Back to Journals » Stem Cells and Cloning: Advances and Applications » Volume 16
Mitochondria in Cancer Stem Cells: From an Innocent Bystander to a Central Player in Therapy Resistance
Authors Garimella SV, Gampa SC, Chaturvedi P ![]()
Received 23 April 2023
Accepted for publication 15 August 2023
Published 23 August 2023 Volume 2023:16 Pages 19—41
DOI https://doi.org/10.2147/SCCAA.S417842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Bernard Binetruy
Sireesha V Garimella,1 Siri Chandana Gampa,1 Pankaj Chaturvedi2
1Department of Biotechnology, School of Science, GITAM (Deemed to be University), Visakhapatnam, Andhra Pradesh, 530045, India; 2Department of Cell and Developmental Biology, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA
Correspondence: Sireesha V Garimella; Pankaj Chaturvedi, Email [email protected]; [email protected]
Abstract: Cancer continues to rank among the world’s leading causes of mortality despite advancements in treatment. Cancer stem cells, which can self-renew, are present in low abundance and contribute significantly to tumor recurrence, tumorigenicity, and drug resistance to various therapies. The drug resistance observed in cancer stem cells is attributed to several factors, such as cellular quiescence, dormancy, elevated aldehyde dehydrogenase activity, apoptosis evasion mechanisms, high expression of drug efflux pumps, protective vascular niche, enhanced DNA damage response, scavenging of reactive oxygen species, hypoxic stability, and stemness-related signaling pathways. Multiple studies have shown that mitochondria play a pivotal role in conferring drug resistance to cancer stem cells, through mitochondrial biogenesis, metabolism, and dynamics. A better understanding of how mitochondria contribute to tumorigenesis, heterogeneity, and drug resistance could lead to the development of innovative cancer treatments.
Keywords: mitochondria, cancer stem cells, drug resistance, therapy, metabolic dysfunction
Introduction
Despite several available therapies cancer is a leading cause of death worldwide. The failure of cancer cells to be eliminated by any kind of chemo or radiation therapy is attributed to a subpopulation of cells in the tumor, referred to as cancer stem cells (CSCs) or tumor-initiating cells (TICs).1–4 In the early nineteenth century, several studies documented the presence of pluripotent stem cells in teratomas and hypothesized their role in tumorigenesis.5,6 However, the debate was rekindled when a study on human acute myeloid leukemia (AML) provided the first evidence for the involvement of stem cells in cancer. This study demonstrated that transplanting a population of cells from AML patients into severe combined immune-deficient (SCID) mice, initiated AML in the mice. These cells were then referred to as the AML-initiating cell population.2 Similarly, CSCs were found in a variety of malignancies, including those of solid tissues.7 In fact, the existence of CSCs in solid tumors was first shown in breast cancer in the early 2000s, where as few as a hundred CSCs were able to form tumors in mice, in contrast to tens of thousands of cells with alternative phenotypes.8 These unusual cell subpopulations that cause tumors in vivo were later discovered in colon and brain malignancies.9,10 To date, CSCs have been isolated from almost all solid tumors, including pancreatic cancer, prostate cancer, melanoma, and ovarian cancer.11–14
CSCs are subpopulations of cancer cells that share characteristics with healthy stem or progenitor cells, such as the ability to self-renew and differentiate into several cell types to aid in the growth and heterogeneity of tumors.15 It is well established that CSCs make up a relatively small fraction of tumor tissues, often between 0.01–2% of the overall tumor mass.4,16 CSCs act as drivers of tumor formation and growth and are frequently associated with aggressive, heterogenous, and therapy-resistant tumors.17–20 CSCs’ resistance to chemotherapy or radiotherapy is linked to various factors, including the pivotal role of the cellular powerhouse – the mitochondrion. Mitochondria contribute to the maintenance of CSCs’ survival and self-renewal, drug resistance, and tumor recurrence. Alterations in mitochondrial structure, function, and location are commonly observed in CSCs.21–24 Consequently, exploring how mitochondrial function regulates CSCs holds promise in facilitating the creation of innovative CSC-targeted treatments to overcome cancer drug resistance.
In this review, we discuss the diverse attributes displayed by CSCs, exploring their connection with mitochondrial biology, and particularly emphasizing the role of mitochondria in CSC drug resistance.
Characteristics of Cancer Stem Cells
A common approach to reduce the tumor burden is to eliminate proliferating cells by chemotherapeutic agents. However, CSCs can undergo quiescence and resist such treatments, triggering a tumor relapse.25,26 Hence, it is essential to understand the basic cellular and molecular factors that influence the functioning and survival of CSCs. In this context, beyond the proliferative and self-renewal capabilities of CSCs, we elucidate several significant traits that govern their tumorigenicity.
Promoting Tumor Recurrence
Despite significant advancements in first-line anti-cancer medication, resection surgery, combination chemotherapy, and radiation, many patients still experience high rates of tumor recurrence and metastasis. The survival of CSCs following conventional therapy is assumed to be the cause of tumor recurrence, which poses a serious clinical problem in the successful treatment of cancer.25,27,28 Current anti-cancer medicines fail to effectively treat CSCs, which contributes to tumor recurrence, diversification, and a poor prognosis.29,30 There are several ways to understand CSCs’ function in promoting cancer recurrence. The foremost cause of tumor recurrence is due to the ability of CSCs to withstand radiation and chemotherapy, thus maintaining a steady supply of tumor-causing cells.25,27 Another viewpoint on recurrence focuses on the significance of epithelial-mesenchymal transition (EMT), which involves the transformation of epithelial cells into mesenchymal phenotypes.31 Overexpression of EMT-related transcription factors (eg Twist and Snail) led to the expression of antigenic markers of neoplastic mammary stem cells in the non-tumorigenic, immortalized human mammary epithelial cells (HMLEs).32 These were able to form mammospheres, a characteristic of CSCs, and also expressed typical CSC markers such as CD44+/CD24−/low.32 In a separate investigation, human breast tumor cells belonging to the claudin-low molecular subtypes demonstrated enrichment of cells that expressed elevated levels of CD44+/CD24−/low markers and exhibited the ability to form mammospheres.33 These cells had high expression of mesenchymal genes like Snail and low expression of cell-cell contact genes such as E-cadherin after treatment with endocrine therapy or chemotherapy.33 Such evidence indicates that the CSCs undergo EMT and escape treatment resulting in tumor recurrence. Moreover, the establishment of CSCs can also occur due to abnormal activation of autocrine and paracrine signaling pathways.34 This phenomenon is corroborated by a study that highlights the coordinated influence of TGFβ-SMAD and Wnt-β catenin pathways in inducing epithelial-mesenchymal transition (EMT) in both normal and tumorigenic human mammary epithelial cells (MECs).35 According to certain studies, stem cell-like subpopulations of mesenchymal circulating tumor cells (CTCs) may serve as markers of micrometastatic status and predictors of the likelihood of tumor recurrence.36
Tumorigenicity and Transplantation Potential
The tumorigenic and metastatic potential of CSC-containing malignancies surpasses that of non-CSC tumor cells, a well-established fact supported by numerous in vitro and in vivo studies. In particular, pancreatic cancer cells expressing CSC markers, such as CD133 and CXCR4, have demonstrated significantly higher tumorigenic and metastatic abilities.37 Additionally, studies involving the transplantation of these CSCs into immunodeficient mice have shown their remarkable capacity to repopulate the original tumor even at low clonal densities, further exemplifying their potent tumorigenic potential.15,38 For instance, in one study, injection of a small number of CD44+/CD24− prostate cells into SCID mice resulted in tumor formation. These cells expressed stem-cell associated BMI1 and OCT-3/4, reinforcing their role as cancer stem cells.38 Various xenograft models, both in vitro and in vivo, have consistently revealed that subpopulations of CSCs from different malignancies exhibit significantly higher proliferative capability, enhanced clonogenic potential, and an increased propensity for tumorigenesis and metastasis. Notably, numerous human malignancies, including leukemia, glioblastoma, breast, and skin cancers, harbor these clonogenic potential cells capable of reforming the parental tumors after transplantation. This underlines the critical role of CSCs in driving tumor initiation, growth, and dissemination, making them an essential target for developing effective cancer therapies.8,38–40
Expression of Specific Markers
Recent advances in single-cell technologies have enabled genomic and proteomic profiling of individual cells. These advancements have also led to robust isolation and characterization protocols to identify CSCs from the rest of the tumor, based on a few molecular markers. However, the CSC isolation protocols are still limited by the cellular heterogeneity within the tumor and the diverse origins of tumors.41,42 It has been demonstrated that several cell surface markers, such as THY1 (THYmocyte differentiation antigen 1), EpCAM (epithelial cell adhesion molecule), ABCB5 (adenosine triphosphate (ATP) -binding cassette B5), CD24, CD133, CD200, CD44, etc may identify populations that are CSC-enriched (Table 1).10,43–45 Other markers have also been used to identify CSCs, like aldehyde dehydrogenase 1 (ALDH1), which is used to characterize CSCs in many types of cancers, including breast, leukemia, colon, liver, pancreatic, lung, prostate, brain and bladder (Table 1).46–51 The expression of CSC markers has been suggested to be associated with certain CSC characteristics like chemoresistance and the recurrence of invasive tumorigenicity.52,53 Numerous studies have also reported the expression of pluripotency factors such as KLF4, NANOG, SOX2, OCT4, and c-MYC as phenotypic markers of CSCs.54–57 A recent study utilizing triple-negative breast cancer cells has established Kruppel-like factor 8 (KLF8) as the master regulator for the expression of these pluripotency markers.58 The study also showed the presence of a positive feedback loop with a metabolic enzyme, O-GlcNAc transferase (OGT). Increased expression of KLF8 correlated with increased resistance to paclitaxel, a commonly used chemotherapeutic agent for breast cancer.58
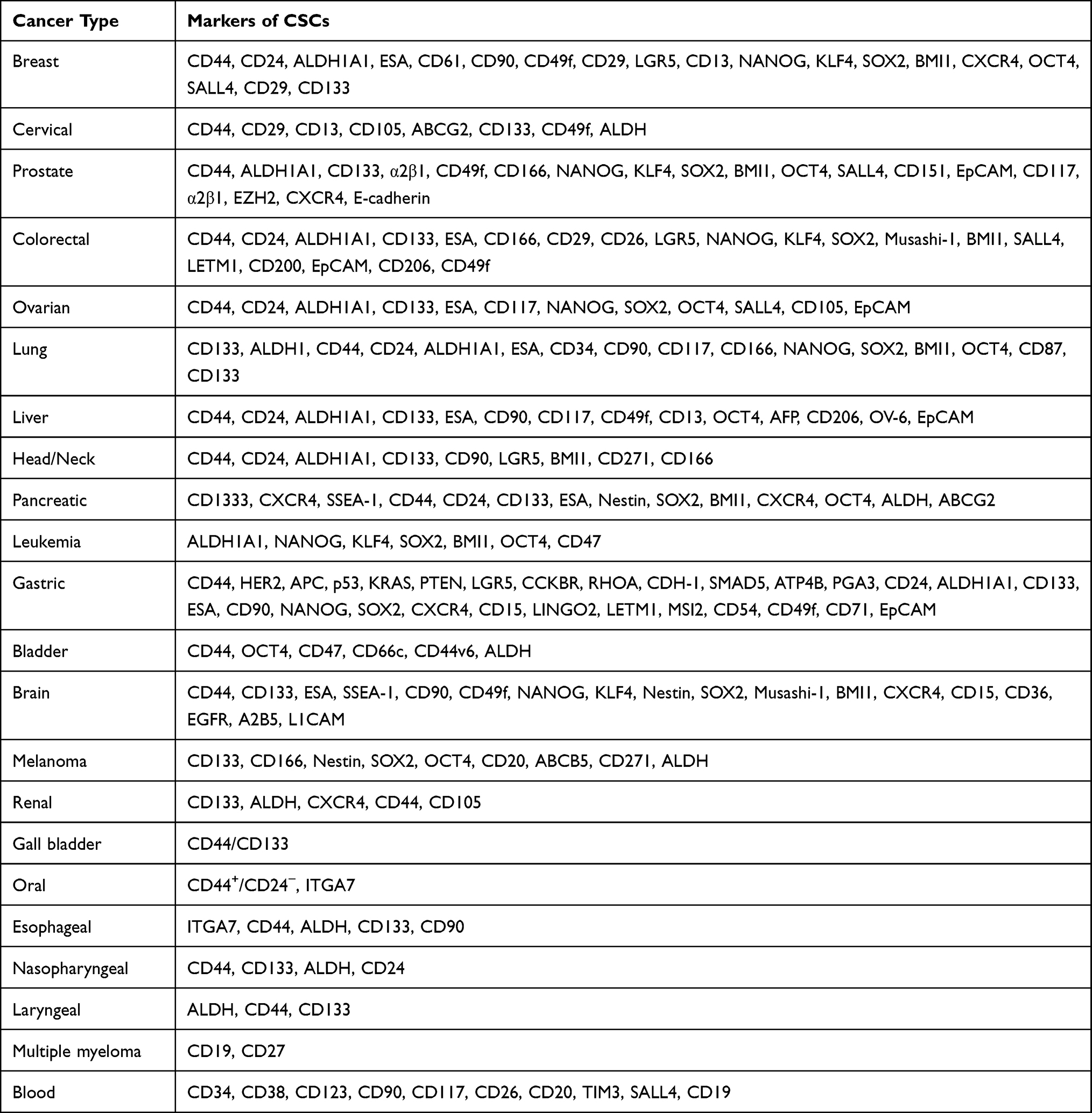 |
Table 1 Cancer Stem Cell Associated Markers Reported in Different Cancers |
CSCs and Acquisition of Therapeutic Resistance
Drug resistance in CSCs is a multifaceted phenomenon involving various mechanisms that enable these cells to survive and persist despite treatment.58,59 This resistance can stem from intrinsic factors, which may be inherited or acquired resistance to medication, as well as extrinsic factors, which result from tumor cells being exposed to chemicals. A few examples of these factors include the hypoxic microenvironment,60–62 disrupted cell cycle regulation,63 increased autophagy,64 epigenetic modifications,30 microRNA dysregulation, interactions with the tumor microenvironment,65 heterogeneity within CSC populations, quiescence,65 interactions with the extracellular matrix, and paracrine signaling. These factors ultimately contribute to drug resistance through downstream processes that include epithelial-mesenchymal transition (EMT), drug efflux through ABC transporters, deregulation of essential signaling pathways, expression of multidrug-resistant (MDR) proteins, upregulation of DNA repair proteins, acquired mutations, evasion of apoptosis, and activation of the DNA damage response (DDR) pathway.66–69 Gaining a comprehensive understanding of these complexities is crucial to develop effective therapies targeting CSCs and ultimately enhancing cancer treatment outcomes. An overview of the processes responsible for drug resistance in CSCs is presented in the following section.
Quiescence
Quiescence is a biological condition in which the cells do not enter the cell cycle, remain in a state of rest but retain the ability to divide. Adult stem cells exhibit quiescence as a part of tissue homeostasis,70 whereas CSCs undergo quiescence to escape drug exposure.71 CSCs can alternate between the phases of proliferation and quiescence, and the latter state is responsible for cancer recurrence and therapy resistance.72,73 CSCs often spend several years in a quiescent state (ie reversible G0 phase) within the body and endure prolonged periods of environmental stress.65 These CSCs in the quiescent state are distinct from active CSCs because they lack unique surface markers and common genotypic and phenotypic traits. They do, however, have certain distinctive traits, such as label retention, low RNA content, and lack of expression of proliferative markers,74 and have been studied in a variety of cancers.75,76 Chemotherapies drive CSCs to enter quiescence through upregulation of hairy and enhancer of split homolog-1 (HES1), a transcriptional repressor of Notch signaling, downregulation of c-MYC resulting in decreased Wnt signaling, increased expression of bone morphogenetic protein 7 (BMP7), which upregulates a metastasis suppressor gene, N-MYC downstream-regulated gene 1 protein (NDRG1) through activation of the p38-MAPK signaling pathway.77–79 Epigenetic modifications like DNA methylation and chromatin remodeling also drive CSCs into quiescence. Through H4K20me3 catalysis, SET domain-containing protein 4 (SETD4) induced quiescence in breast CSCs through tighter heterochromatin formation.75 These genetic and epigenetic alterations act as a switch to regulate the growth arrest and quiescence of CSCs, which are linked to aggressive biology and chemoresistance of malignancies.80,81
Dormancy
Dormancy is a stage in cancer progression in which cells stop proliferating. When the majority of the cancer population exhibits this phenomenon, the result is known as tumor dormancy, and when a single cancer cell exhibits this phenomenon, the process is referred to as quiescence.82 Dormancy is a special case of quiescence and is perhaps a deeper arrested state.83 In contrast to quiescence, where cells resume proliferation more readily, dormancy requires a particular stimulus for cells to proliferate. When cells from the same tumor are disseminated, they have very distinct fates. Most of them experience senescence. Those that survive circulation and extravasation at secondary sites are destined for a period of dormancy but might also enter quiescence based on the signals received from the microenvironment.84 Tumor growth, metastasis, minimal residual disease (MRD), multidrug-resistance (MDR), and tumor expansion are all effects of tumor dormancy.85–88 It is a type of clinical remission in which cancer cells are occult (ie undetectable and asymptomatic), for a lengthy period.89 CSCs and their clonal development are substantially responsible for tumor dormancy and treatment refractoriness in many forms of cancer.90,91 However, it is challenging to identify the precise or overlapping populations responsible for stimulating the processes of dissemination, intravasation, dormancy, and relapse due to the continual refining of the CSCs based on novel markers.91 Numerous malignancies, including pancreatic carcinoma, ovarian cancer, melanoma, lung cancer and chronic myeloid leukemia (CML) have been shown to have cells that combine stemness, drug resistance, and dormancy.92–97
Enhanced ALDH Activity
A family of nicotinamide adenine dinucleotide phosphate [NAD(P)+]-dependent enzymes, the ALDHs detoxify a broad range of aldehydes to weak carboxylic acids, increasing the cell’s resistance to injury from medicines.98 ALDHs play a crucial role in stem cell maintenance and differentiation as well as in healthy development. Accumulating evidence suggests that the expression of ALDH is upregulated as a response to therapeutic intervention, which in turn facilitates the development of resistance to chemotherapy and radiotherapy.99 By metabolizing harmful aldehydes and maintaining low reactive oxygen species (ROS) levels, ALDH enzymes help CSCs survive by regulating their capacity for self-renewal, cell differentiation, and chemoresistance. Through a variety of pathways, they support CSC immune evasion and metabolize retinoic acid, which promotes cancer progression and therapy resistance99–101 and are linked to the self-renewal abilities of stem cells in a variety of cancers, including breast cancer, colon cancer, hepatoma, and lung cancer.99,102–105 For example, increased ALDH gene expression was associated with high Snail expression. Knockdown of Snail decreased ALDH1 expression, inhibited cancer stem-like properties, and tumor formation ability of CD44+CD24−ALDH+ cells of head and neck squamous CSCs.103 High ALDH1 is detected only in CSCs of various tumors like breast, oesophagus, lung, colon, and stomach epithelium and not in the cancer tissues, thus serving as a marker for the identification of CSCs.106 Among the many isoforms, CSCs express high levels of ALDH1A1 and ALDHA3. Normal human and mouse stem cells express high levels of ALDHA1107,108 while normal human mammary cells have high ALDHA3 and low ALDHA1.109 ALDHs mediate drug resistance by converting active 4-hydroperoxycyclophosphamide (4-Hc) to inactivate carboxyphosphamide110 and this effect is reversed by pretreatment with N,N-diethylaminobenzaldehyde (DEAB).111
Apoptosis Evasion Mechanisms
The hallmark features of malignancies are attributed to the intrinsic ability of CSCs to self-renew, proliferate, and disseminate, as well as evade apoptosis via aberrant regulation of signaling pathways involved in programmed cell death.104 Cellular Fas-associated death domain-like IL-1β-converting enzyme (FLICE)-inhibitory protein (c-FLIP) is a negative controller of the death receptor (DR) -initiated apoptotic pathway.112 As a main anti-apoptotic regulator, c-FLIP interacts with Fas-associated death domain (FADD), caspase-8/10, and DR5, preventing the formation of death-inducing signaling complex (DISC) and subsequent activation of the caspase cascade.113 The CSC population was shown to have higher levels of c-FLIP expression than non-CSC-like cancer cells across a variety of malignancies, including leukemia, breast cancer, and glioblastoma.114–116 As a result, compared to their non-CSC-like counterparts, CSCs from these tumors show reduced sensitivity to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Several studies have demonstrated that c-FLIP isoforms sustain the survival and resistance of CSCs against apoptosis and anti-cancer treatments.117–119 Increasing the expression of c-FLIP in CD133+ cells, a marker associated with CSCs involved in metastasis, carcinogenesis, and chemoresistance, can serve as a way to inhibit apoptosis.112
Proteins from the inhibitor of apoptosis (IAP) family, which block apoptosis, are crucial for supporting cell survival. IAPs can directly or indirectly interact with caspases and thwart the apoptotic cascade. As an alternative, certain IAPs take part in signal transduction and activate the nuclear factor kappa B (NF-κB) pathway and promote cell survival. Receptor-interacting protein kinase 1 (RIP1) mediates caspase-dependent activation of cell death. Downregulation of RIP1 levels is mediated by IAPs that recruits inhibitor of nuclear factor- κB (IκB) kinase (IKK) and E3 ligases and drive the ubiquitination/degradation of RIP1, leading to cell survival.120 IAPs are also involved in the maintenance of CSC properties by enhancing the stability of CSC markers like SOX2.121 For example, XIAPs blocked the autophagic degradation of SOX2 by inhibiting the activation of ERK1 in CSCs. In nasopharyngeal CSCs, autophagic degradation of SOX2 was inhibited by XIAPs which negatively regulated the activity of ERK1. SOX2 enhanced the stemness of CSCs, suggesting that IAPs can induce the expression of pluripotency markers.121 Contrary to many oncogenes, B-cell lymphoma-2 (Bcl-2) inhibits cell death and improves tumor cell survival rather than promoting cell proliferation.122 Studies have demonstrated elevated levels of Bcl-2 family proteins in CSCs, and these higher levels have been associated with reduced cell death and treatment resistance in CSCs.123,124 This resistance of cancer cells to treatment and programmed cell death is partially attributed to the balance between anti-apoptotic and pro-apoptotic protein levels, which promotes cell survival.66
High Expression of Drug Efflux Pumps
CSCs have the unique ability to promote tumorigenesis, diversification, and metastasis. According to the CSC model of drug resistance, tumors include a population of pluripotent, drug-resistant cells that may withstand chemotherapeutic shock. CSCs in tumors are protected by ABC efflux pumps, which guard them against the negative effects of chemotherapy. ABCB1, ABCG2, and ABCC1 are among the drug efflux transporter proteins or ABC transporters that have been discovered to be expressed by CSCs. ABC transporters, such as ABCG2, ABCB1, and ABCC1, to mention a few, are linked to drug resistance and are significantly expressed in several malignancies.125–128
Protective Niche
The niche is a term used to describe the unique microenvironment where stem cells divide, differentiate, or stay dormant. Chemokines, immune cells, stromal cells, cytokine networks, growth factors, hypoxic areas, and extracellular matrix (ECM) make up the tumor microenvironment (TME).129 TME promotes CSC self-renewal, angiogenesis, modifying immunity, and other conditions that are favorable for metastasis. Dynamic alterations also contribute to treatment resistance, mostly by assisting CSCs in maintaining their stem-related signaling pathways.112 To maintain CSCs in a stem-like state, the CSC niche modifies the signaling pathways of Wnt-β catenin, Notch, and Sonic Hedgehog (Shh), and/or interferes with the function of key transcriptional regulators such as NANOG, OCT4, and SOX2, among other factors.130 Additionally, studies have also revealed that CSCs possess the ability not only to differentiate but to actively influence the surrounding microenvironment by recruiting niche components.131
Enhanced DNA Damage Response
CSCs are suggested to have an enhanced DDR to resolve DNA damage more effectively than bulk cancer cells.68,132–134 Studies have revealed that CSCs have a greater amount of inherent replicative stress than other types of cancer cells, leading to a constitutively active DDR. For example, in glioblastoma cancer stem-like cells expressing CD133, a CSC marker, increased expression of replicative stress response markers such as replication protein A2 (RPA2) and H2A histone family member X (H2AX) was observed when compared to CD133− cells. This phenomenon is due to the formation of DNA double-stranded breaks in glioblastoma cancer stem-like cells which results in increased DDR.135 Additionally, CSCs share numerous characteristics with normal stem cells. Studies indicate that tissue-specific stem cells employ DNA repair pathways to mediate chemotherapy and radiation therapy resistance, and CSCs may exploit these same processes to their benefit.133,136,137 Also, the remarkable resistance of CSCs to standard chemotherapy and radiotherapy techniques results from their strong capacity to repair DNA damage caused by chemical agents or radiation. This increased DNA repair capacity may be a direct result of improved repair mechanisms or an indirect effect of slowed cell cycle progression.138 Additionally, the CSCs evade therapeutic interventions through modulation of epigenetic marks (eg DNA methylation, promoter methylation/acetylation), long-range chromatin interactions, and altered splicing of nascent transcripts.30
Scavenging of ROS
The physiological and functional activities of a living cell are greatly influenced by its oxidation-reduction (redox) state. Similar to normal stem cells such as hematopoietic stem cells, CSCs also show lower intracellular ROS contents than non-CSCs, which may be due to the increased expression of free radical scavenging systems. Modulation of the level of ROS plays a crucial role in chemoresistance and the upregulation of drug efflux during chemotherapy.139,140 ROS also mediate several processes, such as endoplasmic reticulum (ER) stress, autophagy, and disruption of the cell cycle, which contribute to the acquisition of chemoresistance in CSCs.141–143 For instance, ROS has been demonstrated to shift the ER-stress-mediated apoptosis to autophagy in methotrexate-resistant choriocarcinoma cells,142 highlighting their intricate role in drug resistance mechanisms. Enhanced ROS scavenging mechanism and decreased levels of ROS generation are associated with the increased radioresistance of CSCs in breast carcinoma.132 Upregulation of genes involved in ROS scavenging pathway such as glutathione peroxidase (GPX), superoxide dismutase (SOD), and catalase has been observed in breast cancer CSCs.132 Glutathione is an antioxidant, that plays a critical role in protecting cells against oxidative stress. Buthionine sulfoximine (BSO) inhibits gamma-glutamylcysteine synthetase,144 which results in decreased synthesis of glutathione. In the absence of adequate levels of glutathione, an increase in ROS levels was observed, which significantly reduced the clonogenic characteristics and radiation therapy resistance of CSCs, supporting the hypothesis that ROS scavengers play a role in CSC radioresistance.132
Hypoxic Stability
Hypoxia occurs when tissues receive insufficient oxygen levels, leading to an inability to maintain proper homeostasis. Tumor hypoxia refers to a condition in which the cells within excessively grown tumors receive less than 2% of the oxygen typically available in normal tissues.145 It is strongly associated with CSC’s resistance to radiation and chemotherapy, making the microenvironment an important factor in cancer progression.60,146 The role of hypoxia-inducible factors (HIFs), which function as transcription factors (TFs) in the cell’s oxygen signaling pathways, is gaining increasing recognition due to their involvement in both CSC survival and tumor diversification. Moreover, a growing body of experimental evidence demonstrates that HIFs that are not destroyed in the hypoxic condition of tumor cells take part in the change of CSC phenotypes and regulate tumor radiation or chemotherapy resistance.146 Studies have suggested the role of hypoxia in CSC resistance to radiation and chemotherapy, and that HIFs play a major regulatory role in the hypoxic microenvironment.60–62,146 Hypoxia maintains CSC stemness and promotes resistance through activation of self-renewal signaling pathways such as Notch, Wnt, and Shh.147,148
Stemness Signaling Pathways
CSCs share many characteristics with tissue or embryonic stem cells, including the constant activation of highly conserved signaling pathways involved in tissue homeostasis and development, such as Wnt, Shh, Notch, and Hippo signaling pathways. These pathways have been studied to test potential novel CSC-targeting medications since they are linked to CSC self-renewal.58,149 These and other findings imply that some oncogenic cues can activate CSCs. These signals are followed by a rise in chemotherapeutic treatment resistance and, in certain situations, radiation resistance.58,150 Furthermore, a strong correlation exists between various mitochondrial activities, including mitochondrial biogenesis, metabolism, and dynamics, and the factors contributing to drug resistance in CSCs. A few such factors have been discussed earlier and include ALDH activity, apoptosis evasion mechanisms, ROS scavenging, hypoxic stability, elevated cryoprotective pathways, and more. This observation highlights the pivotal role of mitochondria in the growth and survival of CSCs.151–153 By dissecting the role of mitochondria in CSC survival, we can potentially uncover valuable therapeutic opportunities that could be harnessed for the development of effective cancer treatments and management strategies.
Mitochondrion - A Key Organelle in Cancer Stem Cells
Mitochondria are bioenergetic, metabolic, and signaling organelles that are essential for sensing stress and helping cells adapt to their surroundings. Numerous studies have been conducted on the involvement of mitochondria in the emergence and spread of cancer.154–156 Mitochondria, which are the primary ATP producers, supply the energy required for carrying out cellular functions through a process known as oxidative phosphorylation (OXPHOS).157 In addition to energy production, mitochondria are crucial for the generation of ROS, redox chemicals, and metabolites as well as for controlling cell signaling, cell death, and biosynthetic metabolism.158–160 Due to their wide range of functions, mitochondria play a key role in cells’ capacity to detect stress and adapt to their surroundings.161 Mitochondria in cancer cells adapt to withstand challenging conditions such as hypoxia, nutrient scarcity, and cancer treatments. As a result, they play a pivotal role in tumor formation, necessitating adaptability to counter cellular and environmental changes, as well as the effects of cancer therapies.162 Besides bioenergetics, many other aspects of mitochondrial biology have been implicated in cellular transformation. Some of such processes are mitochondrial biogenesis and turnover, metabolism, fission and fusion dynamics, oxidative stress regulation, cell death susceptibility, and signaling.
The morphology, localization, and functions of mitochondria in CSCs differ from normal cells, normal stem cells, and cancer cells.163–166 CSCs express fewer mitochondrial DNA copies (mtDNA) and low levels of mitochondrial transcription factor A (TFAM) in contrast to normal cells which express many copies of mtDNA and TFAM.163,165,166 Notably, during the process of fibroblast remodeling into iPSCs, a significant reduction in the number of mitochondria takes place, accompanied by decreased mtDNA, mitochondrial mass, and low ROS levels in the stem cells. Conversely, as stem cells differentiate, there is an observable rise in mitochondrial biomass and mtDNA content, resulting in increased ROS and ATP production.167 Structurally, mitochondria in CSCs are small and round in shape and highly perinuclear in localization whereas in normal cells, mitochondria are elongated and tubular in shape and mostly distributed in the cytoplasm.168,169 Cristae within mitochondria appear elongated in regular cells, and spherical in normal stem cells. In CSCs, the cristae become widened and fragmented.170 Due to fragmented mitochondria, CSCs exhibit impaired aerobic function and reduced ETC, which leads to a decrease in ROS levels causing resistance to HIF-1α and subsequent activation of MAPK that helps in the maintenance of stemness.171–173 Also, CSCs exhibit unique metabolic characteristics compared to cancer cells and normal stem cells. They switch between glycolysis and OXPHOS to produce ATP which is required for their activities. CSCs produce oncometabolites like fumarate, succinate, lactate, and 2-hydroxyglutarate which helps in tumor proliferation, angiogenesis, and invasion through accumulation of HIF-1α, production of VEGF through activation of STAT3, activation of p65 via NF-κB pathway and many others.174–176 Metabolically, mitochondria from CSCs vary from non-CSCs in terms of glucose uptake/consumption, ROS levels, ATP contents etc depending on the origin of the cancer.177
In the following section, we discuss the role of different mitochondrial aspects in CSCs and their contribution to drug resistance in CSCs.
Mitochondrial Biogenesis and CSC Resistance
Mitochondrial biogenesis is the process by which cells increase the number and size of mitochondria, an essential process to maintain proper metabolism and the cell cycle. Several mitochondrial proteins involved in biogenesis are encoded in the nucleus and translated to the cytosol. The transport of these proteins from the cytosol to the mitochondria takes place through translocase of the outer membrane (TOM) complex. This process takes place during the M phase of the cell cycle. Thus, mitochondrial biogenesis is linked to cell cycle, thereby enabling proper functioning of the cell.178,179 Each cell contains many copies of mitochondrial DNA (mtDNA). The size of human mtDNA is 16.5 kb and comprises 37 genes responsible for coding 13 polypeptides vital for OXPHOS, along with 2 rRNAs and 22 tRNAs necessary for translating the respiratory subunit mRNAs within the mitochondrial matrix. Other mitochondrial proteins are coded in the nuclear genome.180 Thus, mitochondrial biogenesis is a strictly controlled process that uses “mitonuclear communication” to coordinate a network of both mitochondrial and nuclear DNA (mtDNA and nDNA).181 During the process of mitochondrial biogenesis, a limited number of coactivators and nuclear TFs that are already present in the cell are gradually activated by signaling pathways, leading to the formation of new mitochondria from the pre-existing ones. Mitochondrial biogenesis is stimulated under increased energetic needs by a signaling pathway involving peroxisome proliferator-activated receptor-gamma co-activator 1 (PGC1) family members (such as PGC1α, PGC1β, and PPRC1), nuclear respiratory factors (NRF1 and NRF2), mitochondrial transcription factor A (TFAM), and estrogen-related receptors (ERRs) (ERR -α, -β, and -γ), and to a smaller extent, the peroxisome proliferator-activated receptor (PPAR) family of TFs (Figure 1a).182
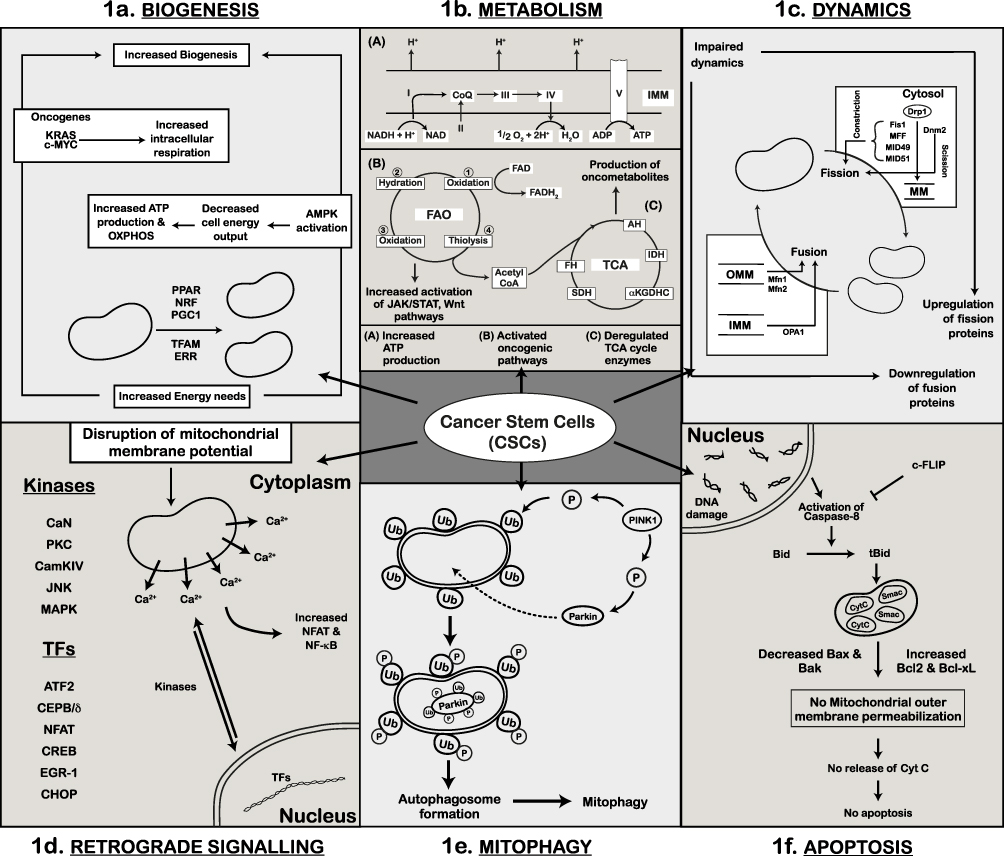 |
Figure 1 Mitochondrial Dysfunction and Cancer Stem Cells (CSCs). (a) Schematic of mitochondrial biogenesis and its regulation by transcription factors (PGC1, NRF, TFAM, PPAR, and ERR). Additionally, depicted are AMPK, oncogenic KRAS, and c-MYC-dependent mechanisms that lead to increase in biogenesis and energy production. This results in elevated oxidative phosphorylation (OXPHOS) and high ATP levels in CSCs. (b) Representation of mitochondrial metabolic dependency in CSCs. Cellular energy is derived through OXPHOS, fatty acid oxidation (FAO), and the TCA cycle within the mitochondria. CSCs exhibit increased oxidative phosphorylation for enhanced ATP production and elevated fatty acid oxidation through activation of oncogenic pathways. Deregulated TCA cycle enzymes in CSCs produce oncometabolites contributing to cancer progression. (c) Representation of altered mitochondrial dynamics in CSCs, where the balance between mitochondrial fission and fusion is disrupted. Upregulation of mitochondrial fission proteins (Drp1) and their regulators (Fis1, MID49, MID51, MFF) and downregulation of mitochondrial fusion proteins (Mfn1, Mfn2, OPA1) leads to impaired mitochondrial dynamics. (d) Increased activity of Ca2+-dependent kinases (PKC, CaN, CAMKIV, JNK, MAPK) due to altered membrane potential in CSCs is shown. Also indicated are the kinases and nuclear transcription factors involved in retrograde signaling. (e) Schematic representation of mitophagy in CSCs. Elevated cytoplasmic PINK1 phosphorylates Parkin and ubiquitinated-OMM proteins. Phosphorylated Parkin is transported into the mitochondria where it ubiquitinates itself and other mitochondrial substrates. These ubiquitin (Ub)-marked mitochondria are degraded by autophagosomes. (f) Mitochondria-mediated apoptosis in CSCs. Cells with damaged DNA activate caspase-8 mediated cell death. In CSCs, activation of caspase-8 is inhibited by high levels of c-FLIP; levels of pro-apoptotic proteins (Bax, Bak) are decreased while levels of anti-apoptotic proteins (Bcl-xL) are increased leading to cell survival and no apoptosis. |
The process of mitochondrial biogenesis is different in CSCs from other cells. CSCs exhibit low levels of TFAM and mtDNA when compared to differentiated cancer cells. This phenomenon has been observed in different cancers like lung, thyroid, and colon.183,184 Reduced number of mtDNA copies helps in the maintenance of the stemness of cancer cells. Stem-cell like characteristics were observed in esophageal squamous cell carcinoma cells exhibiting low copies of mtDNA. Additionally, the knockdown of TFAM in these cells resulted in the formation of spheres.166 Mitochondrial biogenesis and mtDNA alterations are frequently linked to increased tumorigenicity and resistance in CSCs. Ethidium bromide (EtBr) inhibits mtDNA replication. Ovarian cancer cells treated with EtBr showed upregulated proliferation through increased expression of genes like ABCC3, VEGFA, ATF3, etc. They also showed downregulation of mitochondrial-related genes like TMEM165, PDK1, PDK2, etc. The expression of the chemoresistance factor ABCC3, tumorigenicity-related factor HES1 and angiogenesis-related factor VEGFA were upregulated in the cells treated with EtBr. The increased expression of CSC markers CD90 and CD117 was also observed in these cells.21
CSCs exhibit increased energy demands for their survival. This results in the activation of TFs like PGC1, NRFs, etc resulting in increased production of ATP and OXPHOS by the activation of AMPK. Adenosine monophosphate (AMP) - activated protein kinase (AMPK) is frequently activated by decreasing cellular bioenergetic output to create ATP and OXPHOS, which in turn triggers mitochondrial biogenesis.185 AMPK promotes the catabolic pathways performed by the cell resulting in the generation of ATP. The expression of NRF2 was higher in CD44+/CD24− doxorubicin-resistant MCF7 cells. Silencing of NRF2 resulted in higher levels of ROS, decreased tumor growth, and reduced sphere formation and invasion in these cells when compared to controls.186 It is also required for the self-renewal of CSCs. Knockdown of NRF2 decreased the expression of BMI1, SOX2, and Cyclin E in glioma stem cells.187 Increased levels of NRF2 are crucial for the survival of CSCs and attaining drug resistance. In cervical cancer cells with SP phenotype, increased NRF2 expression resulted in enhanced expression of ABC transporter ABCG2 than in the non-SP cells.188 PGC1α plays an important role in causing drug resistance in cancer cells. In ovarian cancer, PGC1α overexpressing cells were resistant to chemotherapy. They expressed drug resistance-related proteins, MDR1 and ABCG2 and this was observed in tumorspheres than differentiated cells. Additionally, the spheres showed elevated mitochondrial mass and fragmented mitochondria at the perinuclear region. Knockdown of PGC1α showed decreased mitochondrial mass, downregulated expression of MDR1 and ABCG2, and sensitized the spheres to cisplatin treatment.189 Oncogenes like KRAS and c-MYC also regulate mitochondrial biogenesis and increase intracellular respiration and biosynthesis, which promotes the development of cancer (Table 2).154,190,191
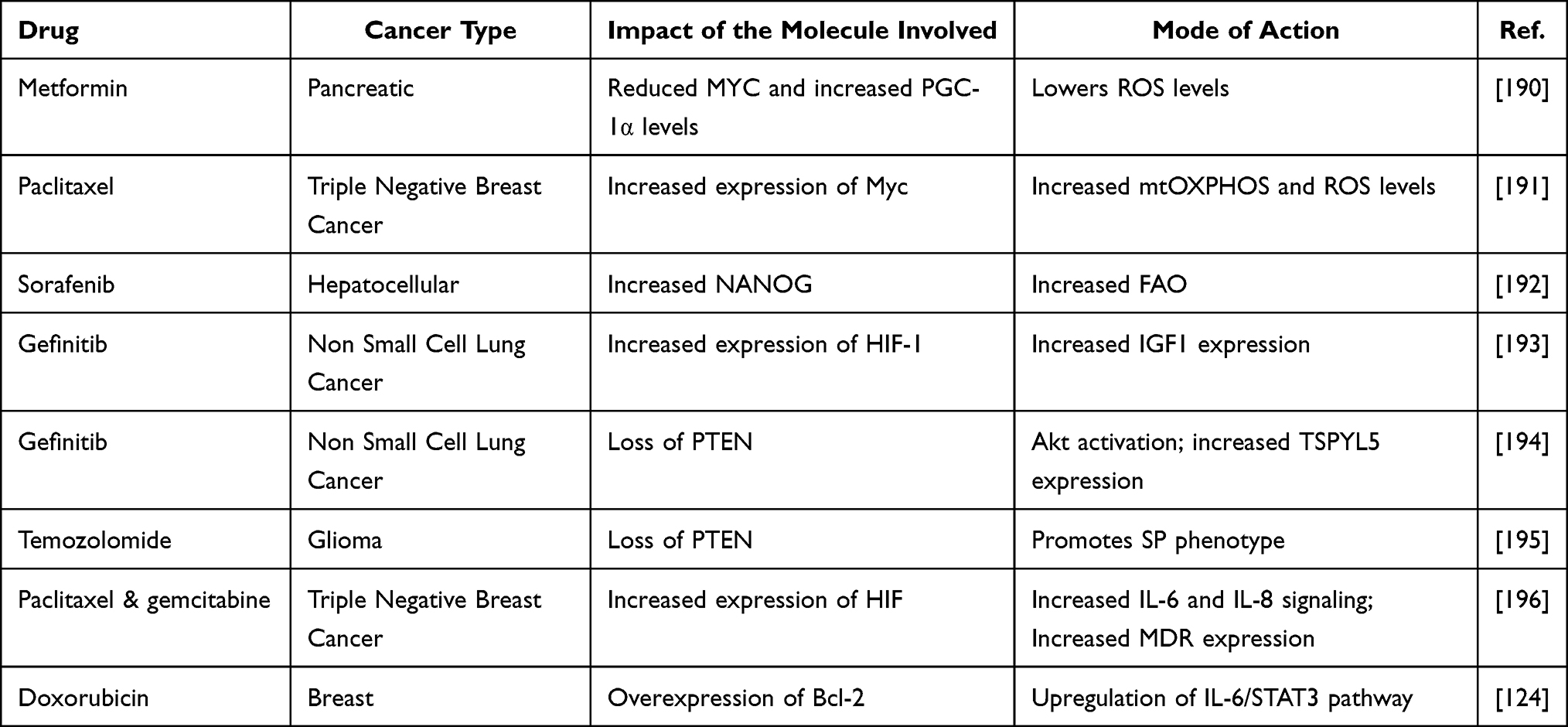 |
Table 2 Drug Category and Mode of Acquired Resistance in Cancer Stem Cells in Different Cancers |
Mitochondrial Metabolism and CSC Resistance
Mitochondria are subcellular organelles that are maternally inherited and are responsible for fundamental mechanisms of ATP production, including OXPHOS and electron transport chain (ETC), fatty acid oxidation (FAO), and tricarboxylic acid (TCA) cycle. In addition to these roles, mitochondria also play a crucial role in other cellular processes such as calcium signaling, apoptosis, and biosynthesis of important molecules such as heme, pyrimidines, and iron-sulfur (Fe-S) clusters. CSCs mostly depend on these processes to meet the energy demands for their survival. Unlike normal stem cells and differentiated cancer cells, CSCs exhibit distinct metabolic characteristics for the maintenance of stemness and self-renewal.172
Oxidative phosphorylation (OXPHOS) plays a critical role in the metabolism of CSCs.197,198 CSCs utilize ATP produced from OXPHOS for their metabolism. It has been observed that CSCs derived from the ovaries of patients exhibited elevated expression of mitochondrial OXPHOS enzymes.199 Unlike differentiated cancer cells, which undergo glycolysis, cancer stem cells depend on OXPHOS for their energy needs. In glioma, the comparison of oxygen consumption rate, glucose uptake, lactate production, and intracellular ATP levels between differentiated cancer cells and CSCs, CSCs showed less glycolytic activity, consumed less glucose, and produced less lactate. The increased levels of ATP were also observed in CSCs than the differentiated cells. Additionally, glioma stem cells were found to be radioresistant.200 In gliomaspheres, OXPHOS is known to be regulated by oncofetal insulin-like growth factor 2 mRNA-binding protein 2 (IMP2, IGF2BP2). IMP2 participates in the assembly and function of mitochondrial respiratory chain complex subunits by binding to mRNAs that code them. Depletion of IMP2 impaired OXPHOS by affecting complex I and complex IV mRNA and protein levels in gliomaspheres.201
Fatty Acid Oxidation (FAO) is required for the maintenance of stemness in CSCs. In a study on liver tumor-initiating stem-like cells (TICs), NANOG was found to be essential for FAO. Knockdown of NANOG resulted in decreased mRNA and protein levels of FAO-associated genes like Echs1, Acads, and AcadvI. The FAO flux analysis with 14C-radiolabeled-palmitic acid to produce acid-soluble 14C metabolites and 14CO2 demonstrated that NANOG+ TICs showed higher levels of FAO activity compared to controls.192 Carnitine palmitoyl transferase I (CPTI) and carnitine palmitoyl transferase II (CPTII) enzymes are crucial in increasing FAO in radioresistant breast cancer cells. Downregulation of the ERK pathway was observed in cells by blocking FAO by CRISPR-mediated CPTI/CPTII knockdown and inhibited the formation of tumorspheres in radioresistant breast CSCs.202 FAO is also regulated by JAK/STAT3 and is critical for CSC self-renewal and chemoresistance. Inhibition of JAK/STAT3 blocked the self-renewal of breast CSCs. It also resulted in reduced expression of the CPT1B gene, which codes for an enzyme involved in FAO.203 The reduced products formed during FAO, FADH2, and NADH, are funnelled back to the respiratory chain where they are oxidized to produce ATP which is required for the survival of CSCs.204 Elevated levels of FAO contribute to chemoresistance in different cancers by increased levels of oncogenic pathways like JAK/STAT3, and Wnt (Table 2).192,203,205,206
The TCA cycle, sometimes referred to as the Krebs cycle or the citric acid cycle, is a sequence of chemical processes that take place in a closed loop and function as an internal metabolic engine in cells oxidizing carbohydrates, proteins, and lipids.204 In a simplistic view, the TCA cycle is a continuous cyclic mitochondrial pathway that is continually oxidizing the acetyl moiety of acetyl-CoA to carbon dioxide (CO2), creating NADH and FADH2, whose electrons power the mitochondrial respiratory chain for ATP production.204
In normal cells, the TCA cycle is fuelled by glucose whereas in CSCs the products of the glutamine pathway fuel the TCA cycle.207 In human malignancies, several mitochondrial enzymes, involved in the TCA cycle like Aconitate Hydratase (AH), Isocitrate Dehydrogenase (IDH), Fumarate Hydratase (FH), Succinate Dehydrogenase (SDH), and α-ketoglutarate dehydrogenase complex (α-KGDHC) are often altered or deregulated (Figure 1b) and have been linked to cancer progression.208 Moreover, these mutations lead to the aberrant accumulation of various metabolites, known as oncometabolites like (R)-2-hydroxyglutarate, fumarate, and succinate. These oncometabolites can interfere with fundamental cellular processes, particularly epigenetic regulation, and contribute to cancer development and progression.209 Oncometabolites can alter epigenetic regulation by inhibiting enzymes involved in the removal of epigenetic marks, such as DNA and histone demethylases, or by promoting the activity of epigenetic writers, such as DNA methyltransferases and histone acetyltransferases. For example, mutations in SDH and FH cause the accumulation of succinate and fumarate and inhibit multiple α-KG-dependent dioxygenases such as histone and DNA demethylases in cancers.210 Also, mutations in SDH and FH result in the stabilization of HIF-1α, a transcription factor responsible for promoting tumor survival and metastasis.211 Another prominent example is the inhibition of the activity of the ten-eleven translocation (TET) methyl-cytosine hydroxylases and Jumonji (JmjC) domain-containing histone demethylases in gliomas and acute myelogenous leukemia due to mutant IDH.30,212 The dysregulation of epigenetic regulation by oncometabolites is thought to play a critical role in the development and progression of several cancers, including renal cell carcinoma and certain types of leukemia. Targeting the metabolism of cancer cells, including the production and accumulation of oncometabolites, is an area of active research for cancer therapy development.
Mitochondrial Dynamics and CSC Resistance
The process of mitochondrial fission (constriction and scission) and fusion, known as mitochondrial dynamics, regulates the shape, quality, and number of mitochondria. In contrast to mitochondrial fusion, which involves joining two mitochondria to form a single mitochondrion, mitochondrial fission is characterized by the division of a single mitochondrion into two daughter mitochondria. Large GTPase proteins from the Dynamin (Dnm) family make up the majority of the core machinery proteins.213 These mechanoenzymes can oligomerize and alter conformation to promote membrane remodeling, constriction, scission, and/or fusion.214 Mitochondrial fission is carried out by the Dnm-related/-like protein 1 (Drp1) that can be recruited to the mitochondrial membrane (MM) from cytoplasm with the help of mitochondrial receptor proteins Fis1, MID49, MID51, MFF (for constriction) and Dnm2 (for scission) (Figure 1c).215 On the other hand, mitochondrial fusion is ensured by mitofusins 1 and 2 (Mfn1 and Mfn2) and optic atrophy 1 (OPA1), which mediate outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) fusion, respectively (Figure 1c).216 Up-regulation of fission-related proteins and down-regulation of fusion-related proteins have been implicated in the onset, development, metastasis, CSC survival, and treatment resistance of several cancers.217–220 The inhibition of Drp1, a fission-related protein using mdivi-1 resulted in the inhibition of cell migration and CSC signaling in breast cancer. The inhibition also reduced the formation of tumorspheres in a dose-dependent manner in breast, lung, and melanoma cells.221 In another study, high expression of OPA1 was observed in the tumorspheres of NSCLC CSCs, which was due to overexpression of SPDEF, a SAM Pointed Domain containing ETS transcription factor.222 Mitochondrial fission and fusion enhance CSC stemness and maintain self-renewal. Knockdown of fission-related genes such as Drp1, and MFF reduces the expression of stemness-associated genes like OCT4, NANOG, etc, and the tumorsphere formation capability of CSCs in brain and prostate cancer.220,223 Moreover, the inhibition of Drp1 by mdivi-1 reduces the capacity of CD44+ CSCs to form tumors in vitro and in vivo in nasopharyngeal cancer.224 Similarly reduced populations of CD133+CD15+ brain tumor-initiating cells and decreased levels of stemness genes in EpCAM+CD133+ liver cancer stem cells were observed upon Drp1 inhibition with mdivi-1.220,225 Phosphorylation of Drp1 induces mitochondrial fragmentation to promote metabolic adaptation and chemoresistance as seen in acute lymphoblastic leukemic T-cells.226
Mitochondrial Retrograde Signaling and CSC Resistance
Mitochondrial retrograde signaling refers to the cellular response to changes in mitochondrial activity and state and is a vital component in maintaining cellular homeostasis. Mitochondrial retrograde signaling enables the transmission of information regarding alterations in mitochondrial bioenergetics and redox potential to the rest of the cell. Altered nuclear gene transcription due to mitochondrial dysfunction opens new avenues in mitonuclear communication.227 Under both normal and pathological circumstances, mitochondria can communicate with the nucleus through mitochondrial retrograde signaling. Disruption of the MM potential and poor absorption of Ca2+ leads to increased intracellular Ca2+. This triggers the activity of Ca2+ dependent kinases such as protein kinase C (PKC), c-Jun N-terminal kinase (JNK), calcium/calmodulin-dependent protein kinase IV (CamKIV), and mitogen-activated protein kinase (MAPK) which then function through various transcription factors like activating transcription factor 2 (ATF2), nuclear factor of activated T-cells (NFAT), CCAAT/enhancer-binding protein delta (CEBP/δ), early growth response protein 1 (Egr-1), cAMP-response element binding protein (CREB), C/EBP homologous protein (CHOP), and NF-κB, to alter the nuclear gene expression (Figure 1d). Additionally, increased Ca2+ levels activate calcineurin (CaN), a calcium-dependent serine-threonine phosphatase that is thought to have developed from RTG-dependent retrograde (RTG) signaling and increases NFAT and NF-κB.228 The ongoing maintenance of the organelle may be viewed as a delicate balance between its biogenesis and the quality control systems (engaged in remodeling and mitophagy) that ensure cell homeostasis and function. Numerous antioxidant enzymes like GPX1, PRDX3, PRDX5, SOD2, chaperones, and quality control proteases work together to maintain this function by promoting protein folding and stability on the mitochondria while degrading accumulating unfolded or misfolded proteins.229
The molecular connection between the nucleus and mitochondria, which involves ATP, calcium, and ROS, is crucial for this regulation.230 Mitochondrial-to-nucleus communication, also activates a coordinated expression of nuclear genes to relieve the stress and/or to compensate for the defect upon organelle dysfunction which are caused by many events, such as mtDNA depletion, deletions, mutations, aggregation of misfolded proteins, oxidative stress, or dramatic changes in morphology and dynamics.231 The dysfunction of mitochondria due to these factors results in the activation of retrograde signaling, which alters the transcription of nuclear genes that encode mitochondrial proteins involved in retrograde signaling. This alteration can lead to the acquisition of stemness, EMT induction, resistance to apoptosis, and drugs in CSCs.232–234 For example, the reduction of mtDNA activated CaN-dependent mitochondrial retrograde signaling and generated breast CSCs. This CaN-mediated mitochondrial retrograde signaling led to the induction of EMT by increased mesenchymal gene expression in mtDNA-reduced cells.235 These changes occur through dysregulation of TFs involved in mitochondrial retrograde signaling. Also, the triggering of the signaling pathways involved in retrograde signaling converges on the upregulation of genes affecting several cellular functions, including apoptosis resistance, MDR, invasion, and EMT.151 In prostate cells depleted of mitochondria, PARP inhibitor AGD14699 activates Ca2+-mediated retrograde signaling and downregulates BRCA2 levels. Decreased levels of BRCA2, a tumor suppressor protein that regulates the homologous DNA repair process, make the prostate cells sensitize to PARP inhibitor, resulting in cell death. This demonstrates that the presence of mitochondria in the cells provides resistance to drugs.233
Increased ROS obtained after mtDNA depletion in hepatocellular carcinoma cells, activates NRF2 signaling pathway and multidrug-resistance proteins MRP1 and MRP2 to help tumor cells fight against ROS and resist cisplatin and doxorubicin treatment.236 Also, mitochondrial stress-related ROS modulates the expression of PGC1α, a key regulator of mitonuclear communication, to promote OXPHOS and confer cisplatin resistance in SKOV3 ovarian cancer cells.237 Porporato et al have shown that dysfunction in the ETS results in ROS overproduction that activates Src, which in turn induces the expression of Pyk2, a FAK family protein tyrosine kinase known to promote cytoskeletal remodeling, migration, and EMT in SiHa cells.238 The above studies indicate a key role for mitochondrial retrograde signaling in maintaining stemness and in drug resistance of cancer cells. However, the role of mitochondrial retrograde signaling in CSC drug resistance is still being explored.
Mitophagy and CSC Resistance
To ensure a robust and healthy mitochondrial population, cells employ a controlled catabolic process known as mitophagy, which serves to eliminate any damaged or defective mitochondria. By doing so, mitophagy plays a crucial role in reducing cell damage, promoting cellular homeostasis, and supporting overall cell survival.239 Mitophagy plays a crucial role in conferring tumor resistance to various cancer therapies (Table 2) by facilitating the degradation of impaired mitochondria, consequently leading to a reduction in mitochondrial ROS levels.240,241 Different routes can be used to activate mitophagy. One such mechanism is through the phosphatase and tensin homolog (PTEN) -induced putative kinase 1 (PINK1) and Parkin signaling pathway. The PINK1/Parkin pathway is in-charge of preparing damaged mitochondria for selective autophagic identification. In general, PINK1 is transported into the IMM by translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM) complexes, where it is digested by the proteasome and cleaved by the mitochondrial protease PARL (presenilin-associated rhomboid-like) (Figure 1e).242 When the mitochondria are depolarized, PINK1 remains connected to the OMM and recruits PARKIN which helps in ubiquitylation of OMM substrates. This ubiquitylation pattern acts as a signal for the sequestration of damaged mitochondria (Figure 1e).243 Thus, depolarization of MM results in increased OMM expression of PINK1 following recruitment of Parkin to the mitochondria allowing selective and effective turnover of damaged mitochondria.244
Another mechanism that contributes to the removal of mitochondria under physiological and diseased conditions is MM receptor-mediated mitophagy. This includes different receptors like BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BNIP3L/NIX, FUN14 domain-containing protein 1 (FUNDC1), an activating molecule in Beclin 1-regulated autophagy (AMBRA1), FK506-binding protein 8 (FKBP8), ATPase family AAA domain-containing protein 3B (ATAD3B), and some kinds of lipids (cardiolipin (CL) and C18-ceramide.245 The key mediators of hypoxia-induced mitophagy include BNIP3 and BNIP3L/NIX. Interestingly, the transcription of BNIP3 and NIX is influenced by HIF-1.
Mitophagy contributes significantly to the mitochondrial stress response through these two pathways, as well as to the regulation of mitochondrial quality and the maintenance of homeostasis.246–249 CSCs utilize mitophagy to promote their survival.248–250 For example, in hepatic CSCs, enhanced mitophagy promoted the recruitment of phosphorylated p53 to the mitochondria thereby increasing the nuclear expression of NANOG and promoting stemness.247 This process facilitates the selective distribution of mitochondria between stem-like and non-stem-like cells. For example, when mammary epithelial stem-like cells divide, the daughter cells with stem cell characteristics inherit fewer older mitochondria, whereas the differentiated cells receive a higher proportion of older mitochondria. Consequently, stem-like cells inherit the newest and most efficient mitochondria, promoting their continued function, while the differentiated daughter cells that receive older mitochondria are eventually eliminated.251 Enhanced mitophagy within the CSC population facilitates the removal of aberrant mitochondria, promoting cell growth and survival across various tumor types.252 The ability of CSCs to enter a state of cell quiescence is tied to mitophagy. Mitophagy results in a decrease of mitochondrial mass and subsequently reduced OXPHOS activity. As a result, cells switch to glycolysis to meet their energy demands.178 Glycolysis drives CSCs to enter a quiescent state and is also crucial to increase antioxidant compensative capacity, enhancing stemness, and improving self-renewal capacity.253–255 As mentioned above, BNIP3 is highly expressed under hypoxic conditions. In glioblastoma cells, growing in hypoxic situations, it has been demonstrated by Jung et al that BNIP3-mediated mitophagy promotes cell survival by clearing ROS levels.249 In oral squamous cell carcinoma, CD44+/ABCB1+/ADAM17+ CSCs exhibited resistance to cisplatin. Higher autophagic flux and mitophagy were observed in drug-resistant FaDu cells compared to parental cells. Mitophagy is a key contributor to doxorubicin resistance in CSCs of HCT8 human colorectal cells. The CD133+/CD44+ cells were more resistant to doxorubicin treatment. Silencing of BNIP3L prevented mitophagy and increased sensitivity to doxorubicin therapy.256 Deletion or mutation of PARK2 and BNIP3 inhibits mitophagy and thereby promotes carcinogenesis. Loss of function mutation in the PARK2 gene has been detected in colorectal cancer.257 Therefore targeting mitophagy in CSCs could sensitize cells to various chemotherapeutic drugs.
Apoptosis and CSC Resistance
Mitochondria play a central role in apoptotic cell death. The intrinsic apoptosis process is triggered by DNA damage, the loss of survival factors, and alterations in cell cycle checkpoints. As part of the intrinsic pathway, BH3-interacting domain death agonist (Bid) is cleaved to truncated Bid (tBid) in the presence of activated caspase-8. This results in tBid translocation to the mitochondria and causes mitochondrial outer membrane permeabilization (MOMP) by activating Bcl-2 associated x -protein (Bax) and Bcl-2 homologous antagonist/killer (Bak), resulting in the release of Cyt C and mitochondria-derived activator of caspase (Smac) from mitochondria which are transported to the cytosol. In the cytosol, Cyt C interacts with ATP, apoptosis peptidase-activating factor-1 (Apaf-1), and initiator pro-caspase-9 to form a signaling complex called apoptosome where caspase-9 is activated, in turn causing the activation of effector caspases-3, −6, and −7 to cause apoptosis (Figure 1f).258 It has been demonstrated that higher levels of Bcl-2 family proteins are related to drug resistance in many cancers. Bcl-2 deregulation hinders the oligomerization of Bax and Bak, preventing MOMP, which in turn blocks the release of Cyt C into the cytosol and thereby inhibits apoptosis (Figure 1f).259 Increased levels of Bcl-2 proteins were detected in many CSCs like breast and colon.123,124 Additionally, mitochondria to nuclear retrograde signaling is related to increased transcription of anti-apoptotic Bcl-2 family members and activation of survival signals like Akt. CSCs also show apoptosis resistance by increased expression of anti-apoptotic proteins like c-FLIP and IAPs (as discussed in the earlier sections) that can block the activation of caspases, thereby inhibiting apoptosis.39,260 Acquired resistance to drugs by CSCs through dysregulation of apoptosis-regulating proteins is a recurrent theme observed in many cancers (Table 2).
Conclusion
Resistance to chemotherapeutic agents has grown into a major issue in the treatment of cancers. CSCs evolve diverse mechanisms to enable this therapeutic evasion of tumors, contributing to poor prognosis. Mitochondria play a central role in imparting drug resistance to the CSCs by altering many pathways involved in biogenesis, metabolism, dynamics, and retrograde signaling. Developing strategies to target different molecules involved in resistance pathways especially those associated with mitochondria, either alone or in combination with various chemotherapeutic agents could help in the sensitization of CSCs, promoting effective treatment.
Mitochondria-targeting therapies for CSCs are a new and promising approach, but still in the preclinical stages. Mitochondrial uncouplers selectively disrupt the proton gradient across the mitochondrial membrane, leading to oxidative stress-induced apoptosis in CSCs. Mitochondrial-targeting drugs, such as elesclomol, induce mitochondrial ROS production and lead to apoptosis in CSCs.261 Targeting mtDNA mutations using drugs or other therapies is another promising strategy for eliminating CSCs. Additionally, targeting mitochondrial dynamics, including fusion and fission, using drugs like mdivi-1 can induce mitochondrial fission, leading to the selective elimination of CSCs. However, it is essential to note that this is still an area of ongoing research, and the development of therapies targeting mtDNA mutations to eliminate CSCs is complex and may face challenges. Understanding the mechanisms and vulnerabilities of CSCs, as well as potential off-target effects of such treatments, will be critical in realizing the full potential of this approach.
Mitochondrial retrograde signaling is a process by which mitochondria communicate with the nucleus to alter gene expression in response to changes in mitochondrial function. Dysregulation of mitochondrial function, such as through mutations or environmental stressors, can lead to the activation of retrograde signaling pathways and alterations in nuclear gene expression that can promote stemness, EMT, drug resistance, and other hallmarks of cancer. Thus, by understanding the link between mitochondrial function and nuclear gene expression, novel strategies to target CSCs and prevent tumor recurrence can be developed. Targeting mitochondrial function or the pathways involved in mitochondrial retrograde signaling could potentially be used to induce apoptosis or differentiation of CSCs, sensitize them to traditional cancer therapies, or prevent the emergence of drug-resistant CSCs. Despite the promising results of these mitochondria-based therapies in preclinical models, there are still several challenges such as potential toxicity to normal cells and the heterogeneity of CSCs that need to be addressed to translate these therapies into clinical applications.
Funding
The authors would like to acknowledge that no external funding was received for the preparation of this review article.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Ebben JD, Treisman DM, Zorniak M, Kutty RG, Clark PA, Kuo JS. The cancer stem cell paradigm: a new understanding of tumor development and treatment. Expert Opin Ther Targets. 2010;14(6):621–632. PMID: 20426697. doi:10.1517/14712598.2010.485186
2. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. PMID: 7509044. doi:10.1038/367645a0
3. Kleinsmith LJ, Pierce GB. Multipotentiality of Single Embryonal Carcinoma Cells. Cancer Res. 1964;24:1544–1551. PMID: 14234000.
4. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. PMID: 9212098. doi:10.1038/nm0797-730
5. Makino S. Further evidence favoring the concept of the stem cell in ascites tumors of rats. Ann N Y Acad Sci. 1956;63:818–830. PMID: 13314436. doi:10.1111/j.1749-6632.1956.tb50894.x
6. Stevens LC. The development of transplantable teratocarcinomas from intratesticular grafts of pre- and postimplantation mouse embryos. Dev Biol. 1970;21:364–382. PMID: 5436899. doi:10.1016/0012-1606(70)90130-2
7. Hermann PC, Bhaskar S, Cioffi M, Heeschen C. Cancer stem cells in solid tumors. Semin Cancer Biol. 2010;20:77–84. PMID: 20371287. doi:10.1016/j.semcancer.2010.03.004
8. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. PMID: 12629218. doi:10.1073/pnas.0530291100
9. Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. PMID: 17122771. doi:10.1038/nature05384
10. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. PMID: 15549107. doi:10.1038/nature03128
11. Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. PMID: 17283135. doi:10.1158/0008-5472.CAN-06-2030
12. Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008;26:2862–2870. PMID: 18539965. doi:10.1200/JCO.2007.15.1472
13. Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. PMID: 18202660. doi:10.1038/nature06489
14. Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–4320. PMID: 18519691. doi:10.1158/0008-5472.CAN-08-0364
15. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25:20. PMID: 29506506. doi:10.1186/s12929-018-0426-4
16. Yang L, Shi P, Zhao G, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8. PMID: 32296030. doi:10.1038/s41392-020-0110-5
17. Capp J-P. Cancer stem cells: from historical roots to a new perspective. J Oncol. 2019;2019:5189232. PMID: 31308849. doi:10.1155/2019/5189232
18. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. PMID: 24522528. doi:10.1038/nature13038
19. Auffinger B, Tobias AL, Han Y, et al. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119–1131. PMID: 24608791. doi:10.1038/cdd.2014.31
20. Hamerlik P, Lathia JD, Rasmussen R, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209:507–520. PMID: 22393126. doi:10.1084/jem.20111424
21. Huang R, Wang J, Zhong Y, et al. Mitochondrial DNA Deficiency in Ovarian Cancer Cells and Cancer Stem Cell-like Properties. Anticancer Res. 2015;35:3743–3753. PMID: 26124317.
22. Huang H, Zhang S, Li Y, et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat Commun. 2021;12:3720. PMID: 34140524. doi:10.1038/s41467-021-24108-6
23. Ren Y, Liang H, Wang X, Cao Z, Ma Y, Liu X. Alterations in mitochondrial function and energy metabolism-related properties in thyroid cancer stem cells. Acta Biochim Pol. 2021;69:11–17. PMID: 34826218. doi:10.18388/abp.2020_5370
24. Kuntz EM, Baquero P, Michie AM, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234–1240. PMID: 28920959. doi:10.1038/nm.4399
25. Chen J, Li Y, T-S Y, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. PMID: 22854781. doi:10.1038/nature11287
26. Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4:e173420. PMID: 29098284. doi:10.1001/jamaoncol.2017.3420
27. Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547:104–108. PMID: 28658204. doi:10.1038/nature22993
28. Chang JC. Cancer stem cells: role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine. 2016;95:S20–5. PMID: 27611935. doi:10.1097/MD.0000000000004766
29. Marzagalli M, Fontana F, Raimondi M, Limonta P. Cancer stem cells-key players in tumor relapse. Cancers. 2021;13:376. PMID: 33498502. doi:10.3390/cancers13030376
30. Sehgal P, Chaturvedi P. Chromatin and cancer: implications of disrupted chromatin organization in tumorigenesis and its diversification. Cancers. 2023;15:466. PMID: 36672415. doi:10.3390/cancers15020466
31. Dave B, Mittal V, Tan NM, Chang JC. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012;14:202. PMID: 22264257. doi:10.1186/bcr2938
32. Mani SA, Guo W, Liao M-J, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. PMID: 18485877. doi:10.1016/j.cell.2008.03.027
33. Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–13825. PMID: 19666588. doi:10.1073/pnas.0905718106
34. Tanabe S, Quader S, Cabral H, Ono R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies. Front Pharmacol. 2020;11:904. PMID: 32625096. doi:10.3389/fphar.2020.00904
35. Scheel C, Eaton EN, SH-J L, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. PMID: 21663795. doi:10.1016/j.cell.2011.04.029
36. Sun Y-F, Xu Y, Yang X-R, et al. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology. 2013;57:1458–1468. PMID: 23175471. doi:10.1002/hep.26151
37. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. PMID: 18371365. doi:10.1016/j.stem.2007.06.002
38. Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98:756–765. PMID: 18268494. doi:10.1038/sj.bjc.6604242
39. Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. PMID: 15466194. doi:10.1158/0008-5472.CAN-04-1364
40. Peitzsch C, Nathansen J, Schniewind SI, Schwarz F, Dubrovska A. Cancer stem cells in head and neck squamous cell carcinoma: identification, characterization and clinical implications. Cancers. 2019;11:616. PMID: 31052565. doi:10.3390/cancers11050616
41. Koh E-Y, You J-E, Jung S-H, Kim P-H. Biological Functions and Identification of novel biomarker expressed on the surface of breast cancer-derived cancer stem cells via proteomic analysis. Mol Cells. 2020;43:384–396. PMID: 32235022. doi:10.14348/molcells.2020.2230
42. Phi LTH, Sari IN, Yang Y-G, et al. Cancer Stem Cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:5416923. PMID: 29681949. doi:10.1155/2018/5416923
43. Walcher L, Kistenmacher A-K, Suo H, et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. 2020;11:1280. PMID: 32849491. doi:10.3389/fimmu.2020.01280
44. Dalerba P, Dylla SJ, Park I-K, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–10163. PMID: 17548814. doi:10.1073/pnas.0703478104
45. Zhang -S-S, Huang Z-W, L-X L, J-J F, Xiao B. Identification of CD200+ colorectal cancer stem cells and their gene expression profile. Oncol Rep. 2016;36:2252–2260. PMID: 27574016. doi:10.3892/or.2016.5039
46. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. PMID: 18784658. doi:10.1038/nrc2499
47. Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. PMID: 18371393. doi:10.1016/j.stem.2007.08.014
48. Jiang F, Qiu Q, Khanna A, et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009;7:330–338. PMID: 19276181. doi:10.1158/1541-7786.MCR-08-0393
49. Li T, Su Y, Mei Y, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab Invest. 2010;90:234–244. PMID: 20010854. doi:10.1038/labinvest.2009.127
50. Rasper M, Schäfer A, Piontek G, et al. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol. 2010;12:1024–1033. PMID: 20627895. doi:10.1093/neuonc/noq070
51. Su Y, Qiu Q, Zhang X, et al. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:327–337. PMID: 20142235. doi:10.1158/1055-9965.EPI-09-0865
52. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16:225–238. PMID: 25748930. doi:10.1016/j.stem.2015.02.015
53. Pearce DJ, Taussig D, Simpson C, et al. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. 2005;23:752–760. PMID: 15917471. doi:10.1634/stemcells.2004-0292
54. Leng Z, Tao K, Xia Q, et al. Krüppel-like factor 4 acts as an oncogene in colon cancer stem cell-enriched spheroid cells. PLoS One. 2013;8:e56082. PMID: 23418515. doi:10.1371/journal.pone.0056082
55. Noh KH, Kim BW, Song K-H, et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest. 2012;122:4077–4093. PMID: 23093782. doi:10.1172/JCI64057
56. Lu H, Lyu Y, Tran L, et al. HIF-1 recruits NANOG as a coactivator for TERT gene transcription in hypoxic breast cancer stem cells. Cell Rep. 2021;36:109757. PMID: 34592152. doi:10.1016/j.celrep.2021.109757
57. Leis O, Eguiara A, Lopez-Arribillaga E, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–1365. PMID: 21822303. doi:10.1038/onc.2011.338
58. Le Minh G, Esquea EM, Dhameliya TT, et al. Kruppel-like factor 8 regulates triple negative breast cancer stem cell-like activity. Front Oncol. 2023;13:1141834. PMID: 37152043. doi:10.3389/fonc.2023.1141834
59. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 2017;7:339–348. PMID: 29071215. doi:10.15171/apb.2017.041
60. Nagaraju GP, Bramhachari PV, Raghu G, El-Rayes BF. Hypoxia inducible factor-1α: its role in colorectal carcinogenesis and metastasis. Cancer Lett. 2015;366:11–18. PMID: 26116902. doi:10.1016/j.canlet.2015.06.005
61. Colwell N, Larion M, Giles AJ, et al. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017;19:887–896. PMID: 28339582. doi:10.1093/neuonc/now258
62. Seidel S, Garvalov BK, Wirta V, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain. 2010;133:983–995. PMID: 20375133. doi:10.1093/brain/awq042
63. Louka M, Boutou E, Bakou V, et al. DNA damage response/repair in cancer stem cells — potential vs controversies. In: Advances in DNA Repair. IntechOpen; 2015.
64. Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: a potential link between chemoresistance, recurrence, and metastasis. Biores Open Access. 2015;4:97–108. PMID: 26309786. doi:10.1089/biores.2014.0035
65. Chen K, Zhang C, Ling S, Wei R, Wang J, Xu X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis. 2021;12:835. PMID: 34482364. doi:10.1038/s41419-021-04116-6
66. Safa AR. Resistance to drugs and cell death in cancer stem cells (CSCs). J Transl Sci. 2020;6:341. PMID: 35330670. doi:10.15761/jts.1000341
67. Yadav AK, Desai NS. Cancer stem cells: acquisition, characteristics, therapeutic implications, targeting strategies and future prospects. Stem Cell Rev Rep. 2019;15:331–355. PMID: 30993589. doi:10.1007/s12015-019-09887-2
68. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. PMID: 17051156. doi:10.1038/nature05236
69. Fang DD, Cao J, Jani JP, et al. Combined gemcitabine and CHK1 inhibitor treatment induces apoptosis resistance in cancer stem cell-like cells enriched with tumor spheroids from a non-small cell lung cancer cell line. Front Med. 2013;7:462–476. PMID: 23820871. doi:10.1007/s11684-013-0270-6
70. Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545. PMID: 20110496. doi:10.1126/science.1180794
71. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. PMID: 21386835. doi:10.1038/nm.2304
72. Luo M, J-F L, Yang Q, et al. Stem cell quiescence and its clinical relevance. World J Stem Cells. 2020;12:1307–1326. PMID: 33312400. doi:10.4252/wjsc.v12.i11.1307
73. Aponte PM, Caicedo A. Stemness in Cancer: stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017;2017:5619472. PMID: 28473858. doi:10.1155/2017/5619472
74. Kurki P, Vanderlaan M, Dolbeare F, Gray J, Tan EM. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp Cell Res. 1986;166:209–219. PMID: 2874992. doi:10.1016/0014-4827(86)90520-3
75. Ye S, Ding Y-F, Jia W-H, et al. SET domain-containing protein 4 epigenetically controls breast cancer stem cell quiescence. Cancer Res. 2019;79:4729–4743. PMID: 31308046. doi:10.1158/0008-5472.CAN-19-1084
76. Gerdes J, Li L, Schlueter C, et al. Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am J Pathol. 1991;138:867–873. PMID: 2012175.
77. Kobayashi A, Okuda H, Xing F, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med. 2011;208:2641–2655. PMID: 22124112. doi:10.1084/jem.20110840
78. Abravanel DL, Belka GK, Pan T, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest. 2015;125:2484–2496. PMID: 25961456. doi:10.1172/JCI74883
79. Nguyen DX, Chiang AC, Zhang XH-F, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. PMID: 19576624. doi:10.1016/j.cell.2009.04.030
80. Yang A, Qin S, Schulte BA, Ethier SP, Tew KD, Wang GY. MYC Inhibition depletes cancer stem-like cells in triple-negative breast cancer. Cancer Res. 2017;77:6641–6650. PMID: 28951456. doi:10.1158/0008-5472.CAN-16-3452
81. Lee SH, Reed-Newman T, Anant S, Ramasamy TS. Regulatory role of quiescence in the biological function of cancer stem cells. Stem Cell Rev Rep. 2020;16:1185–1207. PMID: 32894403. doi:10.1007/s12015-020-10031-8
82. Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, Fisher PB. Dormancy and cancer stem cells: an enigma for cancer therapeutic targeting. Adv Cancer Res. 2019;141:43–84. PMID: 30691685. doi:10.1016/bs.acr.2018.12.002
83. Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4:e83. PMID: 16509772. doi:10.1371/journal.pbio.0040083
84. Truskowski K, Amend SR, Pienta KJ. Dormant cancer cells: programmed quiescence, senescence, or both? Cancer Metastasis Rev. 2023;42:37–47. PMID: 36598661. doi:10.1007/s10555-022-10073-z
85. Jahanban-Esfahlan R, Seidi K, Manjili MH, Jahanban-Esfahlan A, Javaheri T, Zare P. Tumor cell dormancy: threat or opportunity in the fight against cancer. Cancers. 2019;11:1207. PMID: 31430951. doi:10.3390/cancers11081207
86. Hedley BD, Chambers AF. Tumor dormancy and metastasis. Adv Cancer Res. 2009;102:67–101. PMID: 19595307. doi:10.1016/S0065-230X(09)02003-X
87. Manjili MH. Tumor dormancy and relapse: from a natural byproduct of evolution to a disease state. Cancer Res. 2017;77:2564–2569. PMID: 28507050. doi:10.1158/0008-5472.CAN-17-0068
88. Páez D, Labonte MJ, Bohanes P, et al. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clin Cancer Res. 2012;18:645–653. PMID: 22156560. doi:10.1158/1078-0432.CCR-11-2186
89. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697–10711. PMID: 25986923. doi:10.18632/oncotarget.4037
90. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16–27. PMID: 24956577. doi:10.1016/j.semcancer.2014.06.004
91. Kleffel S, Schatton T. Tumor dormancy and cancer stem cells: two sides of the same coin? Adv Exp Med Biol. 2013;734:145–179. PMID: 23143979. doi:10.1007/978-1-4614-1445-2_8
92. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009;26:611–623. PMID: 19421880. doi:10.1007/s10585-009-9260-0
93. Lin W, Rajbhandari N, Liu C, et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013;73:1821–1830. PMID: 23467612. doi:10.1158/0008-5472.CAN-12-2067
94. Gao M-Q, Choi Y-P, Kang S, Youn JH, Cho N-H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29:2672–2680. PMID: 20190812. doi:10.1038/onc.2010.35
95. Roesch A, Fukunaga-Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. PMID: 20478252. doi:10.1016/j.cell.2010.04.020
96. Zeuner A, Francescangeli F, Contavalli P, et al. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-XL inhibition in non-small cell lung cancer. Cell Death Differ. 2014;21:1877–1888. PMID: 25034785. doi:10.1038/cdd.2014.105
97. Holtz MS, Forman SJ, Bhatia R. Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia. 2005;19:1034–1041. PMID: 15815728. doi:10.1038/sj.leu.2403724
98. Zanoni M, Bravaccini S, Fabbri F, Arienti C. Emerging roles of aldehyde dehydrogenase isoforms in anti-cancer therapy resistance. Front Med. 2022;9:795762. PMID: 35299840. doi:10.3389/fmed.2022.795762
99. Clark DW, Palle K. Aldehyde dehydrogenases in cancer stem cells: potential as therapeutic targets. Ann Transl Med. 2016;4:518. PMID: 28149880. doi:10.21037/atm.2016.11.82
100. Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid Proc Natl Acad Sci U S A. 1980;77(5):2936–2940. PMID: 6930676. doi:10.1073/pnas.77.5.2936
101. Lotan R. Different susceptibilities of human melanoma and breast carcinoma cell lines to retinoic acid-induced growth inhibition. Cancer Res. 1979;39(3):1014–1019. PMID: 427741.
102. Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat. 2012;133(1):75–87. PMID: 21818590. doi:10.1007/s10549-011-1692-y
103. Chen Y-C, Chen Y-W, Hsu H-S, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. 2009;385(3):307–313. PMID: 19450560. doi:10.1016/j.bbrc.2009.05.048
104. Safa AR. Resistance to cell death and its modulation in cancer stem cells. Critical Reviews™ in Oncogenesis. 2016;21(3–4):203–219. PMID: 27915972. doi:10.1615/CritRevOncog.2016016976
105. Yang L, Ren Y, Yu X, et al. ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod Pathol. 2014;27(5):775–783. PMID: 24201124. doi:10.1038/modpathol.2013.189
106. Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7(10):11018–11032. PMID: 26783961. doi:10.18632/oncotarget.6920
107. He X, Gonzalez V, Tsang A, Thompson J, Tsang TC, Harris DT. Differential gene expression profiling of CD34 + CD133 + umbilical cord blood hematopoietic stem progenitor cells. Stem Cells Dev. 2005;14(2):188–198. PMID: 15910245. doi:10.1089/scd.2005.14.188
108. Forsberg EC, Prohaska SS, Katzman S, Heffner GC, Stuart JM, Weissman IL. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 2005;1(3):e28. PMID: 16151515. doi:10.1371/journal.pgen.0010028
109. Eirew P, Kannan N, Knapp DJHF, et al. Aldehyde dehydrogenase activity is a biomarker of primitive normal human mammary luminal cells. Stem Cells. 2012;30(2):344–348. PMID: 22131125. doi:10.1002/stem.1001
110. Sahovic EA, Colvin M, Hilton J, Ogawa M. Role for aldehyde dehydrogenase in survival of progenitors for murine blast cell colonies after treatment with 4-hydroperoxycyclophosphamide in vitro. Cancer Res. 1988;48(5):1223–1226.
111. Bunting KD, Townsend AJ. Protection by transfected rat or human class 3 aldehyde dehydrogenases against the cytotoxic effects of oxazaphosphorine alkylating agents in hamster V79 cell lines. J Biol Chem. 1996;271(20):11891–11896. doi:10.1074/jbc.271.20.11891
112. Safa AR. Drug and apoptosis resistance in cancer stem cells (CSCs): a puzzle with many pieces. Cancer Drug Resist. 2022;5(4):850–872. PMID: 36627897. doi:10.20517/cdr.2022.20
113. Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34(3):176–184. PMID: 23070002.
114. Stantic M, Dong L-F, Zobalova R, Prokopova K, Neuzil J. Cancer cells with high expression of CD133 exert FLIP upregulation and resistance to TRAIL-induced apoptosis. Biofactors. 2008;34(3):231–235. PMID: 19734124. doi:10.1002/biof.5520340307
115. Zobalova R, McDermott L, Stantic M, Prokopova K, Dong L-F, Neuzil J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem Biophys Res Commun. 2008;373(4):567–571. PMID: 18590703. doi:10.1016/j.bbrc.2008.06.073
116. Ding L, Yuan C, Wei F, et al. Cisplatin restores TRAIL apoptotic pathway in glioblastoma-derived stem cells through up-regulation of DR5 and down-regulation of c-FLIP. Cancer Invest. 2011;29(8):511–520. PMID: 21877938. doi:10.3109/07357907.2011.605412
117. Gampa SC. Nano-TRAIL: a promising path to cancer therapy. Cancer Drug Resist. 2023;6(1):78–102. PMID: 37065863. doi:10.20517/cdr.2022.82
118. Ivanisenko NV, Seyrek K, Hillert-Richter LK, et al. Regulation of extrinsic apoptotic signaling by c-FLIP: towards targeting cancer networks. Trends Cancer. 2022;8:190–209. PMID: 34973957. doi:10.1016/j.trecan.2021.12.002
119. Yoon MJ, Kang YJ, Kim IY, et al. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis. 2013;34:1918–1928. PMID: 23615398. doi:10.1093/carcin/bgt137
120. Varfolomeev E, Goncharov T, Vucic D. Roles of c-IAP proteins in TNF receptor family activation of NF-κB signaling. Methods Mol Biol. 2015;1280:269–282. PMID: 25736754. doi:10.1007/978-1-4939-2422-6_15
121. Ji J, Yu Y, Z-L L, et al. XIAP Limits autophagic degradation of Sox2 and Is a therapeutic target in nasopharyngeal carcinoma stem cells. Theranostics. 2018;8:1494–1510. PMID: 29556337. doi:10.7150/thno.21717
122. Wang Y-H, Scadden DT. Harnessing the apoptotic programs in cancer stem-like cells. EMBO Rep. 2015;16:1084–1098. PMID: 26253117. doi:10.15252/embr.201439675
123. Lee M-R, S-Y J, Mia-Jan K, Cho M-Y. Chemoresistance of CD133(+) colon cancer may be related with increased survivin expression. Biochem Biophys Res Commun. 2015;463:229–234. PMID: 26002465. doi:10.1016/j.bbrc.2015.05.031
124. Hu Y, Yagüe E, Zhao J, et al. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. 2018;423:47–59. PMID: 29496539. doi:10.1016/j.canlet.2018.02.036
125. Xu M, Gong A, Yang H, et al. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett. 2015;369:124–133. PMID: 26276718. doi:10.1016/j.canlet.2015.08.005
126. Wang J, Liu X, Jiang Z, et al. A novel method to limit breast cancer stem cells in states of quiescence, proliferation or differentiation: use of gel stress in combination with stem cell growth factors. Oncol Lett. 2016;12:1355–1360. PMID: 27446437. doi:10.3892/ol.2016.4757
127. Veringa SJE, Biesmans D, van Vuurden DG, et al. In vitro drug response and efflux transporters associated with drug resistance in pediatric high grade glioma and diffuse intrinsic pontine glioma. PLoS One. 2013;8:e61512. PMID: 23637844. doi:10.1371/journal.pone.0061512
128. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18:452–464. PMID: 29643473. doi:10.1038/s41568-018-0005-8
129. Arneth B. Tumor Microenvironment. Medicina. 2019;56:15. PMID: 31906017. doi:10.3390/medicina56010015
130. Prieto-Vila M, Takahashi R-U, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci. 2017;18:2574. PMID: 29194401. doi:10.3390/ijms18122574
131. Li Y, Wang Z, Ajani JA, Song S. Drug resistance and Cancer stem cells. Cell Commun Signal. 2021;19:19. PMID: 33588867. doi:10.1186/s12964-020-00627-5
132. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. PMID: 19194462. doi:10.1038/nature07733
133. Desai A, Webb B, Gerson SL. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother Oncol. 2014;110:538–545. PMID: 24440048. doi:10.1016/j.radonc.2013.10.040
134. Mathews LA, Cabarcas SM, Hurt EM, Zhang X, Jaffee EM, Farrar WL. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas. 2011;40:730–739. PMID: 21633318. doi:10.1097/MPA.0b013e31821ae25b
135. Carruthers RD, Ahmed SU, Ramachandran S, et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018;78:5060–5071. PMID: 29976574. doi:10.1158/0008-5472.CAN-18-0569
136. Wang Q-E. DNA damage responses in cancer stem cells: implications for cancer therapeutic strategies. World J Biol Chem. 2015;6:57–64. PMID: 26322164. doi:10.4331/wjbc.v6.i3.57
137. Blanpain C, Mohrin M, Sotiropoulou PA, Passegué E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell. 2011;8:16–29. PMID: 21211780. doi:10.1016/j.stem.2010.12.012
138. Liu Y, Zheng C, Huang Y, He M, Xu WW, Li B. Molecular mechanisms of chemo‐ and radiotherapy resistance and the potential implications for cancer treatment. MedComm. 2021;2:315–340. PMID: 34766149. doi:10.1002/mco2.55
139. Cui Q, Wang J-Q, Assaraf YG, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1–25. PMID: 30471641. doi:10.1016/j.drup.2018.11.001
140. Hwang IT, Chung YM, Kim JJ, et al. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem Biophys Res Commun. 2007;359:304–310. PMID: 17537404. doi:10.1016/j.bbrc.2007.05.088
141. Kim E-K, Jang M, Song M-J, Kim D, Kim Y, Jang HH. Redox-mediated mechanism of chemoresistance in cancer cells. Antioxidants. 2019;8:471. PMID: 31658599. doi:10.3390/antiox8100471
142. Shen Y, Yang J, Zhao J, Xiao C, Xu C, Xiang Y. The switch from ER stress-induced apoptosis to autophagy via ROS-mediated JNK/p62 signals: a survival mechanism in methotrexate-resistant choriocarcinoma cells. Exp Cell Res. 2015;334:207–218. PMID: 25912909. doi:10.1016/j.yexcr.2015.04.010
143. Alimbetov D, Askarova S, Umbayev B, Davis T, Kipling D. Pharmacological targeting of cell cycle, apoptotic and cell adhesion signaling pathways implicated in chemoresistance of cancer cells. Int J Mol Sci. 2018;19:1690. PMID: 29882812. doi:10.3390/ijms19061690
144. Bailey HH. L-S,R-buthionine sulfoximine: historical development and clinical issues. Chem Biol Interact. 1998;111–112:239–254. PMID: 9679558. doi:10.1016/s0009-2797(97)00164-6
145. Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18:157. PMID: 31711497. doi:10.1186/s12943-019-1089-9
146. O’Reilly D, Johnson P, Buchanan PJ. Hypoxia induced cancer stem cell enrichment promotes resistance to androgen deprivation therapy in prostate cancer. Steroids. 2019;152:108497. PMID: 31521707. doi:10.1016/j.steroids.2019.108497
147. Qian J, Rankin EB. Hypoxia-induced phenotypes that mediate tumor heterogeneity. Adv Exp Med Biol. 2019;1136:43–55. PMID: 31201715. doi:10.1007/978-3-030-12734-3_3
148. Yan Y, Liu F, Han L, et al. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res. 2018;37:256. PMID: 30340507. doi:10.1186/s13046-018-0925-x
149. Yang Y, Li X, Wang T, Guo Q, Xi T, Zheng L. Emerging agents that target signaling pathways in cancer stem cells. J Hematol Oncol. 2020;13:60. PMID: 32456660. doi:10.1186/s13045-020-00901-6
150. Espinosa-Sánchez A, Suárez-Martínez E, Sánchez-Díaz L, Carnero A. Therapeutic Targeting of Signaling Pathways Related to Cancer Stemness. Front Oncol. 2020;10:1533. PMID: 32984007. doi:10.3389/fonc.2020.01533
151. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 2017;1858:686–699. PMID: 28161329. doi:10.1016/j.bbabio.2017.01.012
152. Bokil A, Sancho P. Mitochondrial determinants of chemoresistance. Cancer Drug Resist. 2019;2:634–646. PMID: 35582564. doi:10.20517/cdr.2019.46
153. Porporato PE, Filigheddu N, Pedro JMB-S, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–280. PMID: 29219147. doi:10.1038/cr.2017.155
154. Jin P, Jiang J, Zhou L, et al. Mitochondrial adaptation in cancer drug resistance: prevalence, mechanisms, and management. J Hematol Oncol. 2022;15:97. PMID: 35851420. doi:10.1186/s13045-022-01313-4
155. Klein K, He K, Younes AI, et al. Role of mitochondria in cancer immune evasion and potential therapeutic approaches. Front Immunol. 2020;11:573326. PMID: 33178201. doi:10.3389/fimmu.2020.573326
156. Grasso D, Zampieri LX, Capelôa T, Van de Velde JA, Sonveaux P. Mitochondria in cancer. Cell Stress. 2020;4:114–146. PMID: 32548570. doi:10.15698/cst2020.06.221
157. Bertram R, Gram Pedersen M, Luciani DS, Sherman A. A simplified model for mitochondrial ATP production. J Theor Biol. 2006;243:575–586. PMID: 16945388. doi:10.1016/j.jtbi.2006.07.019
158. Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. PMID: 22146081. doi:10.1089/ars.2011.4123
159. Tait SWG, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012;125:807–815. PMID: 22448037. doi:10.1242/jcs.099234
160. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426–431. PMID: 28212726. doi:10.1016/j.bbrc.2016.11.088
161. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–754. PMID: 29950572. doi:10.1038/s41556-018-0124-1
162. Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell. 2016;166:555–566. PMID: 27471965. doi:10.1016/j.cell.2016.07.002
163. Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci. 2007;120:4025–4034. PMID: 17971411. doi:10.1242/jcs.016972
164. Wu Q, Tsai H-I, Zhu H, Wang D. The entanglement between mitochondrial DNA and Tumor metastasis. Cancers. 2022;14:1862. PMID: 35454769. doi:10.3390/cancers14081862
165. Liu Z, Shan S, Yuan Z, et al Mitophagy bridges DNA sensing with metabolic adaption to expand lung cancer stem-like cells. EMBO Rep. 2023;24:e54006. PMID: 36416244. doi:10.15252/embr.202154006
166. Masuike Y, Tanaka K, Makino T, et al. Esophageal squamous cell carcinoma with low mitochondrial copy number has mesenchymal and stem-like characteristics, and contributes to poor prognosis. PLoS One. 2018;13:e0193159. PMID: 29447301. doi:10.1371/journal.pone.0193159
167. Armstrong L, Tilgner K, Saretzki G, et al. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells. 2010;28:661–673. PMID: 20073085. doi:10.1002/stem.307
168. Paliwal S, Fiumera HL, Mohanty S. Stem cell plasticity and regenerative potential regulation through Ca2+-mediated mitochondrial nuclear crosstalk. Mitochondrion. 2021;56:1–14. PMID: 33059088. doi:10.1016/j.mito.2020.10.002
169. Cho YM, Kwon S, Pak YK, et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–1478. PMID: 16920071. doi:10.1016/j.bbrc.2006.08.020
170. Spurlock B, Gupta P, Basu MK, et al. New quantitative approach reveals heterogeneity in mitochondrial structure-function relations in tumor-initiating cells. J Cell Sci. 2019;132:jcs230755. PMID: 30910831. doi:10.1242/jcs.230755
171. Chakrabarty RP, Chandel NS. Mitochondria as Signaling organelles control mammalian stem cell fate. Cell Stem Cell. 2021;28:394–408. PMID: 33667360. doi:10.1016/j.stem.2021.02.011
172. Zheng -X-X, Chen -J-J, Sun Y-B, Chen T-Q, Wang J, S-C Y. Mitochondria in cancer stem cells: achilles heel or hard armor. Trends Cell Biol. 2023;33:708–727. PMID: 37137792. doi:10.1016/j.tcb.2023.03.009
173. Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-Inducible Factors and Cancer. Curr Sleep Med Rep. 2017;3:1–10. PMID: 28944164. doi:10.1007/s40675-017-0062-7
174. Mu X, Zhao T, Xu C, et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget. 2017;8:13174–13185. PMID: 28061458. doi:10.18632/oncotarget.14485
175. van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412:477–484. PMID: 18393939. doi:10.1042/BJ20080476
176. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. PMID: 15652751. doi:10.1016/j.ccr.2004.11.022
177. De Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J. 2018;475:1611–1634. PMID: 29743249. doi:10.1042/BCJ20170164
178. García-Heredia JM, Carnero A. Role of mitochondria in cancer stem cell resistance. Cells. 2020;9:1693. PMID: 32679735. doi:10.3390/cells9071693
179. Shiota T, Traven A, Lithgow T. Mitochondrial biogenesis: cell-cycle-dependent investment in making mitochondria. Curr Biol. 2015;25:R78–80. PMID: 25602310. doi:10.1016/j.cub.2014.12.006
180. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. PMID: 20533901. doi:10.1042/bse0470069
181. Praharaj PP, Panigrahi DP, Bhol CS, et al. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: a potential target for anti-CSC cancer therapy. Cancer Lett. 2021;498:217–228. PMID: 33186655. doi:10.1016/j.canlet.2020.10.036
182. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. 2013;5:a015008. PMID: 23818499. doi:10.1101/cshperspect.a015008
183. Liu T, Ma Q, Li W, Hu Y, Yang J, Yao Q. Ubiquilin 1 suppresses the cancer stem cell-like traits of non-small cell lung cancer cells by regulating reactive oxygen species homeostasis. Bioengineered. 2021;12:7143–7155. PMID: 34546848. doi:10.1080/21655979.2021.1979353
184. X-Q Y, Li Q, Wang G-H, et al. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer. 2011;129:820–831. PMID: 21520032. doi:10.1002/ijc.25944
185. Mihaylova MM, Shaw RJ. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy, & metabolism. Nat Cell Biol. 2011;13:1016–1023. PMID: 21892142. doi:10.1038/ncb2329
186. Ryoo I-G, Choi B-H, S-K K, Kwak M-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. 2018;17:246–258. PMID: 29729523. doi:10.1016/j.redox.2018.04.015
187. Zhu J, Wang H, Sun Q, et al. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer. 2013;13:380. PMID: 23937621. doi:10.1186/1471-2407-13-380
188. Jia Y, Chen J, Zhu H, Jia Z-H, Cui M-H. Aberrantly elevated redox sensing factor Nrf2 promotes cancer stem cell survival via enhanced transcriptional regulation of ABCG2 and Bcl-2/Bmi-1 genes. Oncol Rep. 2015;34:2296–2304. PMID: 26324021. doi:10.3892/or.2015.4214
189. Kim B, Jung JW, Jung J, et al. PGC1α induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget. 2017;8:60299–60311. PMID: 28947972. doi:10.18632/oncotarget.19140
190. Sancho P, Burgos-Ramos E, Tavera A, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590–605. PMID: 26365176. doi:10.1016/j.cmet.2015.08.015
191. Lee K-M, Giltnane JM, Balko JM, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017;26:633–647.e7. PMID: 28978427. doi:10.1016/j.cmet.2017.09.009
192. Chen C-L, Uthaya Kumar DB, Punj V, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016;23:206–219. PMID: 26724859. doi:10.1016/j.cmet.2015.12.004
193. Murakami A, Takahashi F, Nurwidya F, et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS One. 2014;9:e86459. PMID: 24489728. doi:10.1371/journal.pone.0086459
194. Kim I-G, Lee J-H, Kim S-Y, Heo C-K, Kim R-K, Cho E-W. Targeting therapy-resistant lung cancer stem cells via disruption of the AKT/TSPYL5/PTEN positive-feedback loop. Commun Biol. 2021;4:778. PMID: 34163000. doi:10.1038/s42003-021-02303-x
195. Bleau A-M, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. PMID: 19265662. doi:10.1016/j.stem.2009.01.007
196. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A. 2014;111:E5429–5438. PMID: 25453096. doi:10.1073/pnas.1421438111
197. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:55. PMID: 27220421. doi:10.1186/s13058-016-0712-6
198. Gao C, Shen Y, Jin F, Miao Y, Qiu X. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS One. 2016;11:e0154576. PMID: 27167619. doi:10.1371/journal.pone.0154576
199. Pastò A, Bellio C, Pilotto G, et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget. 2014;5:4305–4319. PMID: 24946808. doi:10.18632/oncotarget.2010
200. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108:16062–16067. PMID: 21900605. doi:10.1073/pnas.1106704108
201. Janiszewska M, Suvà ML, Riggi N, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–1944. PMID: 22899010. doi:10.1101/gad.188292.112
202. Han S, Wei R, Zhang X, et al. CPT1A/2-mediated FAO enhancement-a metabolic target in radioresistant breast cancer. Front Oncol. 2019;9:1201. PMID: 31803610. doi:10.3389/fonc.2019.01201
203. Wang T, Fahrmann JF, Lee H, et al. JAK/STAT3-regulated Fatty Acid β-Oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27:136–150.e5. PMID: 29249690. doi:10.1016/j.cmet.2017.11.001
204. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. PMID: 31900386. doi:10.1038/s41467-019-13668-3
205. Yi M, Li J, Chen S, et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res. 2018;37:118. PMID: 29907133. doi:10.1186/s13046-018-0784-5
206. Camarda R, Zhou AY, Kohnz RA, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22:427–432. PMID: 26950360. doi:10.1038/nm.4055
207. Cardaci S, Ciriolo MR. TCA cycle defects and cancer: when metabolism tunes redox state. Int J Cell Biol. 2012;2012:161837. PMID: 22888353. doi:10.1155/2012/161837
208. Ghosh P, Vidal C, Dey S, Zhang L. Mitochondria targeting as an effective strategy for cancer therapy. Int J Mol Sci. 2020;21:3363. PMID: 32397535. doi:10.3390/ijms21093363
209. Sciacovelli M, Frezza C. Oncometabolites: unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med. 2016;100:175–181. PMID: 27117029. doi:10.1016/j.freeradbiomed.2016.04.025
210. Xiao M, Yang H, Xu W, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. PMID: 22677546. doi:10.1101/gad.191056.112
211. Gasparre G, Kurelac I, Capristo M, et al. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene MTND1, an oncojanus function. Cancer Res. 2011;71:6220–6229. PMID: 21852384. doi:10.1158/0008-5472.CAN-11-1042
212. Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. PMID: 23558169. doi:10.1126/science.1236062
213. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62:341–360. PMID: 30030364. doi:10.1042/EBC20170104
214. Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13:75–88. PMID: 22233676. doi:10.1038/nrm3266
215. Kraus F, Ryan MT. The constriction and scission machineries involved in mitochondrial fission. J Cell Sci. 2017;130:2953–2960. PMID: 28842472. doi:10.1242/jcs.199562
216. Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–531. PMID: 26667075. doi:10.1146/annurev-physiol-021115-105011
217. Zhao J, Zhang J, Yu M, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–4824. PMID: 23128392. doi:10.1038/onc.2012.494
218. Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017;26:39–48. PMID: 28648983. doi:10.1016/j.cmet.2017.05.016
219. Wei M, Nurjanah U, Li J, et al. YY2-DRP1 axis regulates mitochondrial fission and determines cancer stem cell asymmetric division. Adv Sci. 2023:e2207349. PMID: 37300334. doi:10.1002/advs.202207349
220. Xie Q, Wu Q, Horbinski CM, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–510. PMID: 25730670. doi:10.1038/nn.3960
221. Peiris-Pagès M, Bonuccelli G, Sotgia F, Lisanti MP. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget. 2018;9:13254–13275. PMID: 29568355. doi:10.18632/oncotarget.24285
222. Liu Z, Lei J, Wu T, et al. Lipogenesis promotes mitochondrial fusion and maintains cancer stemness in human NSCLC. JCI Insight. 2023;8:e158429. PMID: 36809297. doi:10.1172/jci.insight.158429
223. Civenni G, Bosotti R, Timpanaro A, et al. Epigenetic control of mitochondrial fission enables self-renewal of stem-like tumor cells in human prostate cancer. Cell Metab. 2019;30:303–318.e6. PMID: 31130467. doi:10.1016/j.cmet.2019.05.004
224. Zhou T-J, Zhang S-L, C-Y H, et al. Downregulation of mitochondrial cyclooxygenase-2 inhibits the stemness of nasopharyngeal carcinoma by decreasing the activity of dynamin-related protein 1. Theranostics. 2017;7:1389–1406. PMID: 28435473. doi:10.7150/thno.17647
225. Tang M, Yang M, Wu G, et al. Epigenetic induction of mitochondrial fission is required for maintenance of liver cancer-initiating cells. Cancer Res. 2021;81:3835–3848. PMID: 34049973. doi:10.1158/0008-5472.CAN-21-0436
226. Cai J, Wang J, Huang Y, et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016;7:e2459. PMID: 27831567. doi:10.1038/cddis.2016.370
227. Cagin U, Duncan OF, Gatt AP, Dionne MS, Sweeney ST, Bateman JM. Mitochondrial retrograde signaling regulates neuronal function. Proc Natl Acad Sci U S A. 2015;112:E6000–6009. PMID: 26489648. doi:10.1073/pnas.1505036112
228. da Cunha FM, Torelli NQ, Kowaltowski AJ. Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxid Med Cell Longev. 2015;2015:482582. PMID: 26583058. doi:10.1155/2015/482582
229. Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. PMID: 22354038. doi:10.1038/emboj.2012.38
230. Biswas G, Adebanjo OA, Freedman BD, et al. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999;18:522–533. PMID: 9927412. doi:10.1093/emboj/18.3.522
231. Arnould T, Michel S, Renard P. Mitochondria retrograde signaling and the upr mt: where are we in mammals? Int J Mol Sci. 2015;16:18224–18251. PMID: 26258774. doi:10.3390/ijms160818224
232. Jiang H-L, Sun H-F, Gao S-P, et al. SSBP1 suppresses TGFβ-driven epithelial-to-mesenchymal transition and metastasis in triple-negative breast cancer by regulating mitochondrial retrograde signaling. Cancer Res. 2016;76:952–964. PMID: 26676758. doi:10.1158/0008-5472.CAN-15-1630
233. Arbini AA, Guerra F, Greco M, et al. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis. 2013;2:e82. PMID: 24336406. doi:10.1038/oncsis.2013.45
234. Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002;21:7839–7849. PMID: 12420221. doi:10.1038/sj.onc.1205983
235. Guha M, Srinivasan S, Ruthel G, et al. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene. 2014;33:5238–5250. PMID: 24186204. doi:10.1038/onc.2013.467
236. Gonzalez-Sanchez E, Marin JJG, Perez MJ. The expression of genes involved in hepatocellular carcinoma chemoresistance is affected by mitochondrial genome depletion. Mol Pharmaceutics. 2014;11:1856–1868. doi:10.1021/mp400732p
237. Shen L, Zhou L, Xia M, et al. PGC1α regulates mitochondrial oxidative phosphorylation involved in cisplatin resistance in ovarian cancer cells via nucleo-mitochondrial transcriptional feedback. Exp Cell Res. 2021;398:112369. PMID: 33220258. doi:10.1016/j.yexcr.2020.112369
238. Porporato PE, Payen VL, Pérez-Escuredo J, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8:754–766. PMID: 25066121. doi:10.1016/j.celrep.2014.06.043
239. Ding W-X, Yin X-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. PMID: 22944659. doi:10.1515/hsz-2012-0119
240. Xie Y, Liu J, Kang R, Tang D. Mitophagy receptors in tumor biology. Front Cell Dev Biol. 2020;8:594203. PMID: 33262988. doi:10.3389/fcell.2020.594203
241. Yan C, Li T-S. Dual role of mitophagy in cancer drug resistance. Anticancer Res. 2018;38:617–621. PMID: 29374684. doi:10.21873/anticanres.12266
242. Arena G, Valente EM. PINK1 in the limelight: multiple functions of an eclectic protein in human health and disease. J Pathol. 2017;241:251–263. PMID: 27701735. doi:10.1002/path.4815
243. Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015;29:989–999. PMID: 25995186. doi:10.1101/gad.262758.115
244. Song L, Huang Y, Hou X, et al. PINK1/Parkin-mediated mitophagy promotes resistance to sonodynamic therapy. Cell Physiol Biochem. 2018;49:1825–1839. PMID: 30231241. doi:10.1159/000493629
245. Denisenko TV, Gogvadze V, Zhivotovsky B. Mitophagy in carcinogenesis and cancer treatment. Discov Oncol. 2021;12:58. PMID: 35201480. doi:10.1007/s12672-021-00454-1
246. Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. PMID: 19273585. doi:10.1128/MCB.00166-09
247. Liu K, Lee J, Kim JY, et al. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell. 2017;68:281–292.e5. PMID: 29033320. doi:10.1016/j.molcel.2017.09.022
248. Im E, Yoo L, Hyun M, Shin WH, Chung KC. Covalent ISG15 conjugation positively regulates the ubiquitin E3 ligase activity of parkin. Open Biol. 2016;6:160193. PMID: 27534820. doi:10.1098/rsob.160193
249. Jung J, Zhang Y, Celiku O, et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 2019;79:5218–5232. PMID: 31488423. doi:10.1158/0008-5472.CAN-19-0198
250. Guan Y, Wang Y, Li B, et al. Mitophagy in carcinogenesis, drug resistance and anticancer therapeutics. Cancer Cell Int. 2021;21:350. PMID: 34225732. doi:10.1186/s12935-021-02065-w
251. Katajisto P, Döhla J, Chaffer CL, et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340–343. PMID: 25837514. doi:10.1126/science.1260384
252. Smith AG, Macleod KF. Autophagy, cancer stem cells and drug resistance. J Pathol. 2019;247:708–718. PMID: 30570140. doi:10.1002/path.5222
253. Shen Y-A, Wang C-Y, Hsieh Y-T, Chen Y-J, Wei Y-H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. 2015;14:86–98. PMID: 25483072. doi:10.4161/15384101.2014.974419
254. Luo M, Shang L, Brooks MD, et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. 2018;28:69–86.e6. PMID: 29972798. doi:10.1016/j.cmet.2018.06.006
255. O’Neill S, Porter RK, McNamee N, Martinez VG, O’Driscoll L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci Rep. 2019;9:3788. PMID: 30846710. doi:10.1038/s41598-019-39789-9
256. Naik PP, Mukhopadhyay S, Panda PK, et al. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018;51:e12411. PMID: 29171106. doi:10.1111/cpr.12411
257. Yan C, Luo L, Guo C-Y, et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017;388:34–42. PMID: 27913197. doi:10.1016/j.canlet.2016.11.018
258. Lopez J, Tait SWG. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112:957–962. PMID: 25742467. doi:10.1038/bjc.2015.85
259. Dlugosz PJ, Billen LP, Annis MG, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006;25:2287–2296. PMID: 16642033. doi:10.1038/sj.emboj.7601126
260. Safa AR, Pollok KE. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers. 2011;3:1639–1671. PMID: 22348197. doi:10.3390/cancers3021639
261. Buccarelli M, D’Alessandris QG, Matarrese P, et al. Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J Exp Clin Cancer Res. 2021;40:228. PMID: 34253243. doi:10.1186/s13046-021-02031-4
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.