()
Received 15 June 2020
Accepted for publication 24 September 2020
Published 23 November 2020 Volume 2020:13 Pages 4235—4248
DOI https://doi.org/10.2147/IDR.S267809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eric Nulens
Mohamed M Adel El-Sokkary,1 Lizaveta Gotina,2,3 Mohammad M Al-Sanea,4 Ae Nim Pae,2,3 Rehab Mohammed Elbargisy1,5
1Department of Microbiology and Immunology, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt; 2Convergence Research Center for Diagnosis, Treatment and Care System of Dementia, Korea Institute of Science and Technology, Seongbuk-gu, Seoul 02792, Republic of Korea; 3Division of Bio-Medical Science & Technology, KIST School, Korea University of Science and Technology (UST), Daejeon, Korea; 4Department of Pharmaceutical Chemistry, College of Pharmacy, Jouf University, Sakaka, Al-Jouf, Saudi Arabia; 5Department of Pharmaceutics, College of Pharmacy, Jouf University, Sakaka, Al-Jouf, Saudi Arabia
Correspondence: Mohamed M Adel El-Sokkary
Department of Microbiology and Immunology, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
Email [email protected]
Ae Nim Pae
Convergence Research Center for Diagnosis, Treatment and Care System of Dementia, Korea Institute of Science and Technology, Hwarangno 14-Gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea
Email [email protected]
Background: Identification and characterization of developed antiviral drug resistance mutations are key to the success of antiviral therapies against hepatitis C virus (HCV), which remains a worldwide highly prevalent pathogenic disease. Although most studies focus on HCV genotypes 1, 2 or 3, the investigation of drug resistance in HCV genotype 4, predominant in North Africa, is especially significant in Egypt.
Methods: We performed mutational and genotypic analysis of the untranslated region (UTR) and nonstructural protein 5B (NS5B) drug resistance-associated regions of HCV for patients in the surrounding villages of Mansoura city, who were not responding to different antiviral treatments (sofosbuvir (SOF), ribavirin, and interferon). Furthermore, molecular modelling approaches (homology modelling and docking studies) were used to investigate the significance of the identified NS5B mutations for SOF and ribavirin binding in the HCV genotype 4a NS5B active site.
Results: Genotypic analysis confirmed all samples to have genotype 4 with sub-genotype 4a predominant. Partial sequencing of the UTR and NS5B resistance-associated regions identified D258E, T282S and A307G mutations in all isolates of NS5B. The UTR mutation site at position 243 was associated with interferon resistance, whereas the NS5B T282S mutation was considered as significant for SOF and ribavirin resistance. Docking studies in the HCV genotype 4a homology model predict SOF and ribavirin to accommodate a nucleotide-like binding mode, in which the T282 residue does interfere with the binding as it would in HCV genotypes 1 and 2. Mutation energy calculations predict T282S to moderately destabilize the binding of SOF and ribavirin by 0.57 and 0.47 kcal/mol, respectively.
Conclusion: The performed study identified and characterized several antiviral drug resistance mutations of HCV genotype 4a and proposed a mechanism by which the T282S mutation may contribute to SOF and ribavirin resistance.
Keywords: hepatitis C virus, drug resistance, T282S mutation, direct-acting antivirals, molecular docking
Introduction
Chronic infection with hepatitis C virus (HCV) is considered a major cause of serious liver diseases, such as liver cirrhosis and liver cancer. It is widely distributed worldwide, but Egypt still has the highest prevalence among all countries affected by HCV infection.1
A high degree of nucleotide sequence variability is found in the HCV coding region. This variability is attributed to errors occurring during replication. As a result, 7 different HCV genotypes have emerged with 31% to 33% nucleotide sequence variation. These genotypes have been further grouped into subtypes that differ by 10–30% in nucleotide sequence.2 Moreover, variation by 5–15% in nucleotide sequence can be detected in isolates of the same subtype.3 These genotypes and corresponding subtypes of HCV have different geographical distributions. Genotype 1 is the most prevalent genotype in North America, South America and Europe, while genotypes 2 and 3 are widely distributed in Asia, Europe and Latin America. Genotype 4 is the predominant genotype in North Africa and the Middle East, chiefly Egypt. Other genotypes5–7 were detected in South Africa, Southeast Asia and Congo, respectively.
Determination of HCV genotype is highly important prior to treatment, as the response to medications and duration of therapy will depend basically on the infecting genotype. For example, genotypes 1 and 4 exhibited more resistance than genotypes 2, 3, and 6 when treated with pegylated interferon-α and ribavirin.4 In Egypt, where genotype 4a is the predominant subtype, effective treatment is directed now to the use of sofosbuvir (SOF) plus ribavirin for a period of 12 to 24 weeks for both untreated and previously treated patients.5
Different techniques have been developed for genotyping HCV. Most of these techniques are based on amplification of specific HCV RNA regions and subsequent analysis of the amplified part through line probe assay, restriction fragment length polymorphism or sequence analysis. The target RNA regions of HCV are the core/E1/NS5B or 5ʹUTR because they provide accurate identification of the circulating subtype of HCV.6
The aim of this study was to classify and investigate the sequence variation in HCV strains from antiviral drug-treated individuals in regions targeted by different antiviral drugs. Molecular modelling was performed to characterize the identified sequence variations and assess their significance for the binding of nucleotide inhibitors such as SOF and ribavirin.
Materials and Methods
Collection of Samples
Blood samples were collected from 48 individuals, including 47 HCV-infected individuals who were treated with sofosbuvir, ribavirin, and interferon in addition to one control (HCV-negative). Samples were obtained from patients in the surrounding villages of Mansoura city. In this study, 5 mL blood samples were collected from the patients, consisting of 26 females and 22 males. After blood clotting, to separate the serum from the blood cells, each sample was centrifuged at 1500 rpm for 10 min. Serum samples were then collected and stored at −80°C in aliquots for genotypic analysis.
Viral RNA Extraction and cDNA Synthesis
RNA extraction was performed by using a GeneJET Viral DNA/RNA Purification Kit (Qiagen) according to the manufacturer’s instructions. Purified RNAs were frozen at −80°C. For further analysis, the concentration of purified RNA was determined using a NanoDrop instrument (OPTIZEN NanoQ, Mecasys). First-strand cDNA synthesis was performed using a Revert Aid First-Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions. The synthesized first-strand cDNAs were stored at −80°C until use.
PCR Amplification of HCV-NS5B
The NS5Bgene was amplified by nested PCR using the primers shown in Table 1, as previously indicated. The first round of each PCR was carried out in a 25 μL volume containing 6 μL of cDNA from each sample, 0.5 μM each NS5BOS1 and NS5BOAS2 primer (Willowfort.UK), 12.5 μL of master mix (Thermo Scientific) and water to 25 μL. Reactions were performed in a Cycler 003 PCR Machine (A & E Lab, UK). PCRs began with 5 min of primary denaturation at 94°C, followed by 35 cycles of 94°C for 30 s, annealing temp (as listed in Table 1) for 30 s, and 72°C for 1 min, and a final extension at 72°C for 10 min. NS5B gene first-round gene amplification was repeated by usual PCR using the NS5B primer as a forward primer and NS5BOAS2 as a reverse primer. All reactions were performed as indicated in one-step PCR amplification. Samples in the first round of amplification producing negative PCR products were subjected to nested PCR using the NS5BIS1 and NS5BIAS2 primers. The second round of amplification was performed in a total volume of 25 mL, as previously indicated.
 |
Table 1 Different Primers Used in This Work |
PCR Amplification of HCV-UTR
UTR gene amplification was performed in a 25 µL volume as previously indicated for the NS5B region. PCRs began with 5 min of primary denaturation at 94°C, followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, and a final extension at 72°C for 10 min (Table 1).
DNA Sequencing
Amplified 17 NS5B and 23 UTR fragments were purified using GeneJET PCR Purification Kit (K0691, Thermo Scientific, USA). Purified amplicons were sequenced on both strands using an ABI PRISM® BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, USA). The reaction mixtures were analysed on an ABI 3730 DNA analyser (Applied Biosystems, Foster City, USA). DNA sequences were analysed by the online BLAST software of the NCBI website http://www.ncbi.nlm.nih.gov/BLAST/). Phylogenetic trees were reconstructed by using the neighbour-joining method with 1000 bootstrap values in the MEGA 4.1 package. Sequences of this study were deposited under the following accession numbers: MN596306-MN596345.
Homology Modelling
The HCV genotype 4a NS5B amino acid sequence was retrieved from the UniProtKB database entry O39929 (reviewed, experimentally validated sequence). Homology modeling was performed based on a template crystal structure of HCV genotype 2a NS5B synthetic construct (a 5-point mutant with deleted delta8 hairpin loop region) in complex with UDP, Mn2+ and a symmetrical primer (PDB ID: 4WTA). This template best represents the nucleotide-binding mode as it is a ternary complex stalled during the elongation process of HCV RNA replication, and also has a satisfactory resolution of 2.8 Å.
Template protein structure preparation included inserting missing atoms in incomplete residues, deleting alternate conformations (disorder), removing all water molecules, standardizing atom names, and protonating titratable residues using pKa predicted by PROPKA.7 Restrained minimization under the OPLS3 forcefield8 within 0.3 Å heavy atom displacement was used to fully optimize and relax the protein structure. Homology modeling was performed using the MODELER program9 incorporated into the Discovery Studio software package (Accelrys, San Diego).10 The homology modeling protocol included copying the coordinates of the RNA primer and the co-crystallized UDP molecule from the template structure and constructing the model loop regions using the DOPE method (Discrete Optimized Protein Energy).11
Out of 10 prepared models, the first 3 with the lowest PDF (probability density function) total energy were examined. The model with the lowest heavy atom RMSD (root-mean-square deviation) value between the model and the template active site residues was chosen as the final homology model. Energy minimization of the final model was performed using the same protein preparation protocol described previously. The 3D structure of the final refined model was validated by its Ramachandran plot, Profiles-3D score and the Maestro Protein Reliability Report,12 which checks such metrics as the average B-factors, steric clashes, protein packing, and peptide planarity, bond angle and length deviations, improper torsions, etc.
Docking Protocol
Molecular docking studies were conducted to predict the binding mode of UDP, SOF, ribavirin, and other known nucleotide inhibitors in the active sites of HCV genotypes 4a. In the absence of a published crystal structure for HCV genotype 4a, docking was performed in the prepared homology model. The appropriate di-phosphate nucleotide form of UDP and the nucleotide inhibitors were first drawn in ChemDraw. Afterward, the ligand 3D conformations were created using Catalyst and fully energy-minimized using the general-purpose all-atom forcefield - CHARMM. Ligands were docked into an 8.05 Å grid using the CDOCKER docking algorithm.13 The crystal structures of HCV genotype 2a with co-crystallized UDP and SOF ligands and an RNA primer (PDB ID: 4WTA and 4WTG, respectfully) were used to estimate the correctness of the dock poses. Heavy atom RMSD between the HCV genotype 2a co-crystal poses and HCV genotype 4a dock poses was calculated as the initial validation parameter. Full validation of the docking protocol’s pose prediction ability was done by assessing the correlation between known nucleotide inhibitor activity and the predicted -CDOCKER Score.
Mutation Energy Calculation
The mutation energy calculations quantify the effect of a specific point mutation on the binding affinity of molecular partners in a protein-ligand complex. This energy effect is equal to the difference between the binding free energy in the mutated structure and wild-type protein. In this case, the binding free energy of each complex is calculated as an empirical weighted sum of van der Waals interactions, electrostatic interactions, an entropy contribution, related to the changes in sidechain mobility, and a non-polar, surface dependent, contribution to solvation energy. The energy terms of this sum were calculated by the CHARMM forcefield and the electrostatic energy – by a Generalized Born implicit solvent model.
The mutation energy was calculated both for the ligand crystal structure poses in HCV genotype 2a NS5B crystal structures (PDB ID: 4WTA and 4WTG) and for the ligand docking poses in the prepared HCV genotype 4a NS5B homology model. For the crystal structure ligand poses, a single mutation energy calculation was performed. For the dock poses, the mean mutation energy value for the top 3 docking poses is reported.
Results
Amplification of the HCV-NS5B Region Using Nested PCR
Amplification of the HCV-NS5B region using PCR and nested RT-PCR confirmed the presence of NS5B in 17 (36%) of the 47 disease samples by using the NS5B primer. However, the NS5BOS1 primer amplified only 6 samples. The remaining 30 samples (64.5%) did not show the presence of NS5B even after a second round of PCR amplification with nested PCR using the NS5BIS1 and NS5BIAS2 primers. All the positive PCR products were of the expected size of approximately 381 bp, which included almost the partial NS5B genomic region.
Amplification of the HCV-UTR Region Using PCR
Amplification of the HCV 5´UTR region using PCR confirmed the presence of the 5´UTR in 23 (49%) of the 47 disease samples. The remaining 24 samples (51%) did not show the presence of the UTR. All the positive PCR products were of the expected size of approximately 302 bp, which included almost the partial UTR genomic region.
Variability of UTR and NS5B Sequences
Identification of HCV Genotypes Based on the Partial Nucleotide Sequence of the NS5B Gene
For the identification of different genotypes using the NS5B partial gene sequence, a phylogenetic tree was constructed based on the 351-bp NS5B partial gene from 17 different samples (Figure 1A). DNA sequences in this study were compared to sequences downloaded from GenBank representing NS5B gene sequences from different areas in the world. These reference HCV strains revealed 4 distinct clusters representing the most related HCV genotype 4 subtypes. As a result, 17 NS5B-positive samples were clustered in one branch and seemed to be more closely related to HCV genotype 4 and subtype a. Similarly, after construction of the phylogenetic tree based on UTR DNA sequences identified in this study, in addition to 21 reference recorded sequences, all 23 samples were found with the highest identities to HCV genotype 4 and subtype a (Figure 1B).
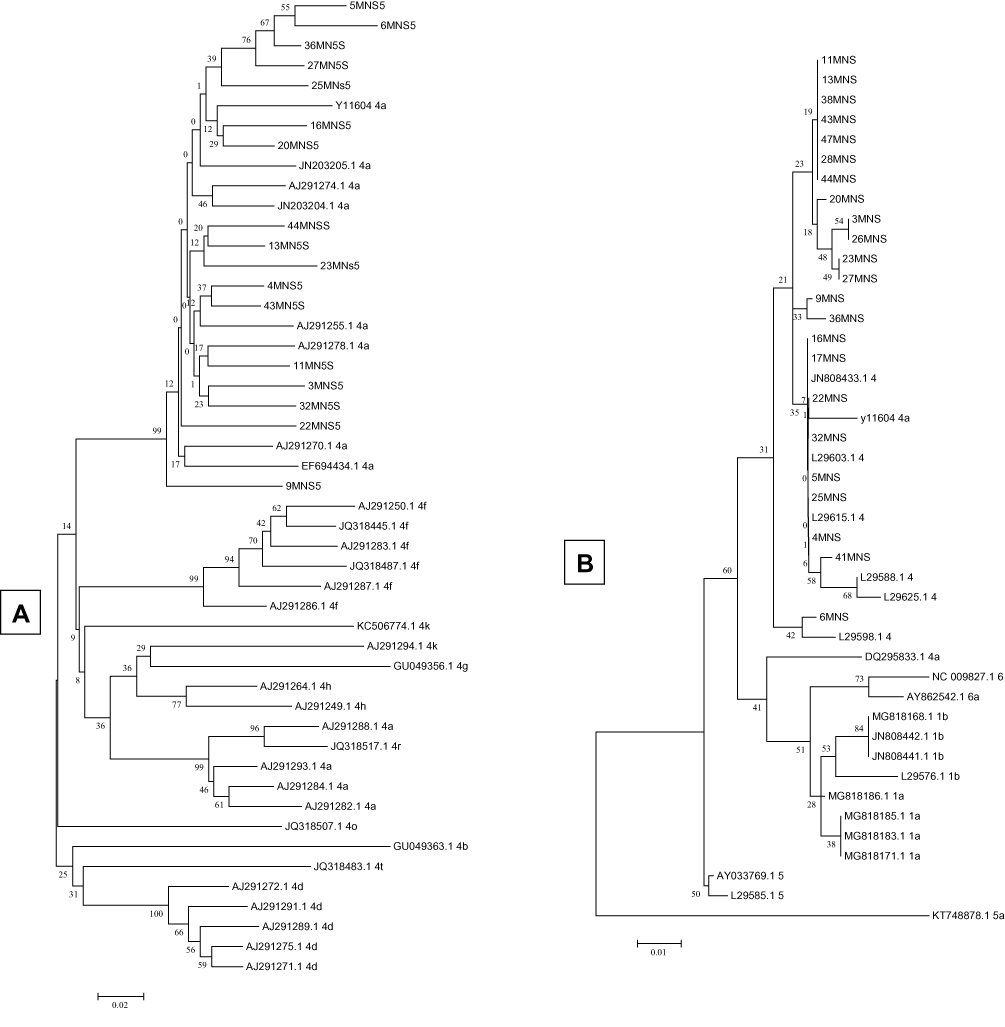 |
Figure 1 Phylogenetic analysis of NS5B (A) and 5ʹUTR (B) sequences illustrating the molecular epidemiology and evolution of HCV genotypes. The phylogenetic tree was constructed in Mega 4 program using the neighbour-joining method and the phylogeny test was 1000 times bootstrapped. |
Amino Acid Sequence Analysis of NS5B and 5´UTR Regions
Amino acid sequence studies of the NS5B region (Table 2) indicated the predominance of E instead of D at position 258, S instead of T at position 282 and G instead of A at position 307, while a wide variation at position 235 could be detected, where V was substituted with A or T. In addition, some mutations were found in the region 303–327, which is close to the region (317–319) targeted by some antiviral drugs such as SOF. Moreover, R309K mutation was detected in only 4 samples. However, D244N and N333R were detected, each in one isolate. Analysis of nucleotide variance in the 5´UTR gene indicated two predominant mutations at the sites T150G and C284T, in addition to one less frequent G nucleotide substitution detected at position 243 in only 11 samples.
 |
Table 2 Amino Acid Sequence Variation Analysis of NS5B Region of 17 Isolates Compared to a Standard Strain |
Homology Model Preparation and Validation
High sequence similarity between HCV NS5B genotype 2a and 4a proteins allowed to perform manual sequence alignment, illustrated in Supplementary Figure 1. The sequence similarity was 83.4% and sequence identity - 72.2%. The active site, defined as a sphere with 8 Å radius around the co-crystallized ligand, was highly conserved between the model sequence and the template with only three sequence differences: the template had S instead of a T at position 282, M instead of F at position 289, and H instead of C in position 223.
The best-prepared homology model had a PDF (probability density function) total energy equal to −15,418.8, and the RMSD to the template was 0.243 Å, indicating close amino acid positions in respect to the model. All of the 3D structure validation methods showed the model to be of high quality and comparable in 3D structure quality to the initial template structure (Supplementary Figure 2). The Ramachandran plotting of non-glycine and non-proline residues showed that the backbone dihedral angles of 97.9% of residues were in the energetically allowed region, 1.7% in the marginal region and only 0.4% in the disallowed region. The Profiles-3D verification score was used to validate whether each residue is in the correct 3D environment, for example, is each residue compatible with its local secondary structure, the occurring polar environment, the fraction of buried area, etc. The homology model showed a total verification score equal to 221.54 out of an expected high score of 247.80 with 96.5% of residues scoring as valid. Invalid residues were more than 15 Å away from the active site and had little effect on the overall model quality. Lastly, the Maestro Protein Reliability Report showed that the homology model was valid by most structural elements: the bond length and bond angle deviations, the average B-factors, the binding site packing, and sidechain dihedrals and planarity. All occurring structural deviations were comparable to and most likely inherited from the initial template crystal structure, which can be seen when comparing Supplementary Figures 2 and 3.
Docking Study Results
The binding poses of UDP and known nucleotide inhibitors in the HCV genotype 4a NS5B homology model were predicted using the small molecule docking method. The CDOCKER method predicts the most favorable binding pose based on CHARMM force field calculations of the ligand interaction energy and ligand strain. A more negative docking score implies a more favorable binding pose.
Although it is known that HCV NS5B polymerase binds tri-phosphate nucleotides, the ligands for docking were prepared in a di-phosphate nucleotide form, which corresponds to the co-crystallized ligand of the published crystal structures 4WTA and 4WTG.
The docking protocol proved to be efficient in reproducing the crystal structure poses for UDF and SOF. The average RMSD between the top 3 ligand docking poses and the crystal structure pose was less than 1 Å (0.855 and 0.967 Å for UDF and SOF, respectively), indicating a correct choice of grid size and protein preparation. Furthermore, the –CDOCKER Score and –CDOCKER Interaction Energy for five different HCV nucleotide inhibitors (Figure 2) were shown to correlate with the drug potency with an R2 equal to 0.93 and 0.90, respectively (Figure 3). Such a correlation implies that the non-bonded interactions occurring in the top docking poses correctly estimate the binding affinity of the inhibitors to the drug target and have a high probability of representing the correct binding pose.
 |
Figure 2 Diphosphate forms of the HCV nucleotide inhibitors chosen for model validation. |
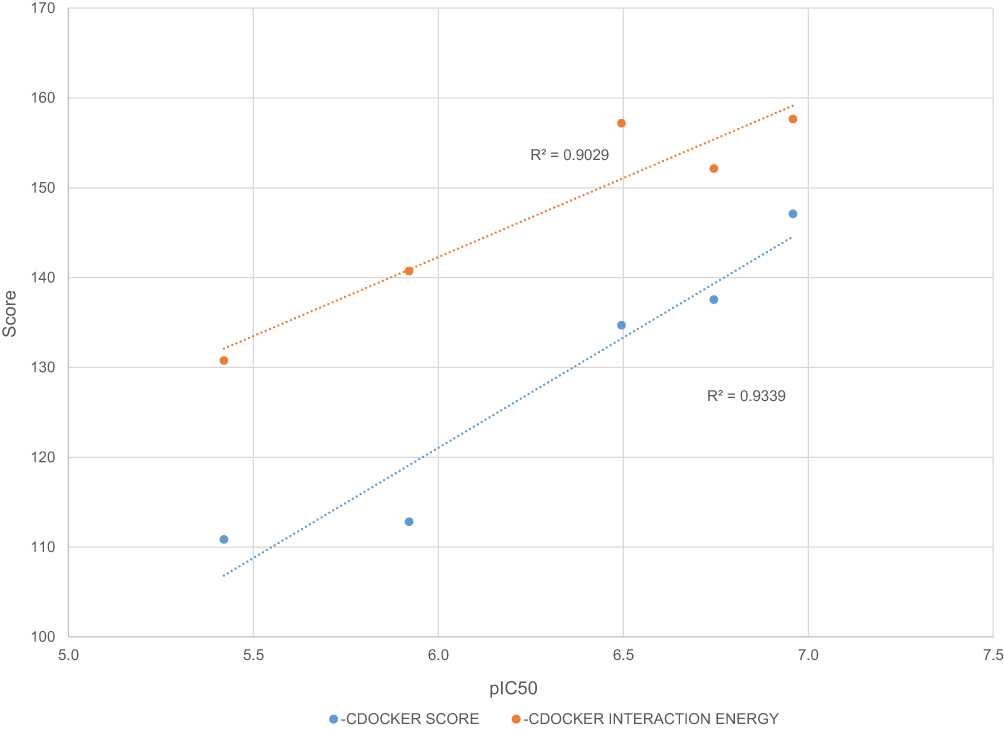 |
Figure 3 Correlation between docking scores and nucleotide inhibitor activity. |
Nucleotides and Nucleotide Inhibitors Binding Mode
Docking studies using the prepared HCV genotype 4a NS5B model confirm a binding mode of nucleotides and nucleotide inhibitors consistent with the HCV genotype 2a crystal structure pose and previously reported structural basis of HCV ribonucleotide substrate binding.14
For ease of description, the common HCV NS5B ligand binding mode can be divided into interactions of the nitrogenous base, pentose sugar, and phosphate group. The nitrogenous base or its analog is involved in nucleoside Watson-Crick pairing interactions with the adjunct RNA template nucleoside and pi-pi stacking interactions with the growing nucleotide chain, as well as H-bonding with K141. The pentose sugar of the nucleoside is oriented by H-bonding with the growing 3ʹ-RNA strand 2ʹ-hydroxyl group, as well as H-bonds with T282 and N291. The phosphate group is mainly coordinated by Mn2+ ions and the catalytic triad (D220, D318, and D319) and additionally by basic R48 and R158 residues on one side of the binding pocket and backbone interactions with C223 and F224 on the other side.
Despite being different in their ribose 2ʹ substitution, according to docking studies, UDP and SOF share a common docking mode. As shown in Figure 4, the 2ʹ-hydroxyl of UDP creates an internal H-bond and acts as an H-bond acceptor for N291. The 2ʹ-hydroxyl of UDP is also in close proximity to form an H-bond with Y282. SOF 2ʹ-fluoro group forms an H-bond with T282, as well as the same internal H-bond and N291 interaction. Ribavirin shares the same common binding pose with the main key interactions: Watson-Crick nucleoside pairing, Mn2+ and Arg-facilitated diphosphate group coordination and H-bonding with T282.
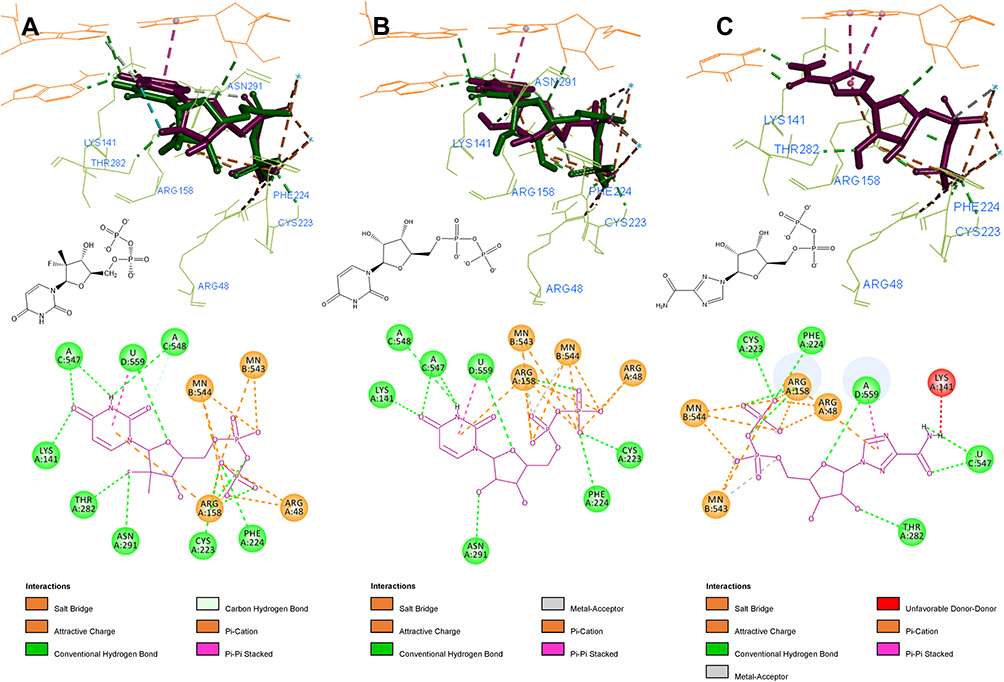 |
Figure 4 3D and 2D interaction patterns for SOF (A), UDP (B) and ribavirin (C). Dock poses (purple) and crystal structure poses (green) shown in stick form, interacting residues (light green) and RNA nucleotides (orange) shown as lines. Mn2+ ions shown as light blue stars. Image prepared in BIOVIA Discovery Studio. |
Structural Analysis of NS5B Amino Acid Variations
At first, to determine the signification of the reported NS5B amino acid variations, their structural mapping was performed and illustrated in Figure 5. All of the detected amino acid variations of NS5B were located in the palm region. The only amino acid variation within 8 Å of the nucleotide-binding site was T282S, for hence it is the only amino acid variation that could affect the SOF or Ribavirin binding affinity. However, the Y285F amino acid variation, located adjunct to the active site (9.2 Å away from SOF), is interestingly close to the RNA strand and, as seen from Figure 6, can be displaced by a nucleoside 2ʹ-substitution when the RNA strand is elongated. All other detected amino acid variations are located in the solvent-exposed outer surface of the palm region further than 15 Å away from the active site.
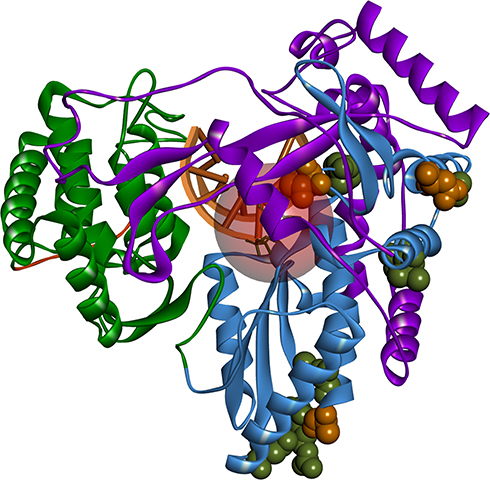 |
Figure 5 Positions of detected amino acid variations in the created homology model. HCV NS5B structure colored according to its domains: fingers – purple, palm – sky blue, thumb – green, C-terminal – orange. Nucleoside binding pocket represented as a red circle (8 Å radius). Occurring amino acid variations represented as spheres; orange – occurring in all of the sequenced isolates; green – occurring in some of the sequenced isolates. Image prepared in BIOVIA Discovery Studio. |
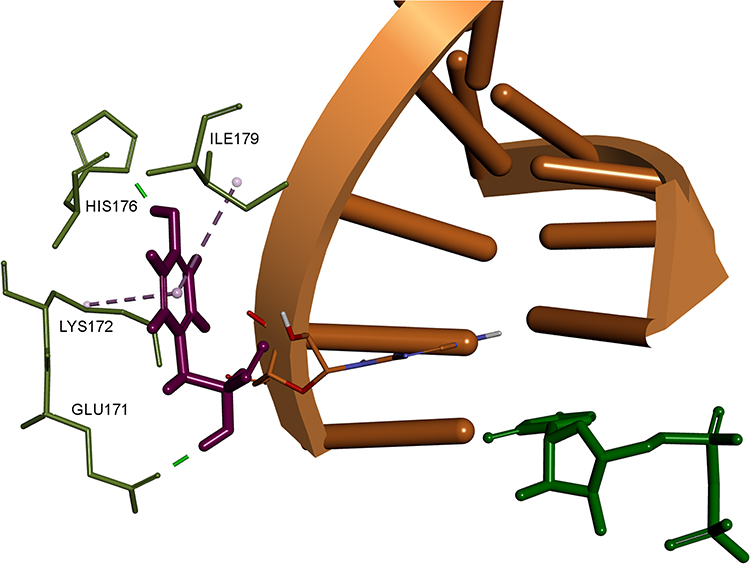 |
Figure 6 3D interaction pattern of F285 (shown as purple stick). Interacting residues colored in light green, RNA strand shown in orange, UDP shown as green stick. Hydrogen atoms and other protein residues hidden for clarity. Image prepared in BIOVIA Discovery Studio. |
Mutation Energy Calculation Results
To quantify the significance of the detected amino acid variations, we performed mutation energy calculations, which estimate the difference in the free binding energy of the ligands in the wild-type protein and the mutant structure. Firstly, we validated our mutation energy calculation method by estimating the change in binding affinity for the well-known S282T resistance mutation in the HCV genotype 2a NS5B protein. The calculation results sufficiently correlate with experimental and crystal structure data. According to our calculations, performed for the crystal structure poses of UDP and SOF inside the prepared 4WTA crystal structure, the S282T mutation results in a favorable mutation effect for UDP binding by −0.71 kcal/mol, whereas for SOF the mutation effect is destabilizing by 1.55 kcal/mol. For SOF, this could result in a 13.7 fold loss in potency, which is in agreement with the experimentally evaluated 20.2-fold shift.15 Interestingly, even for calculations performed in the 4WTG crystal structure, where the S282 side chain is facing the opposite direction, away from the nucleoside binding pocket, the mutation energy calculations resulted in a destabilizing binding effect by 1.00 kcal/mol.
The same binding mutation energy calculations were performed in the HCV genotype 4a NS5B homology model for the top 3 docking poses of UDP, SOF, and Ribavirin, but with the opposite mutation: T282S. The results of the calculations are presented in Table 3. For all three ligands, T282S causes a neutral or moderately destabilizing effect equal to 0.57 kcal/mol for SOF and 0.47 kcal/mol for ribavirin. Given the relationship between the binding affinity and the protein-ligand binding constant, such changes in binding energy could result in only a ~2.3-fold shift in activity.
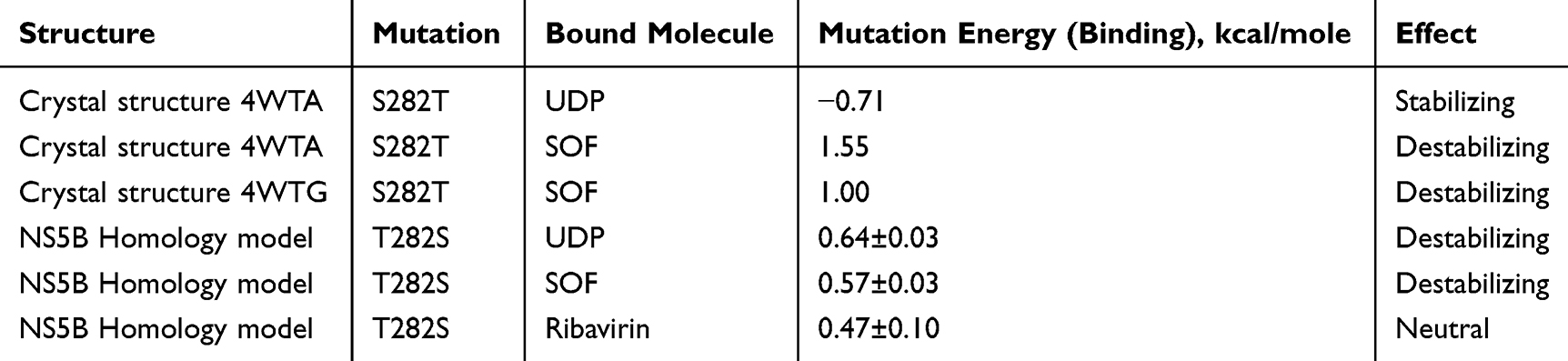 |
Table 3 Results of Mutation Energy of Binding Calculations |
Discussion
Chronic hepatitis C virus (HCV) infection is considered one of the main health problems in Africa, especially in Egypt. Hepatitis C virus (HCV), a blood-borne member of the viral Flaviviridae family, is an enveloped single-stranded RNA virus that can cause a variety of liver-associated diseases, starting from chronic hepatitis and progressing to liver cirrhosis to hepatocellular carcinoma.
In Egypt, different antiviral therapeutic regimens have been prescribed for treating HCV patients. However, not all therapeutic strategies have succeeded in viral elimination. It was concluded that genotype 4, the most predominant genotype in Egypt, was unresponsive to some antiviral agents.
Recently, sequencing analysis has become the most powerful tool for identification and genotyping. Genomic regions of HCV can be classified according to their sequence diversity. Three different regions were resolved, including the highly conserved regions, such as the 5′UTR;16 the relatively variable non-structural regions NS2, 3, and 5b; the 3′UTR; and finally the envelope regions E1 and E2 and NS4 and NS5A, with the maximum sequence diversity. HCV genotyping is usually performed through phylogenetic sequence analysis of the NS5B region, a more variable and more informative region, in addition to the HCV5ʹUTR carrying a highly reduced number of mutations.
To distinguish the genotypic variation, in our study, 47 samples containing both diseased and recovered HCV samples were examined for the presence of different genotypes. Phylogenetic analysis was performed on the sequences of both the NS5B and 5ʹUTR regions. In 17 different resistant samples, subtype 4a was the most prevalent subtype. The predominance of genotype 4a in our study is supported by the results obtained in many previous studies in different Egyptian governorates.17–19
One of the recently approved new treatments for HCV in Egypt is SOF, which can be used alone or in addition to other antiviral drugs, such as ribavirin. Sofosbuvir (Sovaldi, SOF), a newly discovered antiviral drug, is a nucleotide analogue prodrug targeting the NS5B polymerase and inhibiting viral replication. The NS5B polymerase is composed of three major domains: fingers, palm and thumb.20 For efficient therapeutic activity, the nucleotide sequence of the HCV genome can be utilized as an important predictor of each individual response to any antiviral therapeutic agent.
The NS5B variation in antiviral drug-resistant patients targeted by newly approved antiviral drugs was investigated in this study. In the NS5B region, analysis of the amino acid variance revealed the predominance of D258E, T282S and A307G amino acid variants in the 17 sequenced isolates. NS5B mutations at different sites conferring resistance to newly developed nucleoside analogues, such as SOF, have been previously documented in previous studies. These single point mutational changes at C316 and V321 were identified, with possible alteration of the active site21 inhibiting SOF interaction.22 In addition, other single point mutational sites, V321I, L320F and C316N,21 in addition to D220, D318, and D319 have also been previously described.21 Moreover, the NS5B C316N resistance mutation has been reported in patients who failed treatment with tegobuvir.23 Recently, most studies have focused on one catalytic site, which can be inactivated by mutations targeting the 282 amino acid position, in which S is substituted by T or sometimes by C, usually detected in genotypes 1 and 2.21,24–26 However, in this study, a NS5B T282S mutation affecting genotype 4a, was also detected and its association with resistance to SOF was thoroughly investigated using molecular modelling methods.
Concerning Ribavirin, NS5B-associated resistance mutations, previously analysed at different positions (D244N, Q309R, and A333E), located in the mutational active region of genotypes 1 and 2 were usually detected in cases with a high sustained virologic response (SVR).27 Similarly, in this study, R309K was detected in only 4 samples. This mutation site has been previously documented in ribavirin resistance.28 In addition, D244N, which confers resistance to ribavirin,28 was found in only one isolate. Moreover, at the A333 site, which was previously found to confer resistance to ribavirin,29 N333R substitution was detected in only one isolate.
In the case of interferon resistance, some nucleotide substitutions in the HCV 5ʹUTR may also affect viral translation and viral sensitivity.30 Mutations in NS5B did not correlate with the response to interferon therapy.31 One mutation, G160A, in the 5ʹUTR acts as a unique mutation among nonresponders.32 Unfortunately, in the current study, this mutation site was detected at position 243 in 11 isolates, which indicates decreased interferon efficiency as the main drug for the treatment of Egyptian HCV patients.
In order to assess the significance of the identified novel NS5B single point mutations for SOF or Ribavirin binding, structure-based molecular modeling methods were applied. Molecular modelling methods utilize available crystallographic data by mapping the detected amino acid variations onto the structural units of the protein and assessing their relation to structurally significant protein regions. Such methods have been widely applied to investigate the significance of potential SOF resistance-related mutations,21,33–35 hence we applied them in our studies for the NS5B mutations detected in HCV genotype 4a.
In the RCSB Protein Data Bank, the HCV NS5B crystal structures are available only for virus genotypes 1 and 2. Sequence comparison of genotypes 1, 2, and 4 (Figure 7) shows that within 8 Å of the nucleotide-binding site, there are two amino acid differences for genotype 4a: a T instead of S in 282 position and F instead of C or M in 289 position. For genotypes 1 and 2, the S282T mutation is a well-known, infrequently occurring cause of SOF resistance. Previous publications,14,36 have illustrated how the additional methyl group within threonine is capable of causing a steric clash between the T282 side chain and the 2ʹ-methyl group of SOF, as well as possibly changing the orientation of the uracil group, thereby leading to drug resistance (Figure 8).
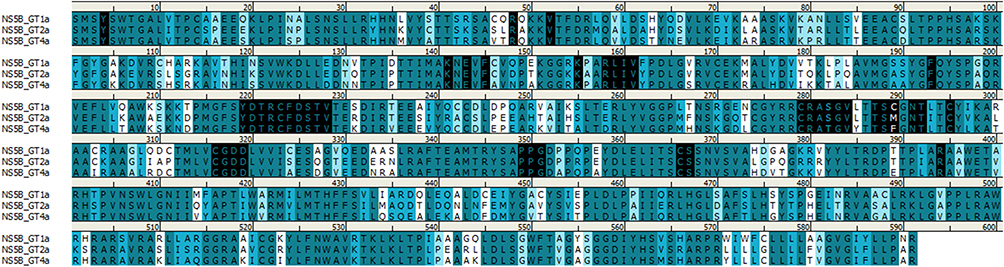 |
Figure 7 Sequence alignment of HCV NS5B sequences for genotypes 1, 2, and 4. Amino acids colored based on similarity. Residues within 8 Å of the nucleotide binding site are highlighted in black. Image prepared using BIOVIA Discovery Studio. |
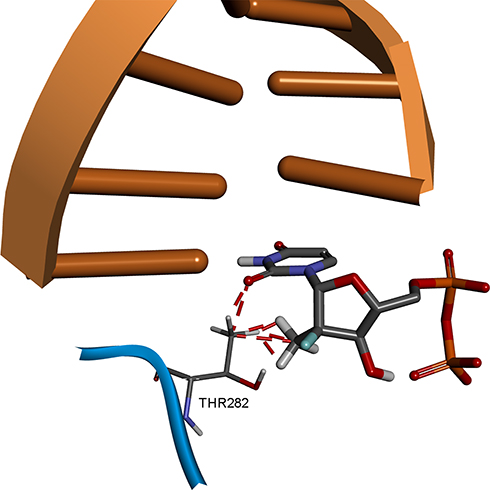 |
Figure 8 Structural basis of S282T SOF resistance mutation in HCV genotype 2a. Image prepared on the basis of 4WTA crystal structure in BIOVIA Discovery Studio. |
However, it appears that in the case of HCV genotype 4a the wild-type T282 residue does not conflict with SOF binding as it would in HCV genotypes 1 and 2. SOF is an FDA approved drug for HCV genotype 4 and has been shown effective in combinational therapy to treat patients in Egypt.5 Together this suggests that the binding pocket of NS5B genotype 4a is large enough to accommodate a T282 residue so that UDP, SOF, and other nucleotide inhibitors could share a common binding mode, in which T282 would not adversely affect the ligand binding.
To investigate this theory, we used the HCV genotype 2a NS5B protein, with co-crystallized UDP and RNA template strand as a template to build the HCV genotype 4a homology model. Several different 3D structure validation methods showed that the structural quality of the prepared model is high and comparable to that of the template crystal structure. Docking studies were performed to generate the energetically favorable binding poses for UDP and several HCV nucleotide inhibitors inside the HCV genotype 4a homology model binding pocket. All docked compounds shared a common interaction pattern: Watson-Crick nucleoside pairing of the nitrogenous base, H-bonding with Y282 or N291 by the 2ʹ-substitution in the pentose sugar sub-structure and Mn2+ and Arg-facilitated diphosphate group coordination. The binding modes of UDP and SOF were very similar to those observed in crystal structures of HCV genotype 2a (heavy atom RMSD less than 1 Å), which was in line with the initial assumption that in the HCV genotype 4a binding pocket T282 does not have an unfavorable effect on SOF binding. Additional certainty in the quality of the generated docking poses was obtained by comparing the known nucleotide inhibitor activity with their calculated docking scores. The high correlation indicates that the dock poses account for all of the important protein–ligand interactions.
Most nucleotide inhibitors differ from RNA nucleotides only in the 2ʹ and 3ʹ ribose substitutions. If the cause of drug resistance is associated directly with the drug binding mode, then it would be because of the inability of the drugs’ 2ʹ and 3ʹ ribose groups to reproduce the network of non-bound interactions of natural nucleotides. In the prepared homology model, these sub-structural elements form H-bonds with residues of the palm region, located in the β5 and αJ subunits of NS5B, as well as the loop in between them (aa 271–304). This region was fully covered in the performed NS5B protein sequencing. So it can be concluded that in this study we investigated the point mutations that can cause drug resistance by interfering with SOF binding to the catalytic site.
Calculation of the mutation binding energy showed that the T282S mutation could reduce the binding energy of SOF and ribavirin by 0.57 and 0.47 kcal/mol, respectively. This absolute value is probably not significant enough to be the sole cause of drug resistance; however, it could contribute to effects caused by other substitutions occurring in the active site, which were not covered in the current study.
Figure 9 illustrates the mechanism of how the substitution of T by S could weaken the hydrogen bonding network of SOF. To be a potent inhibitor, SOF needs to compete with other nucleotides (like UDP), by forming the same H-bonding network. It is known that the H-bond formed by an oxygen atom is generally stronger than that of a fluorine acceptor atom.37 So, the additional methyl group of T could be responsible for favorable positioning of the side chain hydroxyl group to form the correct H-bond with SOF and Ribavirin. The T282S substitution may make the H-bond weaker, which is predicted by the binding mutation energy calculations.
 |
Figure 9 Comparison of SOF hydrogen bonding in HCV genotype 4a model (A) and in T282S mutant structure (B). Image prepared using BIOVIA Discovery Studio. |
Also, it should be noted that the T282S substitution can result in SOF binding to an NS5B conformation where S282 is facing outside of the binding pocket, as was previously reported for HCV genotype 2a.14 In this case, there would be no hydrogen binding between nucleoside inhibitors and the mutated S282 side chain. Given all of the above, molecular modeling shows that a T282S mutation could contribute to drug resistance for nucleotide inhibitors in HCV genotype 4a patients.
Conclusion
Our results showed that among Egyptian patients the most commonly occurring HCV genotype was 4a. In addition, we detected several mutations in the HCV NS5B polymerase and 243 site point mutation in the 5ʹUTR associated with resistance to different antiviral agents (SOF, Ribavirin and interferon), which may affect their therapeutic activities in the future. Molecular modeling shows that a T282S mutation could contribute to drug resistance for nucleotide inhibitors in HCV genotype 4a patients by interfering with inhibitor binding in the NS5B active site. However, further studies, including full sequencing of NS5B genotype 4 samples and their analysis is required to fully assess HCV genotype 4 antiviral agent resistance-related mutations.
Acknowledgments
We would like to thank Mr. Salah El-Hawari, Research lab director, Department of Microbiology and Immunology, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt for providing the HCV diseased samples.
We would like to express our special thanks to Dr. Mahmoud Abd-Elmonem Mahmoud El-Hassab, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Badr University, Cairo, Egypt, who offered us the 3D structure of the generated HCV NS5B4a at the beginning of the current investigation.
The experimental protocol conducted in this study complies with the ethical guidelines adopted by the Code of Ethics of the World Medical Association (The 1975 Declaration of Helsinki) and was approved by The Research Ethics Committee, Faculty of Pharmacy, Mansoura University in October 2019. All patients participated in the study provided informed consent.
Disclosure
The authors report no conflicts of interest for this work.
References
1. El-Tahan RR, Ghoneim AM, Zaghloul H. 5′ UTR and NS5B-based genotyping of hepatitis C virus in patients from Damietta governorate, Egypt. J Adv Res. 2018;10:39–47. doi:10.1016/j.jare.2018.01.004
2. Simmonds P, Bukh J, Combet C, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi:10.1002/hep.20819
3. Shier MK, El-Wetidy MS, Ali HH, Al-Qattan MM. Characterization of hepatitis C virus genotypes by direct sequencing of HCV 5ʹUTR region of isolates from Saudi Arabia. PLoS One. 2014;9:e103160. doi:10.1371/journal.pone.0103160
4. El-Shamy A. Impact of hepatitis C virus heterogeneity on interferon sensitivity: an overview. World J Gastroenterol. 2014;20(24):7555–7569. doi:10.3748/wjg.v20.i24.7555
5. Doss W, Shiha G, Hassany M, et al. Sofosbuvir plus ribavirin for treating Egyptian patients with hepatitis C genotype 4. J Hepatol. 2015;63(3):581–585. doi:10.1016/j.jhep.2015.04.023
6. Kumar A, Rajput MK, Paliwal D, Yadav A, Chhabra R, Singh S. Genotyping & diagnostic methods for hepatitis C virus: A need of low-resource countries. Indian J Med Res. 2018;147(5):445–455. doi:10.4103/ijmr.IJMR_1850_16
7. Olsson M, Søndergard C, Rostkowski M, Jensen J. PROPKA3: consistent Treatment of Internal and Surface Residues in Empirical pKa predictions. J Chem Theor Comput. 2011;7:525–537.
8. Jorgensen W, Maxwell D, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. Journal of the American Chemical Society. 1996;118(45):11225–11236. doi:10.1021/ja9621760
9. Šali A, Blundell T. Comparative Protein Modelling by Satisfaction of Spatial Restraints. Journal of Molecular Biology. 1993;10(3):779–815. doi:10.1006/jmbi.1993.1626
10. Ray SC, Arthur RR, Carella A, Bukh J, Thomas DL. Genetic epidemiology of hepatitis C virus throughout egypt. J Infect Dis. 2000;182(3):698–707. doi:10.1086/315786
11. Shen M, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524.
12. El Hadad S, Al-Hamdan H, Linjawi S, Khudyakov YE. Partial sequencing analysis of the NS5B region confirmed the predominance of hepatitis C virus genotype 1 infection in Jeddah, Saudi Arabia. PLoS One. 2017;12(5):e0178225. doi:10.1371/journal.pone.0178225
13. Wu G, Robertson D, Brooks C, Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER?A CHARMm-based MD docking algorithm.J Computational Chem. 2003;24(13):1549–1562. doi:10.1002/jcc.10306
14. Appleby T, Perry J, Murakami E, et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science. 2015;347(6223):771–775. doi:10.1126/science.1259210
15. Ali S, Leveque V, Le Pogam S, et al. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob Agents Chemother. 2008;52(12):4356–4369. doi:10.1128/AAC.00444-08
16. Shi S, Lai M. HCV ‘5 and ‘3 UTR: when translation meets replication. In: Tan S-L, editor. Hepatitis C Viruses: Genomes and Molecular Biology. horizon bioscience; 2006:49–87.
17. Fakhr AE, Pourkarim MR, Maes P, Atta AH, Marei A, Azab M. Hepatitis C Virus NS5B Sequence-Based Genotyping Analysis of Patients From the Sharkia Governorate, Egypt. Hepat Mon. 2013;13:e12706.
18. Elkady A, Tanaka Y, Kurbanov F, Sugauchi F, Sugiyama M, Khan A. Genetic variability of hepatitis C virus in South Egypt and its possible clinical implication. J Med Virol. 2009;81(6):1015–1023. doi:10.1002/jmv.21492
19. Genovese D, Dettori S, Argentini C, Villano U, Chionne P. Molecular epidemiology of hepatitis C virus genotype 4 isolates in Egypt and analysis of the variability of envelope proteins E1 and E2 in patients with chronic hepatitis. J Clin Microbiol. 2005;43(4):1902–1909. doi:10.1128/JCM.43.4.1902-1909.2005
20. Ranjith-Kumar CT, Kao CC. Biochemical Activities of the HCV NS5B RNA-Dependent RNA Polymerase. 2006.
21. Donaldson EF, Harrington PR, O’Rear JJ, Naeger LK. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology. 2015;61(1):56–65. doi:10.1002/hep.27375
22. Ito J, Suda G, Yamamoto Y, et al. Prevalence and characteristics of naturally occurring sofosbuvir resistance-associated variants in patients with hepatitis C virus genotype 1b infection. Hepatol Res. 2016;46(13):1294–1303. doi:10.1111/hepr.12685
23. Zeuzem S, Buggisch P, Agarwal K, et al. The protease inhibitor, GS-9256, and non-nucleoside polymerase inhibitor tegobuvir alone, with ribavirin, or pegylated interferon plus ribavirin in hepatitis C. Hepatology. 2012;55:749–758. doi:10.1002/hep.24744
24. Migliaccio G, Tomassini JE, Carroll SS, et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol Chem. 2003;278:49164–49170. doi:10.1074/jbc.M305041200
25. Dutartre H, Bussetta C, Boretto J, Canard B. General catalytic deficiency of hepatitis C virus RNA polymerase with an S282T mutation and mutually exclusive resistance towards 2ʹ-modified nucleotide analogues. Antimicrob Agents Chemother. 2006;50(12):4161–4169. doi:10.1128/AAC.00433-06
26. Kulkarni AS, Damha MJ, Schinazi RF, et al. A Complex Network of Interactions between S282 and G283 of Hepatitis C Virus Nonstructural Protein 5B and the Template Strand Affects Susceptibility to Sofosbuvir and Ribavirin. Antimicrob Agents Chemother. 2016;60(4):2018–2027. doi:10.1128/AAC.02436-15
27. Hamano K, Sakamoto N, Enomoto N, et al. Mutations in the NS5B region of the hepatitis C virus genome correlate with clinical outcomes of interferon-alpha plus ribavirin combination therapy. J Gastroenterol Hepatol. 2005;20(9):1401–1409. doi:10.1111/j.1440-1746.2005.04024.x
28. Asahina Y, Izumi N, Enomoto N, et al. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J Hepatol. 2005;43(4):623–629. doi:10.1016/j.jhep.2005.05.032
29. Gededzha MP, Mphahlele MJ, Blackard JT, Selabe SG. Prevalence of NS5B Resistance Mutations in Hepatitis C Virus (HCV) Treatment Naive South Africans. Hepat Mon. 2017;17:e14248.
30. Yasmeen A, Siddiqui AA, Hamid S, Sultana T, Jafri W, Persson MA. Genetic variations in a well conserved 5ʹ-untranslated region of hepatitis C virus genome isolated in Pakistan. J Virol Methods. 2009;160:38–47. doi:10.1016/j.jviromet.2009.04.007
31. Nelson DR, Gray AH, Kolberg JA, et al. Variations of hepatitis C virus NS5B sequence (nucleotides 8261-8566) do not correlate with response to interferon-alpha therapy. J Viral Hepat. 1995;2:285–292. doi:10.1111/j.1365-2893.1995.tb00043.x
32. Zekri AR, El-Din HM, Bahnassy AA, et al. Genetic distance and heterogeneity between quasispecies is a critical predictor to IFN response in Egyptian patients with HCV genotype-4. Virol J. 2007;4(1):16. doi:10.1186/1743-422X-4-16
33. Ji H, Kozak R, Biondi M, et al. Next generation sequencing of the hepatitis C virus NS5B gene reveals potential novel S282 drug resistance mutations. Virology. 2015;477:1–9.
34. Uchida Y, Nakamura S, Kouyama JI, et al. Significance of NS5B Substitutions in Genotype 1b Hepatitis C Virus Evaluated by Bioinformatics Analysis. Sci Rep. 2018;8:8818. doi:10.1038/s41598-018-27291-7
35. Wing PAC, Jones M, Cheung M, et al. Amino Acid Substitutions in Genotype 3a Hepatitis C Virus Polymerase Protein Affect Responses to Sofosbuvir. Gastroenterology. 2019;157(3):692–704 e9. doi:10.1053/j.gastro.2019.05.007
36. Ji H, Kozak R, Biondi M, et al. Next generation sequencing of the hepatitis C virus NS5B gene reveals potential novel S282 drug resistance mutations. Virology. 2015;477:1–9. doi:10.1016/j.virol.2014.12.037
37. Howard J, Hoy V, O’Hagan D, Smith G. How good is fluorine as a hydrogen bond acceptor? Tetrahedron. 1996;52:12613–12622.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.